
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of PVDF/graphene composites with enhanced β phase via conventional melt processing assisted by solid state shear milling technology
Huili Zhang,
Yan Zhu and
Li Li *
*
State Key Laboratory of Polymer Materials Engineering, Polymer Research Institute of Sichuan University, Chengdu 610065, China. E-mail: powerlily@scu.edu.cn; Fax: +86-28-85402465; Tel: +86-28-85405133
First published on 21st January 2020
Abstract
The β-phase crystal, which decides the final electric properties of poly(vinylidene fluoride) (PVDF), is extremely difficult to obtain via conventional melt processing due to its thermal instability. In this work, with the assistance of our independently developed solid state shear milling (S3M) technology, which could provide multiple stresses and form a micro-stretching field on PVDF to promote the transformation of more α phase to β phase, PVDF/graphene (PVDF/GP) composite with relatively higher β phase (42.2%), higher than that directly prepared by melt blending without S3M (33.0%), and dielectric properties was achieved through conventional melt extrusion and injection. When the GP content was 1.0 wt%, the dielectric constant of the composite was 465 at 1000 Hz, about 42 times that of pure PVDF. The special squeezing and shearing forces of S3M also realized the exfoliation of GP as well as the solid grafting of GP layers on PVDF molecules, improving the dispersion of GP layers in PVDF and making them effectively exert their heterogeneous nucleation as well as enhancement effects on PVDF, thus increasing the crystallinity, thermal stability and mechanical properties of the composites.
1 Introduction
With the consistent growth of the energy demand, how to improve energy efficiency has become a research hot spot in recent years.1–3 High dielectric polymer materials have excellent electrical energy storage and good processability, thus having been expected to be applied to many high-tech fields including sensors, brakes, microcapacitors, etc.4–6PVDF is a semi-crystalline polymer with outstanding pyro- and piezoelectric properties.7 For example, its dielectric constant is almost the highest in polymers. The special electric properties of PVDF are directly related to its crystalline phase. Generally, PVDF has three main crystalline phases, i.e. α, β and γ, among which, β phase is of great interest because of its polar crystalline-electro active nature that decides the final electric properties of PVDF.8 However, β phase is extremely unstable in melt state, so PVDF products with good electric properties are difficult to be prepared through conventional melt processing.
To meet the application requirements for electronic products, some fillers with excellent electrochemical properties, such as graphene (GP),9–12 carbon nanotube,13,14 ceramic (BaTiO3, PZT),15–17 metal (Au, Cu),18,19 metal oxide (ZnO),20,21 had been chosen and added into PVDF matrix, and PVDF based composites with relatively higher dielectric constant were achieved. Among these fillers, GP, a new type of two-dimensional carbon nanomaterial with superior mechanical properties, high efficiency electron transfer capability and high thermal conductivity,22,23 has been of interests recent years. By using a small amount of GP, the mechanical, thermodynamic as well as electrical properties of PVDF in theory can all be effectively improved. For example, the addition of 16 wt% surface-modified graphene (SMG) greatly increased the dielectric permittivity of PVDF composite, from 8.3 of pristine PVDF to 83.8 at 1000 Hz;24 while the addition of the reduced graphene oxide (RGO) improved the dielectric constant and piezoelectric coefficient of PVDF by respectively four and two times via promoting the transition of the nonpolar α-phase PVDF to the polar β phase.25 However, to insure the good dispersion of GP with so large specific surface area in PVDF matrix and to obtained more β phase, PVDF and GP are usually mixed in solution and the composite products are always prepared by solution methods, e.g. solution casting or electrospinning, which uses a large amount of toxic solvents, and has complicate and long preparation period as well as high cost, limiting the large-scale manufacture of PVDF/GP composites, no matter how excellent properties they have. Most importantly, the solution methods have no idea on the preparation of three-dimensional products with complex shapes.
