DOI:
10.1039/C9RA09106H
(Paper)
RSC Adv., 2020,
10, 2998-3004
Novel fluorescein polymer-based nanoparticles: facile and controllable one-pot synthesis, assembly, and immobilization of biomolecules for application in a highly sensitive biosensor†
Received
4th November 2019
, Accepted 30th December 2019
First published on 16th January 2020
Abstract
A key aspect of biochip and biosensor preparation is optimization of the optical or electrochemical techniques that combine high sensitivity and specificity. Among them, optical techniques such as the use of fluorescent polymeric nanoparticles have resulted in dramatic progress in the field of diagnostics due to their range of advantages. We herein report a facile approach for the development of novel fluorescein polymeric nanoparticles (FPNPs) with immobilization of specific biomolecules for application in a highly sensitive optical biosensor. A series of three amphiphilic fluorescein polymers (poly(FMA-r-NAS-r-MA)), comprising hydrophobic fluorescein O-methacrylate (FMA), hydrophilic N-acryloxysuccinimide (NAS), and methacrylic acid (MA) monomers were synthesized through radical polymerization. In an aqueous environment, these fluorescein polymers self-assembled into spherical shaped nanoparticles with a well-defined particle size, narrow particle size distribution, and enhanced fluorescence properties. The bio-immobilization properties of the FPNPs were also tunable by control of the activated N-hydroxysuccinimide ester group in the polymer series. Furthermore, the fluorescence sensitivity of bovine serum albumin detection by the FPNPs indicates that the limit of detection and sensitivity were improved compared to conventional fluorescence dye-labelled proteins. These novel FPNPs therefore represent a suitable technology for disease diagnosis and biomarker detection to ultimately improve the sensitivity of existing analytical methodologies in a facile and cost-effective manner.
Introduction
In vitro and in vivo diagnostics including biomarker analysis, cancer diagnosis, diagnostic imaging, and immunoassays, require methods that combine high sensitivity with high specificity.1–3 Among them, optical techniques have resulted in significant progress in the field of diagnostics. In particular, dyes have been employed as popular tools in optical diagnostics, allowing the detection of analytes with good sensitivities, either through color changes or by fluorescence emission.1 However, dyes suffer from photobleaching, and often exhibit asymmetric emission spectra. In this context, photostability is important in the case of prolonged observations, where photobleaching severely impacts the capability of dyes to detect low amounts of an analyte.4–6 Furthermore, dyes such as the fluorescein and rhodamine, suffer from quenching phenomena when present in solution at high concentrations.1,7,8 However, despite these drawbacks, organic dyes are widely employed due to their low costs and ease of use.
Polymeric nanoparticles (NPs), formed through the spontaneous self-assembly of amphiphilic copolymers and control of the hydrophilic-lipophilic balance, have been widely utilized as highly efficient optical diagnostics supporters.9–11 In addition, polymeric NP-containing dyes have the potential to overcome the brightness and photostability limits of fluorescent molecules.12 More specifically, when the dye is loaded into a polymeric matrix, an increased photostability often results due to the “protective” effect of the polymer.8,13,14 It is also possible to embed large amounts of dye molecules into polymeric NPs to enhance the color intensity or the emission brightness.1 Furthermore, the hydrophobic microenvironment created within polymeric NPs can enhance the quantum yields of certain fluorescent dyes.15,16 Moreover, polymeric NPs are considered particularly promising due to their remarkable stabilities in physiological buffers and their well-controlled surface properties.1,14,17 Although the majority of fluorescein-containing fluorescent molecules are poorly soluble in water, the introduction of amphiphilic polymeric NPs can increase the solubility and stability of the fluorescent molecule.18–20 Covalently coupling the dye to the polymer will also decrease the possibility of the leakage of entrapped dyes through diffusion out of the polymer.1,10,14 In addition, to enhance the diagnostic specificity of nanoparticle-based assays, binding of the target biomolecule to the NPs is essential.21,22 For the successful detection of the target analyte, this binding should result in a measurable signal that can be quantified. The presence of reactive functional groups along the polymer backbone therefore allows specific labeling with other molecules to be carried out, in addition to modification of the particle surface for specific applications. Nevertheless, previously developed fluorescent polymeric NPs for in vitro diagnostics applications have a number of limitations.16,23–25 Firstly, the preparation of fluorescent polymers tends to involve low yielding and multi-step processes, and the hydrophobic fluorescein is known to exhibit a poor solubility and photostability.1,12,19,22 Furthermore, the uncontrolled immobilization of large biomolecules through the carboxyl group renders it difficult to directly conjugate a large protein molecule to the sterically hindered tertiary carboxyl group [–C(R)(CH3)–CO2H], and this process often requires an additional surface treatment step using reagents such as 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS).26–29
Thus, we herein provide the first report into the self-assembly of novel fluorescein polymers in a range of molar ratios to ultimately produce NPs for application in a highly sensitive biosensor. Three series of amphiphilic fluorescein polymers, referred to as poly(FMA-r-NAS-r-MA), are synthesized by a facile one-pot radical polymerization process, using the fluorescein O-methacrylate (FMA, a hydrophobic fluorescein residue), N-acryloxysuccinimide (NAS, the pre-activated NHS ester functional group for immobilization of specific biomolecules), and methacrylic acid (MA, a hydrophilic COOH group for increasing the stability of the polymeric NPs) monomers. Ultimately, we aim to prepare novel fluorescein polymeric nanoparticles (FPNPs) with improved solubilities and stabilities using the different poly(FMA-r-NAS-r-MA) copolymers via a nanoprecipitation approach. In addition, the NAS group is introduced for effective immobilization of the biomolecules. The purities and molecular weights of the synthesized fluorescein polymers are then analyzed by proton nuclear magnetic resonance (1H NMR) spectroscopy and gel permeation chromatography (GPC), respectively, while the physicochemical properties of the FPNPs are characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM). The stability of the FPNPs in biological buffers and during resuspension after freeze-drying in the absence of cryoprotectants is evaluated by monitoring changes in the appearance of the NPs. Furthermore, the properties of biomolecules immobilized on the FPNPs are analyzed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) profiles. Finally, to demonstrate the usefulness of these fluorescein polymers, the fluorescence sensitivity toward bovine serum albumin (BSA) detection is assessed and compared with the fluorescein isothiocyanate (FITC)–BSA conjugate.
