
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid polymers bearing oligo-L-lysine(carboxybenzyl)s: synthesis and investigations of secondary structure†
Merve Basak Canalp and
Wolfgang H. Binder *
*
Faculty of Natural Science II (Chemistry, Physics and Mathematics), Martin Luther University Halle-Wittenberg, von-Danckelmann-Platz 4, Halle (Saale) D-06120, Germany. E-mail: wolfgang.binder@chemie.uni-halle.de
First published on 7th January 2020
Abstract
Hybrid polymers of peptides resembling (partially) folded protein structures are promising materials in biomedicine, especially in view of folding-interactions between different segments. In this study polymers bearing repetitive peptidic folding elements, composed of N-terminus functionalized bis-ω-ene-functional oligo-L-lysine(carboxybenzyl(Z))s (Lysn) with repeating units (n) of 3, 6, 12, 24 and 30 were successfully synthesized to study their secondary structure introduced by conformational interactions between their chains. The pre-polymers of ADMET, narrowly dispersed Lysns, were obtained by ring opening polymerization (ROP) of N-carboxyanhydride (NCA) initiated with 11-amino-undecene, following N-terminus functionalization with 10-undecenoyl chloride. The resulting Lysns were subsequently polymerized via ADMET polymerization by using Grubbs’ first generation (G1) catalyst in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) generating the ADMET polymers (A-[Lysn]m) (m = 2–12) with molecular weights ranging from 3 to 28 kDa, displaying polydispersity (Đ) values in the range of 1.5–3.2. After chemical analyses of Lysns and A-[Lysn]ms by 1H-NMR, GPC and MALDI-ToF MS, secondary structural investigations were probed by CD spectroscopy and IR spectroscopy in 2,2,2-trifluoroethanol (TFE). In order to study A-[Lysn]ms with defined molecular weights and low polydispersity values (Đ = 1.03–1.48), the ADMET polymers A-[Lysn=3]m=3 and A-[Lysn=24]m=4 were fractionated by preparative GPC, and subsequently analysed by 1H-NMR, analytical GPC, MALDI-ToF MS and CD spectroscopy. We can demonstrate the influence of chain length of the generated polymers on the formation of secondary structures by comparing Lysns with varying n values to the ADMET-polymers with the help of spectroscopic techniques such as CD and FTIR-spectroscopy in a helicogenic solvent.
Introduction
Advances in the synthesis of biologically active macromolecules have paved the way for understanding the pathogenesis of many health issues such as cancer1 and neurodegenerative diseases, i.e., Alzheimer's2–5 and Parkinson's,6 in order to provide novel pharmaceuticals and drug/gene delivery systems for their treatments.7 In particular, peptides and their conjugates that can meet the requirements of pharmacological activities because they form different reversible secondary structures, for instance α-helix and β-sheet, induced by physicochemical changes, are of special relevance.8–12 Thus, in the field of peptide chemistry, poly-L-lysine apparently holds importance for comprehending the native states of proteins, and thus can influence the drug discoveries for amyloidosis and siRNA/DNA deliveries.13,14 While poly-L-lysine forms various secondary structures depending on the physical and chemical environment, such as e.g., pH, T, solvent, functional end/side groups and chain length,15–22 hybrid polymers consisting of copolymers of this biologically compatible peptide can embody secondary structures with well-defined physical characteristics introducing additional stability, rigidity and flexibility into the overall polymeric system.23–26Various hybrid polymers containing poly-L-lysine/lysine(Z) with defined architectures, i.e., block copolymers, have been synthesized to integrate the biochemical features, e.g. pH responsivity, α-helicity within the synthetic polymer chain, and therewith assess and control functionality of the assembly formation. Diblock copolymers of polyisoprene-block-poly-L-lysine/lysine(Z) have been synthesized by combination of anionic polymerization and ROP methods to obtain ‘‘hybrid’’ rod–coil block copolymers based on a synthetic coil segment and a polypeptide chain, displaying an α-helical conformation for use in nanotechnology.27 Poly(styrene)388-block-poly(L-lysine)138 diblock copolymer changes its assembly to form pH responsive aggregate when mixed with a non-ionic surfactant, such as C12E6.28 The secondary structure of poly(butadiene)107-block-poly(L-lysine)27 has been probed by CD spectroscopy as a function of pH and temperature, suggesting that an α-helix of the poly(L-lysine) block transforms into β-sheets at higher temperature (T = 60 °C) at a pH = 11.2.29 Another study has yet adjoined the secondary structural features of poly(L-lysine), combined with the thermo-responsive behaviour of poly(N-isopropylacrylamide) to achieve amphiphilic hybrid rod-coil block copolymers by combination of atom transfer radical polymerization (ATRP) and ROP.30
In this work it was aimed to combine the variable secondary structure of the oligo-L-lysine(Z) in combination with ethylene chains in a repetitive manner to investigate secondary structural changes such as α-helicity induced assemblies of the so obtained hybrid polymers as represented in Fig. 1. Based on a combination of NCA ROP of N-carboxyanhydrides and ADMET polymerization techniques it is aimed to obtain hybrid polymers with the desired peptide/ethylene units, based on ADMET polymers of oligo-L-lysine(Z)s (green in Fig. 1). As the intermediate oligo-ethylene-segments (shown in grey, Fig. 1) can be considered as noninteracting in the sense of a supramolecular interaction, the so chosen molecular design allows to primarily study the intersegmental interactions of the (partially) folded oligo-L-lysine(Z)s, probed by CD- and IR-methods in TFE. In order to obtain reliable information as to the conformational status of the oligo-L-lysine(Z)s-segment, careful fractionation by preparative GPC was accomplished to enable a detailed study of the intersegmental conformational changes.
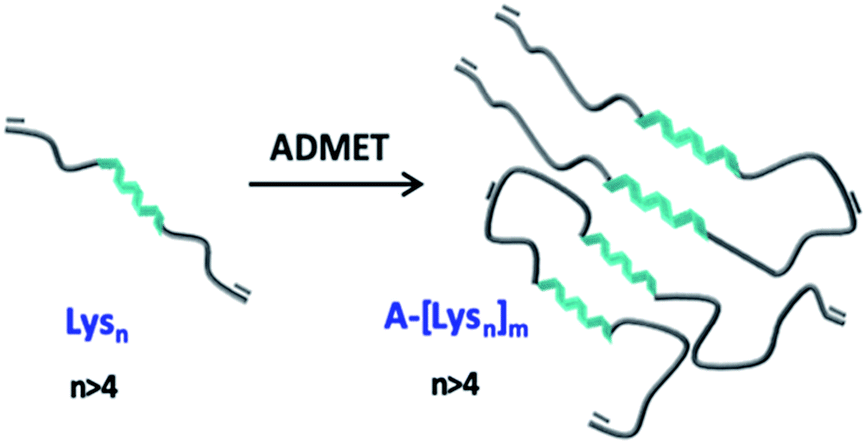 | ||
| Fig. 1 Schematic representation of α-helicity induced assemblies via intersegmental interactions of the oligo-L-lysine(Z)s-segments (green) along the ADMET-polymer, interrupted by oligo-ethylene-segments (grey). | ||
Experimental section
Materials and methods
DMF was dried over CaH2 and freshly distilled by applying vacuum (20 mbar) at 50 °C. Ethyl acetate was dried at 100 °C with P2O5. N-heptane was refluxed over sodium/benzophenone at 120 °C. All of the dried solvents were flushed with N2 gas before usage. All of the reagents were purchased from Sigma-Aldrich. All NMR-spectra were recorded on a Varian spectrometer (Gemini 200, Gemini 2000 and Unity 500) at 400 or 500 MHz at 27 °C. Trimethylsilane was used as internal standard. Deuterated chloroform (CDCl3) and dimethyl sulfoxide (DMSO-d6) were used as solvent. In the case of all polymer samples trifluoroacetic acid (TFA) (15% of volume) was added to the deuterated chloroform (CDCl3). All chemical shifts (δ) were reported in parts per million (ppm) and the coupling constant (J) in Hertz. For the interpretation of the spectra MestReNova v. 6.0.2 5475 was used. For analytical GPC measurements a Viscotek GPCmax VE 2001 with an HHR-H Guard-17369 and a GMHHR-N-18055 column in DMF at 60 °C was used. The sample concentration was 5 mg mL−1, the injection volume was 100 μL. The detection was carried out via the refractive index with a VE 3580 RI detector of Viscotek at a temperature of 35 °C and a flow rate of 1 mL min−1. Polystyrene standards with molecular weights from 1 kDa to 115 kDa were used for external calibration. Preparative GPC was performed by a KD-2002.5 column from Shodex company attached on a VWR Hitachi Chromaster instrument using DMF (HPLC graded) as the eluent at 55 °C with a flow rate of 0.70 mL min−1 injecting sample in a concentration of 15 mg mL−1. Refractive index detector from VWR at 50 °C was employed as the detector. The obtained data were analysed by using EZChrom Elite (version 3.3.2 SP2) software. ESI-ToF measurements were performed on a Focus micro ToF by Bruker Daltonics. The sample (1.00 mg) was dissolved in methanol (1.00 mL, HPLC grade) and directly infused (180.00 μL h−1, positive or negative mode). MALDI-TOF MS measurements were carried out in either linear or reflector modes on a Bruker Autoflex III Smart beam equipped with a nitrogen laser source (λ = 337 nm). The samples were dissolved