Accordingly, in this paper, a simple, environmentally friendly and low cost way, i.e. conventional melt processing assisted by Solid State Shear Milling (S3M) technology, was provided to prepare PVDF/GP composite with GP uniformly dispersed in PVDF matrix and the improved β phase at large scale. S3M is our independently developed technology based on our self-designed pan-mill mechanochemical reactor (Fig. 1), which has special three dimensional scissors and can offer strong squeezing, shear and hoop stress on materials in-between.26,27 Improving the β phase of PVDF via mechanical milling, e.g. ball milling, was first reported by Esterly et al. in 2004,28 but the milling conditions were quite severe, especially that the liquid nitrogen was needed through the whole milling process. Compared with ball milling, which mainly offers impact force, the multiple stresses provided by our pan-milling mechanochemical reactor, in theory, could produce a better micro stretching field on PVDF to induce more α phase to transform to β phase, and simultaneously, promote the solid exfoliation of GP layers as well as the activation of PVDF chains, leading to the good interactions between PVDF and GP as well as the good dispersion of GP in PVDF matrix. The most important of all, the whole milling process was conducted at ambient temperature. Ascribing to the extremely high β-phase PVDF gotten via S3M technology, lots could still be maintained in the final PVDF/GP composites after the conventional melt extrusion and injection without recovering to α phase, endowing the final composite with good comprehensive properties, especially the improved dielectric constant.
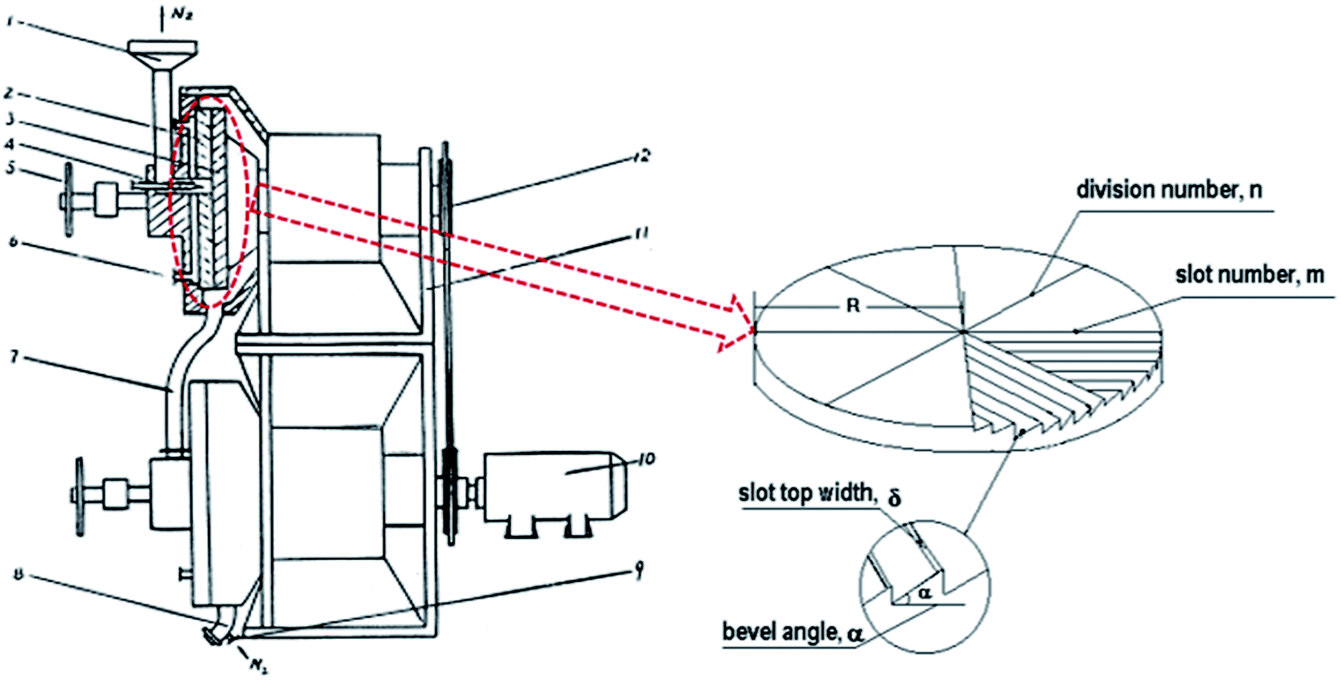 | ||
| Fig. 1 Schematic diagram of the pan-mill equipment.29 (1 – inlet; 2 – stationary pan; 3 – moving pan; 4 – feeding screw; 5 – helve; 6 – entrance of system). | ||
2 Experimental section
2.1 Materials
PVDF (FR906, ρ = 1.76 g cm−3, Tm = 173 °C) was provided by Shanghai 3F New Material Technology Co., LTD (China). GP was purchased from Sixth Element (Changzhou) Materials Technology Co., Ltd (China).2.2 Sample preparation
PVDF/GP co-milled powders were obtained by first mixing certain PVDF powders and GP in a high speed mixer, and then co-milling the mixer at room temperature by using our pan-mill type mechanochemical reactor (Fig. 1). The GP content in the co-milled powders was 0.5 wt%, 1.0 wt%, 1.5 wt%, 2.0 wt% and 2.5 wt%, respectively. The milling cycles were 10 times. The heat generated from the milling process was carried out by circulating cooling water. The co-milled powders were then extruded and granulated on a parallel twin-screw extruder (Nanjing Jayant Co., Ltd, China) to obtain