Experimental
Materials
FMA (95%), MA (99%), tetrahydrofuran (THF, anhydrous, 99.9%), and the inhibitor-removal column were purchased from Sigma-Aldrich (St. Louis, MO, USA). NAS (99%) was purchased from ACROS Organics (Geel, Belgium). 2,2-Azobisisobutyronitrile (AIBN, 99%) was obtained from DAEJUNG (Korea). All commercially-obtained reagents and chemicals were used as received without further purification. BSA (biotechnology grade) was purchased from Bioshop (Burlington, Canada), while the bovine serum albumin antibody (Anti-BSA) was obtained from BioSource (Camarillo, USA). The deionized water (DI water) employed herein was supplied by HyClone (Logan, UT, USA).
Synthesis of the fluorescein polymers (poly(FMA-r-NAS-r-MA))
The novel fluorescein polymers were synthesized via a simple radical polymerization method, as previously reported.30,31 Prior to polymerization, MA was passed over the inhibitor-removal column. FMA (0.2 mmol, 0.08 g), NAS (0.5 mmol 0.085 g), and MA (2 mmol, 0.172 g) were then dissolved in THF (3.4 mL). After the addition of AIBN (0.12 mmol, 0.02 g) as a radical initiator, the resulting mixture was degassed for 5 min by bubbling with a steam of Ar gas, then sealing with Teflon tape. The polymerization reaction was carried out at 70 °C over 24 h under stirring, after which time, the obtained product was cooled to 25 °C and stored at 4 °C prior to use. Various molar ratios were employed for syntheses of the fluorescein polymers (poly(FMA-r-NAS-r-MA)). More specifically, the initial feed ratios of the monomers (FMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NAS:MA) were 0.2:0.5:2 for Poly F1, 0.2:1:1.5 for Poly F2, and 0.2:1.5:1 for Poly F3. The 1H NMR (400 MHz) spectra were recorded on a JEOL JNM-ECZ400S/L1 spectrometer (Tokyo, Japan) using DMSO as a solvent at 25 °C, and the proton chemical shifts are expressed in parts per million (ppm). Poly(FMA-r-NAS-r-MA) with δ = (a) 7.9 (1H of FMA), (b and c) 7.71–7.79 (2H of FMA), (d) 7.27 (1H of FMA), (e) 6.67 (1H of FMA), (g and f) 6.54 (1H of FMA), (h and h′) 2.59 (4H,–CO–CH2–CH2–CO–, of NAS) (ESI Fig. 1†). The molecular weights (g mol−1) and molecular weight distribution indices of the copolymers were determined using GPC (Agilent 1200S/miniDAWN TREOS) with THF as the eluent at a flow rate of 1.0 mL min−1 at 35 °C. GPC results: Poly F1, Mw = 6244 with Mw/Mn = 1.52; Poly F2, Mw = 4961 with Mw/Mn = 1.47; Poly F3, Mw = 3372 with Mw/Mn = 1.39.
:NAS:MA) were 0.2:0.5:2 for Poly F1, 0.2:1:1.5 for Poly F2, and 0.2:1.5:1 for Poly F3. The 1H NMR (400 MHz) spectra were recorded on a JEOL JNM-ECZ400S/L1 spectrometer (Tokyo, Japan) using DMSO as a solvent at 25 °C, and the proton chemical shifts are expressed in parts per million (ppm). Poly(FMA-r-NAS-r-MA) with δ = (a) 7.9 (1H of FMA), (b and c) 7.71–7.79 (2H of FMA), (d) 7.27 (1H of FMA), (e) 6.67 (1H of FMA), (g and f) 6.54 (1H of FMA), (h and h′) 2.59 (4H,–CO–CH2–CH2–CO–, of NAS) (ESI Fig. 1†). The molecular weights (g mol−1) and molecular weight distribution indices of the copolymers were determined using GPC (Agilent 1200S/miniDAWN TREOS) with THF as the eluent at a flow rate of 1.0 mL min−1 at 35 °C. GPC results: Poly F1, Mw = 6244 with Mw/Mn = 1.52; Poly F2, Mw = 4961 with Mw/Mn = 1.47; Poly F3, Mw = 3372 with Mw/Mn = 1.39.
Preparation of the FPNPs
The desired poly(FMA-r-NAS-r-MA) (5 mg) was dissolved in THF (1 mL) at 25 °C, then added dropwise into DI water (5 mL) using a syringe pump (LEGATO100, Kd Scientific, Korea) at a rate of 0.075 mL s−1 under gentle stirring.32 Subsequently, the resulting solution was subjected to vacuum drying for 30 min to remove the THF. The final concentration of the FPNPs in DI water was 1.0 mg mL−1. DLS measurements were carried out using a Zetasizer instrument (ELSZ-2000, Otsuka electronic, Tokyo, Japan). The morphologies of the nanoparticles were obtained at 25 °C using TEM (JEM-2100PlusHR, JEOL, Tokyo, Japan). Samples for the TEM observations were prepared by dropping the NP solutions onto a carbon-coated copper grid and drying at 25 °C.
Immobilization of BSA onto the FPNPs
The possibility of directly immobilizing biomolecules on FPNPs was tested by their treatment with BSA (at the desired concentration in DI water) for 2 h at 25 °C with gentle mixing. Following immobilization of the BSA, the FPNPs were immediately immersed in DI water and incubated for 4 h to hydrolyze any unreacted NHS esters. After this time, the un-immobilized BSA was removed by centrifugation at 4000g for 30 min. TEM was employed to visualize the BSA-immobilized FPNPs. Electrophoresis, i.e., SDS-PAGE, was performed to assess the conjugation process. Electrophoresis was carried out by regular SDS-PAGE with 12% polyacrylamide gel and 5% stacking gel according to the manufacturer's instructions (Bio-Rad, Hercules, CA, USA). UV-vis spectra were recorded on a Mega-900 UV-vis spectrophotometer (SCINCO, Seoul, Korea).