in DMF (HPLC grade) (c = 5 or 10 mg mL−1) with the ration of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:1 (matrix:analyte:salt). As matrix dithranol and/or DCTB with c = 10 mg mL−1 in THF, and as salt potassium trifluoroacetate (KTFA), sodium trifluoroacetate (NaTFA) and/or lithium trifluoroacetate (LiTFA) with c = 5 mg mL−1 in THF were used. For the data evaluations and simulation of the mass spectra of the polymer samples the computer programme Flex analysis (version 3.0) was used. CD spectroscopy measurements were performed with the instrument, JASCO Corp., J-810, Rev. 1.00, at a constant temperature (20 °C). The UV absorption was measured in CD units of millidegrees in the wavelength range of 260–190 nm. The cuvette cell used had a diameter of 0.1 cm. The correction of the measurements was done by subtraction of the absorption of the solvent, i.e., 2,2,2-trifluoroethanol (TFE), from the absorbance of the sample which had a concentration of 0.2 mg mL−1. FTIR experiments were performed on a VERTEX 70 IR spectrometer from Bruker. For solid state investigations by FTIR spectroscopy a single reflex-diamond attenuated total reflectance unit was utilized. Samples prepared in TFE at different concentrations were investigated with a Specac Omni Cell demountable cell purchased from Sigma-Aldrich with CaF2 windows and a poly(tetrafluoroethylene) spacer which is 0.1 mm by measuring the pure solvent (TFE) as the background. The absorption bands were recorded in cm−1 in an area of 1500–1800 cm−1. For our data interpretation OPUS software was used.
:10:1 (matrix:analyte:salt). As matrix dithranol and/or DCTB with c = 10 mg mL−1 in THF, and as salt potassium trifluoroacetate (KTFA), sodium trifluoroacetate (NaTFA) and/or lithium trifluoroacetate (LiTFA) with c = 5 mg mL−1 in THF were used. For the data evaluations and simulation of the mass spectra of the polymer samples the computer programme Flex analysis (version 3.0) was used. CD spectroscopy measurements were performed with the instrument, JASCO Corp., J-810, Rev. 1.00, at a constant temperature (20 °C). The UV absorption was measured in CD units of millidegrees in the wavelength range of 260–190 nm. The cuvette cell used had a diameter of 0.1 cm. The correction of the measurements was done by subtraction of the absorption of the solvent, i.e., 2,2,2-trifluoroethanol (TFE), from the absorbance of the sample which had a concentration of 0.2 mg mL−1. FTIR experiments were performed on a VERTEX 70 IR spectrometer from Bruker. For solid state investigations by FTIR spectroscopy a single reflex-diamond attenuated total reflectance unit was utilized. Samples prepared in TFE at different concentrations were investigated with a Specac Omni Cell demountable cell purchased from Sigma-Aldrich with CaF2 windows and a poly(tetrafluoroethylene) spacer which is 0.1 mm by measuring the pure solvent (TFE) as the background. The absorption bands were recorded in cm−1 in an area of 1500–1800 cm−1. For our data interpretation OPUS software was used.
Synthesis of N-carboxyanhydride (NCA) monomer
The synthesis of NCA of N-carboxybenzyl(Z)-L-lysine has been reported in the literature and was performed similar to our previously reported work.22,31Ring opening polymerization (ROP) of NCA monomer
ROP of NCA monomer was performed similar to the procedure with small modifications on the initiator to monomer ratio in order to achieve different degree of polymerizations.22,31N-terminus functionalization of mono-functional oligo-L-lysine(carboxybenzyl(Z))s (lysn)
The amidation reactions of N-terminus of oligo-L-lysine(Z)s were performed similar to the previously reported work (see ESI†).22ADMET polymerization of N-terminus functionalized oligo-L-lysine(carboxybenzyl(Z))s (A-[Lysn]m)
A general procedure was followed for the synthesis of ADMET polymers. A pre-dried Schlenk flask equipped with a stirrer filled with the respective pre-polymer Lysn. As a sample case, the pre-polymer Lysn=3 (1000 mg) was dried under high vacuum for 72 hours before addition of small amount of HFIP (≈2 mL) under nitrogen atmosphere, just enough to dissolve Lysn=3 at 45 °C. After 30 minutes, a trace amount of G1 (≈0.1 mg) was added to the reaction mixture under nitrogen atmosphere (N2 atm) and let stir for 16 hours. Addition of trace amounts of G1 was repeated and after 2 hours of stirring under N2 atm at 45 °C, gradual vacuum of 850 mbar to 700 mbar was applied continuously. Following this, the reaction medium was let stir under stable vacuum at 700 mbar for another 16 hours. Eventually the stirring was stopped due to high viscosity, therefore the viscous dark yellow/green substance was dissolved in DMF (≈1–2 mL) and subsequently precipitated in cold methanol (≈30 mL). The resulting dark yellow precipitate was washed several times with methanol and centrifuged until light yellow/white solid was obtained as the final product (A-[Lysn=3]m) (795 mg, yield: 70–80 wt%). 1H-NMR analysis of polymer was prepared in a mixture of CDCl3 and TFA (volume of 15%). Analytical GPC and MALDI-ToF MS samples were prepared in HPLC graded DMF (see Table 1, and ESI†).| Entry | Pre-polymers (Lysn) | ADMET polymers (A-[Lysn]m) | Solvent | Catalyst | DPNMR (n, m) | Mn, GPC [kDA] | Đ |
|---|---|---|---|---|---|---|---|
| I | Lysn=3 | A-[Lysn=3]m=3 | HFIP | G1 | 4, 3 | 3.7 | 2.2 |
| II | Lysn=3 | A2-[Lysn=3]m=2 | HFIP | G1 | 4, 2 | 2.9 | 1.5 |
| III | Lysn=6 | A-[Lysn=6]m=12 | HFIP | G1 | 6, 12 | 11.2 | 3.2 |
| IV | Lysn=6 | A2-[Lysn=6]m=2 | HFIP | G1 | 6, 2 | 3.7 | 1.4 |
| V | Lysn=6 | A3-[Lysn=6]m=18 | TFE | G1 | 6, 18 | 20.9 | 4.8 |
| VI | Lysn=12 | A-[Lysn=12]m=8 | HFIP | G1 | 12, 8 | 15.7 | 2.4 |
| VII | Lysn=24 | A-[Lysn=24]m=4 | HFIP | G1 | 24, 4 | 10.6 | 1.7 |
| VIII | Lysn=24 | A2-[Lysn=24]m=3 | HFIP | G1 | 24, 3 | 7.5 | 1.5 |
| IX | Lysn=30 | A-[Lysn=30]m=7 | HFIP | G1 | 30, 7 | 27.8 | 2.4 |
Results and discussion
Our synthetic strategy is based on a combination of ring opening polymerization (ROP) of N-carboxyanhydride (NCA), followed by ADMET polymerization of appropriately bis-ene-functionalized oligomers to place the folding-elements of Lysns along a polymer chain, subsequently studying their intra- and interchain secondary structures. Acyclic diene metathesis (ADMET) polymerization32–34 opens up possibilities to introduce different kinds of functional groups35–38 into a polyethylene chain, introducing even anti-inflammatory drugs, such as ibuprofen and naproxen, into a polyethylene backbone.39The ROP of NCA monomers is a viable and proven strategy to obtain artificial homo- and block co-peptides, where end group functionalized polymers can be obtained, given that the ROP mechanism follows the aspects of “livingness”.40,41 Firstly, mono-functional oligo-L-lysine(Z)s with varying repeating units (n = 3, 6, 12, 24, 30) were synthesized via ROP of the lysine-NCA monomer using 11-aminoundece as the primary initiator in DMF. It has been known that livingness and end group fidelity during ROP of NCAs can be achieved via the “Normal Amine Mechanism (NAM)” using primary or secondary amines as initiators.20,42–44 Therefore the NCA monomer of N-carboxybenzyl(Z)-L-lysine amino acid, NCA(Z), was initiated via the primary amine, 11-aminoundecene, to obtain the mono-functional oligo-L-lysine(carboxybenzyl(Z))s in the first step. After proving the chain length and end groups via NMR- and MS-analyses (see ESI†), the N-terminal end groups were functionalized by direct amidation with 10-undecenoyl chloride in situ to achieve the respective bis-ω-ene-functional pre-polymers, Lysns.22
Relying on the excellent possibilities of ADMET polymerization to include amino acids onto a polyolefin chain,45–47 the Lysns (n = 3, 6, 12, 24, 30) were further polymerized by ADMET polymerization in HFIP by addition of Grubb's first generation catalyst (G1) (see Scheme 1 and Table 1). All obtained Lysns and A-[Lysn]ms were characterized by 1H-NMR, MALDI-ToF MS and analytical GPC (see Table 2 and ESI†). Additionally, the ADMET polymers A-[Lysn=3]m=3 and A-[Lysn=24]m=4 were fractionated by preparative GPC and then selectively investigated by MALDI-ToF MS, analytical GPC, 1H-NMR (see Table 2, Fig. 2, 3 and ESI†). Structural investigations of Lysns, A-[Lysn]ms and the selected fractions of A-[Lysn]ms were mainly done by CD spectroscopy in TFE (see Table 2, Fig. 4, 5 and also ESI†). IR spectroscopy in TFE was also probed to analyse secondary structure features of Lysns and A-[Lysn]ms (see Fig. 6 and ESI†).