the composite masterbatch, and then injected into dumbbell-type standard samples (length × width × thickness: 150 mm × 10 mm × 4 mm) for mechanical property test on a single screw injection molding machine (Ningbo Haitian Group Co., Ltd, China). The 6 heating sections of the parallel twin-screw extruder were set as 170 °C, 180 °C, 185 °C, 185 °C, 180 °C and 175 °C, respectively; while the temperature of the 4 heating sections of the injection molding machine were 180 °C, 190 °C, 190 °C and 185 °C, respectively. The un-milled PVDF/GP composite (GP content: 1.0 wt%) was directly extruded and injected as a control sample.2.3 Characterization
Scanning electron microscope (SEM) was performed to observe the morphologies of the composites by using an INSPECTF scanning electron microscope (FEI, USA) at a voltage of 20 kV. The samples were sputtered with gold for testing after being quenched and fractured in liquid nitrogen.Transmission electron microscope (TEM) images were recorded on Tecnai G2 F20 transmission electron microscope (FEI, USA) with acceleration voltage of 200 kV. The PVDF/GP films with 50–100 nm thickness for TEM analysis were prepared by microtoming on an ultramicrotome (UC6&FC6, Leica, Germany) at −60 °C.
Melting and crystallization behaviors were recorded on a TA-Q20 differential scanning calorimeter (TA instruments, USA) from room temperature to 200 °C at a heating or cooling rate of 10 °C min−1 under nitrogen atmosphere.
Thermal stability was studied using TA-Q50 thermogravimetric analyzer (TA instruments, American) from room temperature to 700 °C at a heating rate of 10 °C min−1 under nitrogen atmosphere.
X-ray Diffraction (XRD) analyses were conducted on a DX-1000 X-ray diffractometer (Danfangyuan, China) using CuKα radiation of wavelength. The diffraction intensity of Cu Kα radiation was assessed at a voltage of 40 kV and 25 mA. The samples were measured from 5° to 50° with a scanning speed of 0.02° min−1.
Fourier transform infrared (FTIR) spectra were obtained using a Nicolet 6700 spectrophotometer (Thermo Scientific, USA) from 4000 cm−1 to 400 cm−1 at 128 scans with a transmission scanning mode.
The dynamic mechanical properties were studied using TA's Q800 dynamic mechanical analyzer (TA instruments, USA) from −100 °C to 150 °C at a heating rate of 3 °C min−1.
Mechanical performances were carried out using Instron 5567 testing machine (Instron, USA) at room temperature with a speed of 50 mm min−1. Five specimens were tested for each group and the average values were calculated.
The dielectric properties were measured by an Agilent HP4294A system (Agilent Technologies, USA) in the frequency range of 100 Hz to 10 MHz with voltage amplitude of 500 mV.