Fluorescence sensitivity for BSA detection
The fluorescence sensitivity for BSA detection was examined using a 96-well plate according to standard protocols.29 The anti-BSA was diluted to a final concentration of 0.01 ng mL−1 and an aliquot (50 μL) was added to each well and stored overnight at 4 °C to achieve immobilization. After washing well with PBS buffer, well-blocking was performed using 1% gelatin (200 μL) per well over 2 h at 25 °C. In addition, FPNPs–BSA and FITC–BSA (0.1 mg mL−1) were blocked using a 1:1 ratio to gelatin over 10 min at 25 °C. The FPNPs–BSA and FITC–BSA were added to each well, and incubation allowed to proceed for 1 h at 37 °C. After this time, the microplate well was washed several times with PBS, and fluorescence detection was carried out by measuring the fluorescence at 480 nm using a microplate reader (VICTOR X5, PerkinElmer, Waltham, MA, USA).
Results and discussion
Synthesis and characterization of the novel fluorescein polymers (poly(FMA-r-NAS-r-MA))
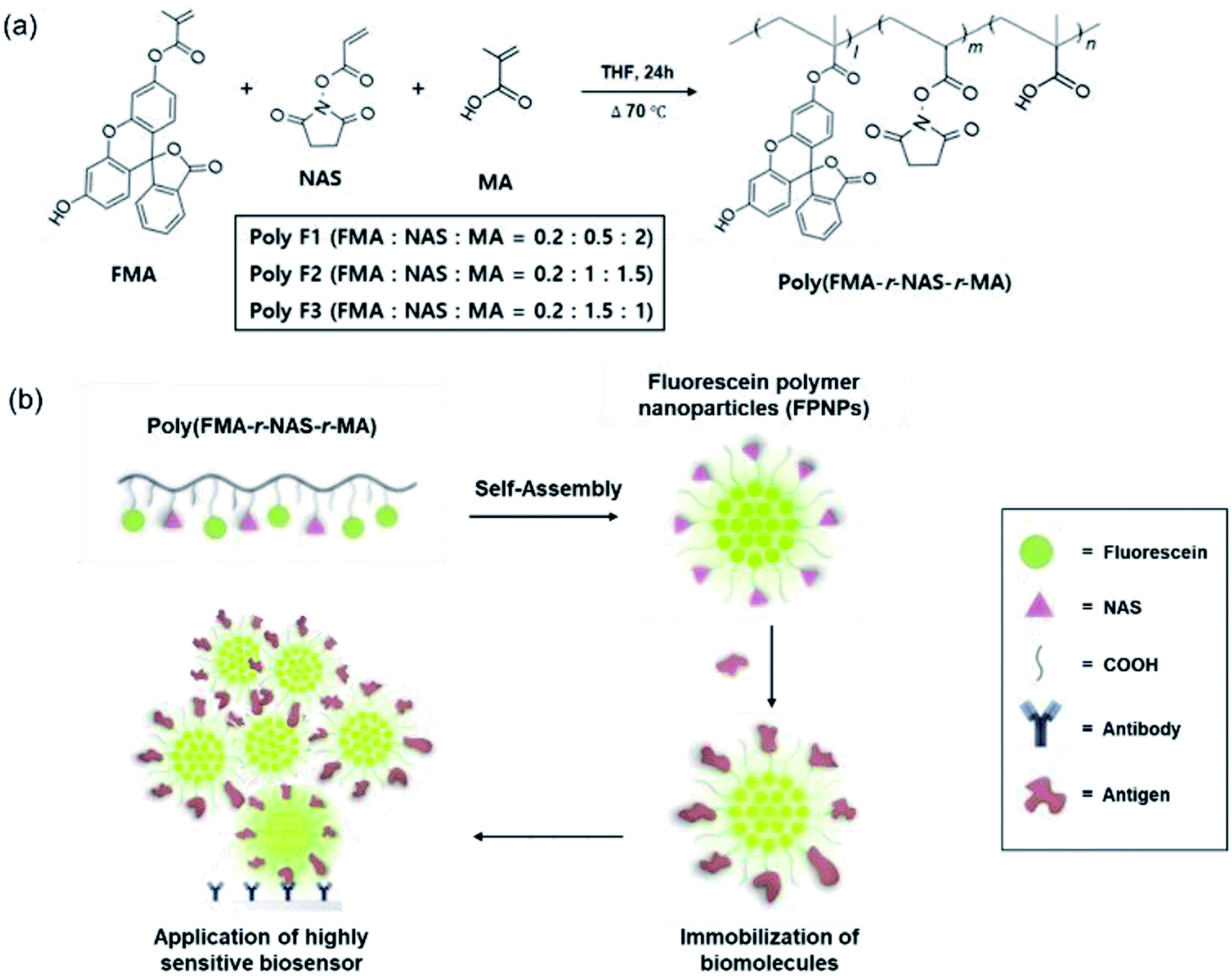
There are several drawbacks associated with conventional methods for the preparation of FPNPs, including low polymer yields due to multi-step preparation processes, the poor stability of the hydrophobic fluorescein, and uncontrolled immobilization of the biomolecules.23,33 To address such issues, we attempted the design of novel fluorescein polymers via a facile and controllable one-pot synthetic route for application in a highly sensitive biosensor. The chemical structures of the newly designed fluorescent copolymers are shown in Fig. 1a, where the copolymer, referred to as poly(FMA-r-NAS-r-MA), was synthesized from three different monomers (i.e., FMA, NAS, and MA). One key difference from the conventionally used polymers is the inclusion of an activated NHS ester of acrylic acid as a monomer. The resulting polymers therefore exhibit reduced steric hindrances (quaternary → tertiary carboxylic acid) than previously reported polymers, and so are more reactive toward the amine-containing biomolecules.29
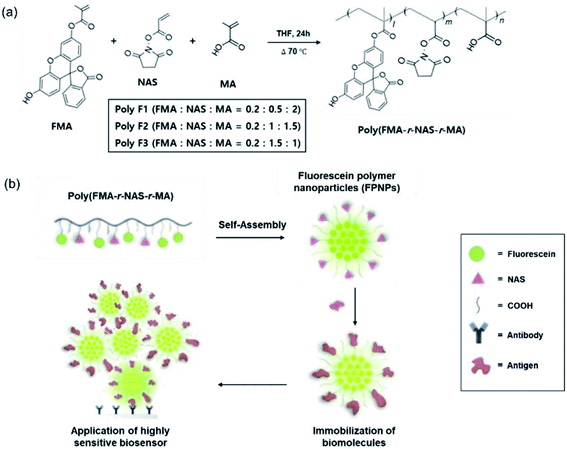 |
| | Fig. 1 (a) Controllable one-pot synthesis of the fluorescein polymers, poly(FMA-r-NAS-r-MA), referred to as Poly F1, Poly F2, and Poly F3, obtained through variation in the molar ratios of the FMA, NAS, and MA monomers. (b) Schematic representation of the preparation of FPNPs and biomolecule immobilization for application in a highly sensitive biosensor. | |
