
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReduction-responsive molecularly imprinted nanogels for drug delivery applications†
Y. Zhao,
C. Simon,
M. Daoud Attieh,
K. Haupt * and
A. Falcimaigne-Cordin*
* and
A. Falcimaigne-Cordin*
Sorbonne Universités – Université de Technologie de Compiègne, CNRS Enzyme and Cell Engineering Laboratory, CS 60319, 60203 Compiègne Cedex, France. E-mail: aude.cordin@utc.fr; karsten.haupt@utc.fr
First published on 5th February 2020
Abstract
Degradable molecularly imprinted polymers (MIPs) with affinity for S-propranolol were prepared by the copolymerization of methacrylic acid as functional monomer and a disulfide-containing cross-linker, bis(2-methacryloyloxyethyl)disulfide (DSDMA), using bulk polymerization or high dilution polymerization for nanogels synthesis. The specificity and the selectivity of DSDMA-based molecularly imprinted polymers toward S-propranolol were studied in batch binding experiments, and their binding properties were compared to a traditional ethylene glycol dimethacrylate (EDMA)-based MIP. Nanosized MIPs prepared with DSDMA as crosslinker could be degraded into lower molecular weight linear polymers by cleaving the disulfide bonds and thus reversing cross-linking using different reducing agents (NaBH4, DTT, GSH). Turbidity, viscosity, polymer size and IR-spectra were measured to study the polymer degradation. The loss of specific recognition and binding capacity of S-propranolol was also observed after MIP degradation. This phenomenon was applied to modulate the release properties of the MIP. In presence of GSH at its intracellular concentration, the S-propranolol release was higher, showing that these materials could potentially be applied as intracellular controlled drug delivery system.
Introduction
During the recent decades, stimuli-responsiveness has been playing an increasingly important role in materials development for a diverse range of biological applications.1–5 Among the most extensively explored applications is smart drug delivery, for intracellular gene delivery systems or for small anti-cancer molecules, where their capacity of responding to a variation of the cellular environment or physiological conditions can advantageously be used to specifically trigger the drug release.6–10 Especially, disulfide-based polymers and conjugates have recently attracted considerable interest as effective delivery vehicles, as proven by the many papers and reviews focusing on reduction-sensitive polymers and their possible application in the biomedical field.11 Reduction sensitivity of materials containing disulfide bonds has been exploited to develop specific intracellular drug delivery systems for various therapeutic agents such as anti-cancer drugs, peptides, proteins, siRNA, DNA, that require to be delivered inside the cell to exert their therapeutic effects.Indeed, the disulfide bonds can be converted into thiol groups under reductive environment. This results in carrier degradation and the subsequent release of the encapsulated molecules. The material degradation also facilitates the carrier removal after drug release.12 As the intracellular concentration of glutathione (GSH, 1–10 mM), a biological reductive peptide, is 100 to 1000 times higher than its level in extracellular environment (20–40 μM),13 disulfide reductive reactions can occur only after cell internalization of the materials. A stable delivery system outside the cell can thus be provided in contrast to other systems such as pH-responsive or hydrolysable polymers that can be degraded in the blood circulation or in the extracellular matrix. Furthermore, disulfide-based materials could also be applied for the development of anti-cancer drug delivery systems, due to the higher GSH concentration in cancer cells and the abnormal physiological conditions of the cancerous or inflamed tissues.14
Micro/nano gels are often preferred for biomedical applications, owing to their tunable size, their internal cross-linked network for the incorporation of payload molecules, and a large specific surface area for modification. Microgels incorporating disulfide bonds in the polymer backbone were synthesized by several polymerization methods15 and with the use of appropriate disulfide-functionalized initiators,16 monomers or cross-linkers.17 Especially, gels containing disulfide functionalized dimethacrylate cross-linkers such as DSDMA have been widely synthesized by copolymerization with different monomers. They have been applied for DNA, anti-cancer drug, fluorescent dye delivery,18 for self healing materials19 or scaffolds.20 Selective reduction of the disulfide groups in DSDMA cross-linked copolymers with phosphine or thiol-containing molecules leads to the cleavage of the intermolecular branching and to the formation of low molecular weight polymer chains inducing dissolution or swelling of the gel.
Molecularly imprinted polymers (MIPs) are biomimetic synthetic receptors that possess the most important feature of natural receptors like antibodies – the ability to recognize and bind a target molecule.21 MIPs are generally synthesized by thermo- or light-initiated radical polymerization, wherein a self-assembled complex is formed through non-covalent interactions between the functional monomer(s) and a template, the target molecule or a derivative, which is then co-polymerized with a cross-linker. After template removal from the polymer, specific cavities, which are complementary in both shape and chemical functionality to the template molecule, are formed. Due to their high affinity and selectivity, MIPs are widely used in affinity technology, in the food, environment and biomedical domains, reviewed elsewhere.22 MIPs also emerged recently in the biomedical field for cell and tissue imaging23 or as controlled delivery systems for bio-active substances.24 In the latter application, the high affinity between polymers and template leads to a sustained drug delivery, controlled release sometimes depending on the surrounding environment. Few works reported the synthesis of stimuli-responsive MIPs; they were either sensitive to pH, light25 or temperature.26 Design of multifunctional materials both targeting a tissue or cells and capable of controlling the drug delivery, are highly relevant to increase the pharmaceuticals efficiency. Although, MIP are promising drug delivery system, their application as pharmaceuticals is still at its early stage compared to some other traditional formulations. Indeed, highly relevant aspects of materials designed for medical sciences such as biocompatibility, cytotoxicity, bioavailability, behavior in biological environment (in vivo experiments) have been little studied or were not considered at all for MIPs, thus hampering their development in this field.27 In the emerging application of MIPs in biomedical field, redox-sensitive MIP are highly interesting as carriers targeting a specific cell or tissue or for intracellular drug delivery by responding to a biological environment change. In addition, the degradation of the MIP matrix could improve the biocompatibility of this carrier.
