
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeven new metabolites of drostanolone heptanoate by using Beauveria bassiana, and Macrophomina phaseolina cell suspension cultures†
Zahid Hussaina,
Atia-tul-Wahab *b,
Nusrat Hussainac,
Shabbir Hussainad,
Atta-ur-Rahmana and
M. Iqbal Choudhary*abe
*b,
Nusrat Hussainac,
Shabbir Hussainad,
Atta-ur-Rahmana and
M. Iqbal Choudhary*abe
aH. E. J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi-75270, Pakistan. E-mail: iqbal.choudhary@iccs.edu; Fax: +922134824924-5; Tel: +922134824924-5
bDr. Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences, University of Karachi, Karachi-75270, Pakistan
cDepartment of Chemistry, University of Baltistan Skardu, 16100, Pakistan
dDepartment of Chemistry, Karakoram International University, Gilgit-15100, Gilgit-Baltistan, Pakistan
eDepartment of Biochemistry, Faculty of Science, King Abdulaziz University, Jeddah-21412, Saudi Arabia
First published on 2nd January 2020
Abstract
The present study reports the biotransformation of an anabolic-androgenic steroid (AAS) drostanolone heptanoate (1) by using two microbial cultures, Beauveria bassiana, and Macrophomina phaseolina. Fermentation of 1 with B. bassiana yielded five new transformed products 2–6, while with M. phaseolina it afforded two new 7–8, and two known 9–10 metabolites. The main sites of hydroxylation in the steroidal skeleton of 1 were at C-5, C-7, C-11, C-14, C-15, and C-20, hydrolysis of the ester moiety at C-17, and reduction of the carbonyl group at C-3. The structures of the transformed products were determined by using mass, NMR, and other spectroscopic techniques.
Introduction
Steroids constitute an important class of biologically active compounds due to their anabolic, anti-inflammatory, anti-microbial, anti-leishmanial, and anti-cancer properties. Therefore, there have been sustained efforts for the synthesis of new, and derivatizations of existing steroidal drugs with desired pharmacodynamic profiles. Currently a bio-catalytic approach has largely been employed for the structural modifications of steroids, particularly due to the ability of microorganisms to catalyze functionalization at almost every site of the inert steroidal skeleton.1–3 Moreover, microorganisms can grow, and replicate quickly, and produce a variety of enzymes under ambient conditions. Biotransformations have been successfully employed where chemical methods were found to be difficult and expensive. In addition, the products obtained are highly regio-, enantio-, and stereo-selective, and the procedures are eco-friendly, and economical.4,5Drostanolone heptanoate (1) (C27H44O3) is a dihydrotestosterone (DHT)-derived anabolic androgenic steroid (AAS). Athletes, and body builders use it to increase muscles strength, and body mass without gaining fat.6 However, due to its adverse effects on liver, serum lipid, and reproductive system, the use of drostanolone heptanoate (1), and other anabolic androgenic steroids have now been discontinued by the International Olympic Committee (IOC).7 In contrary, many research groups have studied the treatment of estrogen-dependent breast carcinoma using 1 as an aromatase inhibitor.8–11
Several bio-transformed products of drostanolone from the urine of drostanolone-dosed rabbits have been reported in literature, including hydroxylations at C-15, C-16, and C-17, reduction at C-3, and oxidation at C-17.12 Similarly, several metabolites of drostanolone on incubation with cryopreserved human hepatocytes are also reported.13
In continuation of our structural transformation studies on steroidal drugs,14–20 biotransformation of drostanolone heptanoate (1) was carried out. Based on small scale screening results, compound 1 was subjected to biotransformation by using two microbial cell cultures, Beauveria bassiana, and Macrophomina phaseolina, for the first time, yielding seven new, and two known compounds. The purpose of this study was to synthesize structurally diverse analogues of drostanolone heptanoate (1) for their potential use in the biomedical research, by employing mild, and low-cost biotransformation procedures.
Experimental
General
All the chemicals used for microbial transformation were purchased from the Sigma Aldrich (Germany), and Oxoid Limited (UK), whereas drostanolone heptanoate (1) was purchased from the Shanghai Bettersyn Biotech Co., Ltd., Shanghai (China). Silica gel pre-coated thin layer chromatography (TLC) plates (E. Merck, Germany), and silica gel column chromatography (Germany) were used for analysis, and fractionations (initial purification), while preparative recycling reverse phase HPLC-LC-908, fitted with JAIGEL-ODS-L-80 columns (Japan), was used for the final purification of transformed products. The FAB-, and HRFAB-MS (JEOL HX110) (Japan), and ESI-, and HRESI-MS (Bruker, Maxis II) (Germany) techniques were used to deduce the molecular formulae of all compounds. The 1H-, and the 13C-NMR spectra were recorded on Bruker Avance spectrometers (300–600 MHz) (Switzerland). The melting points were recorded using Stuart SMP-10 instrument (Switzerland). The JASCO P-200 polarimeter was used to measure the optical rotations, while the IR data were recorded using KBr disks in CHCl3 on the Bruker Vector 22 FT-IR spectrometer (Japan). Molibdophosphoric acid (MPA) was used as a staining reagent to detect the transformed products on TLCs.Microbial cultures, and media preparation
Beauveria bassiana (ATCC-7159) was purchased from the American Type Culture Collection, while Macrophomina phaseolina (KUCC-730) was obtained from the Karachi University Culture Collection. Both cultures were grown on Sabouraud dextrose agar slants, and maintained at 4 °C.The culture media for the growth of fungi was prepared by adding 50 g glucose, 25 g peptone, 25 g yeast extract, 25 g KH2PO4, 25 g NaCl, and 50 mL glycerol, in distilled water. The quantity, and recipe for both cultures were the same.
General fermentation, extraction, and purification protocol
The media for the growth of microorganisms was prepared by adding the specific amounts of media ingredients in distilled water. Media was then transferred to 100 mL Erlenmeyer flasks, cotton plugged, and autoclaved at 121 °C, followed by inoculation of spores of fungi from mycelia on SDA slants under sterilized conditions. Flasks were placed on a rotary shaker at 26 °C for the maximum growth of fungi. After suitable growth, substrate 1 was dissolved in acetone, and equally distributed in each fungus-containing flasks, and again placed on a rotary shaker at 26 °C with 125 rpm for the fermentation process. A positive control (only substrate in media), and a negative control (microbial culture in media) were prepared to identify the degradation of substrate in media, and fungal metabolites, respectively. The fermentation reaction was stopped by adding dichloromethane to each flask, followed by filtration of fungal biomass. The filtrate was then extracted with dichloromethane, dried by adding anhydrous sodium sulfate, filtered, and evaporated under high vacuum to afford a solid crude material. This crude material was then fractionated through silica gel column chromatography with gradient hexanes, and acetone elution. After TLC analysis, the fractions of similar polarities were combined together into ten main fractions. These combined fractions were then subjected to preparative recycling RP-HPLC, using an isocratic methanol and water solvent systems. A constant flow rate of 4 mL min−1 was maintained throughout the analysis to isolate pure metabolites 2–10.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); retention time (min): 17 (MeOH
O); retention time (min): 17 (MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O; 75:25); HRFAB-MS m/z 337.2356 ([M + H] +; calcd. 336.2301); FAB-MS m/z 337 [M + H]+; 1H-NMR (CD3OD, 300 MHz): Table 1; 13C-NMR (CD3OD, 100 MHz): Table 2.