In our system, the density of the hydrophobic fluorescein and hydrophilic NAS, and the COOH residues in the fluorescein polymers could be controlled by simply changing the initial molar ratios of FMA, NAS, and MA to yield the three fluorescein polymer series (i.e., Poly F1, F2, and F3). Chemical structures of fluorescein polymer series were analyzed before and after polymerization by 1H NMR spectroscopy (ESI Fig. 1†). Before synthesis, general methacrylate monomer peaks were observed at 5.58–6.15 ppm, and the peaks in the spectra were generally sharp. However, the monomer peaks disappeared and broad polymer alkyl chain peaks appeared after polymerization, implying that the polymers were successfully synthesized in the THF solution. The actual FMA content in Poly F1, Poly F2, and Poly F3, obtained on the basis of integration values in the 1H NMR spectrum, were found to be 8%, respectively. Furthermore, the true NAS contents of Poly F1, Poly F2, and Poly F3 were confirmed to be 21%, 41%, and 60%, respectively. These results indicated that the compositions of the copolymers were almost well matched to the initial feed ratios. To determine the purities of the synthesized fluorescein polymers, the molecular weights (Mw) and polydispersity indices (PDI; Mw/Mn) of the fluorescein polymers were evaluated by GPC, as outlined in Table 1. The molecular weights of the fluorescein polymers ranged from 3 to 6 kDa, indicating an increase as the component proportion of MA was increased from Poly F3 to Poly F1. The polydispersity indices of the fluorescein polymers also showed a relatively narrow distribution of <1.6, and the GPC chromatogram showed a bimodal distribution (ESI Fig. 2†). Hence, the fluorescein polymers were successfully synthesized.
Table 1 GPC results for the synthesized fluorescein polymers (poly(FMA-r-MA-r-NAS)). The molecular weights (Mw) and polydispersity indices (PDIs) of the polymer groups were analyzed using a polystyrene standard
| Polymers groups |
Mn (g mol−1) |
Mw (g mol−1) |
PDI |
| Poly F1 |
4100 |
6200 |
1.52 |
| Poly F2 |
3400 |
4900 |
1.47 |
| Poly F3 |
2400 |
3400 |
1.39 |
Preparation and characterization of the FPNPs
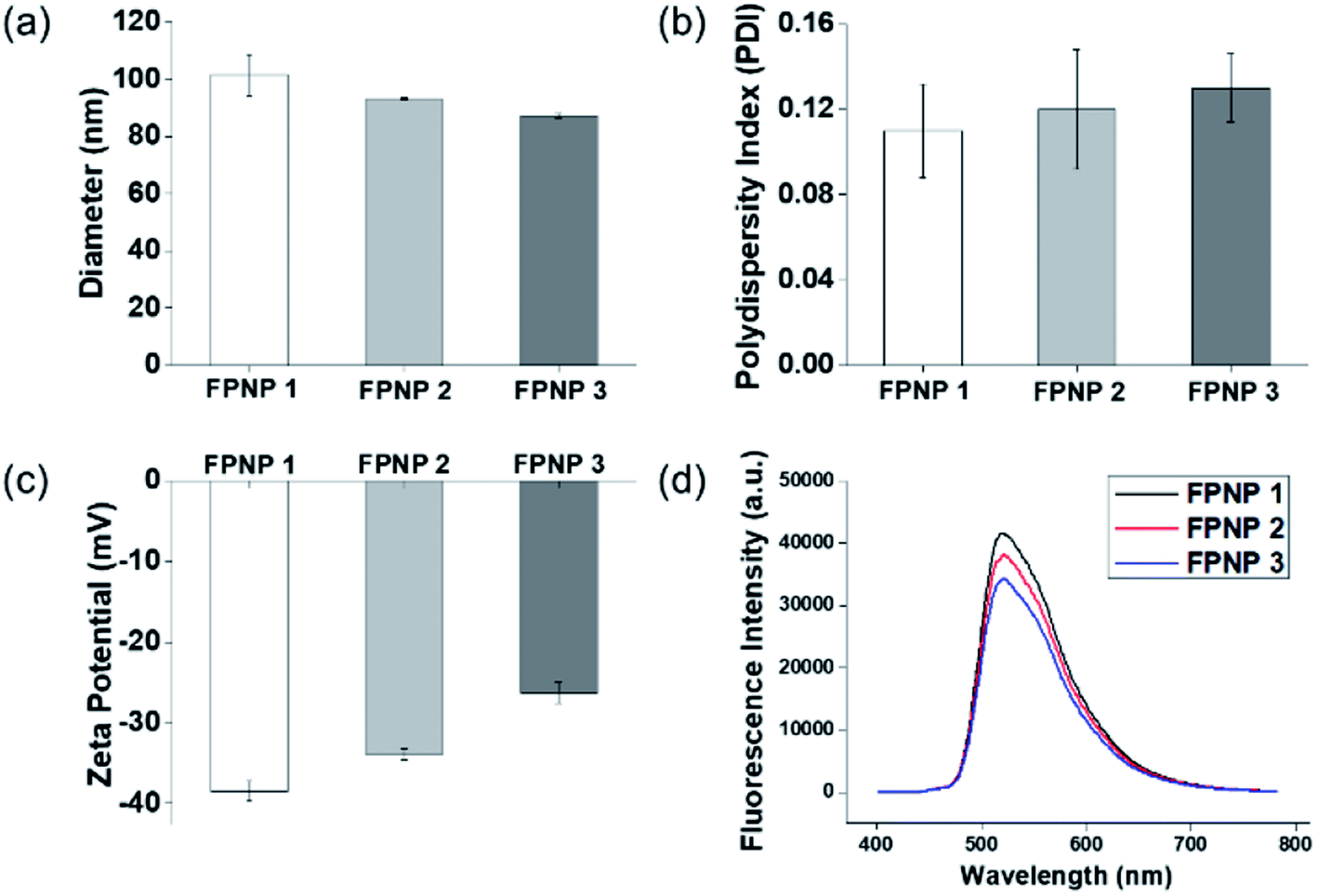
The preparation of polymer NPs with a controllable size and zeta potential in addition to the incorporation of stable fluorescent molecules could allow optimization for their application in in vitro diagnostics. Thus, the FPNPs, namely FPNP1, FPNP2, and FPNP3, were spontaneously prepared through self-assembly of the synthesized fluorescein polymers into stable nano-aggregates in aqueous solution, where the resulting NPs contained a hydrophobic fluorescein core, a hydrophilic NAS shell, and the carboxyl group. As shown in Fig. 2, the hydrodynamic diameters, PDIs, and surface charges of the FPNPs could be controlled