3 Results and discussion
3.1 Dispersion of GP layers in PVDF/GP composites
SEM images of pure PVDF and PVDF/GP composites were shown in Fig. 2. Clearly, PVDF/GP composites presented the quite different microstructure from pure PVDF. Different from the smooth fracture surface of pure PVDF (Fig. 2(a)), the fracture sections of the composites became rough with the introduction of GP layers (Fig. 2(b) and (c)), and no obvious interfaces were observed ascribing to the good interactions between milled GP and PVDF. Usually, GP layers tend to aggregate due to their nanometer size effect,30 and the strong van der Waals force formed among the layers are quite difficult to be broken up by the shearing force produced during the conventional melt blending process. However, under the strong shearing and pulverization forces of our independently developed pan-mill equipment, the van der Waals force among GP layers were gradually destroyed and part GP layers were exfoliated, producing more thin layers with large specific surface area and exposing more oxygen-containing functional groups on the surface of GP, thus improving the hydrogen bonding between GP layer and the hydrogen or/and fluorine atoms in PVDF.31 The good interactions between milled GP and PVDF greatly promoted the dispersion of GP layers in PVDF matrix, which was reflected in the SEM images by that no apparent aggregations of GP layers could be seen (Fig. 2(b) and (c), white dots), while obvious aggregations were seen in the un-milled composite with the same GP content (1 wt%), as circled in Fig. 2(d). | ||
| Fig. 2 SEM images of pure PVDF (a), PVDF/0.5 wt% GP (b), PVDF/1.0 wt% GP (c) and un-milled PVDF/1.0 wt% GP (d) composites. | ||
The exfoliation of GP layers and their dispersion in PVDF matrix were further identified by TEM, as shown in Fig. 3. Apparently, single-layer GP or GP with reduced layers (In red circle) could be seen, confirming that GP was exfoliated to some extent. The thinning GP layers were uniformly dispersed in the matrix, presenting the folded scaly shape, without apparent orientation.
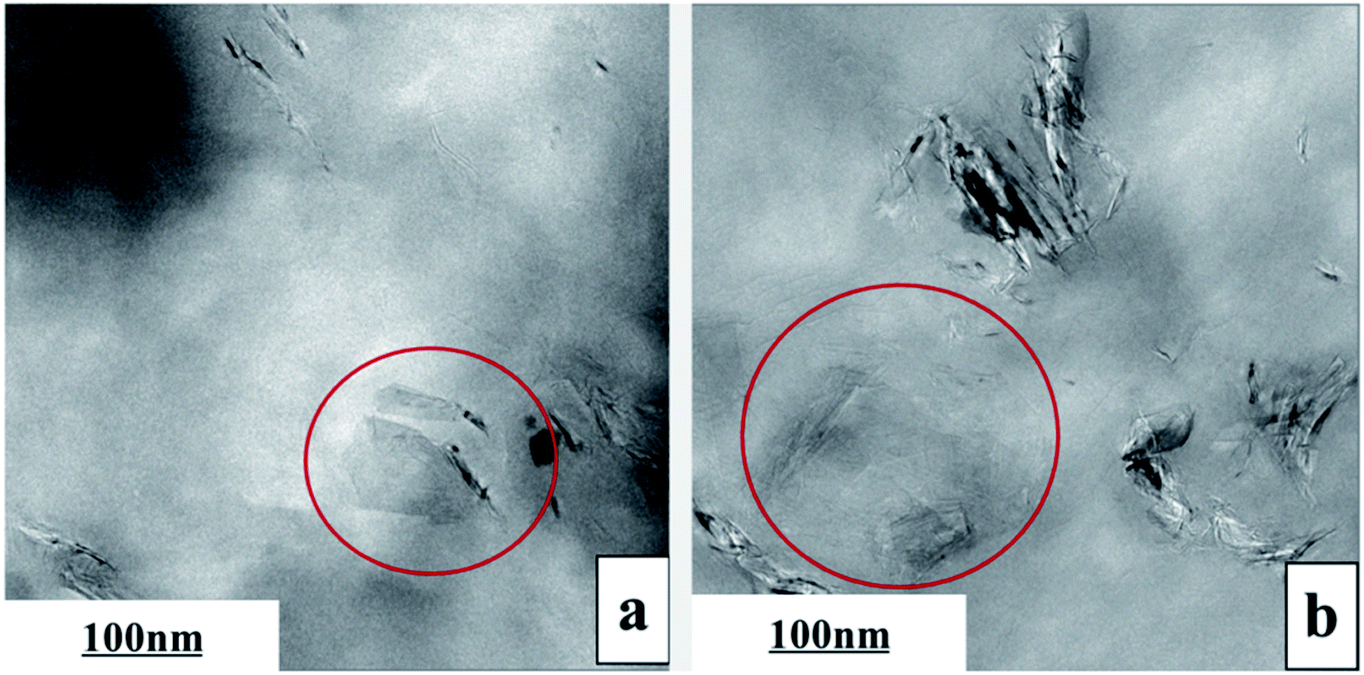 | ||
| Fig. 3 TEM images of PVDF/0.5 wt% GP (a) and PVDF/1.0 wt% GP (b) composites. | ||
3.2 Interactions between GP and PVDF
