
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorene benzothiadiazole co-oligomer based aqueous self-assembled nanoparticles†
J. Schilla,
L. Ferrazzano b,
A. Tolomellib,
A. P. H. J. Schenning*c and
L. Brunsveld*a
b,
A. Tolomellib,
A. P. H. J. Schenning*c and
L. Brunsveld*a
aLaboratory of Chemical Biology, Department of Biomedical Engineering and Institute for Complex Molecular Systems Eindhoven University of Technology, P. O. Box 513, 5600MB Eindhoven, The Netherlands. E-mail: l.brunsveld@tue.nl
bDepartment of Chemistry, University of Bologna, Via Selmi 2, 40126 Bologna, Italy
cFunctional Organic Materials and Devices, Institute for Complex Molecular Systems, Eindhoven University of Technology, P. O. Box 513, 5600MB, Eindhoven, The Netherlands. E-mail: a.p.h.j.schenning@tue.nl
First published on 2nd January 2020
Abstract
Self-assembled π-conjugated nanoparticles with tunable optical characteristics are appealing for sensing and imaging applications due to their intrinsic fluorescence, supramolecular organization and dynamics. Here we report on the facile synthesis of fluorene benzothiadiazole co-oligomers in which structural backbone alterations induce bathochromically shifted optical characteristics. Moreover, the nature of the oligomer side-chains revealed the role of bulkiness and polarity on the optical and self-assembly behavior. Co-assemblies were prepared that showed energy transfer between the different oligomers which allows for tuning of the emission color. These compounds thus extend the π-conjugated-oligomer toolbox from which nanoparticles can be prepared with tailored physicochemical properties that may result in supramolecular materials for biosensing.
Introduction
Nano-sized architectures – which are self-assembled from molecular building blocks – have advantageous features for applications in a biological setting such as e.g. imaging and sensing and have thus been developed in many forms.1–3 In contrast to molecular probes, nanoparticles are often non-cytotoxic and inert to nonspecific interactions with surrounding biomolecules.4 Moreover, surface modification of nanoparticles with moieties as high affinity ligands and sensitive dyes can vastly benefit the detectability and the ability to aim for specific targets.5 A versatile manufacturing process, by which different monomers are mixed, can readily yield modular nanostructures with a controlled morphology and a decoration topology upon design. While many different kinds of nanoparticles are prepared,6–9 fluorescent nanoparticles commonly allow for a high spatial and temporal resolution and relatively straightforward detection.10,11 The fluorescent systems constructed from π-conjugated chromophores generally present key features such as e.g. high photostability, large Stokes shift and high quantum yield and have, therefore, attracted much attention.12–15 Most of these oligomers were derived from their corresponding polymers16 and show an increased dynamic behavior and more controllability yet retaining the polymers' key properties including outstanding optical characteristics.17 While there are many examples of π-conjugated structures in this field,18,19 fluorene co-oligomers particularly present an attractive and delicate interplay between chemical structure and photophysical properties.20 Their π-conjugated backbone commonly represents a low band gap donor–acceptor type co-oligomer by direct covalent linkage of the donor and acceptor fragment through aromatic stabilization of the quinoid structure.21,22 The donating fluorene fragment allows for a high absorption cross section and accommodates efficient through-bond energy transfer to the acceptor fragment, which results in bright chromophores that emit at long wavelengths.23–25 The central π-conjugated backbone is typically flanked by a combination of hydrophobic and hydrophilic side-chains. The design of the central core largely dictates the fluorescence emission profile,26,27 while the nature of these side-chains influences the self-assembly behavior in aqueous solution.28,29 The concomitant unimolecular fluorene-based assemblies, which are commonly prepared through reprecipitation, have shown to allow for the encapsulation of guest molecules,17,30 cellular uptake31,32 and cargo delivery.33,34 Moreover, multicomponent assemblies have become vastly more prominent for biological applications due to their modularity and hence tunability of their intrinsic characteristics. These co-assemblies showed to allow for a tunable fluorescence output35,36 and controllable dynamics depending on the molecular design and oligomer blend.37,38 The additional tunable surface modulation has led to multivalent nanoparticles that bear a high potential as supramolecular imaging and sensing probes.39,40Nanostructures that display a large Stokes shift, preferably emitting in the infrared region to reduce optical interference, are advantageous for bioimaging applications.41 We report here a systematic route towards a novel class of fluorene benzothiadiazole donor–acceptor π-conjugated co-oligomers with red emitting properties and a high quantum yield. Co-assemblies were prepared with these co-oligomers that showed efficient energy transfer between different monomeric building blocks. We envision that these results elucidate the interplay between molecular design and physicochemical properties of fluorene co-oligomers and present a straightforward method to prepare red-emitting nanoparticles in water with a high quantum yield.