by varying the composition of fluorescein polymers. More specifically, the FPNP size reduced slightly from 100 to 85 nm upon increasing the amount of the hydrophilic NAS group between FPNP1 and FPNP3. Likewise, the surface charges of the FPNPs increased from −38 to −27 mV upon decreasing the number of carboxyl groups. In addition, the FPNPs exhibited a narrow particle size distribution with a PDI of <0.15, indicating that they could undergo efficient self-assembly due to a suitable hydrophilic/hydrophobic balance in the amphiphilic fluorescein polymers. Furthermore, Fig. 2d shows the range of emission of the FPNPs when excited at 495 nm (λmax of FITC). As indicated, the fluorescence intensity of the FPNPs increased from FPNP3 to FPNP1, demonstrating that the polymers in the self-assembled nanoparticles that contained fewer NAS groups could contain greater numbers of fluorescein moieties. Moreover, the morphologies of the FPNPs were observed by TEM, as presented in Fig. 3a. It should be noted here that the sizes observed by TEM were similar or smaller than those determined by DLS, likely due to the FPNPs observed by TEM having undergone dehydration, which leads to NP shrinkage.
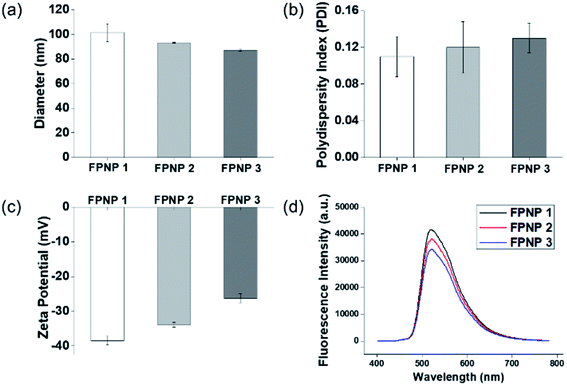 |
| | Fig. 2 (a) Hydrodynamic diameters, (b) polydispersity indices (PDIs), (c) surface charges, and (d) fluorescence intensities of the prepared FPNPs. | |
 |
| | Fig. 3 TEM images of the prepared FPNP1. (a) FPNP1, scale bar = 100 nm, (b) FPNP1–BSA clusters, scale bar = 500 nm. (c) UV-vis spectra of BSA (black line), the bare FPNP1 (red line), and the BSA-immobilized FPNP1 (blue line). | |
Control and optimization of biomolecule immobilization on the FPNPs
In the cases of previously synthesized carboxyl-functionalized amphiphilic polymers, small biomolecules such as biotin are easily immobilized; however, the attachment of large biomolecules is difficult due to the presence of steric hindrance at the quaternary carboxylic acid moiety. Thus, to determine whether larger protein molecules can be directly conjugated through the pre-activated NHS ester group of FPNPs, we employed BSA as a model protein. After purification, the BSA-immobilized FPNPs were visualized by TEM to determine their morphologies (Fig. 3b), and it was found that aggregates and nanoclusters were formed. The FPNPs–BSA clusters were also characterized by UV-vis analysis to confirm significant spectral changes (Fig. 3c). Characteristic individual peaks corresponding to the FPNPs (at 452 nm) and BSA (at 278 nm) decreased in intensity following BSA immobilization, thereby confirming the successful conjugation of BSA to the FPNPs.