Raman spectrum is an effective way to study the interactions between polymer and fillers. The Raman spectrum of pure GP, PVDF and PVDF/GP composite powders were shown in Fig. 4. As could be seen, pure GP only presented two characteristic peaks, i.e. G band at about 1580 cm−1 and D band at about 1340 cm−1; while 2D band of GP at about 2690 cm−1 appeared and the intensity ratio of D band and G band (ID/IG) increased in PVDF/GP composite powders.32 2D bond reflects the layer number of GP. Its appearance confirmed the exfoliation of S3M on GP layers, which was consistent with the results of SEM and TEM. ID/IG is usually used as a measure of defect density of GP.33 ID/IG of pure GP was 1.19, but greatly decreased after being co-milled with PVDF, i.e. 0.64 for the composite with 0.5 wt% GP, and further decreased with the increase of GP content in the milled composite powders, i.e. 0.27 for the composite with 2.5 wt% GP. The decreased ID/IG implied the defects healed of GP, possibly caused by the interactions formed between the polar groups of PVDF and GP layers. The higher the GP content, the more interactions formed between PVDF and GP, leading to the continuous decrease of ID/IG. It should be noted that pure PVDF showed three main peaks, respectively at 794 cm−1 (the characteristic peak of α phase), 2984 cm−1 and 3028 cm−1 (the stretching vibration peak of CH2).34,35 With the addition of GP, the α-phase peak at 794 cm−1 became more and more weak, even disappeared when GP content reached 2.5 wt%, indicating the transformation of α phase to β phase and the induced effect of GP on this transformation.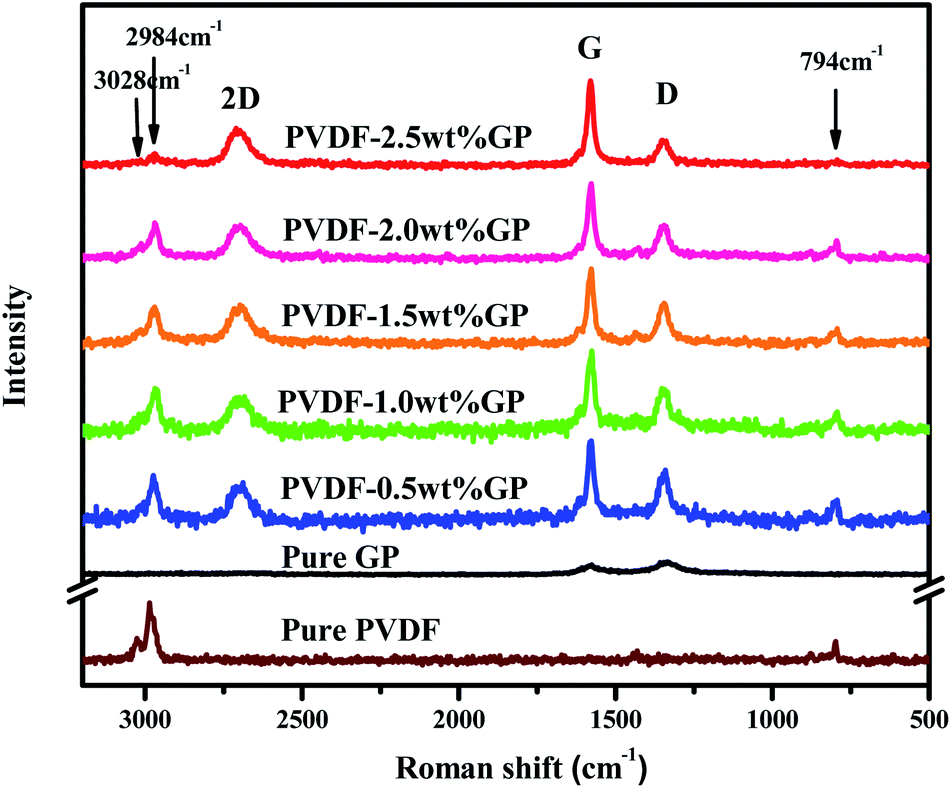 | ||
| Fig. 4 Raman spectra of pure GP, PVDF and PVDF/GP composite powders. | ||
The effects of S3M technology on the interface interactions between PVDF and GP layers were analyzed by comparing the FTIR spectra of GP and the residue of the milled composite powders first extracted by dimethyl formamide (DMF) for 48 h and then dried at 70 °C for 10 h, as shown in Fig. 5. It could be seen from figure that after being extracted, the residual powders presented a medium strong peak at 877 cm−1 and a small peak at 840 cm−1, respectively corresponding to the C–C skeleton vibration of PVDF molecular chains and the CH2 sway vibration in β phase of PVDF, while the infrared spectrum of GP had no such