In the present work, we have synthesized reduction-sensitive molecularly imprinted polymers by using a cross-linker containing a disulfide group such as bis(2-methacryloyloxyethyl) disulfide (DSDMA) or N,N′-bis(acryloyl)cystamine (BAC), which can be cleaved in reductive media (Scheme 1). These cross-linkers are not used traditionally for MIP synthesis. As the cross-linker represents usually 80% of the MIP composition, the nature of the cross-linker thus has a strong effect on the structure and binding properties of MIP. Molecular imprinting with unusual cross-linkers is thus challenging. S-Propranolol, a beta-blocker used especially in cardiac disorders, has been selected as a model template for this study. Indeed, S-propanol is being employed frequently as template to develop new MIP systems.28 S-Propranolol MIPs were obtained both in bulk format and as nanogels by high dilution polymerization. Further, we studied and characterized the MIPs' degradation in the presence of different reducing agents (NaBH4, DTT, GSH) and the effect of this treatment on the binding properties and release of S-propranolol to demonstrate that the degradation triggers controllable release of encapsulated molecules.
 | ||
| Scheme 1 Schematic representation of syntheses and reduction-induced degradation of S-propranolol molecularly imprinted polymers for drug delivery. | ||
Experimental part
Materials
All chemicals and solvents were of analytical grade and purchased from Sigma-Aldrich, unless stated otherwise. 2,2′-Azobis(2,4-dimethyl)valeronitrile (ABDV) was obtained from DuPont Chemicals (Wilmington, USA). [3H]-S-Propranolol (23.2 Ci mmol−1) was from Sigma. S-Propranolol hydrochloride was converted into the free base by extraction from a sodium carbonate solution at pH 9 with chloroform.Synthesis of reduction-responsive MIPs
Molecularly imprinted micro- and nanoparticles were synthesized by bulk or high dilution polymerization, respectively, under the conditions described in Table 1. In a typical procedure, the template molecule (S-propranolol) was dissolved in acetonitrile or DMSO in a vial sealed with a silicon rubber septum. DMSO was used as solvent for nanogel synthesis to produce homogenous nanoparticles. The polymerization initiator (ABDV), the functional monomer (MAA) and the cross-linking monomer (EDMA, DSDMA or BAC) were added with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.88:8:40 molar ratio to the template, respectively. The solution was purged with a gentle flow of nitrogen for 10 min. Polymerization was carried out in a water bath at 50 °C for 24 hours for bulk polymerization or 4 days for nanogels. After polymerization, bulk polymers were crushed and ground in a mortar. To remove the template and any unreacted monomers, bulk polymers were washed by incubation–centrifugation cycles in methanol/acetic acid 9:1 mixtures (4×) and methanol (3×). Nanogels were dialyzed against water/acetic acid 19:1 mixtures (3×) followed by water (3×) and collected by freeze-drying. Non-imprinted control polymers (NIP) were synthesized in the same way but in the absence of the template S-propranolol.
:0.88:8:40 molar ratio to the template, respectively. The solution was purged with a gentle flow of nitrogen for 10 min. Polymerization was carried out in a water bath at 50 °C for 24 hours for bulk polymerization or 4 days for nanogels. After polymerization, bulk polymers were crushed and ground in a mortar. To remove the template and any unreacted monomers, bulk polymers were washed by incubation–centrifugation cycles in methanol/acetic acid 9:1 mixtures (4×) and methanol (3×). Nanogels were dialyzed against water/acetic acid 19:1 mixtures (3×) followed by water (3×) and collected by freeze-drying. Non-imprinted control polymers (NIP) were synthesized in the same way but in the absence of the template S-propranolol.
| Polymer | Polymerization method | Solvent | Cma (%) | S-Propranolol (mmol) | MAA (mmol) | Cross-linker (mmol) | IF | ||
|---|---|---|---|---|---|---|---|---|---|
| DSDMA | EDMA | BAC | |||||||
| a Abbreviation Cm, monomer concentration calculated as the mass ratio of functional monomer and cross-linker to the total mass of polymer formulation.b Imprinting factor determined for a polymer concentration of 0.08 g L−1.c Imprinting factor determined for a polymer concentration of 0.8 g L−1. | |||||||||
| M1 | Bulk | ACN | 46.9 | 0.015 | 0.12 | 0.6 | — | — | 7.2b |
| N1 | Bulk | ACN | 46.9 | — | 0.12 | 0.6 | — | — | — |
| M2 | Bulk | ACN | 46.9 | 0.015 | 0.12 | — | 0.6 | — | 6b |
| N2 | Bulk | ACN | 46.9 | — | 0.12 | — | 0.6 | — | — |
| M3 | Bulk | ACN | 46.9 | 0.015 | 0.12 | — | — | 0.6 | 1.5b |
| N3 | Bulk | ACN | 46.9 | — | 0.12 | — | — | 0.6 | — |
| M4 | High dilution | DMSO | 0.25 | 0.015 | 0.12 | 0.6 | — | — | 2.4c |
| N4 | High dilution | DMSO | 0.25 | — | 0.12 | 0.6 | — | — | — |
| M5 | High dilution | DMSO | 0.25 | 0.015 | 0.12 | — | 0.6 | — | 1.4c |
| N5 | High dilution | DMSO | 0.25 | — | 0.12 | — | 0.6 | — | — |
Characterization
where η: viscosity of polymer suspension and η0: viscosity of solvent.

Evaluation of MIPs binding properties
For batch binding experiments, MIP and NIP (0.02 to 0.1 g L−1) were incubated in a solution of [3H]-S-propranolol (0.6 pmol in acetonitrile) for 2 hours on a rocking table. The tubes were centrifuged at 17500 rpm and 0.5 mL of supernatant was transferred into scintillation vials containing 4 mL scintillation fluid (Ultima Gold, PerkinElmer). The concentration of free radioligand was measured with a liquid scintillation counter (Beckman LS-6000 IC) and used to determine the percentage of [3H]-S-propranolol bound to the polymer. The imprinting factor (IF) was used to evaluate the efficiency of the molecular imprinting by comparing the amount of [3H]-S-propranolol bound to the MIP and the NIP.