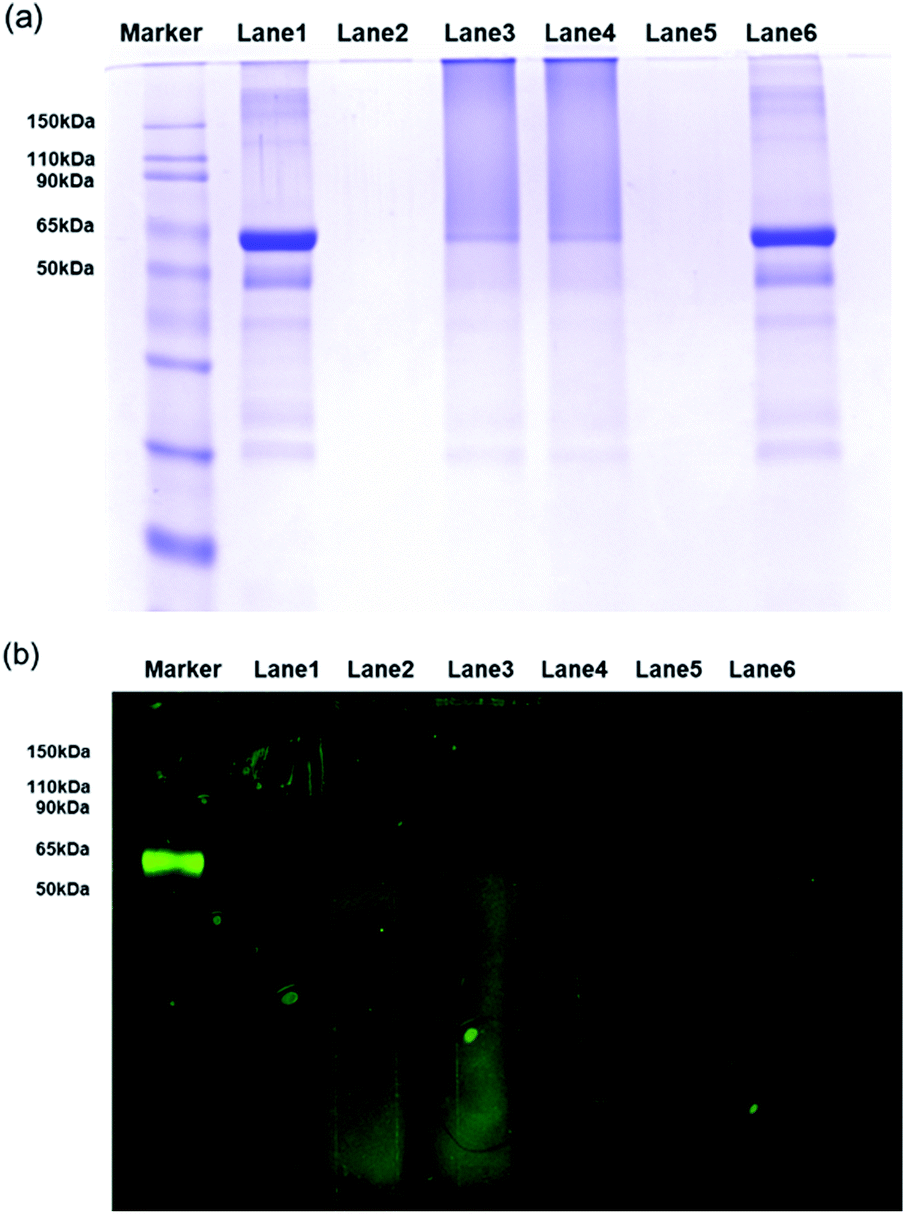
Furthermore, SDS-PAGE was conducted to examine the bio-conjugation and fluorescence properties of the FPNPs (see Fig. 4a). Thus, the protein BSA standard exhibited a band at 62 kDa (lane 1), while the FPNPs moved rapidly to the bottom of the gel and so were not observed (lane 2). In addition, the BSA-immobilized FPNPs (lane 3) showed upper BSA bands and tailing, which may be attributed to the fact that FPNPs–BSA form nanoclusters larger than the bare BSA. To remove any residual BSA, centrifugation was carried out at 4000 rpm for 30 min. The resulting pellet (lane 4) corresponded to FPNPs–BSA clusters, while the supernatant (lane 5) indicated that no residual BSA was present. A simple mixture of BSA and the FPNPs was also examined (lane 6) to examine the ionic bonding between the FPNPs and BSA. Moreover, Fig. 4b shows the fluorescent scanning image of Fig. 4a. In this case, the FPNPs (lane 2) exhibited fluorescence under the 50 kDa band while BSA (lane 1) showed no fluorescence, and FPNPs–BSA clusters (lanes 3 and 4) showed a strong fluorescence above 62 kDa. Subsequently, to examine the properties of the prepared FPNPs, the three samples were run at same concentration of overdosed BSA (Fig. 5). Upon moving from FPNP1 to FPNP3, it was confirmed that a greater quantity of BSA was conjugated (lanes 2, 4, and 6), and the amount of residual BSA was lower (lanes 3, 5, and 7). The amount of immobilized BSA on the FPNPs clusters was quantified by comparison of the intensities of the SDS-PAGE using Image J software. As shown in Fig. 5, upon moving from FPNP1 to FPNP3, the amount of conjugated BSA increased from 18% (90 μg BSA/1 mg of FPNP1) to 30% (150 μg of BSA/1 mg of FPNP2) to 47% (235 μg of BSA/1 mg of FPNP3) compared to BSA alone (lane 1; 500 μg ml−1). These results indicate that upon increasing the number of NAS functional groups from Poly F1 to Poly F3, the ability to bind proteins increases. More importantly, the dispersion stability of the FPNPs-BSA clusters was monitored, and it was found that FPNP1 was not aggregated, but instead maintained a good dispersion after 5 d, while FPNP2 and FPNP3 precipitated in the form of aggregates (ESI Fig. 3†). These results suggest that FPNP1 exhibits a higher stability due to the presence of additional negative charges and an optimized NAS content, thereby implying that FPNP1 bearing an appropriate immobilization degree and an excellent stability could be considered a potential platform for the sensitive optical biosensor.