characteristic peaks. Because unreacted PVDF had been completely extracted before FTIR test, the new appeared peaks indicated that solid-phase grafting of GP layers onto PVDF molecular chains occurred with the proceeding of the pan-milling process, in which more and more squeezing and shearing forces were exerted on PVDF, making some PVDF radicals released and reacted with the oxygen functional groups on GP layers, so improved the compatibility between PVDF and GP as well as the dispersion of GP in PVDF. Compared with the traditional grafting methods, solid-phase grafting method showed lower reaction temperature, simple post-treatment, high efficiency, energy saving and small pollution, so would have a good application prospect.36
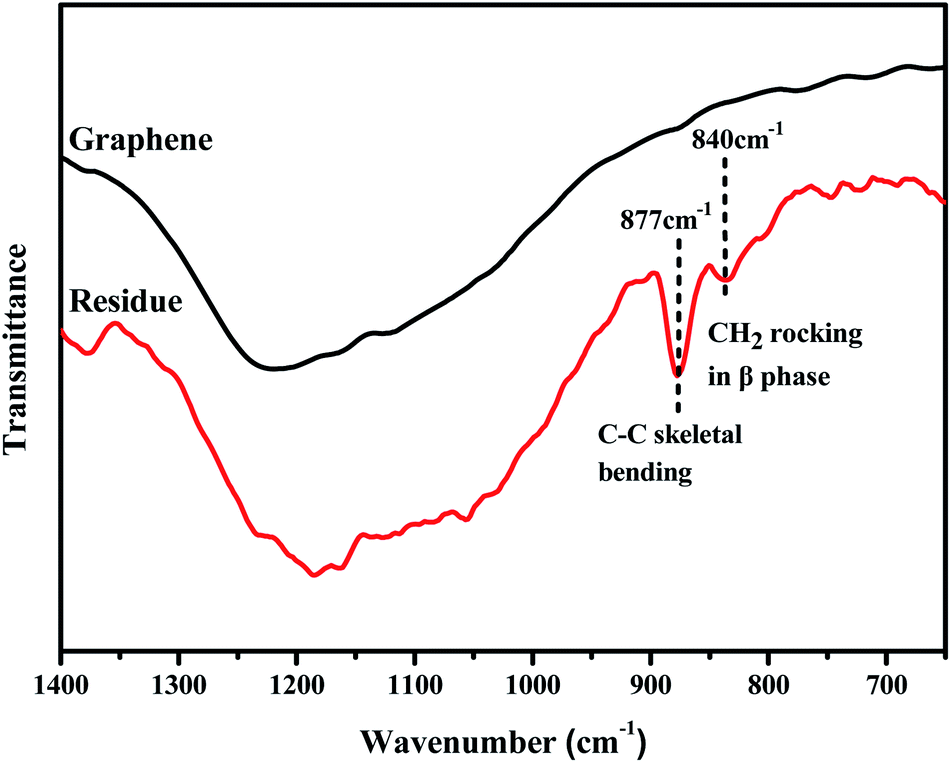 | ||
| Fig. 5 FTIR spectra of GP and the residue after extraction. | ||
3.3 Crystalline behaviors of PVDF/GP composites
PVDF is a semi-crystalline polymer with three main crystal types formed in different crystallization conditions, i.e. α phase, β phase and γ phase, which can be transformed into each other. The X-ray diffraction characteristic peaks of α phase, which has TGTG′ conformation and nonpolarity, locate at 2θ = 17.7°, 18.3°, 19.20° and 26.6°, respectively corresponding to (100), (020), (110) and (021) planes, while β phase, which has TTTT conformation and high polarity, usually presents a characteristic peak at 2θ = 20.3°, corresponding to (110) and (200) planes.7X-ray diffraction patterns of pure GP, PVDF and PVDF/GP composite powders obtained by S3M technology were shown in Fig. 6. Obviously, the pure PVDF pellet mainly presented α-phase peaks, respectively at 2θ = 17.7°, 18.3°, 19.20° and 26.6°, while the milled pure PVDF powders only had one peak in the range of 2θ = 5° to 30°, i.e. the overlapping peak of α phase at 2θ = 17.7°, 18.3° and 19.20° as well as β phase at 2θ = 20.3°. Further adding GP to co-mill, the position of this overlapping peak didn't change, but its strength at about 2θ = 20.3° apparently enhanced, indicating the increase of β phase in milled PVDF and also PVDF/GP composite powders. When suffering the strong stress force of pan mill, PVDF molecular chains were forced to move and extend, producing similar effect as the solid-phase stretching, thus resulting in the