Langmuir binding isotherms were recorded to determine the dissociation constant (Kd) and maximum binding capacity (Bmax). The polymer at a fixed concentration of 0.1 g L−1 was incubated with 0.6 pmol [3H]-S-propranolol and S-propranolol with a concentration ranging from 0.171 μM to 60 μM in acetonitrile. Binding isotherms were then fitted with the Langmuir model according to the following equation:

To evaluate the selectivity of the bulk MIP, competitive binding assays with the two enantiomers of propranolol were performed to determine their IC50. 0.1 g L−1 polymer were incubated with 0.6 pmol [3H]-S-propranolol and with varying concentrations (from 2 pM to 0.2 mM) of S-propranolol or R-propranolol as competitors in anhydrous acetonitrile. The cross-reactivity is defined as the IC50 ratio of the two enantiomers:
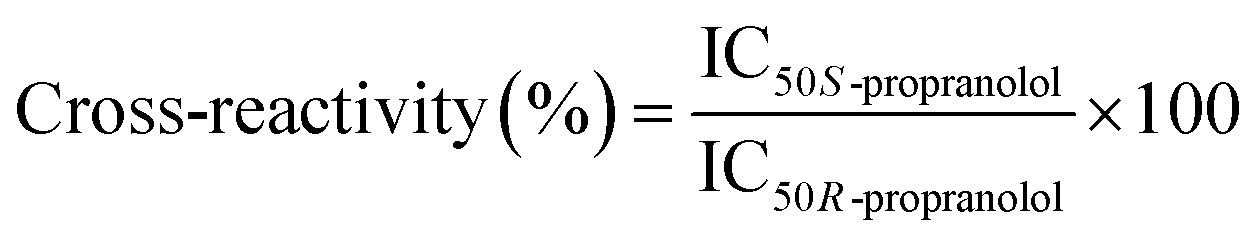
Degradation tests of nanogels
The degradation tests of nanogels were performed both in organic and aqueous media according to the reducing agent. For the test in organic media, a 1 g L−1 solution of polymer nanogels cross-linked with DSDMA was prepared in DMSO, then 50 mM NaBH4 was added. The reaction was allowed to proceed for 5 min where after the unreacted NaBH4 was inactivated by adding 10 μL of 2 mM HCl. To determine the size of the polymer particles after degradation, 50 μL polymer suspension was withdrawn and mixed with 950 μL ethanol to adjust the final concentration to 0.05 g L−1, then one drop was deposited on a glass substrate and dried under N2 flow prior to SEM measurements. The polymer composition after degradation was analyzed by FTIR.The degradation test in aqueous media was carried out using DTT or GSH as reducing agent instead of NaBH4. A DSDMA cross-linked nanogel solution (1 g L−1) was prepared in oxygen-free Tris–HCl buffer (100 mM, pH 8.0), DTT (20 or 40 mM) was added and reacted under nitrogen for 10 min at 37 °C. After the degradation, the polymers were analyzed by dynamic light scattering, size exclusion chromatography and viscosimetry. For nanogel degradation by GSH, DSDMA-based nanogel solutions (1 g L−1) were mixed with GSH (20 mM) in oxygen-free sodium acetate buffer (100 mM, pH 5.0) during 14 hours. Control experiments were carried out in the same way but without GSH.
In vitro release of S-propranolol from nanogels
S-Propranolol (2 mM) and MIPs or NIPs (1 g L−1) were incubated in anhydrous acetonitrile overnight with gentle agitation, then the particle suspension in acetonitrile was put in a dialysis tube and dialyzed against sodium acetate buffer (0.1 M, pH 5.0) to remove the unbound S-propranolol. The release study was carried out with or without the presence of GSH (10 mM) in sodium acetate buffer (0.1 M, pH 5.0) at 37 °C. The concentration of the released S-propranolol was determined by spectrofluorimetry (Fluorolog, HORIBA) with excitation and emission wavelengths, respectively at 290 nm and 357 nm.Results and discussion
Synthesis and characterization of reduction-sensitive MIPs
In the molecular imprinting field, the functional monomer, cross-linker and solvent have to be carefully selected to obtain MIPs with highly specific and selective binding properties for a given target molecule. While the cross-linker is thought not to be directly involved in the interaction with the template (in fact it often is), as a major component in the MIPs formulation it still plays a determining role for the MIP recognition properties. Indeed, the cross-linking monomer and degree have to be controlled to tune the polymer's inner morphology such as porosity, and polymeric chain flexibility or matrix rigidity for good template accessibility to the binding sites and to find an appropriate rigidity of the binding cavities allowing a high affinity for the template.29 By introducing a cleavable bond in the cross-linker structure, the 3D-network could be degraded under the appropriate trigger into linear polymer chains, leading to an enhancement of the matrix flexibility and subsequently to a loss of the molecular recognition properties. If MIPs are loaded with a target molecule as a cargo, the degradation of the polymer induced by a stimulus can provoke the release of the entrapped molecule. This feature can be especially relevant for applications as drug delivery systems. As shown in Scheme 1, we propose to apply this concept for the production of MIPs that can be degraded in a reductive environment by using a cleavable cross-linker containing a disulfide bond and S-propranolol as model drug. The synthesis of reduction-sensitive molecularly imprinted polymers for S-propranolol was done using methacrylic acid as functional monomer and disulfide-based cross-linker with the ratio 8:40:1 respectively to the template, as previously reported in the literature.30 Two disulfide-based cross-linkers, bis(2-methacryloyl)oxyethyl disulfide (DSDMA) and N,N′-bis(acryloyl)cystamine (BAC) were tested in this study to replace the commonly used EDMA for MIP synthesis (Scheme 1). In addition, EDMA-based MIPs for S-propranolol imprinting were also prepared in the same manner as a control to study the effect of the reducing agent on morphological, structural and binding properties of the MIPs.
Disulfide and EDMA-based MIP particles were synthesized in the form of bulk polymers and nanogels by different polymerization methods. Fig. 1 shows the SEM images of MIP particles obtained by bulk polymerization, as well as the TEM images and DLS analysis of MIP nanogels prepared by high dilution polymerization with different cross-linkers. Bulk polymer particles resulting from the grinding of monolithic materials present an irregular and rough morphology with a particles size greater than 1 μm, whereas nanoparticles were obtained by high dilution polymerization.
 | ||
| Fig. 1 SEM images of bulk polymer cross-linked by EDMA (A), DSDMA (B) or BAC (C). TEM photographs of nanogels cross-linked by EDMA (D) or DSDMA (E) and their corresponding size distribution (F) determined by DLS in DMSO. | ||
One can also observe a strong effect of the nature of the cross-linker on the polymer structure and porosity. Whereas the morphologies of EDMA and DSDMA-based polymers seem similar, the shape and surface rugosity of BAC-based polymers are very different with a cauliflower-like structure. Although the binding properties of bulk polymers are easily evaluated, their irregular particle form and size hinders their applications in many domains. Therefore, we also synthesized smaller MIP nanogels with a diameter in the lower nm range. Radical polymerization in highly diluted solution is the simplest way to prepare nanogels, since no surfactants or other additives are required.31 Polymerization yields for M4 and M1 synthesized by high dilution and bulk polymerization were 50% and 83%, respectively. The morphology of the DSDMA and EDMA nanogels are quite similar with a size (determined by DLS in DMSO) of around 30 nm.