:H2O; 75:25); HRFAB-MS m/z 337.2356 ([M + H] +; calcd. 336.2301); FAB-MS m/z 337 [M + H]+; 1H-NMR (CD3OD, 300 MHz): Table 1; 13C-NMR (CD3OD, 100 MHz): Table 2.
| Positions | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.08 dd (J = 12.6, 5.7); 1.05 m | 2.44 dd (J = 13.8, 5.8); 1.98 m | 2.10 m; 1.00 m | 2.91 dd (J = 13.8, 6.0); 1.32 m | 2.54 br. t (J = 13.8); 1.88 dd (J = 13.8, 3.0) | 2.44 br. t (J = 13.8); 1.16 m | 1.61 m; 1.04 m | 1.35 m; 1.00 br. t (J = 12.8) | 2.08 dd (J = 13.2, 6.0); 1.04 m | 1.34 m; 1.05 m |
| 2 | 2.55 m | 2.58 m | 2.54 m | 2.63 m | 2.58 m | 2.56 m | 1.42 overlap | 1.41 m | 2.55 m | 1.51 m |
| 3 | — | — | — | — | — | — | 3.67 m | 3.70 m | — | 3.69 m |
| 4 | 2.42 br. t (J = 13.8); 2.00 dd (J = 14.1, 3.3) | 2.93 d (J = 6.0); 1.19 m | 2.46 m; 2.08 m | 2.44 br. t (J = 14.1); 2.00 overlap | 2.82 dd (J = 3.8, 4.8); 1.25 m | 2.87 dd (J = 13.8, 5.7); 1.20 m | 1.50 overlap; 1.45 m | 1.51 m; 1.39 m | 2.87 br. t (J = 13.5); 1.99 m | 1.82 m; 1.54 m |
| 5 | 1.54 m | 1.60 m | 1.57 m | 1.57 m | 1.49 m | 1.55 m | — | 1.33 m | 0.91 m | 0.75 m |
| 6 | 1.75 m; 1.31 overlap | 1.56 m; 1.42 m | 1.63 m; 1.44 m | 1.40 m; 1.37 m | 1.57 overlap; 1.43 m | 1.37 m; 1.33 m | 1.21 m; 1.18 m | 1.41 m; 1.25 m | 1.33 m; 1.30 m | 1.61 m; 1.28 m |
| 7 | 1.58 m; 1.32 overlap | 3.24 m | 3.30 m | 1.55 m; 1.26 m | 3.77 m | 2.06 m; 1.13 m | 1.63 m; 1.53 m | 1.53 m; 1.36 m | 1.74 m; 0.88 m | 1.21 m; 1.16 m |
| 8 | 1.50 m | 1.66 m | 1.60 m | 1.52 m | 1.52 m | 1.67 m | 1.43 m | 1.71 m | 1.46 overlap | 1.42 m |
| 9 | 0.77 m | 0.88 br. t (J = 10.2) | 0.82 m | 0.86 br. t (J = 10.2) | 1.42 m | 0.86 br. t (J = 10.2) | 1.73 m | 1.48 m | 1.47 overlap | 1.66 m |
| 10 | — | — | — | — | — | — | — | — | — | — |
| 11 | 1.40 m; 1.33 overlap | 3.92 dt (J = 10.5, 5.1) | 1.67 m; 1.66 m | 3.93 dt (J = 10.8, 5.0) | 3.88 dt (J = 10.8, 5.1) | 3.87 m | 1.51 overlap; 1.41 overlap | 1.56 m; 1.32 m | 1.56 m; 1.40 m | 1.67 m; 0.89 m |
| 12 | 1.18 dt (J = 12.9, 3.9); 1.71 m | 2.08 dd (J = 12.0, 4.8); 1.10 m | 1.82 m; 1.10 m | 2.11 dd (J = 12.0, 4.8); 1.10 m | 2.06 overlap; 1.07 br. t (J = 11.7) | 2.07 overlap; 1.16 m | 1.57 m; 1.44 m | 1.43 m; 1.25 m | 1.85 m; 1.01 m | 1.42 m; 1.00 m |
| 13 | — | — | — | — | — | — | — | — | — | — |
| 14 | 1.08 m | 1.20 m | 1.03 m | 1.08 m | 1.40 m | 1.05 m | 1.55 m | — | 0.91 m | 0.95 m |
| 15 | 2.12 m; 1.47 m | 1.88 m; 1.67 m | 4.06 dt (J = 9.0, 4.8) | 1.73 m; 1.70 m | 1.68 m; 1.26 m | 3.90 m | 1.37 m; 1.33 m | 1.64 m; 1.51 m | 3.90 m | 1.57 m; 1.24 m |