Results and discussion
Molecular design
An oligo-fluorene benzothiadiazole co-oligomer (OF) approach38 was taken as a molecular scaffold for the design of novel π-conjugated donor–acceptor co-oligomers. Two bridging vinyl moieties were installed through a sequential Stille and Heck coupling between the donor and acceptor fragments, respectively, to allow for planarization and hence conjugation by an in-plane twist of the entire π-conjugated backbone (Scheme 1). Moreover, different benzothiadiazole (B) acceptor fragments were introduced to systematically study the influence of an increased hydrophobicity and enlarged electronic nature of the fluorene co-oligomer backbone on their physicochemical properties. An additional di-benzothiadiazole (dB) moiety was used to enlarge the acceptor capacity that may allow for a higher quantum yield. A naphthobisthiadiazole (NB) acceptor was also proposed as it has a much greater electron-accepting character which may intensify the donor–acceptor conjugation, that would consequently reduce the band gap of these quadrupolar chromophores and would thus result in longer emission wavelengths.42 The effect of polarity on the optical characteristics, through for instance solvation, as well as on the self-assembly behavior was probed by the installation of flanking side-chain with altered size and hydrophobicity (a–d). Side-chain substitution of fluorene co-oligomers with a similar backbone were prepared in a one-pot fashion in which straightforward purification of the statistical distribution yielded the symmetrical and unsymmetrical derivatives in satisfactory yields. A detailed description of the synthesis of OFs 11–22 can be found in the ESI.†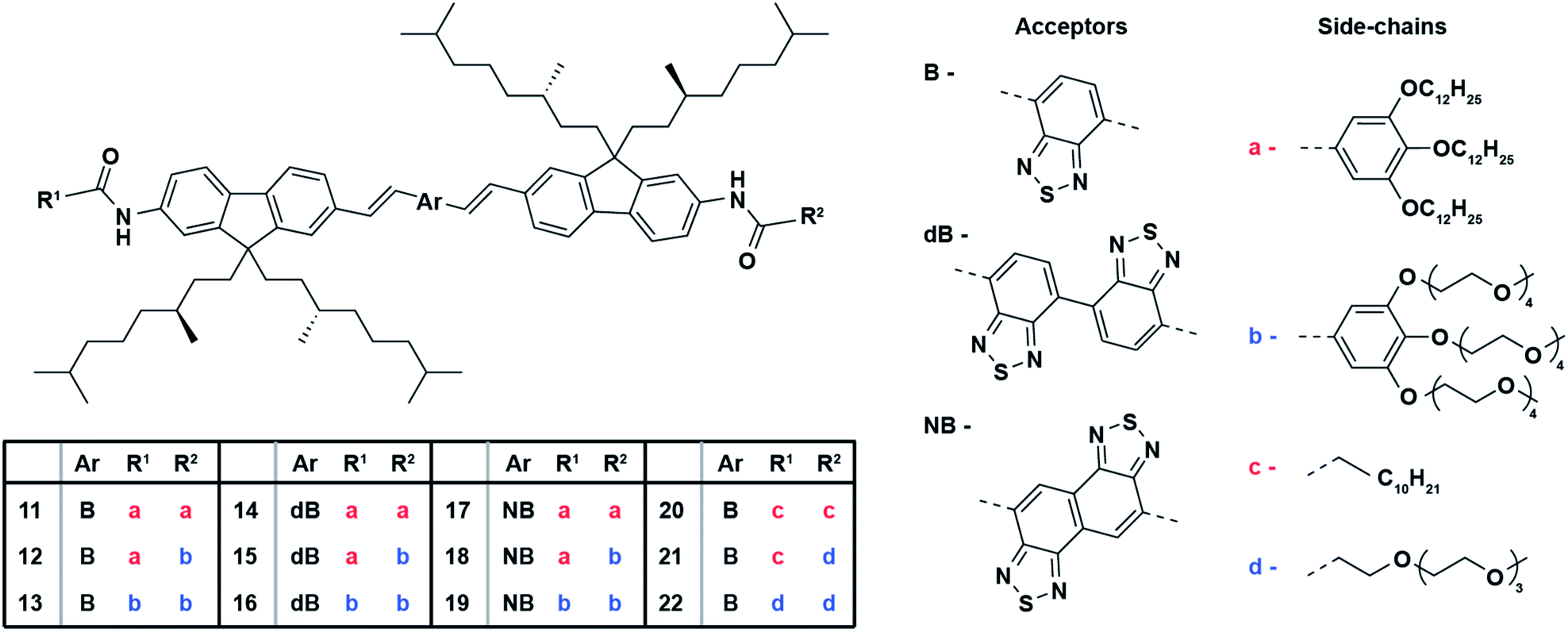 | ||
| Scheme 1 The molecular design of fluorene benzothiadiazole co-oligomers in which the central acceptor element and the side-chains are systematically altered. The color of the side chain refers to polarity (red = apolar; blue = polar). | ||
Optical characteristics and self-assembly
The optical properties of the fluorene benzothiadiazole co-oligomers were studied in their molecularly dissolved state in anhydrous tetrahydrofuran (THF) in order to evaluate the influence of different aromatic core moieties and varying side-chains. Nano-sized unimolecular architectures in water were prepared by means of reprecipitation; rapid injection of a stock solution of each fluorene benzothiadiazole co-oligomer in organic solvent into filter sterilized demi-water followed by manual stirring yielded nanostructures. These nano-sized architectures, stable in terms of size and optical properties over at least several days, were characterized to study the influence of molecular design on their self-assembly behavior and their photophysical properties. The exact values of their maximum absorption and emission wavelengths, including the extinction coefficient, quantum yield and hydrodynamic radius are tabulated for comparison in Table 1 (see also Fig. S1†). In organic solvent, benzothiadiazole acceptor bearing OFs 11–13 show two absorption bands around 360 nm and 490 nm (Fig. 1A). The first band likely results from the π–π transition, whereas the latter can be attributed to the charge-transfer transition that arises from the donor–acceptor system. A strong polarity dependency of the extinction coefficients at higher absorption maxima can be observed. Concomitant excitation provides fluorescence spectra that show their maximum emission wavelength around 595 nm with an apparent diminished peak intensity yet without affecting the fluorescence quantum yield. The unimolecular nano-sized architectures of OFs 11–13 in water displayed only minor changes in absorption as compared to their molecularly dissolved state (Fig. 1B). However, a clear hypsochromic shift of especially the second absorption maxima can be observed. It should be noted that this blue-shift is different for each of the three co-oligomers, reflecting the role of the side-chains on the aggregation behavior. Hydrophobic side-chains seemingly induce a larger shift of the absorption wavelength, suggesting that these nanostructures may be more tightly packed into H-type aggregates.43 The fluorescence spectra show larger differences, including a clear bathochromic shift and a diminished intensity of the emission maxima for the most polar derivative OF 13. The shift from 590 nm for the most nonpolar derivative OF 11 to 612 nm for OF 13 is most