Binding properties of reduction-sensitive MIPs
After template removal, MIP selectivity and specificity were studied by radioligand binding assays in acetonitrile using 3H-labeled S-propranolol. Binding isotherms were obtained by incubating varying concentrations of MIP and NIP prepared by bulk or high dilution polymerization with a constant amount of 3H-labeled S-propranolol (Fig. 2). For bulk polymers, the adsorption on DSDMA-MIP are higher than on EDMA-MIP with a better imprinting factor, whereas the non-imprinted control polymers have very low S-propranolol adsorption. BAC-MIP has a very low S-propranolol binding, probably resulting from the strong morphological difference compared to DSDMA- and EDMA-MIP. For nanogels, the same behavior can be observed with a higher imprinting factor for DSDMA-MIP than EDMA-MIP (2.4 compared to 1.4 for EDMA-MIP at a polymer concentration of 0.1 mg mL−1), but with a lower adsorption capacity than for bulk MIPs. Furthermore, the affinity and enantioselectivity of EDMA and DSDMA cross-linked MIPs have been assessed as shown in Table 2. The dissociation constant (Kd) and maximum binding capacity (Bmax) of S-propranolol were determined by fitting the binding isotherms to a Langmuir model (see ESI Fig. S1†). Similar affinities and binding capacities were obtained for both materials. The enantioselectivity of the MIPs was then determined by competitive radioligand binding experiments. A fixed amount of polymer was incubated with a fixed amount of radiolabeled S-propranolol and increasing concentrations of competing ligand (R-propranolol or S-propranolol) in anhydrous acetonitrile to obtain the competition curves for EDMA- or DSDMA-based MIPs (see ESI Fig. S1†) and their corresponding cross-activities. The EDMA-based MIP shows an excellent enantioselectivity with a cross-reactivity of 1.4%, as previously reported.30 The cross-reactivity of the R enantiomer is somewhat higher with the DSDMA-based MIP. However, the MIP is nevertheless highly specific and selective. This shows that DSDMA can be an interesting cross-linker alternative to EDMA for MIP synthesis.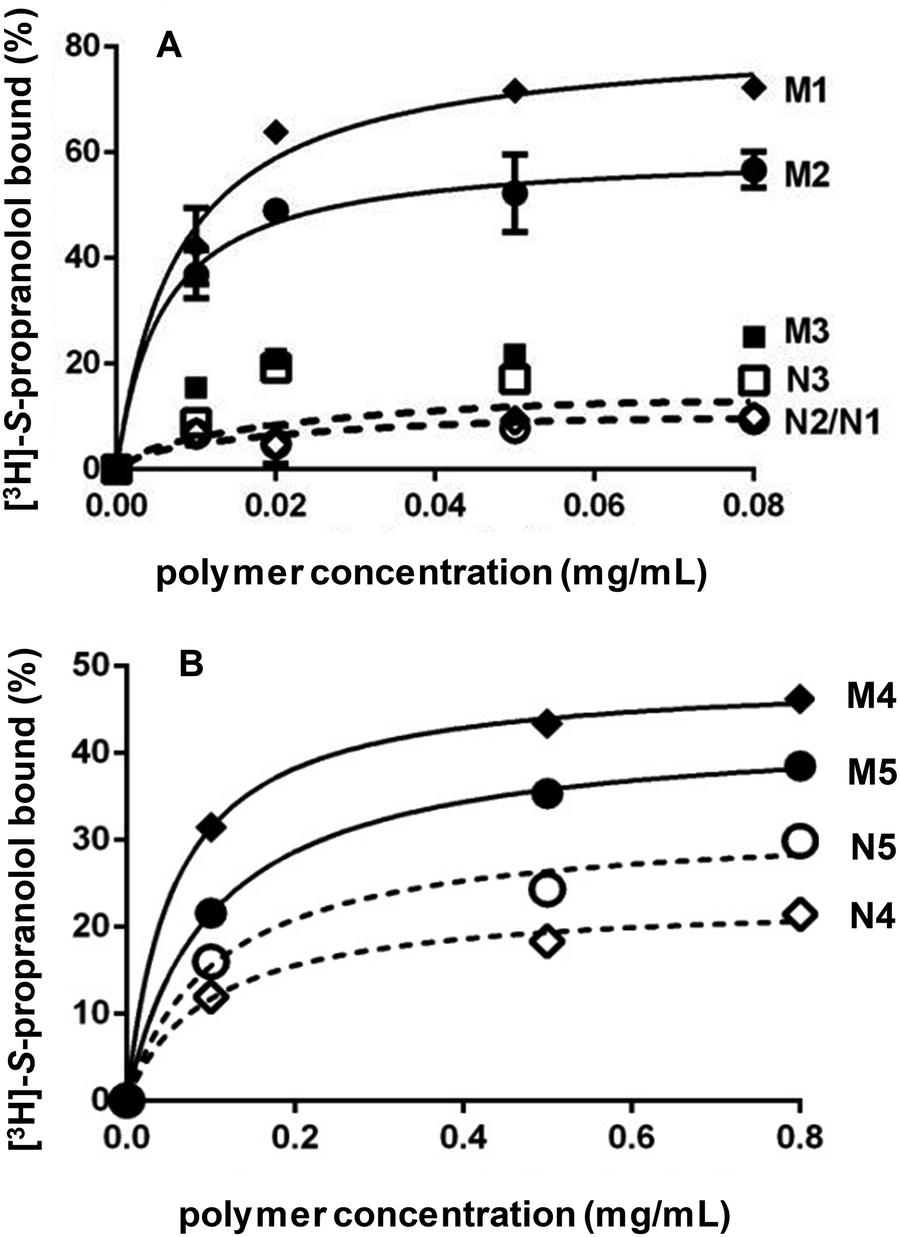 | ||
| Fig. 2 Binding of [3H]-S-propranolol in anhydrous acetonitrile by MIP and NIP synthesized by bulk polymerization (A) with DSDMA (M1/N1), EDMA (M2/N2) or BAC (M3/N3) as cross-linker and by high dilution polymerization (B) with DSDMA (M4/N4) or EDMA (M5/N5) as cross-linker. | ||
Reductive cleavage of disulfide-containing polymers
Chemical modifications after treatment with the reducing agent were observed by FTIR spectroscopy (Fig. 3). FTIR spectra of EDMA and DSDMA-based MIPs are very similar relating to their close chemical structures and contain all the characteristic bands that are present in FTIR spectra of MAA and cross-linkers. A number of characteristic peaks were observed at 2950 cm−1 (aliphatic CH2, CH, CH3 groups), 1440 cm−1 (aliphatic CH2), 1380 cm−1, 1240 cm−1, 1144 cm−1 (C–O) associates with the ethylene glycol unit. On the other hand, the strong bands at 1730 cm−1 and the weaker at 1650 cm−1 were assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the ester/acid group and CC vibration band of the residual vinylic groups confirming a successful co-polymerization of the cross-linkers with the functional monomer MAA. In the spectra of degraded materials, a new band at 2250 cm−1, which corresponds to the presence of free thiol groups, is observed only for the DSDMA-based polymer indicating that the disulfide bonds in this polymer have been successfully reduced by NaBH4. However, the EDMA-based polymer spectrum is also slightly modified after its incubation with NaBH4. A decreased peak intensity of the CO group is observed and a strong band at 3440 cm−1 corresponding to O–H stretching vibration appears. As suggested above for the reduction experiments with DSDMA, these modifications have been attributed to a potential concomitant ester reduction by the strong reducing agent. Therefore, soft reducing agents, namely DTT or GSH, described in the literature for their specific disulfide bond reduction properties, were also evaluated for the degradation of nanogels.