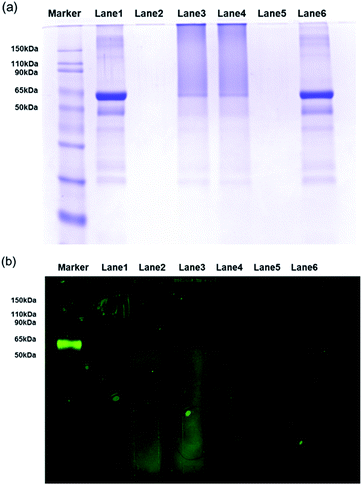 |
| | Fig. 4 (a) SDS-PAGE profiles following Coomassie brilliant blue staining. (b) Fluorescent scanning image of part (a). Lane 1: BSA; lane 2: FPNP1; lane 3: BSA-immobilized FPNP1; lane 4: pellet of lane 3 after centrifugation; lane 5: supernatant of lane 3 after centrifugation; and lane 6: mixture of BSA and FPNP1. | |
 |
| | Fig. 5 SDS-PAGE profiles following Coomassie brilliant blue staining. Lane 1: BSA; lane 2: FPNP1–BSA clusters; lane 3: supernatant of FPNP1; lane 4: FPNP2–BSA clusters; lane 5: supernatant of FPNP2; lane 6: FPNP3–BSA clusters; and lane 7: supernatant of FPNP3. | |
Fluorescence sensitivity for BSA detection
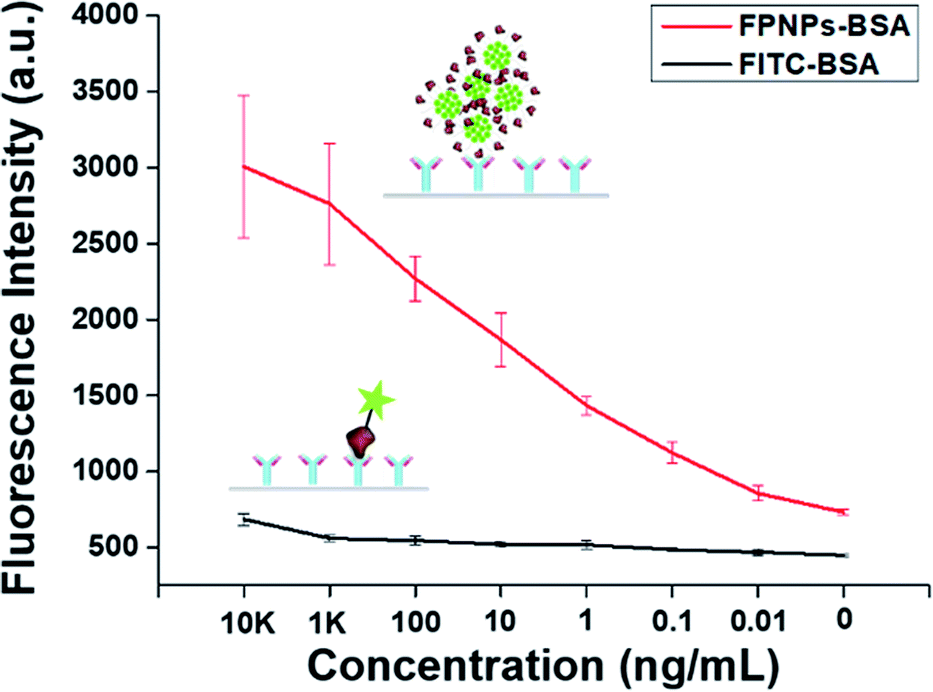
To demonstrate the applicability of the optical biosensor in enhancing the sensitivity of immunoassays, we customized the fluorescence sensitivity using the prepared FPNPs-based BSA detection section and compared our results with those obtained using FITC–BSA with BSA as a model analyte. The fluorescence intensity following anti-BSA coating was measured as a function of the anti-BSA concentration. Thus, Fig. 6a and b show calibration plots and the data table for the FPNP–BSA clusters and FITC–BSA based fluorescence sensitivities, and for BSA detection, respectively. As indicated, the fluorescence sensitivity of the FPNPs showed an obvious difference between the fluorescence intensity and the anti-BSA concentration over a concentration range of 0.01 to 104 ng mL−1, where the detection limit for BSA was 0.01 ng mL−1. In comparison, the FITC–BSA based fluorescence sensitivity for BSA detection exhibited a slight difference in the fluorescence intensity over a concentration range of 1 to 104 ng mL−1, giving a BSA detection limit of 1 ng mL−1. These results therefore indicate that the prepared FPNPs exhibited highly sensitive BSA detection over a large concentration range, thereby confirming that they could be applicable in highly sensitive optical immunoassays for detecting low concentrations of specific biomarkers.
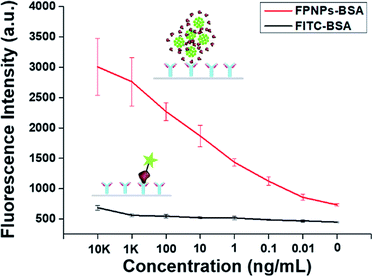 |
| | Fig. 6 Fluorescence sensitivity for BSA detection: calibration curve and results obtained for anti-BSA concentrations ranging from 0 to 10 μg mL−1. Error bars represent the standard deviation determine from at least three replicate measurements. | |
Conclusions
In summary, we note that this study is the first of its kind in reporting the self-assembly of novel fluorescein polymers with different molar ratios to produce a range of novel fluorescein polymeric nanoparticles (FPNPs) with tunable sizes and surface charges, in addition to biomolecule immobilization and high sensitivities, for application as optical biosensors. We demonstrated that novel amphiphilic fluorescein polymers, i.e., poly(FMA-r-MA-r-NAS), were successfully synthesized via a simple radical polymerization process, and that these polymers could self-assemble into NPs exhibiting a good stability and an enhanced fluorescence intensity, via a one-step synthetic process. The physicochemical properties of the prepared FPNPs were optimized using poly(FMA-r-MA-r-NAS) random copolymers with different monomer ratios under mild nanoprecipitation conditions. Due to the incorporation of the active N-acryloxysuccinimide (NAS) group, the NPs easily conjugated to the bovine serum albumin (BSA) biomolecule in an aqueous environment. More importantly, the fluorescence emission intensity was significantly enhanced, and was found to exhibit a good stability in solution. Moreover, the fluorescence sensitivity for BSA detection by the prepared FPNPs exhibited a significantly higher sensitivity compared to the conventional fluorescein isothiocyanate (FITC)–BSA conjugate, with a BSA detection limit of 0.01 ng mL−1 being achieved. As such, their facile preparation method and high fluorescence sensitivity render these FPNPs practically appealing. More importantly, this work provides new insights into the highly sensitive self-assembly behavior of fluorescein polymers, thereby indicating that the developed novel FPNPs exhibit potential for use as platforms in biomarker analysis, cancer detection, and immunoassays in a range of diagnostic fields.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the grant of Korea Institute of Ceramic Engineering and Technology (KICET).