likely due to additional water that is encapsulated by nano-architectures of the latter. It was shown for fluorene–benzothiadiazole copolymers that their emission is dependent on the polarity of the environment,44 which is in this case clearly dictated by the polarity of the side-chains, similar to the observations here. Satisfactory fluorescence quantum yields (over 10%) are observed for these nanostructures which are decreasing with increasing polarity which could be explained by water encapsulation, similar as mentioned before. These numbers are remarkably high as compared to the corresponding fluorene-based polymers44 and also gratifying as compared to other red-emitting fluorene co-oligomers.35 Dynamic light scattering (DLS) measurements showed a strong scattering correlation for OFs 11–13. These correlations could be best fitted with a first order exponential decay, revealing a single population of architectures. The concomitant scattering intensity distributions revealed hydrodynamic radii of around 80 nm (Fig. 1C and S2†) and thus these OFs clearly formed self-assembled nanostructures in water. The hydrodynamic radius of the OFs increases with side-chain hydrophobicity, which can intuitively be explained by a reduced solubility. The sizes of the nanostructures were also confirmed by transmission electron microscopy (TEM). Fig. 1D and S3† show the TEM images of OFs 11–13 that reveal nanostructure sizes that are slightly smaller than the observed sizes in DLS, that is, the difference between the actual and hydrodynamic size. TEM analysis also revealed regular spherical particles with a fairly monodisperse distribution and an amorphous internal morphology.| OF | UV-Vis absorption λ/nm (ε/103 M−1 cm−1) | Fluorescencea λmax/nm (φPL/%) | Stokes shift/nm (cm−1) | Radius Rh/nm | |||
|---|---|---|---|---|---|---|---|
| THF | H2O | THF | H2O | THF | H2O | H2O | |
| a N,N′-Bis(pentylhexyl)perylene-3,4:9,10-tetracarboxylic diimide (φ = 0.99) in methylene chloride was used as a reference. For full spectra see Fig. S1. | |||||||
| 11 | 363 (59.4); 492 (35.1) | 360 (37.6); 468 (16.9) | 594 (78) | 590 (23) | 102 (3490) | 122 (4418) | 92 |
| 12 | 342 (59.8); 479 (20.2) | 338 (43.4); 459 (13.2) | 594 (75) | 590 (17) | 116 (4070) | 131 (4837) | 86 |
| 13 | 373 (60.1); 494 (41.8) | 360 (43.6); 481 (22.4) | 596 (80) | 612 (10) | 102 (3464) | 131 (4450) | 67 |
| 14 | 356 (36.8); 488 (22.1) | 330 (29.9); 468 (13.5) | 600 (81) | 590 (59) | 109 (3741) | 122 (4418) | 140 |
| 15 | 356 (36.1); 489 (21.9) | 350 (23.4); 477 (10.5) | 601 (78) | 597 (64) | 109 (3727) | 120 (4214) | 70 |
| 16 | 356 (35.4); 488 (21.1) | 345 (25.3); 472 (10.9) | 600 (77) | 607 (51) | 111 (3797) | 135 (4712) | 85 |
| 17 | 354 (58.4); 527 (30.2) | 357 (29.1); 507 (13.9) | 617 (79) | 610 (48) | 73 (2309) | 103 (3330) | 134 |
| 18 | 355 (52.8); 527 (29.4) | 355 (52.7); 511 (18.0) | 618 (80) | 627 (38) | 74 (2336) | 116 (3621) | 125 |
| 19 | 355 (55.1); 526 (27.7) | 355 (47.3); 515 (16.4) | 618 (76) | 630 (54) | 76 (2400) | 115 (3544) | 104 |
| 20 | 355 (52.0); 488 (27.4) | 356 (43.5); 481 (21.2) | 596 (82) | 612 (29) | 115 (3908) | 131 (4450) | 80 |
| 21 | 356 (44.4); 487 (25.8) | 351 (40.6); 461 (18.8) | 596 (78) | 602 (30) | 117 (3978) | 141 (5081) | 82 |
| 22 | 357 (41.9); 486 (25.1) | 347 (37.9); 459 (16.5) | 596 (72) | 608 (27) | 119 (4047) | 149 (5339) | 75 |
 | ||
| Fig. 1 Optical properties of benzothiadiazole bearing fluorene co-oligomers. UV-Vis absorption spectra (dashed lines) and corresponding fluorescence spectra (solid lines) of OFs 11–13 in (A) THF and (B) water (c = 1.5 × 10−5 M, λexc = 480 nm). (C) DLS data indicating diameters of nanoparticles formed in water and (D) a representative TEM images of OF 11 in water (c = 1.5 × 10−5 M, scale bar: 0.5 μm, insets: magnified TEM image of the same sample, scale bar: 100 nm). | ||