O stretching vibration of the ester/acid group and CC vibration band of the residual vinylic groups confirming a successful co-polymerization of the cross-linkers with the functional monomer MAA. In the spectra of degraded materials, a new band at 2250 cm−1, which corresponds to the presence of free thiol groups, is observed only for the DSDMA-based polymer indicating that the disulfide bonds in this polymer have been successfully reduced by NaBH4. However, the EDMA-based polymer spectrum is also slightly modified after its incubation with NaBH4. A decreased peak intensity of the CO group is observed and a strong band at 3440 cm−1 corresponding to O–H stretching vibration appears. As suggested above for the reduction experiments with DSDMA, these modifications have been attributed to a potential concomitant ester reduction by the strong reducing agent. Therefore, soft reducing agents, namely DTT or GSH, described in the literature for their specific disulfide bond reduction properties, were also evaluated for the degradation of nanogels.
 | ||
| Fig. 3 FTIR spectra of bulk polymers cross-linked by EDMA (a) or DSDMA (b) and after 4 h incubation with the reducing agent NaBH4. EDMA cross-linked polymers with NaBH4 (c) and DSDMA cross-linked polymers with NaBH4 (d). | ||
Characterization of the nanogels degradation
By decreasing the particles size, a fully degradable system might be obtained. DSDMA-based nanogels were incubated in Tris-buffer (pH 8.0, 0.1 M) with DTT. As shown in Scheme 1 and Fig. 4A, the turbidity of DSDMA-based polymer solution decreases rapidly after the addition of DTT and a transparent solution is obtained in less than one hour, due to the degradation of DSDMA-based nanogel into smaller more soluble polymeric chains. No modification of the size distribution and the turbidity was observed for EDMA-based polymer, which suggests that the EDMA cross-linker is not cleavable by DTT. | ||
| Fig. 4 Turbidity evolution (A) and particle size (B) of nanogel (EDMA or DSDMA) solutions (1g L−1) in 100 mM Tris–HCl buffer pH 8 with or without DTT (40 mM), measured by UV-Vis spectroscopy at 600 nm and DLS, respectively. Specific viscosity of DSDMA cross-linked nanogel in 100 mM Tris–HCl buffer pH 8 with (full circles) or without DTT (open circles) (C). SEC-RI chromatogram of DSDMA-based nanogels before and after incubation with different concentrations of DTT in Tris–HCl buffer 100 mM (D). | ||
The degradation of the nanogel under reductive environment was also demonstrated by the modification of the particles size characterized by DLS analysis, viscosimetry and SEC. DLS analysis of the hydrodynamic diameter of DSDMA-based nanogel (Fig. 4B) after incubation with DTT showed a large decrease in particles size from 615 nm to 295 nm, whereas EDMA-based particles are less affected. The nanoparticles have larger diameters in aqueous media than after their synthesis, depending also on buffer composition. This is attributed to a different polymer swelling according to the solvent composition (pH, buffer nature) and the possible formation of nanogel clusters in aqueous media. The DSDMA-based nanogel degradation into smaller polymer chains is also confirmed by the difference of the specific viscosity of DSDMA-based polymer solutions in the presence of DTT (Fig. 4C). Indeed, viscosity allows to estimate the molecular weight and the shape of a polymer.33 The decrease in specific viscosity under reductive environment for DSDMA-based polymer solution can be attributed to a modification of the polymer molecular weight or to its structure. Finally, SEC analysis was performed to study the polymer molecular weight modification. After treatment with DTT, the reduced polymer was extracted with chloroform and analyzed by SEC. As shown in Fig. 4D, a noticeable difference in retention time was observed for DSDMA-based polymer incubated with different DTT concentrations indicating a decrease of the polymer molecular weight with a higher amount of reducing agent. Thus, the disulfide links in the nanogels matrix can be cleaved in the presence of a soft reducing agent inducing a degradation of the polymer network. The degradation of DSDMA cross-linked polymer was also examined under a physiological reducing environment using GSH as reducing agent at the intracellular concentration (10 mM) at 37 °C. DLS analysis and turbidity (see ESI Fig. S3†) showed a sharp decrease of polymer size from 955 nm to 255 nm and the solubilization of the polymer as with DTT after 4 h incubation, suggesting that GSH can also degrade DSDMA-based nanogels by reducing the disulfide bonds incorporated into the polymer.