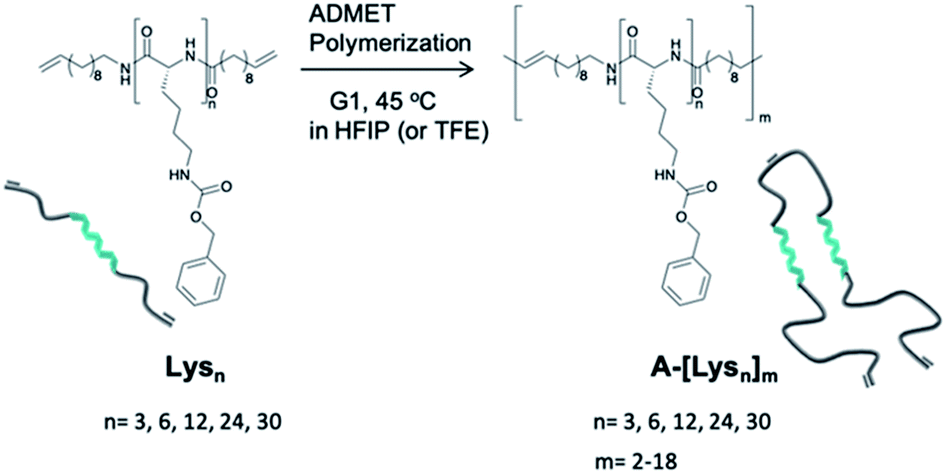 | ||
| Scheme 1 Schematic representation of the synthetic pathway of ADMET polymerization of Lysn22 by using Grubb's first catalyst (G1). | ||
| Entry | Samples | DPtheo. (n) | DPNMR (n, m) | Mn, MALDI [kDA] | Mn, GPC in DMF [kDA] | Đ | CD-Spec. (in TFE) |
|---|---|---|---|---|---|---|---|
| Oligo-L-lysine(Z)s | |||||||
| I | Lysn=3 | 3 | 4 | 2.2 ± 1.8 | 1.9 | 1.06 | β II turn |
| II | Lysn=6 | 6 | 6 | 3.2 ± 1.6 | 2.8 | 1.17 | 25% α-helicity |
| III | Lysn=12 | 12 | 11 | 3.3 ± 1.7 | 3.3 | 1.36 | 27% α-helicity |
| IV | Lysn=24 | 24 | 23 | 3.6 ± 1.2 | 6.3 | 1.40 | 53% α-helicity |
| V | Lysn=30 | 30 | 30 | 5.6 ± 4 | 8.0 | 1.33 | 65% α-helicity |
|
|||||||
| ADMET polymers of oligo-L-lysine(Z)s & the selected fractions | |||||||
| VI | A-[Lysn=3]m=3 | 3 | 3 | 2.5 ± 1.2 | 3.7 | 2.20 | β II turn |
| VII | F10 + 11 + 12 | 3 | NA | NA | 23.2 | 1.21 | 18% α-helicity |
| VIII | F13 + 14 | 3 | NA | 8.1 ± 3 | 12.8 | 1.12 | 15% α-helicity |
| IX | F15 | 3 | NA | 6.5 ± 2 | 9.2 | 1.08 | β II turn |
| X | F17 | 3 | NA | 2.8 ± 1.7 | 5.8 | 1.05 | β II turn |
| XI | F18 | 3 | NA | 2.6 ± 1.6 | 4.7 | 1.05 | β II turn |
| XII | F21 + 22 + 23 | 3 | NA | 2.6 ± 0.5 | 2.6 | 1.04 | β II turn |
| XIII | F24 + 25 | 3 | NA | 2.1 ± 0.5 | 2.1 | 1.03 | β II turn |
| XIV | A-[Lysn=6]m=12 | 6 | 12 | 4.3 ± 2.5 | 11.2 | 3.21 | 32% α-helicity |
| XV | A-[Lysn=12]m=8 | 12 | 8 | 4.7 ± 3.2 | 15.7 | 2.43 | 44% α-helicity |
| XVI | A-[Lysn=24]m=4 | 24 | 4 | 4.1 ± 2.4 | 10.6 | 1.74 | 69% α-helicity |
| XVII | F12 | 24 | NA | NA | 23.2 | 1.16 | 77% α-helicity |
| XVIII | F13 | 24 | 4 | NA | 19.1 | 1.15 | 62% α-helicity |
| XIX | F15 | 24 | NA | 14.2 ± 2 | 12.1 | 1.07 | 53% α-helicity |
| XX | F16 | 24 | NA | 13.7 ± 2 | 10.9 | 1.07 | 52% α-helicity |