| 16 | 1.52 m; 1.47 m | 1.95 m; 1.44 m | 2.04 m; 1.93 m | 2.02 overlap; 1.48 m | 2.01 m; 1.56 overlap | 2.05 overlap; 1.84 m | 1.98 m; 1.46 m | 2.14 m; 1.52 m | 1.53 m; 1.26 m | 1.94 m; 1.46 m |
| 17 | 4.58 t (J = 8.7) | 3.55 t (J = 8.4) | 3.74 t (J = 9.0) | 3.59 t (J = 8.7) | 3.60 t (J = 8.4) | 3.78 t (J = 9.0) | 3.61 t (J = 8.4) | 4.20 t (J = 7.2) | 3.55 t (J = 8.7) | 3.54 t (J = 8.4) |
| 18 | 0.83 s | 0.77 s | 0.78 s | 0.76 s | 0.75 s | 0.76 s | 0.72 s | 0.81 s | 0.74 s | 0.71 s |
| 19 | 1.12 s | 1.24 s | 1.14 s | 1.22 s | 1.20 s | 1.23 s | 0.94 s | 0.83 s | 1.11 s | 0.82 s |
| 20 | 0.96 d (J = 6.3) | 0.94 d (J = 6.6) | 0.97 d (J = 6.3) | 3.75 dd (J = 10.8, 5.1); 3.47 dd (J = 11.1, 5.7) | 0.93 d (J = 6.3) | 0.93 d (J = 6.3) | 0.91 d (J = 5.4) | 0.91 d (J = 6.8) | 0.94 d (J = 6.6) | 0.91 d (J = 6.9) |
| 21 | — | — | — | — | — | |||||
| 22 | 2.29 t (J = 7.2) | — | — | — | — | |||||
| 23 | 1.59 m; 1.32 overlap | — | — | — | — | |||||
| 24 | 1.38 overlap; 1.33 overlap | — | — | — | — | |||||
| 25 | 1.30 overlap; 1.29 overlap | — | — | — | — | |||||
| 26 | 1.34 overlap; 1.30 overlap | — | — | — | — | |||||
| 27 | 0.90 t (J = 6.9) | — | — | — | — |
| Carbons | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 49.8 | 45.6 | 49.3 | 45.7 | 45.6 | 46.1 | 35.1 | 42.3 | 49.9 | 42.2 |
| 2 | 42.1 | 42.2 | 42.0 | 49.9 | 42.8 | 42.3 | 31.3 | 33.1 | 42.1 | 40.0 |
| 3 | 215.4 | 215.5 | 215.0 | 214.3 | 215.5 | 215.9 | 71.3 | 71.4 | 215.6 | 71.4 |
| 4 | 45.5 | 51.2 | 45.0 | 46.3 | 51.4 | 51.7 | 38.0 | 37.8 | 45.6 | 38.1 |
| 5 | 36.3 | 47.4 | 46.6 | 50.2 | 42.8 | 50.5 | 77.0 | 48.8 | 55.2 | 56.1 |
| 6 | 32.4 | 40.2 | 37.8 | 30.3 | 38.0 | 30.4 | 29.3 | 29.2 | 29.8 | 21.5 |
| 7 | 26.2 | 75.0 | 74.8 | 24.2 | 67.4 | 33.0 | 27.5 | 27.0 | 32.5 | 29.4 |
| 8 | 49.4 | 43.3 | 44.8 | 36.1 | 40.2 | 35.9 | 33.2 | 40.2 | 36.6 | 36.6 |
| 9 | 55.2 | 59.1 | 58.5 | 60.9 | 52.9 | 61.0 | 49.6 | 39.7 | 49.6 | 33.1 |
| 10 | 37.3 | 38.8 | 45.8 | 39.0 | 44.1 | 39.3 | 42.4 | 37.6 | 37.7 | 37.4 |
| 11 | 29.8 | 69.6 | 22.1 | 69.5 | 69.7 | 69.2 | 24.1 | 20.6 | 22.2 | 32.9 |
| 12 | 38.2 | 49.4 | 38.2 | 49.0 | 48.5 | 49.6 | 33.4 | 30.0 | 38.0 | 37.8 |
| 13 | 43.9 | 44.9 | 58.4 | 44.4 | 49.6 | 45.6 | 44.1 | 49.0 | 44.1 | 44.1 |
| 14 | 51.9 | 51.1 | 52.1 | 51.3 | 45.9 | 58.5 | 44.8 | 84.8 | 52.2 | 52.4 |
| 15 | 28.5 | 27.2 | 73.1 | 32.4 | 23.4 | 72.5 | 26.2 | 32.7 | 72.5 | 24.3 |
| 16 | 23.5 | 31.1 | 40.5 | 30.7 | 30.6 | 43.0 | 30.6 | 30.1 | 24.3 | 30.6 |