Notes and references
- F. Canfarotta, M. J. Whitcombe and S. A. Piletsky, Biotechnol. Adv., 2013, 31, 1585–1599 CrossRef CAS PubMed.
- A. Reisch and A. S. Klymchenko, Small, 2016, 12, 1968–1992 CrossRef CAS PubMed.
- P. Mi, F. Wang, N. Nishiyama and H. Cabral, Macromol. Biosci., 2017, 17, 1600305 CrossRef PubMed.
- J. U. Menon, P. Jadeja, P. Tambe, K. Vu, B. Yuan and K. T. Nguyen, Theranostics, 2013, 3, 152–166 CrossRef CAS PubMed.
- K. Yokoyama, H. Cho, S. P. Cullen, M. Kowalik, N. M. Briglio, H. J. Hoops, Z. Zhao and M. A. Carpenter, Int. J. Mol. Sci., 2009, 10, 2348–2366 CrossRef CAS PubMed.
- X. Zhang, K. Wang, M. Liu, X. Zhang, L. Tao, Y. Chen and Y. Wei, Nanoscale, 2015, 7, 11486–11508 RSC.
- S. Takalkar, K. Baryeh and G. Liu, Biosens. Bioelectron., 2017, 98, 147–154 CrossRef CAS PubMed.
- K. Li and B. Liu, Chem. Soc. Rev., 2014, 43, 6570–6597 RSC.
- G. M. Soliman, Int. J. Pharm., 2017, 523, 15–32 CrossRef CAS PubMed.
- X. Zhang, X. Zhang, B. Yang, J. Hui, M. Liu, Z. Chi, S. Liu, J. Xu and Y. Wei, Polym. Chem., 2014, 5, 683–688 RSC.
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- R. Jenkins, M. K. Burdette and S. H. Foulger, RSC Adv., 2016, 6, 65459–65474 RSC.
- H. Peng, X. Liu, G. Wang, M. Li, K. M. Bratlie, E. Cochran and Q. Wang, J. Mater. Chem. B, 2015, 3, 6856–6870 RSC.
- M. P. Robin and R. K. O'Reilly, Polym. Int., 2015, 64, 174–182 CrossRef CAS.
- Y. Chang, Y. Li, S. Yu, J. Mao, C. Liu, Q. Li, C. Yuan, N. He, W. Luo and L. Dai, Nanotechnology, 2015, 26, 025103 CrossRef CAS PubMed.
- Z. Wu, J. Appl. Polym. Sci., 2008, 110, 777–783 CrossRef CAS.
- B. Sierra-Martin and A. Fernandez-Barbero, Adv. Colloid Interface Sci., 2016, 233, 25–37 CrossRef CAS PubMed.
- H. Lu, F. Su, Q. Mei, Y. Tian, W. Tian, R. H. Johnson and D. R. Meldrum, J. Mater. Chem., 2012, 22, 9890–9900 RSC.
- F. Thielbeer, S. V. Chankeshwara and M. Bradley, Biomacromolecules, 2011, 12, 4386–4391 CrossRef CAS PubMed.
- C. Kaewsaneha, P. Tangboriboonrat, D. Polpanich and A. Elaissari, ACS Appl. Mater. Interfaces, 2015, 7, 23373–23386 CrossRef CAS PubMed.
- W. Ma, A. Saccardo, D. Roccatano, D. Aboagye-Mensah, M. Alkaseem, M. Jewkes, F. Di Nezza, M. Baron, M. Soloviev and E. Ferrari, Nat. Commun., 2018, 9, 1489 CrossRef PubMed.
- Y. H. Han, R. K. Kankala, S. B. Wang and A. Z. Chen, Nanomaterials, 2018, 8, 360 CrossRef PubMed.
- D. Sung, S. Yang, J. W. Park and S. Jon, Biomed. Microdevices, 2013, 15, 691–698 CrossRef CAS PubMed.
- M. Mitra, M. Kandalam, J. Rangasamy, B. Shankar, U. K. Maheswari, S. Swaminathan and S. Krishnakumar, Mol. Vision, 2013, 19, 1029 CAS.
- S. K. Singh, S. Shrivastava, M. K. Nayak, A. S. K. Sinha, M. V. Jagannadham and D. Dash, J. Bionanosci., 2009, 3, 88–96 CrossRef CAS.
- D. Sung, D. H. Shin and S. Jon, Biosens. Bioelectron., 2011, 26, 3967–3972 CrossRef CAS PubMed.
- J. Y. Oh, H. S. Kim, L. Palanikumar, E. M. Go, B. Jana, S. A. Park, H. Y. Kim, K. Kim, J. K. Seo, S. K. Kwak, C. Kim, S. Kang and J. H. Ryu, Nat. Commun., 2018, 9, 4548 CrossRef PubMed.
- A. Pollak, H. Blumenfeld, M. Wax, R. L. Baughn and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 6324–6336 CrossRef CAS.
- D. Sung, S. Park and S. Jon, Langmuir, 2012, 28, 4507–4514 CrossRef CAS PubMed.
- X. Wu, Y. Qiao, H. Yang and J. Wang, J. Colloid Interface Sci., 2010, 349, 560–564 CrossRef CAS PubMed.
- D. Sung and S. Yang, Electrochim. Acta, 2014, 133, 40–48 CrossRef CAS.
- W. I. Choi, N. Kamaly, L. Riol-Blanco, I.-H. Lee, J. Wu, A. Swami, C. Vilos, B. Yameen, M. Yu and J. Shi, Nano Lett., 2014, 14, 6449–6455 CrossRef CAS PubMed.
- F. Xu, H. Li, Y.-L. Luo and W. Tang, ACS Appl. Mater. Interfaces, 2017, 9, 5181–5192 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09106h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Won Il Choi
a,
Won Il Choi