| XXI | F18 | 24 | NA | 7.7 ± 2 | 7.1 | 1.08 | 40% α-helicity |
| XXII | A-[Lysn=30]m=7 | 30 | 7 | 4.3 ± 2 | 27.8 | 2.41 | 79% α-helicity |
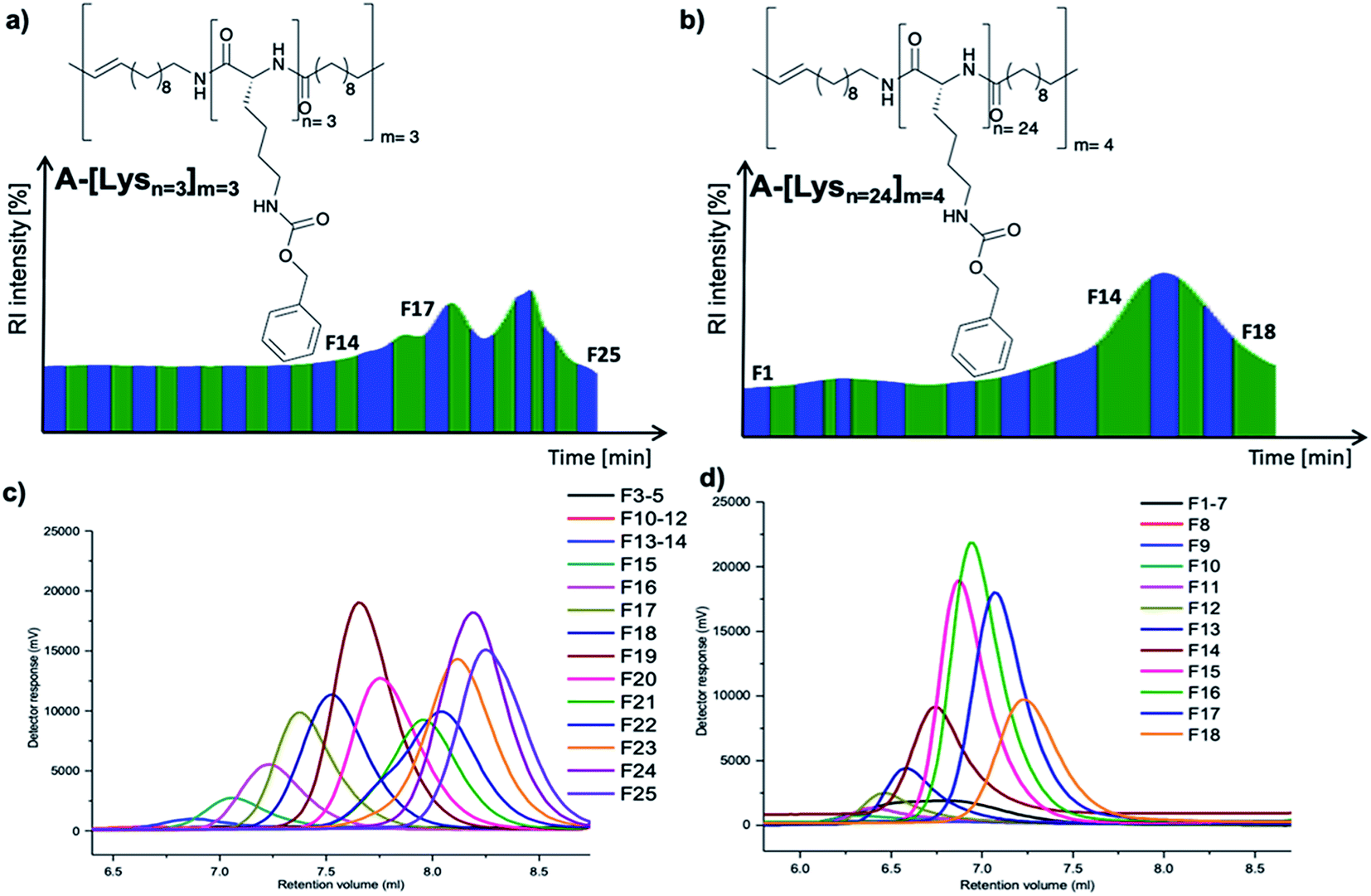 | ||
| Fig. 2 Preparative GPC graphs of (a) A-[Lysn=3]m=3 (b) A-[Lysn=24]m=4 along with their analytical GPC results of respective (c) fractions of A-[Lysn=3]m=3 and (d) fractions of A-[Lysn=24]m=4. | ||
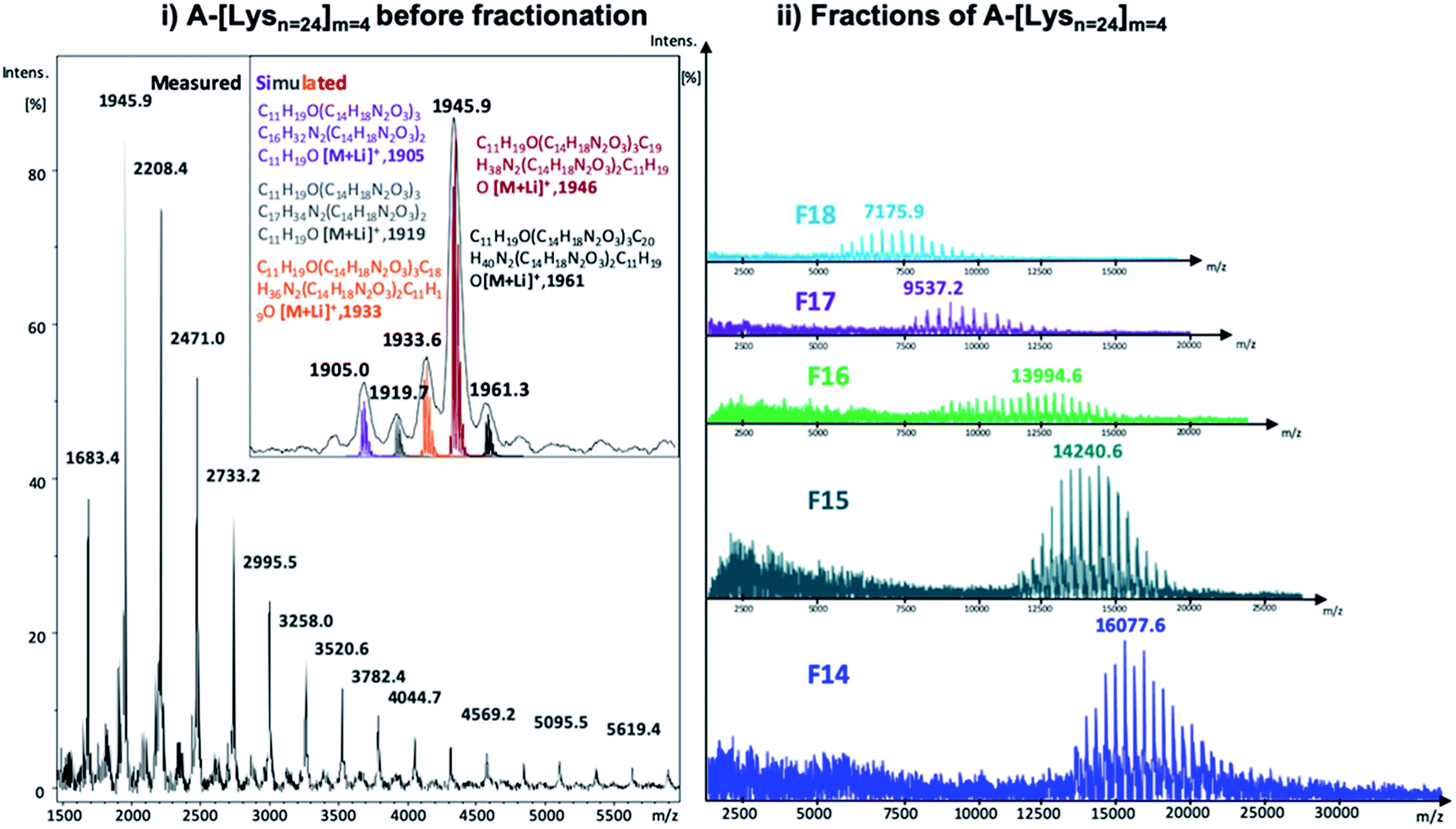 | ||
| Fig. 3 MALDI-ToF MS spectra of (i) A-[Lysn=24]m=4 before fractionation along with the simulated isotopic patterns (reflector mode) and (ii) the selected fractions of A-[Lysn=24]m=4 (linear mode) (also see ESI†). | ||
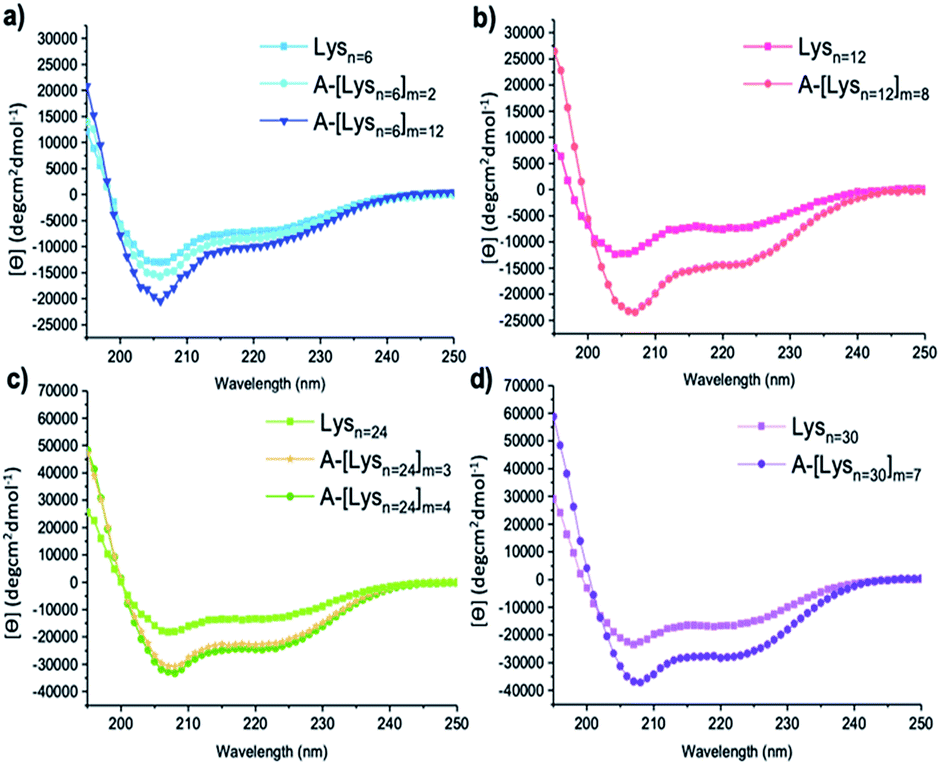 | ||
| Fig. 4 CD spectra of (a) Lysn=6, A-[Lysn=6]m=2, A-[Lysn=6]m=12 (b) Lysn=12, A-[Lysn=12]m=8 (c) Lysn=24, A-[Lysn=24]m=3, A-[Lysn=24]m=4, (d) Lysn=30 and A-[Lysn=30]m=7. (c = 2 mg mL−1 in TFE). | ||
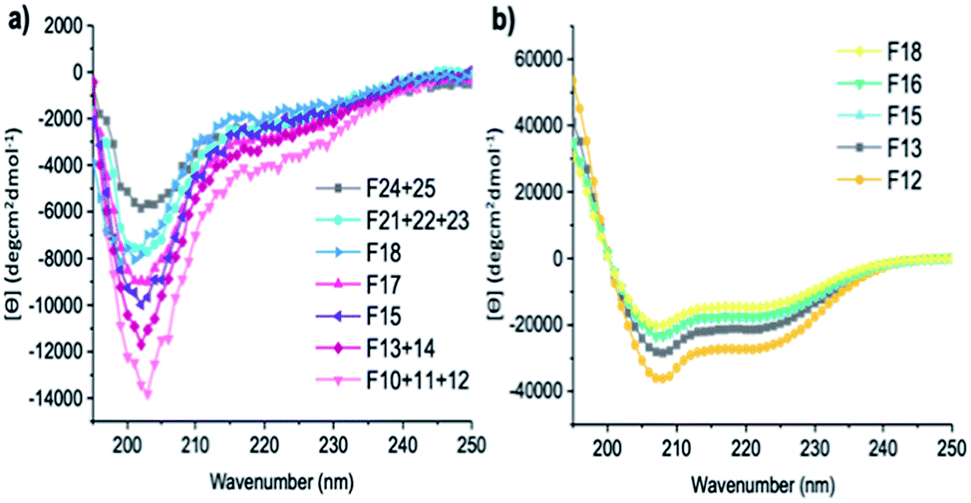 | ||
| Fig. 5 CD spectra of fractions of (a) A-[Lysn=3]m=3 (F10 + 11 + 12, F13 + 14, F15, F17, F18, F21 + 22 + 23, F24 + 25) and (b) A-[Lysn=24]m=4 (F12, F13, F15, F16, F18). (c = 2 mg mL−1 in TFE). | ||
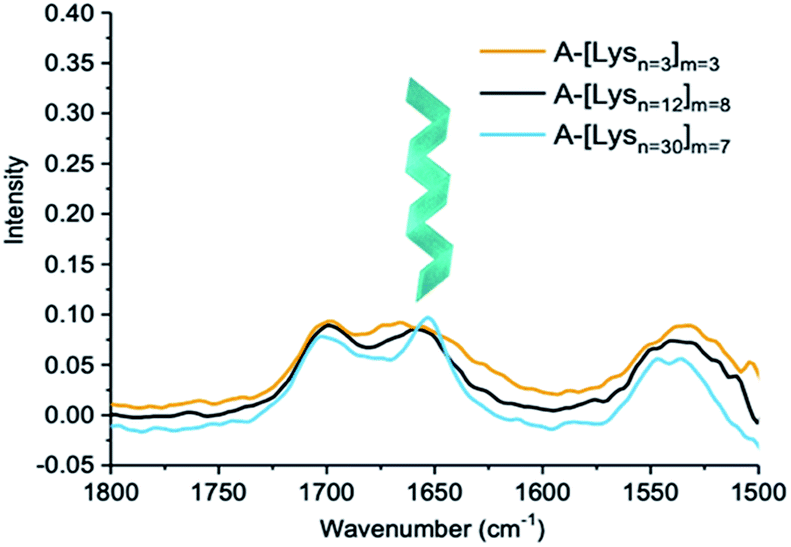 | ||
| Fig. 6 IR spectra of ADMET polymers A-[Lysn=3]m=3, A-[Lysn=12]m=8 and A-[Lysn=30]m=7 (c = 5 mg mL−1 in TFE). | ||
Fractionation of ADMET polymers by preparative GPC
With ADMET being a polycondensation reaction, the obtained polymers display polydispersity (PDI) values in the range of 1.5–3.2. As the chain length dependent folding studies requires smaller PDIs, the fractional analyses of selected ADMET polymers were accomplished with the help of preparative GPC of these hybrid polymers, proving their low polydispersity (Đ) values (see Table 2, and also ESI Tables 1S and 2S†).Thus, ADMET polymers