Binding property change after the reduction cleavage of the disulfide bonds
Binding properties of DSDMA cross-linked bulk and nanogel MIPs were also investigated after treatment with different reducing agents (NaBH4 or DTT). A loss of their binding properties was observed for both MIP formats and reducing agents as shown in Fig. 5. However, a stronger effect of the reducing agent on the S-propranolol binding properties have been obtained with nanogels than with bulk polymer with a binding decrease of 89% and 70%, respectively. This difference is more significant when DTT was used as reducing agent. The binding loss was 75% for nanogel but only 14% for bulk polymer. With the NIPs, virtually no change in binding properties was observed after the treatment with the reducing agents. This was expected since the non-specific binding to the NIP is much less dependent on the integrity of the 3D polymer network than the binding to the specific binding sites in the MIP.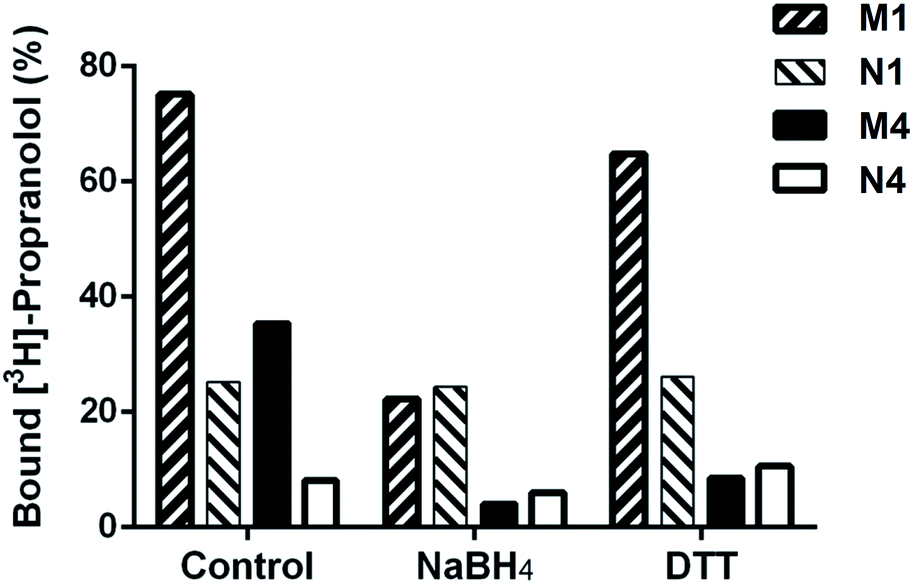 | ||
| Fig. 5 Degradation effect with different reducing agents (500 mM NaBH4 or 40 mM DTT) on binding of [3H]-S-propranolol to MIP (M1) and NIP (N1) synthesized by bulk polymerization and to MIP (M4) and NIP (N4) synthesized by high dilution polymerization, all cross-linked by DSDMA. | ||
Release of S-propanolol from disulfide-based MIP nanogels
MIPs have particular release properties compared to other drug delivery systems due to their specific molecular recognition.34 In order to explore the potential application of these redox-sensible MIPs as intracellular drug delivery system, the effect of the polymer network cleavage under physiological reductive environment on the drug release properties have been examined. Indeed, the degradation of the polymer network is one of the possible release mechanisms of the encapsulated molecule in drug delivery systems.35 The release experiment was performed with 10 mM GSH in sodium acetate buffer (pH 5.0, 0.1 M), since this pH and GSH concentration are usually found in endosomes and lysosomes.13,36First, we investigated the release of the 8-anilino-1-naphthalenesulfonic acid (ANS) fluorescent dye from a fluo-tagged DSDMA-based polymer. Indeed, ANS is a solvatochromic dye, sensitive to polarity change of the environment. It exhibits a decrease in quantum yield accompanied by a red shift of fluorescence maximum as the polarity of the solvent increases.37 Thus the release of ANS can be monitored directly by spectrofluorimetry. ANS fluo-tagged DSDMA-based polymer nanogels were synthesized by free radical polymerization using the same formulation as for the MIP nanogels, without the template (see ESI†). Fluo-tagged nanogels with 28 nm particles size have been obtained. The cytotoxicity of these particles was estimated on HaCat cells by in vitro MTT assays (see ESI Fig. S4†). No cytotoxicity for DSDMA-based particles were observed up to the concentration of 200 mg L−1, suggesting the low toxicity of these nanogels. This result indicates that DSDMA-based nanogel could be a promising pharmaceuticals carrier. The nanogels' fluorescence spectrum and the release of ANS from DSDMA cross-linked nanogels with or without addition of GSH in the medium are presented in Fig. S5 (ESI†). After 96 h incubation, only 30% ANS was released from DSDMA-based nanogel in a media without GSH, while the released amount of ANS was 51% in the presence of 10 mM GHS. The release of ANS from the nanogels is accelerated in the presence of GSH due to the cleavage of the disulfide bonds in the polymer network, thus facilitating the diffusion of ANS within the nanogels.
The effect of the polymer network cleavage on the MIP release properties was then examined under physiological reductive environment. For this purpose, DSDMA-based MIP and NIP nanogels were first incubated with S-propranolol (0.59 mg adsorbed S-propranolol per mg MIP, 0.51 mg adsorbed S-propranolol per mg NIP), then release experiments of S-propranolol from the nanogels in the presence/absence of GSH were carried out by a dialysis method (Fig. 6). Without GSH, the S-propranolol release from NIP or MIP reached an equilibrium after 2 h with a maximum amount of S-propranolol released of 33% and 22% from NIP and MIP, respectively, and no-burst effect was observed. The S-propranolol release from NIP is faster and higher than from MIP (1.7 and 1.5 times, respectively). This may be explained by the lower affinity of S-propranolol for the NIP. In the presence of GSH, the amount of S-propranolol released from MIP and the release rate were increased by 1.5 and 2 times, respectively, due to the degradation of the polymer network. More interestingly, the amount released at the end was comparable with NIP, which confirms the loss of molecular recognition of MIP when reducing agent was present. S-Propanolol release from MIP and NIP were also fitted by the Peppas model38 to study the release mechanism (see ESI Table S1†). The release exponent n was determined to be 0.44, 0.87 and 0.52, respectively for NIP, MIP and MIP in presence of GSH, suggesting clearly different release mechanisms between these systems. These results indicated that DSDMA cross-linked MIP permits control over the drug delivery with a limited release of the drug under physiological condition as in bloodstream but triggered a rapid and sustained drug release in reductive environment as the one found in cells or in tumor tissues. Thus, reduction-responsive molecularly imprinted polymers are promising materials as an intracellular drug delivery system.