OFs 14–16 have an additional benzothiadiazole moiety installed at the core of the π-conjugated backbone. In organic solution, these fluorene co-oligomers showed similar optical characteristics as compared to OFs 11–13, despite the extended π-conjugated system. However, the side-chains of this class of oligomers seem to have less influence on the optical properties as absorption and fluorescence spectra completely overlap in organic solvent (Fig. S1†). The side-chain polarity has contrarily a large influence on the maximum emission wavelength in aqueous solution, similar to OFs 11–13 (Fig. 1B). A difference of approximately 20 nm can be observed comparing the most apolar OF 14 to the most polar OF 16, with the highest wavelength for the latter. The fluorescence quantum yield of this class of oligomers is remarkably high and superior to OFs 11–13. The enhanced quantum yield may be explained by an enhanced acceptor capacity through the extension of the acceptor fragment by an additional benzothiadiazole moiety. Interesting to note is that the hydrodynamic radii of the self-assembled structures from these oligomers is largely different with double the size of the apolar nanoparticles as compared to the polar ones (Fig. S2†). These size differences were less strongly observed in TEM (Fig. S3†).
Naphthobisthiadiazole was introduced as acceptor fragment bridging the two donor fluorene moieties in OFs 17–19. These oligomers show a bathochromically shifted second absorption band as compared to OFs 11–16, most likely due to the greater electron-accepting character of naphthobisthiadiazole and thus the enhanced charge-transfer transition. In organic solution, the optical properties of OFs 17–19 showed to be independent of side-chain polarity, similar to the co-oligomers discussed before (Fig. S1†). In contrast, the optical characteristics in water showed to be highly dependent on side-chain polarity for these co-oligomers. The extinction coefficient was found to be significantly lower for OF 17 as compared to the more polar derivatives. Moreover, the maximum emission wavelength shifted 20 nm depending on the side-chain polarity showing the highest emission maxima for the most polar derivative, while retaining satisfactory fluorescence quantum yields that approach 50%. Fig. S2† shows the size distribution of these core-enlarged oligomers measured with DLS. Although the hydrodynamic radii of the self-assembled structures seem to be rather similar, a slight polarity dependency can be observed. The largest size was observed for the most apolar derivative and the smallest for the most polar. Interestingly, structures of a broader size-range were found in DLS for derivative OF 18. TEM images showed, however, similar architectures for all three derivatives (Fig. S3†). The morphological difference of OF 18, as indicated by the shoulder in its DLS trace, is thus most likely in size rather than in shape.
While the aforementioned fluorene co-oligomers all bear large flanking gallic acid wedges, single chain side-chains were also introduced to reduce the bulkiness of the oligomers. Benzothiadiazole bearing OFs 20–22 only show minor differences in their optical properties compared to OFs 11–13. The side-chain polarity does not seem to influence the optical properties of these co-oligomers in both organic and aqueous solution (Fig. S1†). Interestingly, the spectra of the molecularly dissolved and self-assembled state are remarkably similar, which is in contrast to the observations made for the fluorene co-oligomers featuring the gallic acid bearing side-chains and highlight the role of side-chain size on the physicochemical characteristics. The fluorescence quantum yield of these nanostructures is also slightly higher, possibly due to less encapsulated water molecules as a result of the lower side-chain component. DLS analysis revealed self-assembled architectures with hydrodynamic radii of around 80 nm for all three OFs (Fig. S2†), which was confirmed by TEM analysis (Fig. S3†).
All the fluorene co-oligomers studied here formed stable self-assembled fluorescent nanostructures in water. The physical properties of the fluorene co-oligomers bearing the large gallic acid wedges (OFs 11–19) can be modulated by the nature of the side-chains. Nanostructures which are self-assembled from fluorene co-oligomers with polar side chains were generally found to be smaller in size accompanied by a lower fluorescence quantum yield as their respective apolar derivatives. In contrast, OFs 20–22, which are equipped with smaller single chain side-chains, are less prone to changes in their physical properties upon varying side-chain polarity, while bearing similar morphological properties. These side-chains may, intuitively, be too small to (i) accommodate large amounts of water that can influence the optical properties of the aromatic core – which was shown to be sensitive to polarity changes – and (ii) to alter the self-assembly characteristics which mainly are dominated by the aromatic core and apolar fluorene side chains.