A-[Lysn=3]m=3 (Table 2, entry VI) and A-[Lysn=24]m=4 (Table 2, entry XVI) were separated into fractions (Table 2, entries VII–XIII for the former and Table 2, entries XVII–XXI for the letter) according to their molecular weights, thus displaying significantly lower Đ values than their native unfractionated samples via preparative GPC technique in DMF at a column temperature of 55 °C.
Preparative GPC traces of A-[Lysn=3]m=3 (Fig. 2a) and A-[Lysn=24]m=4 (Fig. 2b) along with the analytical GPC results of their fractions (Fig. 2c and d respectively) are demonstrated. As depicted in Fig. 2a and b, all fractions are named as Fx, i.e., x is the alignment number of the fraction trace in the preparative GPC graph. The complete analytical GPC results of all the fractions of A-[Lysn=3]m=3 (Table 1S†) and A-[Lysn=24]m=4 (Table 2S†) are present within the ESI.†
As can be seen from the obtained analysis results, molecular weights of fractions of ADMET polymer A-[Lysn=3]m=3 (Table 2, entry VI) increases with the decreasing number of the fraction trace number, e.g. F25 (Mn ≈ 2 kDa) and F15 (Mn ≈ 10 kDa) (see ESI, Table 1S†). All obtained fractions of A-[Lysn=3]m=3 measured by analytical GPC in DMF display low Đ values (1.20 > Đ > 1.03) which proves the successful separation of the fractions.
In the case of the high molecular weight ADMET polymer, A-[Lysn=24]m=4 (Table 2, entry XVI), molecular weight of the fractions also increases with the decreasing number of the fraction trace number, e.g. F18 (Table 2, entry XXI) (Mn ≈ 7.2 kDa) and F10 (Mn ≈ 23 kDa) (see ESI, Table 2S†). All obtained fractions of A-[Lysn=24]m=4 measured by analytical GPC in DMF have low Đ values (1.48 > Đ > 1.07) when they are compared with ADMET polymers which have Đ values over 2.0 in general.