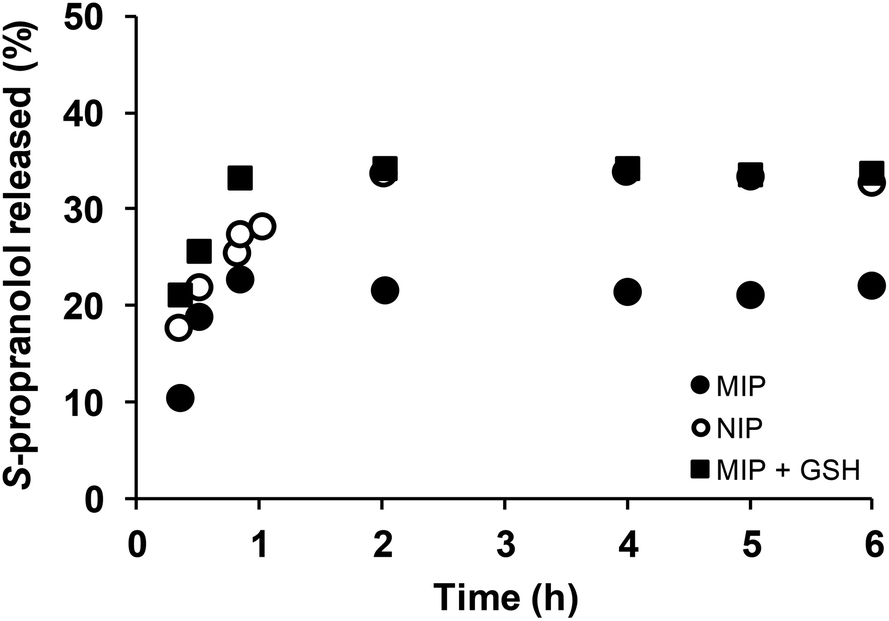 | ||
| Fig. 6 Release of S-propranolol from MIP nanogels with or without GSH. MIP nanogel (full circle), NIP nanogel (empty circle) and MIP nanogel (full square) with presence of 10 mM GSH in sodium acetate buffer (0.1 M, pH 5.0). | ||
Conclusion
Reduction-responsive molecularly imprinted polymers were synthesized by using a disulfide-containing cross-linker, which can be cleaved in a reductive environment. Different forms of reduction-responsive MIPs were synthesized with size ranges from 30 nm to 2 μm. The reduction-responsive MIPs demonstrate similar or even better binding property for a β-blocker (S-propranolol) compared to conventional MIPs synthesized using the cross-linker EDMA. In a reductive environment, disulfide bonds can be cleaved to produce less cross-linked materials and thus induce morphological and chemical modification, associated with the modulated binding and the release of template molecules. This suggests the possibility of this reduction-responsive MIPs being used for intracellular drug delivery applications.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the European Regional Development Fund ERDF for co-funding of the equipment under CPER 2014–2020, the Conseil Régional de Picardie (BIOMIP) and from the EU FP7 Marie Curie Programme (ITN CHEBANA) for financial support, Franck Merlier from the Enzyme and Cell Engineering Laboratory for SEC measurements and Frederic Nadaud from the Physico-Chemical Analysis Laboratory of Université de Technologie de Compiègne for SEM and TEM measurements.References
- T. M. Reineke, ACS Macro Lett., 2016, 5, 14 CrossRef CAS; A. S. Hoffman, Adv. Drug Delivery Rev., 2013, 65, 10 CrossRef.
- J. N. Anker, W. P. Hall, O. Lryandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442 CrossRef CAS.
- S. Y. Lee, H. Lee, I. In and S. Y. Park, Eur. Polym. J., 2014, 57, 1 CrossRef CAS; H. Y. Yang, M.-S. Jang, Y. Li, J. H. Lee and D. S. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 19184 CrossRef PubMed.
- Q. Xu, C. He, Z. Zhang, K. Ren and X. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 30692 CrossRef CAS PubMed.
- Z. Liu and P. Calvert, Adv. Mater., 2000, 12, 288 CrossRef CAS.
- M. Motornov, S. Minko, K. J. Eichhorn, M. Nitschke, F. Simon and M. Stamm, Langmuir, 2003, 19, 8077 CrossRef CAS; E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866 CrossRef PubMed.
- C. L. Bayer and N. A. Peppas, J. Controlled Release, 2008, 132, 216 CrossRef CAS; A. K. Bajkai, S. K. Shukla, S. Bhanu and S. Kankane, Prog. Polym. Sci., 2008, 33, 1088 CrossRef; Y. Kakizawa, A. Harada and K. Kataoka, Biomacromolecules, 2001, 2, 491 CrossRef; Y. C. Wang, F. Wang, T. M. Sun and J. Wang, Bioconjugate Chem., 2011, 22, 1939 CrossRef PubMed.
- J. Zhang, Z. Cui, R. Field, M. G. Moloney, S. Rimmer and H. Ye, Eur. Polym. J., 2015, 67, 346 CrossRef CAS.
- R. de la Rica, D. Aili and M. M. Stevens, Adv. Drug Deliv. Rev., 2012, 64, 967 CrossRef CAS PubMed; J. Hu, G. Zhang and S. Liu, Chem. Soc. Rev., 2012, 41, 5933 RSC; K. B. Fonseca, D. B. Gomes, K. Lee, S. G. Santos, A. Sousa, E. A. Silva, D. J. Mooney, P. L. Granja and C. C. Barrias, Biomacromolecules, 2014, 15, 380 CrossRef PubMed.
- T. Manouras and M. Vamvakaki, Polym. Chem., 2017, 8, 74 RSC.