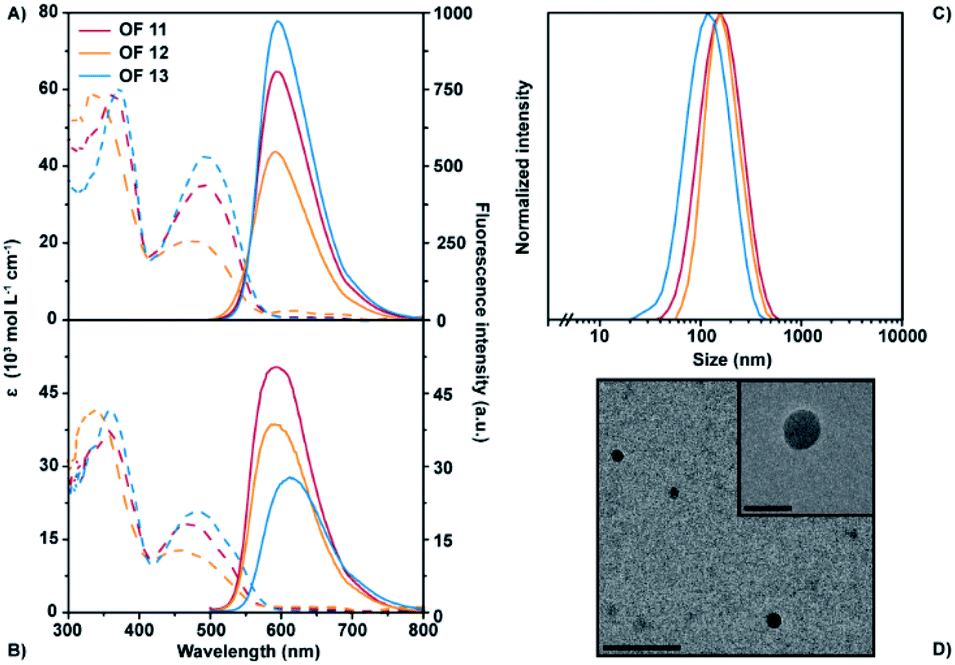
The aromatic backbone of OFs 11–13 are by design most analogous to previously reported38 fluorene benzothiadiazole co-oligomers 23 (Fig. 2A) and by comparison solely modified by the introduction of two vinyl moieties between the donor and acceptor fragments. This modification introduces multiple degrees of rotational freedom through reduction of steric hindrance, which in turn allows for planarization of the entire aromatic backbone. The extended π-conjugated system hence results in an overall bathochromic shift of the optical spectra (Fig. 2B). The absorption maxima of OF 12 are bathochromically shifted as compared to OF 23, even though the extinction coefficient seems to be slightly lower. More importantly, the maximum emission wavelength has shifted with over 40 nm from 551 nm to 594 nm while comparable quantum yields were obtained. A superior enhancement of the fluorescence quantum yield was obtained by the installation of an additional benzothiadiazole acceptor moiety. The quantum yields of OFs 14–16 exceeded values of 50%, which are the highest found to date for red-emitting fluorene co-oligomers. An additional bathochromic shift of the optical spectra was obtained by replacement of the acceptor fragment by a naphthobisthiadiazole moiety resulting in OFs 17–19. The absorption and emission maxima shifted by an additional 20–40 nm, depending on the side-chain substitution topology, while retaining satisfactory quantum yields approaching 50%.
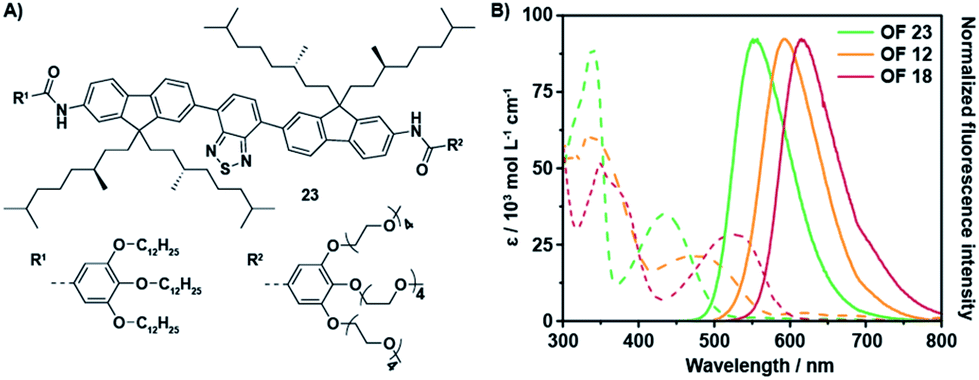 | ||
| Fig. 2 Comparison of the optical properties between fluorene co-oligomers with and without the bridging vinyl moieties. (A) Molecular structure of OF 23.38 (B) UV-Vis absorption spectra (dashed lines) and corresponding fluorescence spectra (solid lines) of OFs 12, 18 and 23 in THF (c = 1.5 × 10−5 M, λexc = 480, 510 and 430 nm, resp.). | ||
The ensemble of backbone alterations, including the introduction of the naphthobisthiadiazole core and the two bridging vinyl moieties, resulted in a maximum emission wavelength of 630 nm (OF 18). That is a total bathochromic shift of 80 nm as compared to a previously reported fluorene co-oligomer, such as e.g. OF 23 (Fig. 2B).38 These classes of fluorene co-oligomers also span the entire 80 nm emission range, which may allow for tuning of the optical characteristics of the final material upon desire. Moreover, a significant enhancement in fluorescence quantum yields is obtained as compared to their corresponding polymers44 and other red-emitting fluorene co-oligomers.33,35
Co-assemblies
The absorption spectrum of OF 18 partially overlaps with the emission spectrum of OF 23, which would potentially allow for FRET from the donor (OF 23) to the acceptor (OF 18) co-oligomer. Co-assemblies of these two co-oligomers were prepared by straightforward mixing both stock solutions in THF, in which the donor concentration was retained constant over all samples, prior to reprecipitation in water. Stable fluorescent nanoparticles were obtained after annealing at 90 °C for 5 minutes, to ensure full mixing of the co-oligomers. The fluorescence spectra of these co-assemblies are shown in Fig. 3A in which the ratio of these two derivatives were systematically varied. FRET occurred for all the mixtures, which is shown by the reduction in donor intensity and enhanced emission of the acceptor upon introduction of the latter. The observed general enhancement of acceptor emission and the rapid quenching of the donor emission, even at low acceptor concentrations, shows intra-nanoparticle energy transfer. A representative non-linear evolution of the fluorescence intensity of the mixed structures with varied ratios can be clearly observed in the normalized spectrum, with the highest donor intensity for the highest incorporation ratio (Fig. 3B). Together with the observed gradual bathochromic