In Fig. 3, the measured MALDI-ToF MS spectra of A-[Lysn=24]m=4 before fractionation with the simulated isotopic patterns (Fig. 3i) and after fractionation of selected fractions of A-[Lysn=24]m=4, i.e., F14–F18 (Fig. 3ii) are shown. The measured MALDI spectra and the respective simulated isotopic patterns of A-[Lysn=24]m=4 (Fig. 3i) prove the ADMET polymer structure, having –CH2 characteristic difference due to the fragmentated isomers of ADMET polymers, where the simulated and the measured spectra match. Similar to the analytical GPC investigations, the measured MALDI spectra of selected fractions of A-[Lysn=24]m=4 in Fig. 3ii verify the increasing molecular weight of the fractions with decreasing fraction trace number obtained by preparative GPC (see Fig. 2 and also ESI, Table 2S†). According to the measured MALDI spectra in Fig. 3ii, F14 has a molecular weight of ≅16 ± 3 kDa whereas the low molecular weight fraction F18 has a molecular weight of ≅7 ± 3 kDa, which evidences an average molecular weight difference of 9 kDa. Therefore, all these fractional analyses of ADMET polymers are suited for the subsequent secondary structural investigations.
Conformational investigations by spectroscopic methods in TFE
In this work, the analyses of the secondary structures of the oligomers and their ADMET derivative polymers were conducted in an α-helix promoting solvent 2,2,2-trifluoroethanol (TFE) which stabilizes α-helix structures due to its high polarity.48,49 We aimed to investigate the effects of number of repeating units (n) present in oligomers and in the ADMET polymers on the formation of secondary structures, i.e., their α-helix propensities, as measured by CD spectroscopy in TFE. The summary of all CD spectroscopy measurements along with their analytical GPC results of oligomers, ADMET polymers and their selected fractions are presented within Table 2. The percentages of α-helicities are presented in Table 2 and were calculated according to literature.50 (see ESI, eqn (1S)†) The secondary structural investigations of Lysns along with their ADMET polymer derivatives A-[Lysn]ms are shown in Fig. 4. Selected fractions of A-[Lysn=3]m=3 and A-[Lysn=24]m=4 are depicted in Fig. 5.Enhanced α-helicity with increasing n number was observed for oligo-L-lysine(Z)s Lysns (see Table 2, and ESI Fig. 20S†) as also supported by our previous investigations.22 As an example, Lysn=6 (Table 2, entry II) displays 25% α-helicity, while Lysn=30 (Table 2, entry V) with its higher degree of polymerization displays 65% α-helicity. Similarly, structural investigations utilizing CD spectroscopy have also been probed by Huesmann et al.20 where mono-functional poly-L-lysine(Z)s prepared by hexylamine with n = 24, 57, 87, 196 (Đs ∼ 1.70) have been investigated in HFIP. According to their investigations, poly-L-lysine(Z) with n = 5–15 did not display an ordered secondary conformation, however only at a chain length of n ≈ 60 α-helicity was observed, thus showing an increase in helicity with increasing n.
Secondly, the effects resulting from repetition of the helical segments on the secondary structure formation was investigated by the comparison of the oligomers and their respective ADMET polymer derivatives. Thus, the measured CD spectra of the oligomers along with their respective ADMET polymers are given in Fig. 4. In this regard, since the helical segments are repetitively present within the hybrid polymers of A-[Lysn]ms (n > 4), it is observed from the increasing intensities of [θ]222 from Fig. 4 that their α-helicity values increase from around 30 to 80 percentage (see Table 2, entries XIV, XV, XVI and XXII). To exemplify, Lysn=24 (Table 2, entry IV) displays ∼ 53% α-helicity, and on the other hand the respective ADMET polymer of Lysn=24, A-[Lysn=24]m=4 (Table 2, entry XVI), displays around 69% α-helicity. ADMET polymer of Lysn=30, A-[Lysn=30]m=7 (Table 2, entry XXII), in a similar way, shows a higher tendency to form helical structure which increases from 65 to 79% α-helicity. Both results point at an interference of the individual Lysn – blocks along the ADMET-polymers, leading to a distinct increase of their helicity.
It also was of interest to investigate the effects of the chain lengths of the ADMET polymers on the formation of α-helices. Therefore, selected fractions of ADMET polymers, namely A-[Lysn=3]m=3 (Table 2, entry VI) and A-[Lysn=24]m=4, were examined by CD spectroscopy in TFE. From the CD measurements represented in Fig. 5 and the calculations of α-helicity percentage values given in Table 2, one can deduce that the overall molecular weight of the hybrid polymers, and thus, the repeating unit of L-lysine(Z) moieties plays a decisive role in forming α-helices. Before fractional analyses, as it was expected, ADMET polymer A-[Lysn=3]m=3, did not show any α-helicity due to its low number of L-lysine(Z) units, devoid of a stabilization of a proper helical turn.51,52 However, after fractionation of A-[Lysn=3]m=3, α-helicity could be observed from CD spectroscopy measurements done in TFE, where the obtained fractions F10–12 (Table 2, entry VII) and F13–14 (Table 2, entry VIII) show around 15–18% α-helicity as it is also shown in Fig. 5a. Furthermore, some selected fractions of A-[Lysn=24]m=4 were also investigated by CD as shown in Fig. 5b. The signal intensities at 222 nm, [θ]222, get even more significant for the fractions of A-[Lysn=24]m=4 as the molecular weight of the fraction increases from F18 (Table 2, entry XXI) to F12 (Table 2, entry XVII), again proving that α-helicity propensities increase with the increasing molecular weight of the hybrid polymer.
Last but not least, the molecular weight dependence on the secondary structural features of the selected fractions of both A-[Lysn=3]m=3 and A-[Lysn=24]m=4 could also be compared. For instance, F10 + 11 + 12 (Table 2, entry VII) and F12 (Table 2, entry XVII) display the same Mn values (23.2 kDa), however the former one shows only around 18% α-helicity whereas the letter has around 77% α-helicity. Similarly, comparing the α-helicity percentage values of F13 + 14 (Table 2, entry VIII) with F15 (Table 2, entry XIX) and F15 (Table 2, entry IX) with F16 (Table 2, entry XX) indicates that not only the molecular weights of the hybrid polymers but also the number of repeating units of L-lysine(Z) along the whole chain plays a significant role in their secondary structural properties, i.e., their α-helicity propensities.
Furthermore, infrared (IR) spectroscopy, which is a well-known and versatile experimental technique to analyse chemical as well as biological samples either in solution or in the solid state, was additionally employed.19,53,54 With the help of IR spectroscopy measurements, it was intended to gain insight into the presence of the other common secondary structures, e.g., β-sheets and β-turns. However, owing to the sufficient solubility of only some of the samples, we could obtain structural investigations by IR spectroscopy in only partial depth (see ESI†).
With the help of IR, one can observe secondary structures which produce characteristic electron densities in the amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, giving rise to distinctive amide I frequencies (1620–1700 cm−1) in the IR spectra. Since the length of the hydrogen bond resulting from α-helix conformation formed by the CO group of the residue n positioned to interact intramolecularly with the N–H group of the residue n + 4 will be slightly longer (and thus weaker) than that of in a β-sheet structure, it gives further increase in amide I frequency.53,55 Concordantly, α-helix structures contribute to amide I frequency signals at around 1648–1660 cm−1, while β-sheet structures are observed at around 1625–1640 cm−1.56
O groups, giving rise to distinctive amide I frequencies (1620–1700 cm−1) in the IR spectra. Since the length of the hydrogen bond resulting from α-helix conformation formed by the CO group of the residue n positioned to interact intramolecularly with the N–H group of the residue n + 4 will be slightly longer (and thus weaker) than that of in a β-sheet structure, it gives further increase in amide I frequency.53,55 Concordantly, α-helix structures contribute to amide I frequency signals at around 1648–1660 cm−1, while β-sheet structures are observed at around 1625–1640 cm−1.56
IR spectra of ADMET polymers, A-[Lysn=3]m=3 are shown with a yellow line, A-[Lysn=12]m=8 with a black line and A-[Lysn=30]m=7 with a blue line are shown in Fig. 6. As previously observed from CD investigations in TFE, α-helicity structure formation tendency increases with the increasing number of repeating units (n) which could be also observed from the intense sharp peak of the blue line at ≈1650 cm−1 proving α-helicity of the ADMET polymer (A-[Lysn=30]m=7) in IR spectra. Since the number of repeating units of peptide decreases, as seen in the cases of ADMET polymers, A-[Lysn=12]m=8 and A-[Lysn=3]m=3, the IR signal at ≈1650 cm−1 get weaker and broader most probably because of the presence of other unordered structure. The signals at around 1700 cm−1 stem from the dynamic nature of secondary structure formation resulting in different secondary structural features such as β-sheets and β-turns.