| 17 | 84.0 | 81.8 | 79.5 | 81.9 | 82.0 | 79.2 | 82.3 | 79.6 | 79.2 | 82.5 |
| 18 | 12.6 | 12.6 | 13.3 | 12.5 | 12.3 | 13.9 | 10.8 | 15.7 | 11.6 | 11.7 |
| 19 | 14.2 | 12.9 | 12.7 | 12.9 | 11.9 | 13.0 | 14.7 | 12.3 | 12.6 | 12.6 |
| 20 | 14.9 | 15.0 | 14.9 | 62.5 | 15.0 | 15.0 | 19.2 | 19.0 | 15.0 | 19.0 |
| 21 | 175.5 | — | — | — | — | — | — | — | — | — |
| 22 | 35.3 | — | — | — | — | — | — | — | — | — |
| 23 | 26.2 | — | — | — | — | — | — | — | — | — |
| 24 | 29.8 | — | — | — | — | — | — | — | — | — |
| 25 | 32.6 | — | — | — | — | — | — | — | — | — |
| 26 | 23.5 | — | — | — | — | — | — | — | — | — |
| 27 | 14.3 | — | — | — | — | — | — | — | — | — |
O); retention time (min): 19 (MeOH:H2O; 75:25); HRFAB-MS m/z 337.2372 ([M + H]+; calcd. 336.2301); FAB-MS m/z 337 [M + H]+; 1H-NMR (CD3OD, 300 MHz): Table 1; 13C-NMR (CD3OD, 150 MHz): Table 2.O); retention time (min): 18 (MeOH:H2O; 70:30); HRFAB-MS m/z 337.2366 ([M + H]+; calcd. 336.2301); FAB-MS m/z 337 [M + H]+; 1H-NMR (CD3OD, 300 MHz): Table 1; 13C-NMR (CD3OD, 100 MHz): Table 2.O); retention time (min): 26 (MeOH:H2O; 70:30); HRFAB-MS m/z 337.2365 ([M + H]+; calcd. 336.2301); FAB-MS m/z 337 [M + H]+; 1H-NMR (CD3OD, 300 MHz): Table 1; 13C-NMR (CD3OD, 100 MHz): Table 2.O); retention time (min): 24 (MeOH:H2O; 70:30); HRFAB-MS m/z 337.2372 ([M + H]+; calcd. 336.2301); FAB-MS m/z 337 [M + H]+; 1H-NMR (CD3OD, 300 MHz): Table 1; 13C-NMR (CD3OD, 100 MHz): Table 2.:H2O; 75:25); HRESI-MS m/z 340.2851 ([M + NH4]+; calcd. 322.2508); ESI-MS m/z 340.3 ([M + NH4]+; 304 [M−H2O]+); 1H-NMR (CD3OD, 300 MHz): Table 1; 13C-NMR (CD3OD, 150 MHz): Table 2.:H2O; 75:25); HRESI-MS m/z 323.2586 ([M + H] +; calcd. 322.2508); ESI-MS m/z 323.3 [M + H]+; 1H-NMR (CD3OD, 400 MHz): Table 1; 13C-NMR (CD3OD, 100 MHz): Table 2.O); HRESI-MS m/z 305.2480 ([M + H]+; calcd. 304.2402); ESI-MS m/z 305.2 [M + H]+; 287.2 ([M + H]+–H2O); 1H-NMR (CD3OD, 300 MHz): Table 1; 13C-NMR (CD3OD, 100 MHz): Table 2.Results and discussion
Biotransformation of drostanolone heptanoate (1) with cell suspension cultures of B. bassiana, and M. phaseolina produced nine transformed products 2–10. B. bassiana yielded five new metabolites 2–6 (Scheme 1), whereas M. phaseolina afforded two new 7 and 8, and two known 9 and 10 metabolites (Scheme 2). The structures of new compounds were deduced by using different spectroscopic techniques, and by comparing their spectral data with substrate 1. The structures of known metabolites were also determined by comparing their spectroscopic data with the reported data.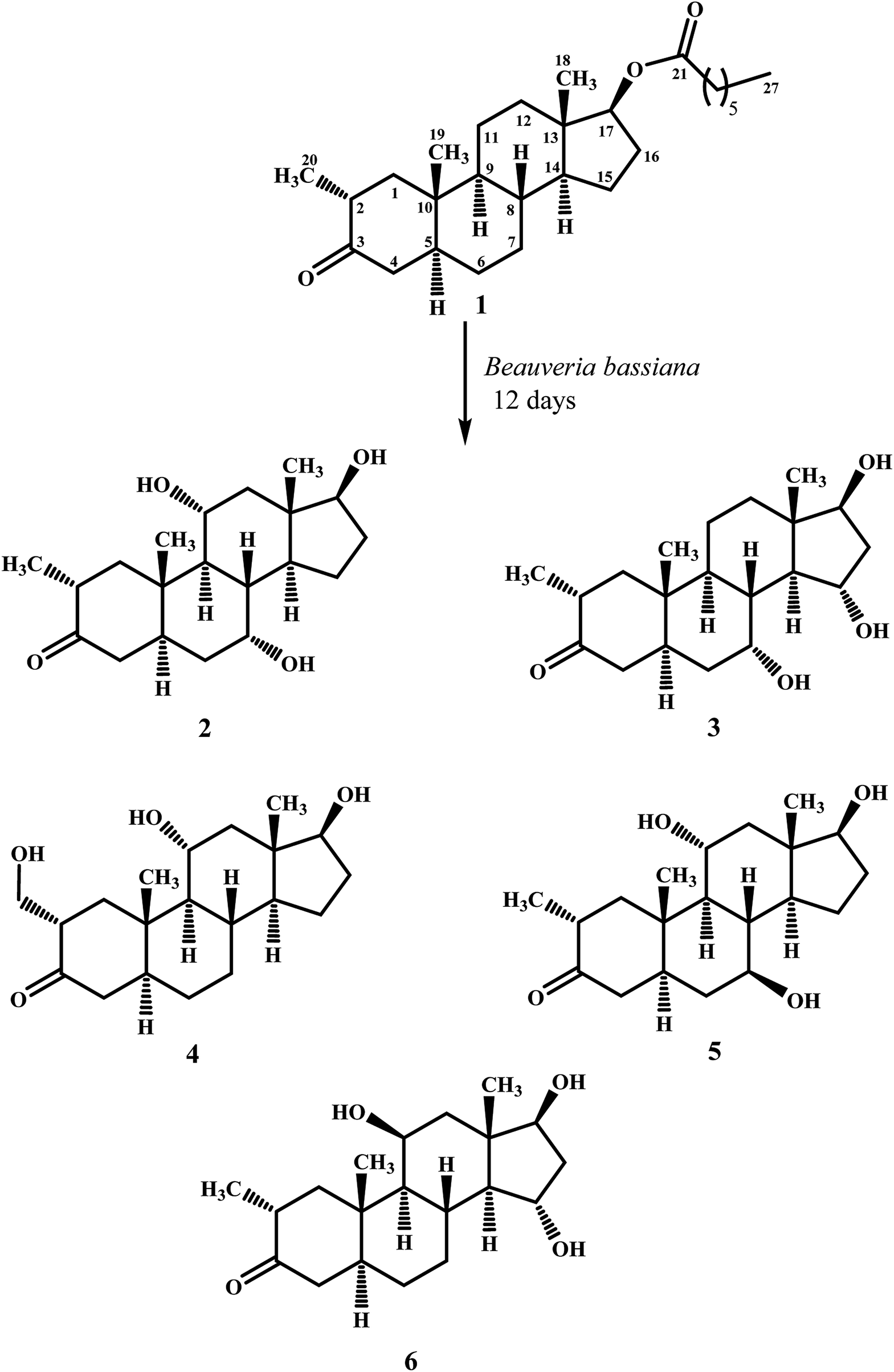 | ||