shift with increasing incorporation ratios of OF 18, this suggests the formation of a blend at low incorporation ratios and of homogeneous domains at high incorporation ratios.45 This phase separation phenomenon is referred to as self-sorting and was previously observed for fluorene co-oligomer mixtures.37 This is most likely caused by the different torsion angles between the fluorene moieties and the central benzothiadiazole, via the introduction of the vinyl moieties, resulting in a preferred self-sorting behavior rather than perfect mixing.38 These two-component nanoparticles exert energy transfer at low incorporation ratios due to the high quantum yield of the acceptor co-oligomers as compared to other multi-component systems including red-emitting fluorene co-oligomers.35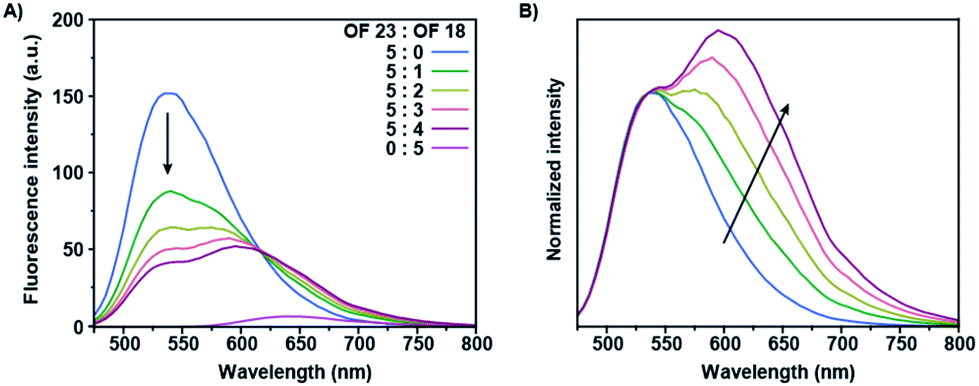 | ||
| Fig. 3 Intra-nanoparticle energy transfer in dual-colour co-assemblies. (A) Fluorescence spectrum of co-assemblies of OF 18 and 23 in water at different acceptor incorporation ratios (c = 1.5 × 10−5 M for OF 23 in all the samples and proportionally for OF 18, λexc = 430 nm). (B) Normalized fluorescence spectrum to guide the eye on the evolution of the FRET-ratio at different incorporation ratios. The arrows indicate the progression of the fluorescence spectra upon higher incorporation ratios. | ||
Conclusions
In summary, we have shown the facile synthesis of a new series of red-emitting fluorene benzothiadiazole co-oligomers, and their nanoparticle assembly in water, driven by the lipophilicity of the aromatic core and specifically incorporated hydrophobic side chains, with large stokes shifts >100 nm and high quantum yields. Planarization of the π-conjugated backbone was induced by the introduction of two vinyl moieties bridging the fluorene moieties and the central benzothiadiazole core providing donor–acceptor conjugation. Together with the substitution of the single benzothiadiazole acceptor fragment for an extended naphthobisthiadiazole with a greater electron-accepting character, a total bathochromic shift of the emission maxima of 80 nm was realized as compared to previously reported fluorene co-oligomers. Moreover, a significant increase in fluorescence quantum yield of the nanoparticles in water, as compared to the corresponding polymers and other red-emitting fluorene co-oligomers, was obtained with values around 50%. The side-chain polarity was shown to render the optical properties and is thus an important parameter to identify the preferred molecular structure per application. In general, apolar co-oligomers showed slightly blue-shifted emission maxima and an enhanced fluorescence quantum yield as compared to their corresponding polar co-oligomers. The straightforward introduction of smaller side-chains revealed a reduced influence on the physicochemical properties.Co-assemblies of a previously reported fluorene co-oligomer and a core-extended fluorene co-oligomer as synthesized here showed efficient intra-nanoparticle energy transfer, especially due to the enhanced fluorescence quantum yield of the latter. The FRET efficiency could even be optimized by mixing the most bathochromically shifted co-oligomer, which shows significant overlap of the optical spectra and an even higher quantum yield. Moreover, the ensemble of new fluorene co-oligomers contains derivatives that display emission maxima over an optical range between 590 and 630 nm, which may allow for tuning of the fluorescence output of multi-component co-assemblies. The evolution of FRET-ratio depending on acceptor incorporation ratios may thus allow these co-assemblies to be further developed into an optically responsive system. We consider these novel fluorene-based derivatives as a promising class of donor–acceptor π-conjugated co-oligomers for the preparation of multicomponent nanomaterials with tailored physicochemical properties.