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180 CrossRef CAS PubMed; R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2 CrossRef PubMed; B. Gyarmati, A. Nemethy and A. Szilagyi, Eur. Polym. J., 2013, 49, 1268 CrossRef.
- J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2017, 8, 97 RSC.
- F. Q. Schafer and G. R. Buettner, Free Radic. Biol. Med., 2001, 30, 1191 CrossRef CAS.
- P. Kuppusamy, H. Li, G. Ilangovan, A. J. Cardounel, J. L. Zweier, K. Yamada, M. C. Krishna and J. B. Mitchell, Cancer Res., 2002, 62, 307 CAS.
- Y. Wang, L. Zhang, X. Zhang, X. Wei, Z. Tang and S. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 5833 CrossRef CAS PubMed.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 3087 CrossRef CAS.
- X. Jiang, S. Liu and R. Narain, Langmuir, 2009, 25, 2576 Search PubMed; J. Rosselgong, W. R. S. Barton, D. Price and S. P. Armes, Macromolecules, 2010, 43, 2145 CrossRef CAS; J. Zeng, P. Du, L. Liu, J. Li, K. Tian, X. Jia, X. Zhao and P. Liu, Mol. Pharm., 2015, 12, 4188 CrossRef PubMed; H. Yang, Q. Wang, S. Huang, A. Xiao, F. Li, L. Gan and X. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 7729 CrossRef.
- H. Y. Cho, A. Srinivasan, J. Hong, E. Hsu, S. Liu, A. Shrivats, D. Kwak, A. K. Bohaty, H.-J. Paik, J. O. Hollinger and K. Matyjaszewski, Biomacromolecules, 2011, 12, 3478 CrossRef CAS; Y. Wang, C.-Y. Hong and C.-Y. Pan, Biomacromolecules, 2012, 13, 2585 CrossRef PubMed.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolay, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142 CrossRef CAS.
- H.-W. Chien, W.-B. Tsai and S. Jiang, Biomaterials, 2012, 33, 5706 CrossRef CAS PubMed.
- K. Haupt, A. V. Linares, M. Bompart and B. Tse Sum Bui, Top. Curr. Chem., 2012, 325, 1 CrossRef CAS PubMed.
- I. Chianella, A. Guerreiro, E. Moczko, J. S. Caygill, E. V. Piletska, I. M. P. De Vargas Sansalvador, M. J. Whitcombe and S. A. Piletsky, Anal. Chem., 2013, 85, 8462 CrossRef CAS PubMed; J. Wackerlig and P. A. Lieberzeit, Sens. Actuators, B, 2015, 207, 144 CrossRef; Z. Iskierko, P. S. Sharma, K. Bartold, A. Pietrzyk-Le, K. Noworyta and W. Kutner, Biotechnol. Adv., 2016, 34, 30 CrossRef PubMed.
- M. Panagiotopoulou, Y. Salinas, S. Beyazit, S. Kunath, L. Duma, E. Prost, A. G. Mayes, M. Resmini, B. Tse Sum Bui and K. Haupt, Angew. Chem., Int. Ed., 2016, 55, 8244 CrossRef CAS PubMed; Z. El-Schich, M. Abdullah, S. Shinde, N. Dizeyi, A. Rosen, B. Sellergren and A. Wingren, Tumor Biol., 2016, 37, 13763 CrossRef PubMed; S. Kunath, M. Panagiotopoulou, J. Maximilien, N. Marchyk, J. Sänger and K. Haupt, Adv. Healthcare Mater., 2015, 4, 1322 CrossRef PubMed.
- C. F. van Nostrum, Drug Discovery Today: Technol., 2005, 2, 119 CrossRef CAS PubMed.
- L. Fang, S. Chen, X. Guo, Y. Zhang and H. Zhang, Langmuir, 2012, 28, 9767 CrossRef CAS PubMed; C. Gong, K.-L. Wong and M. H. Lam, Chem. Mater., 2008, 20, 1353 CrossRef.
- N. Li, L. Qi, Y. Shen, J. Qiao and Y. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 17289 CrossRef CAS PubMed.
- P. Lulinski, Mater. Sci. Eng., C, 2017, 76, 1344 CrossRef CAS.
- E. III Paruli, T. Griesser, F. Merlier, C. Gonzato and K. Haupt, Polym. Chem., 2019, 10, 4732 RSC.
- N. Holland, J. Frisby, E. Owens, H. Hughes, P. Duggan and P. McLoughlin, Polymer, 2010, 51, 1578 CrossRef CAS; I. Iturralde, M. Paulis and J. R. Leiza, Eur. Polym. J., 2014, 53, 282 CrossRef.
- L. I. Andersson, Anal. Chem., 1996, 68, 111 CrossRef CAS PubMed; L. Ye, I. Surugiu and K. Haupt, Anal. Chem., 2002, 74, 959 CrossRef.
- N. Perez, M. J. Whitcombe and E. N. Vulfson, J. Appl. Polym. Sci., 2000, 77, 1851 CrossRef CAS.
- S. R. S. Ting, E. H. Min, P. B. Zetterlund and M. H. Stenzel, Macromolecules, 2010, 43, 5211 CrossRef CAS.
- M. Sun, M.-X. Qiao, J. Wang and L.-F. Zhai, ACS Sustainable Chem. Eng., 2017, 5, 7832 CrossRef CAS.
- C. Bodhibukkana, T. Srichana, S. Kaewnopparat, N. Tangthong, P. Bouking, G. P. Martin and R. Suedee, J. Controlled Release, 2006, 113, 43 CrossRef CAS PubMed; R. Suedee, T. Srichana and G. P. Martin, J. Controlled Release, 2000, 66, 135 CrossRef PubMed.
- Z.-Y. Qiao, R. Zhang, F.-S. Du, D.-H. Liang and Z.-C. Li, J. Controlled Release, 2011, 152, 57 CrossRef CAS PubMed.
- D. Schmaljohann, Adv. Drug Deliv. Rev., 2006, 58, 1655 CrossRef CAS PubMed.
- J. Slavik, Biochim. Biophys. Acta, 1982, 694, 1 CrossRef CAS.
- G. W. Sinclair and N. A. Peppas, J. Membr. Sci., 1984, 17, 329 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07512g |
| This journal is © The Royal Society of Chemistry 2020 |