| Scheme 1 Biotransformation of drostanolone heptanoate (1) with B. bassiana. | ||
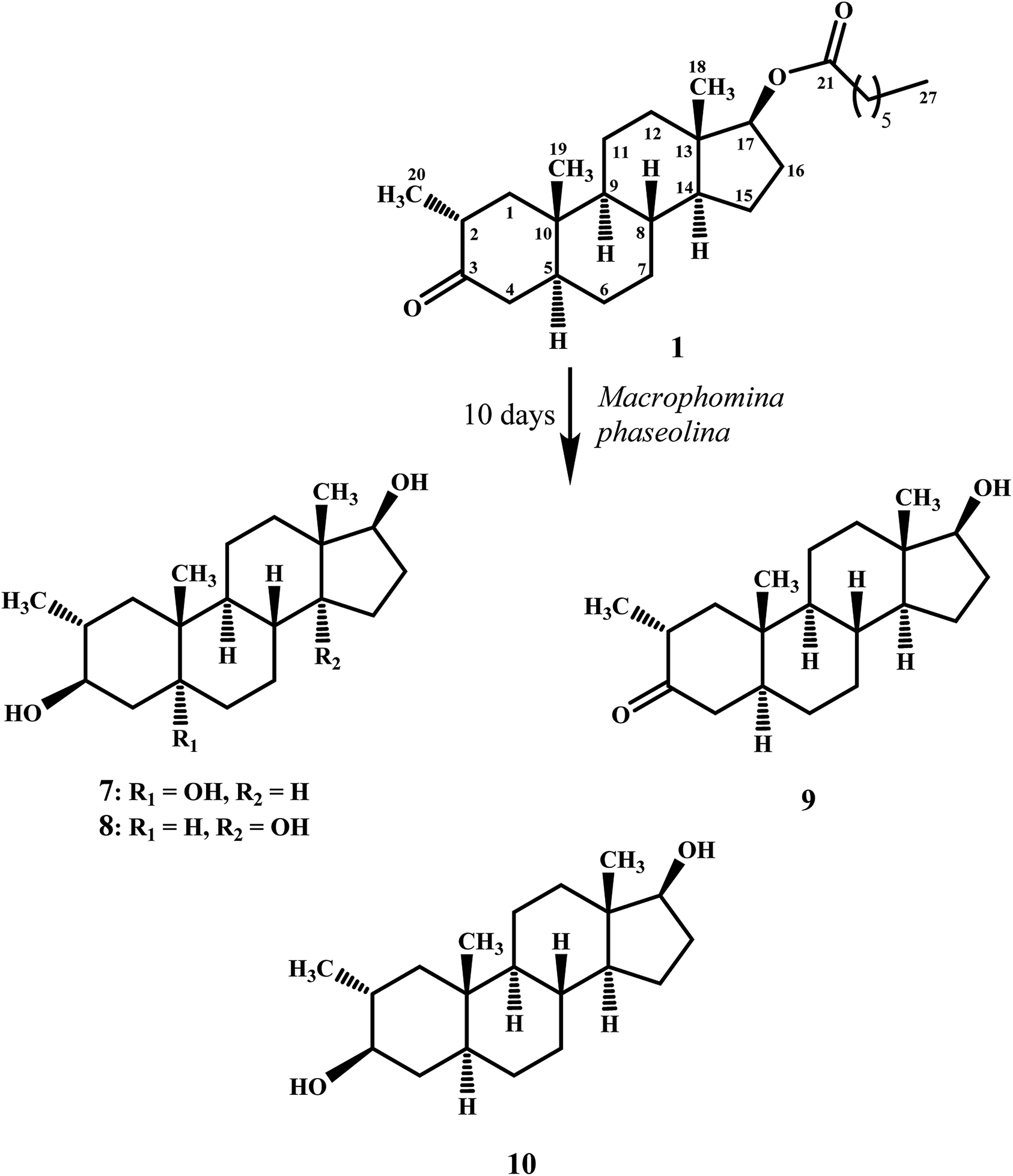 | ||
| Scheme 2 Biotransformation of drostanolone heptanoate (1) with M. phaseolina. | ||
Metabolite 2 was purified as optically active, colorless solid through recycling RP-HPLC. HRFAB-MS showed the [M + H]+ at m/z 337.2356 (calcd. 336.2301, C20H32O4), 24 amu lesser than substrate 1. This suggested hydrolysis of ester group, and addition of two oxygen atoms. Absorption bands at νmax (cm−1) 3475, 3330, 3326, and 1687 were observed in the IR spectra for three OH, and a carbonyl groups. In the 1H-NMR spectrum, three downfield methine signals at δ 3.92 (dt, J11a,9a/12a = 10.5 Hz, J11a,12e = 5.1 Hz), 3.55 (t, J17α,16α/β = 8.4 Hz), and 3.24 (m) were appeared, indicating presence of hydroxyl groups. Similarly, downfield signals for oxymethine carbons were observed in the 13C-NMR spectrum at δ 81.8, 75.0, and 69.6. In the HMBC spectrum of compound 2 (Fig. 1), H-12 showed HMBC interactions with C-9, C-11, C-14, C-17, and C-18, supporting the positions of both OH groups at C-11, and C-17. Similarly, H-9 showed cross peaks with C-7, C-8, C-10, C-11, and C-14, suggested OH groups at C-7, and C-11. The configuration of H-7, H-11, and H-17 were deduced as β, β, and α (Fig. 2), based on NOESY correlations of H-7 with β-oriented H-8, and H-11 with β-oriented H3-19, and H3-18 (Fig. 2). Thus the structure of new metabolite was determined as 2α-methyl-7α,11α,17β-trihydroxy-5α-androstan-3-one (2).
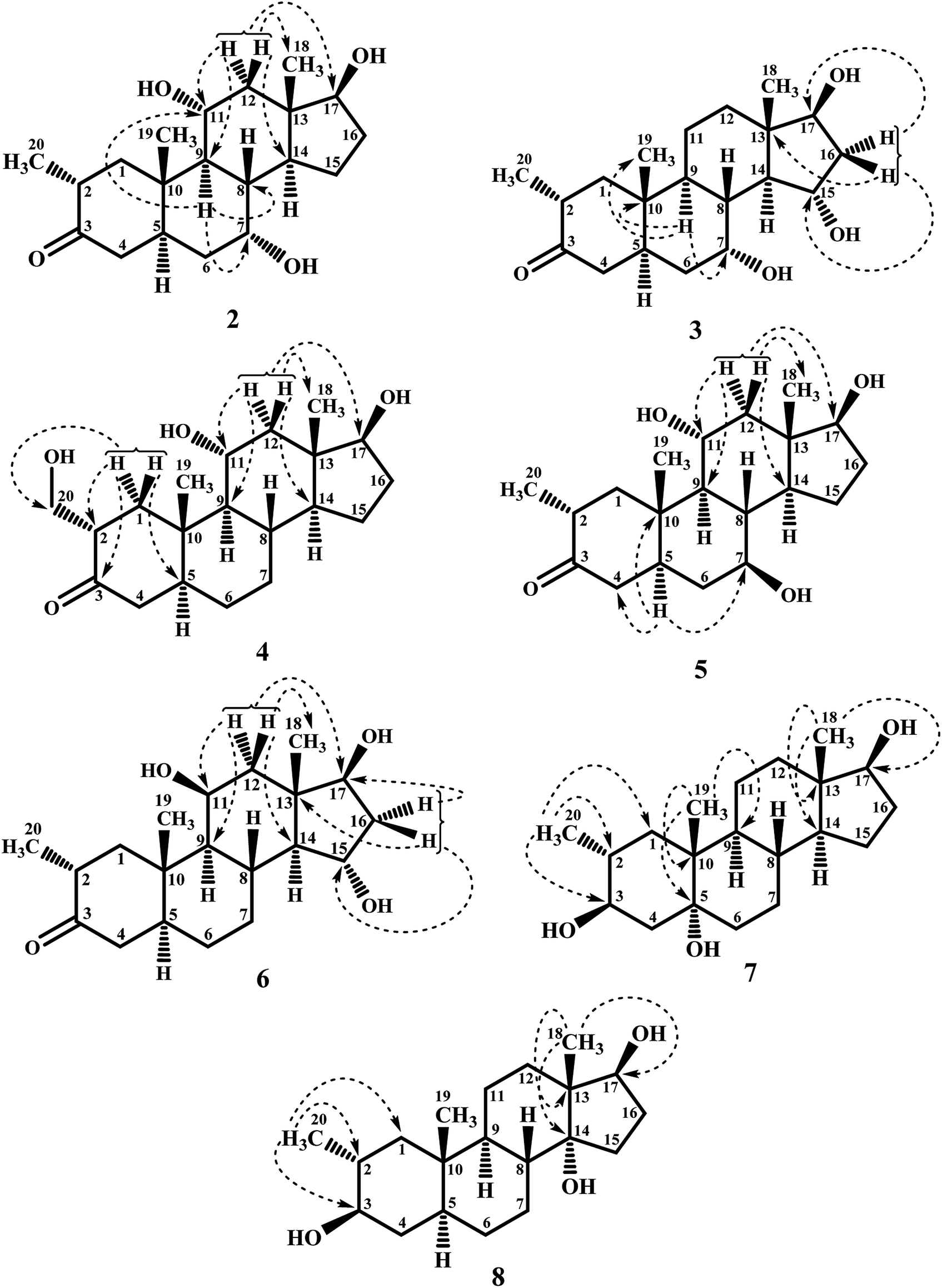 | ||
| Fig. 1 Key HMBC interactions of metabolites 2–8. | ||
 | ||
| Fig. 2 Key COSY, and NOESY interactions in metabolites 2–8. | ||
Compound 3 was obtained as optically active, colorless solid by using recycling RP-HPLC. HRFAB-MS showed the [M + H]+ at m/z 337.2372 (calcd. 336.2301, C20H32O4), suggesting hydrolysis of ester group, and addition of two oxygen atoms. This was 24 amu lesser than substrate 1. IR showed absorption bands at νmax (cm−1) 3733, 3375, 3278, and 1687 for three OH, and a carbonyl groups. The 1H-, and the 13C-NMR spectra of compounds 2, and 3 were found to be distinctly similar. In the 1H-NMR spectrum, three downfield methine signals at δ 4.06 (dt, J15β,16α/β = 9.0 Hz, J15β,14α = 4.8 Hz), 3.74 (t, J17α,16α/β = 9.0 Hz), and 3.30 (m) were assigned to the protons