Experimental section
General remarks
The general remarks as well as the synthesis details can be found in the ESI.†Particle formation
Nano-sized architectures were prepared from unimolecular building blocks by means of reprecipitation. This includes rapid injection of 15 μL of a 1 × 10−3 M OF stock solution dissolved in anhydrous tetrahydrofuran (≥99.9%) into 1 mL filter sterilized demineralized water followed by manual stirring, yielding a 1.5 × 10−5 M nanoparticle solution.Optical measurements
UV-Vis spectra were measured on a Jasco V-650 spectrophotometer equipped with a PerkinElmer PTP-1 Peltier temperature control system. The spectra were measured in quartz cuvettes and extinction coefficients were calculated from Lambert–Beer's law. Fluorescence spectra were recorded on a Varian Cary Eclipse fluorescence spectrophotometer. Fluorescence quantum yields (φ) were calculated from the integrated intensity under the emission band (I) using eqn (1), where OD is the optical density of the solution at the excitation wavelength and n is the refractive index. N,N′-Bis(pentylhexyl)perylene-3,4:9,10-tetracarboxylic acid bisimide (φ = 0.99) in methylene chloride was used as a reference.
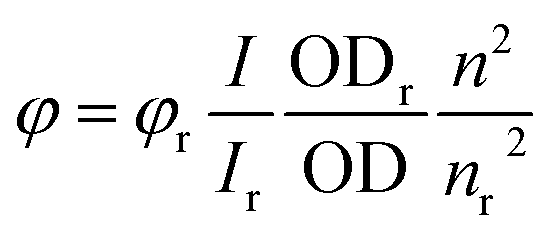 | (1) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by funding from HTSM through STW grant 12859-FluNanoPart and the Netherlands Organization for Scientific Research (NWO) via Gravity program 024.001.035 and VICI grant 016.150.366.Notes and references
- C. T. Matea, T. Mocan, F. Tabaran, T. Pop, O. Mosteanu, C. Puia, C. Iancu and L. Mocan, Int. J. Nanomed., 2017, 12, 5421–5431 CrossRef CAS PubMed.
- J.-H. Lee, H.-Y. Cho, H. K. Choi, J.-Y. Lee and J.-W. Choi, Int. J. Mol. Sci., 2018, 19, 2021 CrossRef CAS PubMed.
- X. Wu, C. Hao, J. Kumar, H. Kuang, N. A. Kotov, L. M. Liz-Marzán and C. Xu, Chem. Soc. Rev., 2018, 47, 4677–4696 RSC.
- A. B. Chinen, C. M. Guan, J. R. Ferrer, S. N. Barnaby, T. J. Merkel and C. A. Mirkin, Chem. Rev., 2015, 115, 10530–10574 CrossRef CAS PubMed.
- Y. Chen, Y. Xianyu and X. Jiang, Acc. Chem. Res., 2017, 50, 310–319 CrossRef CAS PubMed.
- S. K. Nune, P. Gunda, P. K. Thallapally, Y.-Y. Lin, M. L. Forrest and C. J. Berkland, Expert Opin. Drug Delivery, 2009, 6, 1175–1194 CrossRef CAS PubMed.
- L. K. Bogart, G. Pourroy, C. J. Murphy, V. Puntes, T. Pellegrino, D. Rosenblum, D. Peer and R. Lévy, ACS Nano, 2014, 8, 3107–3122 CrossRef CAS PubMed.
- J. Estelrich, M. J. Sánchez-Martín and M. A. Busquets, Int. J. Nanomed., 2015, 10, 1727–1741 CAS.
- P. Padmanabhan, A. Kumar, S. Kumar, R. K. Chaudhary and B. Gulyás, Acta Biomater., 2016, 41, 1–16 CrossRef CAS PubMed.
- O. Tagit and N. Hildebrandt, ACS Sens., 2017, 2, 31–45 CrossRef CAS PubMed.
- Z. Farka, T. Juřík, D. Kovář, L. Trnková and P. Skládal, Chem. Rev., 2017, 117, 9973–10042 CrossRef CAS PubMed.
- A. Kaeser and A. P. H. J. Schenning, Adv. Mater., 2010, 22, 2985–2997 CrossRef CAS PubMed.
- D. Görl, X. Zhang and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6328–6348 CrossRef PubMed.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- I. Fischer and A. P. H. J. Schenning, in Organic Electronics, ed. F. Cicoira and C. Santato, Wiley-VCH Verlag GmbH & Co. KGaA, 1st edn, 2013, pp. 1–25 Search PubMed.
- Y. Braeken, S. Cheruku, A. Ethirajan and W. Maes, Materials, 2017, 10, 1420 CrossRef PubMed.
- I. Fischer, A. Kaeser, M. A. M. Peters-Gumbs and A. P. H. J. Schenning, Chem.–Eur. J., 2013, 19, 10928–10934 CrossRef CAS PubMed.
- A. Kaeser and A. P. H. J. Schenning, Adv. Mater., 2010, 22, 2985–2997 CrossRef CAS PubMed.