In order to work up a connection in between secondary structures of different types of hybrid polymers such as linear peptides with β-turn motifs in TFE57 and amyloid-type fibrils with β-sheet structures in solution, CD and IR methods have been used jointly.58 Consequently, IR measurements assist the secondary structural analyses performed by CD spectroscopy, while the latter provides more accurate estimations of α-helix content and the former one is more sensitive to β-sheets.59 In comparison to IR measurements, our CD investigations performed in TFE help us quantify α-helicity of the oligomers and the ADMET polymers straightforwardly as shown in Fig. 4 and 5b and Table 2. Hence, in an attempt to indicate the presence of β-sheets and β-turns, especially in the case of highly α-helical polymers, e.g., A-[Lysn=30]m=7, we employed IR as shown in Fig. 6. Similarly, another study has utilized both IR and CD methods and reported that at native pH, lipopeptides form β-sheet nanofibers in aqueous solution with a strong IR peak at 1616–1620 cm−1 and CD spectra displaying similar characteristic signals of a β-sheet secondary structure, i.e., minima at ∼216 nm and a maximum at ∼197 nm.60
To this date, the features determining the folding behaviours of helix bundles, e.g., four-helix bundle motifs, have been drawn upon as a model for understanding the forces governing functional protein folding such as in the case of transmembrane protein GLUT1.61–63 According to the findings the burial of hydrophobic residues, conformational entropy, packing constraints, helix dipole and interhelical turns have an impact on forming such helix–helix bundles. Additionally, it has been known that the perfectly ordered biologically active three-dimensional structures of proteins form such functional assemblies by the interplay of varying interactions, e.g., noncovalent interactions.64,65 These noncovalent cooperative interactions are mainly interpreted from their hydrogen bonding affinities.66,67 Similar to our current study model helical polymers, studied under different conditions to examine their helix–coil transitions, also showed changes in secondary structure induced by the repeating number of helical chains, i.e., length and strength of helices.68–70 Based on CD and IR-measurements in our study we can also prove a change of helical structures induced by chain length (n), end chain moieties and helicogenic solvent effects. In summary, our secondary structural investigations in TFE revealed that ADMET polymers have higher propensities than the oligomers to exhibit and stabilize α-helical structure, proposed as a result of the packing of their repetitive helical segments present within the hybrid polymer chain that form intramolecular interactions of the hydrogen bonding moieties, namely L-lysine(Z)s, once the α-helix structure has been provided (n > 4).
Conclusions
We here aimed to generate bio-mimicking macromolecules with repetitive helical segments that could serve as models to investigate their structural properties in a reductionist approach manner. Therefore, we successfully synthesized and characterized hybrid polymers, ADMET polymers (A-[Lysn]m) of bis-ω-ene-functional oligo-L-lysine(Z)s (Lysn), with different repeating units (n = 3, 6, 12, 24, 30). We separated ADMET polymers, A-[Lysn=3]m=3 and A-[Lysn=24]m=4, into their fractions by using preparative GPC technique in DMF to access lower polydispersity values with controlled molecular weights. All obtained oligo-L-lysine(Z)s, ADMET polymers, and selectively chosen fractions of A-[Lysn=3]m=3 and A-[Lysn=24]m=4 were analysed by 1H-NMR, analytical GPC and MALDI-ToF MS.The secondary structural investigations were conducted with the help of CD spectroscopy and IR spectroscopy in TFE. Throughout our secondary structural investigations, we intended to draw attention to the effects of number of chains (n) of Lysns, as reported in our preceding work, and of their ADMET derivatives, i.e., A-[Lysn]ms, in an α-helix promoting solvent. Our secondary structural investigations have shown that an increasing number of peptide unit increases the propensity to form α-helices. This phenomenon can be better understood by comparing the CD spectra of the fractions of A-[Lysn=3]m=3 and A-[Lysn=24]m=4 selectively investigated by CD spectroscopy in TFE, proving the effects of the controlled molecular weights with low Đ values of the hybrid polymers on the formation of α-helices. Thus, the individual oligomers, the Lysn segments, do interact along the main chain, inducing increased helicity with increasing chain length of the overall ADMET polymer. It can be proposed that this type of cooperative phenomenon is similar to other multi-folded proteins, where close proximity between individual segments can change the overall conformation of individual part – e.g. in globular proteins with helix–helix interaction motifs, as common for many transmembrane proteins. We think that these polymers can serve as a model-system to study influences from distance-related folding and refolding phenomena.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the grant Project A03 nr. 189853844-TRR 102 (Polymers under multiple constraints) for financial support from the German Research Foundation (DFG) via the research centre SFB/TRR 102 (Project A3 and A12). We also thank a grant from the “Leistungszentrum Chemie und Biosystemtechnik des Landes Sachsen-Anhalt” for financial support, internal project S1 (Binder). We would like to thank Prof. Hauke Lilie from the Institute of Biochemistry and Biotechnology, Martin Luther University Halle-Wittenberg for allowing to work with the CD spectroscopy instrument.Notes and references
- Y. K. Sung and S. W. Kim, Cancer Med., 2019, 2, 6–13 Search PubMed.
- R. N. Kalaria and S. I. Harik, J. Neurochem., 1989, 53, 1083–1088 CrossRef CAS PubMed.
- M. M. M. P. W. Hruz, Mol. Membr. Biol., 2001, 18, 183–193 CrossRef PubMed.
- M. Mueckler and C. Makepeace, Biochemistry, 2009, 48, 5934–5942 CrossRef CAS PubMed.
- D. Deng, C. Xu, P. Sun, J. Wu, C. Yan, M. Hu and N. Yan, Nature, 2014, 510, 121 CrossRef CAS PubMed.
- C. Giordano, D. Albani, A. Gloria, M. Tunesi, S. Batelli, T. Russo, G. Forloni, L. Ambrosio and A. Cigada, Int. J. Artif. Organs, 2009, 32, 836–850 CrossRef CAS PubMed.
- N. Alasvand, S. Kargozar, P. B. Milan, N. P. S. Chauhan and M. Mozafari, in Adv. Func. Polym. for Biomed. Apps., ed. M. Mozafari and N. P. Singh Chauhan, Elsevier, 2019, pp. 275–299 Search PubMed.
- M. Fändrich and C. M. Dobson, EMBO J., 2002, 21, 5682–5690 CrossRef PubMed.
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169–190 CrossRef CAS PubMed.
- A. Lalatsa, A. G. Schätzlein, M. Mazza, T. B. H. Le and I. F. Uchegbu, J. Controlled Release, 2012, 161, 523–536 CrossRef CAS PubMed.
- A. Makino, Polym. J., 2014, 46, 783 CrossRef CAS.
- N. Liu, B. Li, C. Gong, Y. Liu, Y. Wang and G. Wu, Colloids Surf., B, 2015, 136, 562–569 CrossRef CAS PubMed.
- J. S. Choi, E. J. Lee, Y. H. Choi, Y. J. Jeong and J. S. Park, Bioconjugate Chem., 1999, 10, 62–65 CrossRef CAS PubMed.
- P. Liu, H. Yu, Y. Sun, M. Zhu and Y. Duan, Biomaterials, 2012, 33, 4403–4412 CrossRef CAS PubMed.
- A. I. Arunkumar, T. K. S. Kumar, T. Sivaraman and C. Yu, Int. J. Biol. Macromol., 1997, 21, 299–305 CrossRef CAS PubMed.
- A. I. Arunkumar, T. K. S. Kumar and C. Yu, Int. J. Biol. Macromol., 1997, 21, 223–230 CrossRef CAS PubMed.
- O. Kambara, A. Tamura, A. Naito and K. Tominaga, Phys. Chem. Chem. Phys., 2008, 10, 5042–5044 RSC.
- Ł. Szyc, S. Pilorz and B. Czarnik-Matusewicz, J. Mol. Liq., 2008, 141, 155–159 CrossRef.
- A. Mirtič and J. Grdadolnik, Biophys. Chem., 2013, 175–176, 47–53 CrossRef PubMed.