geminal to the OH groups. Consistent with this observations, the 13C-NMR spectrum showed methine carbons resonances at δ 79.5, 74.8, and 73.1. However, the HMBC, COSY, and NOESY spectra of both compounds were found different. Compound 3 displayed HMBC interactions (Fig. 1) of H-9 with C-5, C-7, C-10, and C-19, suggesting one of the OH groups at C-7. Similarly, the other two OH groups were placed at C-15, and C-17, based on the HMBC correlations of H-16 with C-13, C-15, and C-17. The configuration of H-7, H-15, and H-17 were deduced as β, β, and, based on NOESY correlations of H-7 with β-oriented H-8, H-15 with axially-oriented H3-18, and H-17 with α-oriented H-14 (Fig. 2). Thus the structure of new compound was deduced as 2α-methyl-7α,15α,17β-trihydroxy-5α-androstan-3-one (3).
Metabolite 4 was obtained as a white solid using recycling RP-HPLC. The HRFAB-MS of 4 showed the [M + H]+ at m/z 337.2366 (calcd. 336.2301, C20H32O4) suggesting the addition of two oxygen atoms, along with hydrolytic loss of ester moiety. The IR spectrum revealed the presence of three OH (3737, 3410, and 3369 cm−1), and a carbonyl (1703 cm−1) groups. Signal for the C-20 methyl protons in the 1H-NMR spectrum was also found missing. An additional downfield signal at δ 62.5 for C-20 in the 13C-NMR spectrum also indicated the oxidation of the methyl group as CH2–OH. New downfield signals for H-11 at δ 3.93 (dt, J11a,9a/12a = 10.8 Hz, J11a,12a = 5.0 Hz), H-17 at δ 3.59 (t, J17α,16α/β = 8.7 Hz), and H2-20 at δ 3.75 (dd, J20a,20b = 10.8 Hz, J20a,2β =5.1 Hz), and δ 3.47 (dd, J20b,20a = 11.1 Hz, J20b,2β = 5.7 Hz) were appeared in the 1H-NMR spectrum. This was further established through the HMBC correlations (Fig. 1). The HMBC correlations between H-1 and C-2, C-3, C-5, C-10, and C-20, and from H-12 to C-9, C-11, C-13, C-14, and C-17 were observed. These correlations supported the OH groups at C-11, C-17, and C-20. In the COSY spectrum (Fig. 2), the homonuclear 1H–1H couplings were observed between H-11, and H-9, and H-12, while H-2 showed the COSY cross peaks with H-1, and H2-20. The coupling constant of H-11 (dt, J11a,9a/12a = 10.8 Hz, J11a,12a = 5.0 Hz), and its NOESY correlations with H3-18 (δ 0.76 s), implied that OH group at C-11 was equatorial (α) (Fig. 2). Likewise, H-17 showed NOESY cross peaks with H-14, indicating a β-orientation of the OH group at C-17. This new metabolite was thus identified as 2α-hydroxymethyl-11α,17β-dihydroxy-5α-androstan-3-one (4).