- X. Xu, R. Liu and L. Li, Chem. Commun., 2015, 51, 16733–16749 RSC.
- R. Abbel, A. P. H. J. Schenning and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4215–4233 CrossRef CAS.
- Y. Wang and T. Michinobu, J. Mater. Chem. C, 2016, 4, 6200–6214 RSC.
- K. Yamamoto, Y. Ie, M. Nitani, N. Tohnai, F. Kakiuchi, K. Zhang, W. Pisula, K. Asadi, P. W. M. Blom and Y. Aso, J. Mater. Chem. C, 2018, 6, 7493–7500 RSC.
- S. Yao, H.-Y. Ahn, X. Wang, J. Fu, E. W. Van Stryland, D. J. Hagan and K. D. Belfield, J. Org. Chem., 2010, 75, 3965–3974 CrossRef CAS PubMed.
- J. Sun, X. Li, K. Du and F. Feng, Chem. Commun., 2018, 54, 9194–9197 RSC.
- W. Sheng, S. T. Nick, E. M. Santos, X. Ding, J. Zhang, C. Vasileiou, J. H. Geiger and B. Borhan, Angew. Chem., Int. Ed., 2018, 57, 16083–16087 CrossRef CAS PubMed.
- R. Abbel, R. van der Weegen, W. Pisula, M. Surin, P. Leclère, R. Lazzaroni, E. W. Meijer and A. P. H. J. Schenning, Chem.–Eur. J., 2009, 15, 9737–9746 CrossRef CAS PubMed.
- B. A. D. Neto, A. A. M. Lapis, E. N. da Silva Júnior and J. Dupont, Eur. J. Org. Chem., 2013, 2013, 228–255 CrossRef CAS.
- Y. Koizumi, S. Seki, S. Tsukuda, S. Sakamoto and S. Tagawa, J. Am. Chem. Soc., 2006, 128, 9036–9037 CrossRef CAS PubMed.
- L. Zhu, C. Yang and J. Qin, Chem. Commun., 2008, 6303–6305 RSC.
- B. Balan, C. Vijayakumar, S. Ogi and M. Takeuchi, J. Mater. Chem., 2012, 22, 11224–11234 RSC.
- F. Tang, C. Wang, J. Wang, X. Wang and L. Li, ACS Appl. Mater. Interfaces, 2014, 6, 18337–18343 CrossRef CAS PubMed.
- A. H. A. M. van Onzen, L. Albertazzi, A. P. H. J. Schenning, L.-G. Milroy and L. Brunsveld, Chem. Commun., 2017, 53, 1626–1629 RSC.
- J. Pennakalathil, E. Jahja, E. S. Özdemir, Ö. Konu and D. Tuncel, Biomacromolecules, 2014, 15, 3366–3374 CrossRef CAS PubMed.
- X. Wang, F. He, L. Li, H. Wang, R. Yan and L. Li, ACS Appl. Mater. Interfaces, 2013, 5, 5700–5708 CrossRef CAS PubMed.
- R. Abbel, R. van der Weegen, E. W. Meijer and A. P. H. J. Schenning, Chem. Commun., 2009, 1697–1699 RSC.
- Y. Xiao, R. Zhang, H. Gao, H. Zhao and X. Cheng, J. Mater. Chem. C, 2019, 7, 1237–1245 RSC.
- A. L. Stevens, A. Kaeser, A. P. H. J. Schenning and L. M. Herz, ACS Nano, 2012, 6, 4777–4787 CrossRef CAS PubMed.
- A. Kaeser, I. Fischer, R. Abbel, P. Besenius, D. Dasgupta, M. A. J. Gillisen, G. Portale, A. L. Stevens, L. M. Herz and A. P. H. J. Schenning, ACS Nano, 2013, 7, 408–416 CrossRef CAS PubMed.
- K. Petkau, A. Kaeser, I. Fischer, L. Brunsveld and A. P. H. J. Schenning, J. Am. Chem. Soc., 2011, 133, 17063–17071 CrossRef CAS PubMed.
- T. Yang, Y. Zuo, Y. Zhang, Z. Gou, X. Wang and W. Lin, J. Mater. Chem. B, 2019, 7, 4649–4654 RSC.
- B. A. D. Neto, P. H. P. R. Carvalho and J. R. Correa, Acc. Chem. Res., 2015, 48, 1560–1569 CrossRef CAS.
- S. Griesbeck, E. Michail, C. Wang, H. Ogasawara, S. Lorenzen, L. Gerstner, T. Zang, J. Nitsch, Y. Sato, R. Bertermann, M. Taki, C. Lambert, S. Yamaguchi and T. B. Marder, Chem. Sci., 2019, 10, 5405–5422 RSC.
- Y. Deng, W. Yuan, Z. Jia and G. Liu, J. Phys. Chem. B, 2014, 118, 14536–14545 CrossRef CAS.
- K.-Y. Pu, L. Cai and B. Liu, Macromolecules, 2009, 42, 5933–5940 CrossRef CAS.
- C. F. Huebner, R. D. Roeder and S. H. Foulger, Adv. Funct. Mater., 2009, 19, 3604–3609 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures; supporting Fig. S1–S3. See DOI: 10.1039/c9ra09015k |
| This journal is © The Royal Society of Chemistry 2020 |