- D. Huesmann, A. Birke, K. Klinker, S. Türk, H. J. Räder and M. Barz, Macromolecules, 2014, 47, 928–936 CrossRef CAS.
- K. Cieślik-Boczula, Biochimie, 2017, 137, 106–114 CrossRef PubMed.
- M. B. Canalp, A. Meister and W. H. Binder, RSC Adv., 2019, 9, 21707–21714 RSC.
- A. Carlsen and S. Lecommandoux, Curr. Opin. Colloid Interface Sci., 2009, 14, 329–339 CrossRef CAS.
- J.-P. Chen, I. M. Chu, M.-Y. Shiao, B. R.-S. Hsu and S.-H. Fu, J. Ferment. Bioeng., 1998, 86, 185–190 CrossRef CAS.
- M. Metzke, N. O'Connor, S. Maiti, E. Nelson and Z. Guan, Angew. Chem., 2005, 44, 6529–6533 CrossRef CAS PubMed.
- K. T. Al-Jamal, W. T. Al-Jamal, J. T. W. Wang, N. Rubio, J. Buddle, D. Gathercole, M. Zloh and K. Kostarelos, ACS Nano, 2013, 7, 1905–1917 CrossRef CAS PubMed.
- J. Babin, J. Rodriguez-Hernandez, S. Lecommandoux, H.-A. Klok and M.-F. Achard, Faraday Discuss., 2005, 128, 179–192 RSC.
- G. Orts Gil, M. Łosik, H. Schlaad, M. Drechsler and T. Hellweg, Langmuir, 2008, 24, 12823–12828 CrossRef PubMed.
- K. E. Gebhardt, S. Ahn, G. Venkatachalam and D. A. Savin, J. Colloid Interface Sci., 2008, 317, 70–76 CrossRef CAS PubMed.
- C.-J. Huang and F.-C. Chang, Macromolecules, 2008, 41, 7041–7052 CrossRef CAS.
- G. J. M. Habraken, M. Peeters, C. H. J. T. Dietz, C. E. Koning and A. Heise, Polym. Chem., 2010, 1, 514–524 RSC.
- K. B. Wagener, J. M. Boncella and J. G. Nel, Macromolecules, 1991, 24, 2649–2657 CrossRef CAS.
- H. Li, L. Caire da Silva, M. D. Schulz, G. Rojas and K. B. Wagener, Polym. Int., 2016 DOI:10.1002/pi.5188.
- J. A. Smith, K. R. Brzezinska, D. J. Valenti and K. B. Wagener, Macromolecules, 2000, 33, 3781–3794 CrossRef CAS.
- F. Marsico, M. Wagner, K. Landfester and F. R. Wurm, Macromolecules, 2012, 45, 8511–8518 CrossRef CAS.
- T. Lebarbé, A. S. More, P. S. Sane, E. Grau, C. Alfos and H. Cramail, Macromol. Rapid Commun., 2014, 35, 479–483 CrossRef PubMed.
- S. Reimann, U. Baumeister and W. H. Binder, Macromol. Chem. Phys., 2014, 215, 1963–1972 CrossRef CAS.
- J. T. Offenloch, J. Willenbacher, P. Tzvetkova, C. Heiler, H. Mutlu and C. Barner-Kowollik, Chem. Commun., 2017, 53, 775–778 RSC.
- J. K. Leonard, D. Turek, K. B. Sloan and K. B. Wagener, Macromol. Chem. Phys., 2010, 211, 154–165 CrossRef CAS.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS PubMed.
- Y. Shen, X. Fu, W. Fu and Z. Li, Chem. Soc. Rev., 2015, 44, 612–622 RSC.
- H. H. James, in Ring-Opening Polymerization, American Chemical Society, 1985, ch. 5, vol. 286, pp. 67–85 Search PubMed.
- W. Zhao, Y. Gnanou and N. Hadjichristidis, Biomacromolecules, 2015, 16, 1352–1357 CrossRef CAS PubMed.
- W. Vayaboury, O. Giani, H. Cottet, A. Deratani and F. Schué, Macromol. Rapid Commun., 2004, 25, 1221–1224 CrossRef CAS.
- T. E. Hopkins, J. H. Pawlow, D. L. Koren, K. S. Deters, S. M. Solivan, J. A. Davis, F. J. Gómez and K. B. Wagener, Macromolecules, 2001, 34, 7920–7922 CrossRef CAS.
- T. E. Hopkins and K. B. Wagener, Macromolecules, 2003, 36, 2206–2214 CrossRef CAS.
- J. K. Leonard, T. E. Hopkins, K. Chaffin and K. B. Wagener, Macromol. Chem. Phys., 2008, 209, 1485–1494 CrossRef CAS.
- P. Pengo, L. Pasquato, S. Moro, A. Brigo, F. Fogolari, Q. B. Broxterman, B. Kaptein and P. Scrimin, Angew. Chem., 2003, 115, 3510–3514 CrossRef.
- R. Rajan and P. Balaram, Int. J. Pept. Protein Res., 1996, 48, 328–336 CrossRef CAS PubMed.
- K.-S. Krannig, J. Sun and H. Schlaad, Biomacromolecules, 2014, 15, 978–984 CrossRef CAS PubMed.
- K. Olsen and J. Bohr, Theor. Chem. Acc., 2010, 125, 207–215 Search PubMed.
- B. Haimov and S. Srebnik, Sci. Rep., 2016, 6, 38341 Search PubMed.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- M. Rozenberg and G. Shoham, Biophys. Chem., 2007, 125, 166–171 CrossRef CAS PubMed.
- W. K. Surewicz, H. H. Mantsch and D. Chapman, Biochemistry, 1993, 32, 389–394 CrossRef CAS PubMed.
- M. Jackson and H. H. Mantsch, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 95–120 CrossRef CAS PubMed.
- M. Hollósi, Z. Majer, A. Z. Rónai, A. Magyar, K. Medzihradszky, S. Holly, A. Perczel and G. D. Fasman, Biopolymers, 1994, 34, 177–185 CrossRef PubMed.
- M. J. Krysmann, V. Castelletto and I. W. Hamley, Soft Matter, 2007, 3, 1401–1406 RSC.
- K. A. Oberg, J. M. Ruysschaert and E. Goormaghtigh, Eur. J. Biochem., 2004, 271, 2937–2948 CrossRef CAS PubMed.
- V. Castelletto, A. Kaur, R. M. Kowalczyk, I. W. Hamley, M. Reza and J. Ruokolainen, Biomacromolecules, 2017, 18, 2013–2023 CrossRef CAS PubMed.
- J. M. Pascual, D. Wang, R. Yang, L. Shi, H. Yang and D. C. De Vivo, J. Biol. Chem., 2008, 283, 16732–16742 CrossRef CAS PubMed.
- S. Kamtekar and M. H. Hecht, FASEB J., 1995, 9, 1013–1022 CrossRef CAS PubMed.
- A. Kauko, L. E. Hedin, E. Thebaud, S. Cristobal, A. Elofsson and G. von Heijne, J. Mol. Biol., 2010, 397, 190–201 CrossRef CAS PubMed.
- M. J. Williams and M. Bachmann, Polymers, 2016, 8, 245 CrossRef PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS PubMed.
- Y. Zhou, G. Deng, Y.-Z. Zheng, J. Xu, H. Ashraf and Z.-W. Yu, Sci. Rep., 2016, 6, 36932 CrossRef CAS PubMed.
- Y. Li, Y. Wang, G. Huang and J. Gao, Chem. Rev., 2018, 118, 5359–5391 CrossRef CAS PubMed.
- Y. Ren, H. Fu, R. Baumgartner, Y. Zhang, J. Cheng and Y. Lin, ACS Macro Lett., 2017, 6, 733–737 CrossRef CAS.
- Y. Ren, R. Baumgartner, H. Fu, P. van der Schoot, J. Cheng and Y. Lin, Biomacromolecules, 2017, 18, 2324–2332 CrossRef CAS PubMed.
- Y.-D. Wu and Y.-L. Zhao, J. Am. Chem. Soc., 2001, 123, 5313–5319 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR, MALDI-ToF, CD, IR, etc. See DOI: 10.1039/c9ra09189k |
| This journal is © The Royal Society of Chemistry 2020 |