Compound 5 was isolated as colorless solid through recycling RP-HPLC with a retention time of 26 min. It showed the [M + H]+ at m/z 337.2365 (calcd. 336.2301, C20H32O4) in the HRFAB-MS. The IR spectrum showed absorption bands at υmax (cm−1) 3743, 3429, 3375, and 1699, for OHs, and a carbonyl, respectively. The 1H-, and 13C-NMR spectra of compound 5, were found to be distinctly similar to compounds 2, and 3. Three new downfield signals for oxymethine protons were observed at δ 3.88 (dt, J11a,9a/12a = 10.8 Hz, J11a,12e =5.1 Hz), 3.77 (m), and 3.60 (t, J17α,16α/β = 8.4 Hz) in the 1H-NMR spectrum. Similarly, 13C-NMR spectrum of 5 showed downfield signals for oxymethine carbons at δ 69.7, 67.4, and 82.0. The positions of OH groups at C-11, and C-17 were deduced through the HMBC correlations (Fig. 1) of H-12 (δ 2.06, overlap; δ 1.07 br. t; J12a,12e/11a = 11.7 Hz) with C-9 (δ 52.9), C-11 (δ 69.7), C-13 (δ 49.6), C-14 (δ 45.9), and C-17 (δ 82.0) indicated the OH groups at C-11, and C-17. Similarly, OH group at C-5 was determined through the HMBC correlations of H-5 (δ 1.52, m) with C-4 (δ 51.4), C-7 (δ 67.4), and C-10 (δ 44.1) (Fig. 1). An OH group at C-11 was deduced as α on the basis of NOESY interactions (Fig. 2) of H-11 (δ 3.88 dt, J11a,9a/12a = 10.8 Hz, J11a,12e = 5.1 Hz) with axially-oriented H3-18 (δ 0.75 s), and H3-19 (δ 1.20 s). Similarly, the β-orientations of OH groups at C-7, and C-17 in compound 5 were deduced through the key NOESY correlations of axially-oriented H-5 with H-7, and axially-oriented H-14 with H-17 (δ 3.60 t, J17α,16α/β = 8.4 Hz), respectively. The structure of a new metabolite was thus deduced as 2α-methyl-7β,11α,17β-trihydroxy-5α-androstan-3-one (5).
Metabolites 6 was obtained as colorless solid by using RP-HPLC with a retention time of 24 min. The molecular formula of compound 6 was consistent with C20H32O4, deduced from the HRFAB-MS at m/z 337.2365 (calcd. 336.2301; [M + H]+). Absorption bands in the IR spectrum at υmax (cm−1) 3733, 3421, 3305, and 1689, were due to the presence of three OHs, and a carbonyl, respectively. The 1H-, and 13C-NMR spectra of compounds 5, and 6 were found to be distinctly similar to compounds 2, and 3. The 1H-NMR spectrum showed three new downfield signals for methine protons at δ 3.78 (t, J17α,16α/β = 9.0 Hz), 3.87 (m), and 3.90 (m). Signals for deshielded methine carbons at δ 69.2, 72.5, and 79.2 were appeared in the 13C-NMR spectrum of 6. This suggested the presence of three OH groups. OH Groups at C-11, C-17 were deduced through the HMBC correlations (Fig. 1) of H-12 (δ 2.07 overlap; δ 1.16 m) with C-9 (δ 61.0), C-11 (δ 69.2), C-13 (δ 45.6), C-14 (δ 58.5), and C-17 (δ 79.2), whereas, OH group at C-15 was determined through the HMBC correlations of H-16 with C-13, C-14, C-15, and C-17. In the NOESY spectrum (Fig. 2) of compound 6, cross peaks were observed between axially-oriented H-14, and H-17 suggesting β-orientation of OH group at C-17. Similarly, β-stereochemistry of OH groups at C-11, and C-15 were also deduced from the NOESY correlations of H-11 with axially-oriented H-9, and H-15 with axially-oriented H-8 and H3-18. The structure of a new metabolite was thus deduced as 2α-methyl-11β,15α,17β-trihydroxy-5α-androstan-3-one (6).
Compound 7 was purified as a white solid through recycling RP-HPLC with a retention time of 21 min. The HRESI-MS of metabolite 7 showed the [M + NH4]+ at m/z 340.2851 (calcd. 322.2508, C20H32O3). This suggested dihydroxylation, along with hydrolysis of side chain ester group. The IR spectrum showed broad absorbances at υmax 3373 cm−1 for OH groups. Absence of the bridge methine signals in the 1H-, and the 13C-NMR spectra of 7 suggested the hydroxylation at tertiary carbon. An OH group at C-5 was deduced through the HMBC correlations of H-3, H3-19, H2-6, and H-9 with C-5 (Fig. 1). Stereochemistry of OH group at C-3 was assigned as β, based on NOESY correlations of axially-oriented H-3 with equatorially-oriented H3-20 (Fig. 2). An OH group at C-5 (δ 77.0) was placed as α, based on the comparison of the reported chemical shifts for 5α-OH in steroids.15,20 Thus the structure of a new metabolite was identified as 2α-methyl-3β,5α,17β-trihydroxy-androstane (7).
Metabolite 8 was obtained as a white solid by using RP-HPLC with a retention time of 27 min. The HRESI-MS of 8 showed the [M + H]+ at m/z at 323.2586 (calcd. 322.2508), indicating hydrolysis of side chain ester group, along with dihydroxylation. Broad absorption bands at υmax 3377 cm−1 in the IR spectrum were due to the OH groups. The 1H-, and 13C-NMR spectra of metabolite 7 were apparently identical to 8. Absence of the bridge methine signals in the 1H-, and 13C-NMR spectra of 8 suggested the hydroxylation at tertiary carbon. An OH group at C-14 was deduced through the HMBC correlations of H-17, H-8, H2-15, H2-16, and H3-18 with C-14 (Fig. 1). The relative stereochemistry at C-3 in compound 8 was deduced from the NOESY interactions between axially-oriented H-3, and equatorially-oriented H3-20 (Fig. 2). Similarly, OH group at C-14 was deduced as α on the basis of reported chemical shift for 14α-OH in similar steroidal compound.18 Thus the structure of a new compound was deduced as 2α-methyl-3β,14α,17β-trihydroxy-5α-androstane (8).
Metabolites 9, and 10 were obtained as white solids by repeated column chromatography. HRESI-MS of compound 9 showed the [M + H]+ at m/z 305.2480 (calcd. 304.2402; C20H30O2) 47 amu less than the substrate 1, suggesting hydrolysis of side chain ester group. Whereas the HRESI-MS of metabolite 10 at m/z 324.2902 (calcd. 306.2599; [M + NH4]+) suggested a molecular composition of C20H34O2. This indicated the reduction of ketonic carbonyl group, along with hydrolysis of ester side chain of substrate 1. Metabolites 9, and 10 were previously reported by Templeton and Kim from the urine of drostanolone dosed rabbits.12 The spectroscopic data of metabolites 9, and 10 was consistent with the literature data. Their structures were identified as 2α-methyl-17β-hydroxy-5α-androstan-3-one (9), and 2α-methyl-3β,17β-dihydroxy-5α-androstane (10).
Conclusion
Microbial biotechnology has wide applications in the synthesis of libraries of structurally diverse analogues of bioactive natural products, and drugs. The present study afforded seven new, and two known metabolites of drostanolone heptanoate (1) by using two microbial cultures, Beauveria bassiana, and Macrophomina phaseolina. Incubation of 1 with B. bassiana led to the synthesis of five new metabolites 2–6, whereas M. phaseolina afforded two new 7, and 8, and two known transformed products 9, and 10. Hydroxylation, reduction, and hydrolysis were the main reactions during structural transformation of 1. The transformed products will be evaluated for potential biological activities in the future. In addition, metabolites 2–10 will also be studied at enzymatic level to understand the mechanism involved during biocatalysis.Conflicts of interest
Authors has no conflicts of interest.Acknowledgements
Authors acknowledge the financial support of the Higher Education Commission Pakistan through a project of National Research Program for Universities (NRPU) (# 7993) entitled, “Synthesis of new analogues of anti-inflammatory agents via microbial biotechnological methods”.References
- K. Kontula, O. Janne, R. Vihko, E. de Jager, J. de Visser and F. Zeelen, Progesterone-binding proteins, in vitro binding and biological activity of different steroidal ligands, Acta Endocrinol., 1975, 78, 574–592 CAS.
- T. Horiike, M. Ohshiro and M. Kuroyanagi, Biotransformation of the germacrane type sesquiterpene curdione by suspension cultured cells of Lonicera japonica, Phytochemistry, 1997, 44, 627–632 CrossRef CAS.
- M. I. Choudhary, S. Sultan, S. Jalil, S. Anjum, A. A. Rahman and H. K. Fun, Atta-ur-Rahman. Microbial transformation of mesterolone, Chem. Biodiversity, 2005, 2, 392–400 CrossRef CAS PubMed.
- J. E. Leresche and H. P. Meyer, Chemocatalysis and biocatalysis (biotransformation): Some thoughts of a chemist and of a biotechnologist, Org. Process Res. Dev., 2006, 10(3), 572–580 CrossRef CAS.
- M. Rabinovich, A. Bolobova and L. Vasil'chenko, Fungal decomposition of natural aromatic structures and xenobiotics: a review, Appl. Biochem. Microbiol., 2004, 40(1), 1–17 CrossRef CAS.
- F. Delbeke, N. Desmet and M. Debackere, The abuse of doping agents in competing body builders in Flanders (1988-1993), Int. J. Sports Med., 1995, 16(1), 66–70 CrossRef CAS.
- M. S. Bahrke and C. E. Yesalis, Abuse of anabolic androgenic steroids and related substances in sport and exercise, Curr. Opin. Pharmacol., 2004, 4(6), 614–620 CrossRef CAS.
- M. Chowdhury, A. Banks, W. Bond, W. Jones and H. Ward, A comparison of drostanolone propionate (masteril) and nandrolone decanoate (deca-durabolin) in the treatment of breast carcinoma, Clin. Oncol., 1976, 2(3), 203–211 CAS.
- B. Clavel, J. Cappelaere, J. Guerin, T. Klein, E. Pommatau and J. Berlie, Management of advanced breast cancer in postmenopausal women. A comparative trial of hormonal therapy, chemotherapy, and a combination of both, La semaine des hopitaux: organe fonde par l'Association d'enseignement medical des hopitaux de Paris, 1982, 58(34), 1919–1923 CAS.
- L. Marinov, V. Tsekova, K. Koinov, M. Velikova and D. Micheva, Drostanolone propionate (masteril) in disseminated breast cancer in women. Immediate results, Khirurgiia, 1986, 40(6), 80–86 Search PubMed.
- G. Trams, Effect of drostanolone propionate on the binding of oestradiol and dihydrotestosterone by normal and malignant target tissues, Eur. J. Cancer, 1965, 13(2), 149–153 CrossRef.
- J. F. Templeton and R. S. S. Kim, Metabolism of 17β-hydroxy-2α-methyl-5α-androstan-3-one in the rabbit, Steroids, 1977, 29(3), 371–381 CrossRef CAS PubMed.
- J. Gauthier, D. Goudreault, D. Poirier and C. Ayotte, Identification of drostanolone and 17-methyldrostanolone metabolites produced by cryopreserved human hepatocytes, Steroids, 2009, 74(3), 306–314 CrossRef CAS PubMed.
- Z. Hussain, N. Dastagir, S. Hussain, A. Jabeen, S. Zafar, R. Malik, S. Bano, A. Wajid and M. I. Choudhary, Aspergillus niger-mediated biotransformation of methenolone enanthate, and immunomodulatory activity of its transformed products, Steroids, 2016, 112, 68–73 CrossRef CAS PubMed.
- Atia-tul-Wahab, M. Siddiqui, I. Ibrahim, A. Hussain, E. H. Ajandouz, A. Hijazi, E. Baydoun and M. I. Choudhary, Cunninghamella blakesleeana-mediated biotransformation of a contraceptive drug, desogestrel, and anti-MDR-Staphylococcus aureus activity of its metabolites, Bioorg. Chem., 2018, 77, 152–158 CrossRef CAS PubMed.
- M. Siddiqui, M. S. Ahmad, Atia-tul-Wahab, S. Yousuf, N. Fatima, N. N. Shaikh, Atta-ur-Rahman and M. I. Choudhary, Biotransformation of a potent anabolic steroid, mibolerone, with Cunninghamella blakesleeana, C. echinulata, and Macrophomina phaseolina, and biological activity evaluation of its metabolites, PLoS One, 2017, 12(2), e0171476 CrossRef.
- S. Bano, Atia-tul-Wahab, S. Yousuf, A. Jabeen, M. A. Mesaik, Atta-ur-Rahman and M. I. Choudhary, New anti-inflammatory metabolites by microbial transformation of medrysone, PLoS One, 2016, 11(4), e0153951 CrossRef PubMed.
- M. I. Choudhary, M. Siddiqui, Atia-tul-Wahab, S. Yousuf, N. Fatima, M. S. Ahmad and H. Choudhry, Bio-catalytic structural transformation of anti-cancer steroid, drostanolone enanthate with Cephalosporium aphidicola and Fusarium lini, and cytotoxic potential evaluation of its metabolites against certain cancer cell lines, Front. Pharmacol., 2017, 8, 900 CrossRef PubMed.
- E. Baydoun, Atia-tul-Wahab, S. Iqbal, C. Smith and M. I. Choudhary, Biotransformation of drospirenone, a contraceptive drug, with Cunninghamella elegans, Steroids, 2017, 126, 30–34 CrossRef CAS PubMed.
- M. S. Ahmad, S. Yousuf, Atia-tul-Wahab, A. Jabeen, Atta-ur-Rahman and M. I. Choudhary, Biotransformation of anabolic compound methasterone with Macrophomina phaseolina, Cunninghamella blakesleeana, and Fusarium lini, and TNF-α inhibitory effect of transformed products, Steroids, 2017, 128, 75–84 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05878h |
| This journal is © The Royal Society of Chemistry 2020 |