destruction of the original PVDF spherical crystalline structure and the formation of the unidirectional microcrystalline arrangement structure. In this way, the sawtooth chain dipole with polarity in the crystal cell would arrange along the stress direction and induce the conformational transformation. With the addition of GP, its synergistic action with the external stress field on PVDF molecules promoted the further transition of non-polar α phase to polar β phase, leading to the further increase of the content of β-phase PVDF and the enhancement of the strength of β-phase peak in Fig. 5. It should be noted that the peak of the composite at about 2θ = 26° was the overlapping peak of α-phase PVDF at 2θ = 26.6° and pure GP at 2θ = 25.5° ((020) plane). However, ascribing to the decreased α phase in milled PVDF and the small GP content, this peak was almost invisible.
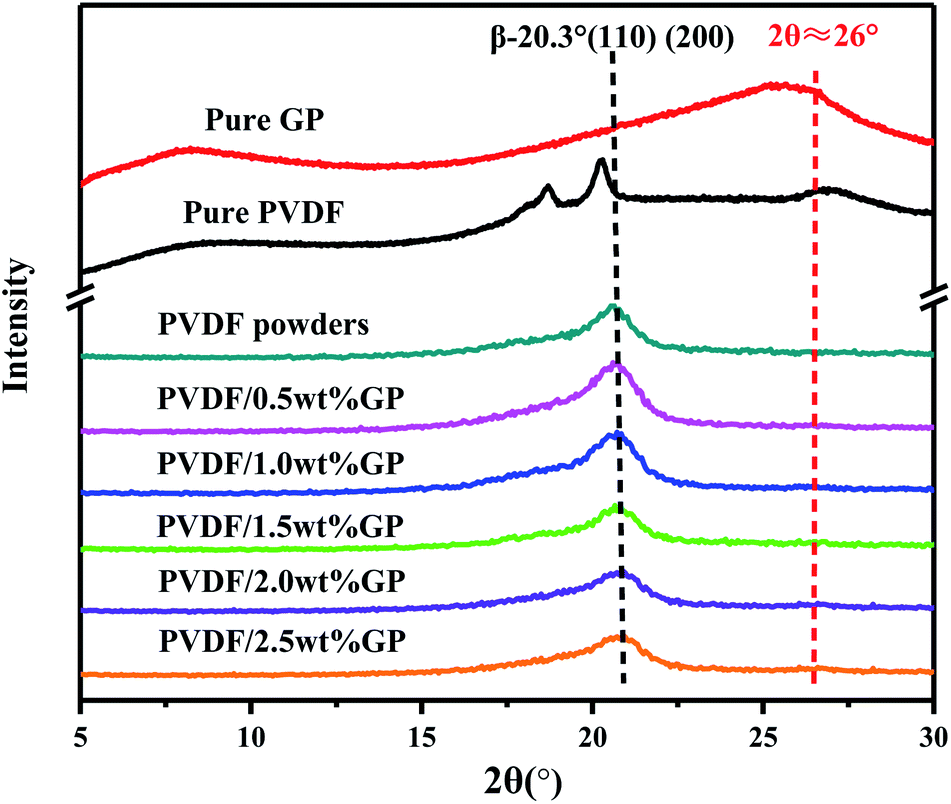 | ||
| Fig. 6 XRD curves of pure GP, PVDF and PVDF/GP composite powders. | ||
FTIR is another effective way to study the crystal types of PVDF. The characteristic peaks of the three crystal types of PVDF in FTIR spectra had been appointed as follows:34,37,38 α-phase peaks at 766 cm−1, 795 cm−1, 855 cm−1 and 976 cm−1; β-phase peaks at 510 cm−1, 840 cm−1 and 1279 cm−1; and γ-phase peaks at 812 cm−1 and 1234 cm−1. Apparently, compared with the peak at 880 cm−1, both the β-phase characteristic peaks at 840 cm−1 and 1279 cm−1 of milled PVDF powders enhanced, further confirming the induced transformation effect of S3M technology on PVDF. When co-milling with GP, these two β-phase characteristic peaks became stronger, and stronger with the increase of GP content, indicating again that the addition of GP promoted the crystallization transformation of PVDF and induced the formation of more β-phase crystals. As known, GP has strong restraint effect, so can be used as nucleating agent. According to the density functional theory, TTTT full trans conformation (β phase) is easier to be absorbed on the surface of GP compared with TGTG′ conformation (α phase), i.e. the addition of GP was in favor of the formation of β-phase crystal,39 as shown in Fig. 8.40
The relative content of β phase in PVDF could be calculated by the following eqn (1),41 and the results were shown in Fig. 9:
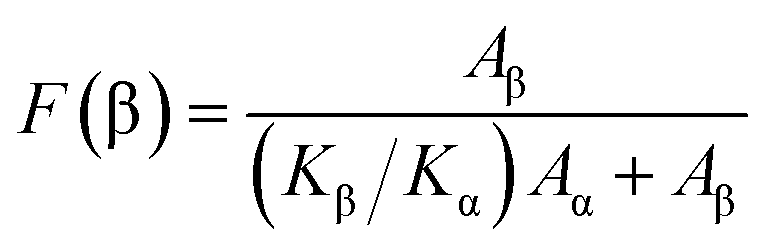 | (1) |
It should be noted that α phase has the lowest free energy and is the most stable in the three crystal types of PVDF, so is the most likely to be formed during the melting and cooling process.42 Accordingly, when suffering melt processing, the unstable TTTT all-trans conformation (β phase) would return to the stable TGTG′ conformation (α phase) due to the thermal motion of PVDF molecules, thus decreasing the polarity and electrical properties of PVDF based material. To confirm the advantages of our S3M technology, the crystalline behaviors of PVDF/GP composites obtained by first melt extrusion and then injection were analyzed via XRD and FTIR, as shown in Fig. 10(a) and (b). By comparing the XRD curves of the composite powders (Fig. 6) and the composites (Fig. 10(a)), it could be seen that ascribing to the strong squeezing and shearing forces produced during pan-milling process, β-phase crystal dominated in PVDF/GP composite powders, so XRD curve mainly presented an obvious β-phase peak at 2θ = 20.3°; while for the composite subjected to melt processing, the characteristic peaks of α phase at 2θ = 17.7° (100), 18.3° (020) and 26.6° (021) appeared. The same phenomenon had also been observed in FTIR spectra (Fig. 7 and Fig. 10(b)), i.e. the vibration peaks of β phase at about 840 cm−1 and 1279 cm−1, which were apparently existed in the composite powders, greatly weakened in the composite. Both identified the transformation of β phase to α phase during the melt processing. The relative β-phase content in PVDF/GP composite was calculated from FTIR, as shown in Fig. 10(c). Compared with the β-phase content in Fig. 9, it could be easy to see that with the same GP content, PVDF/GP composite showed lower β-phase content than PVDF/GP powders. For example, when GP content was 1.0 wt%, the β-phase in the milled powder was 88.0%, while in the composite was 42.2%, intuitively confirming the loss of β phase during melt processing. However, for the extremely high content of β phase, induced by pan mailing and GP, in our milled PVDF/GP composite powders, a lot of β phase were still be maintained after melt processing, leading to the relatively high β phase in the final composite, about 9.2% higher than that in the un-milled composite (33.0%).
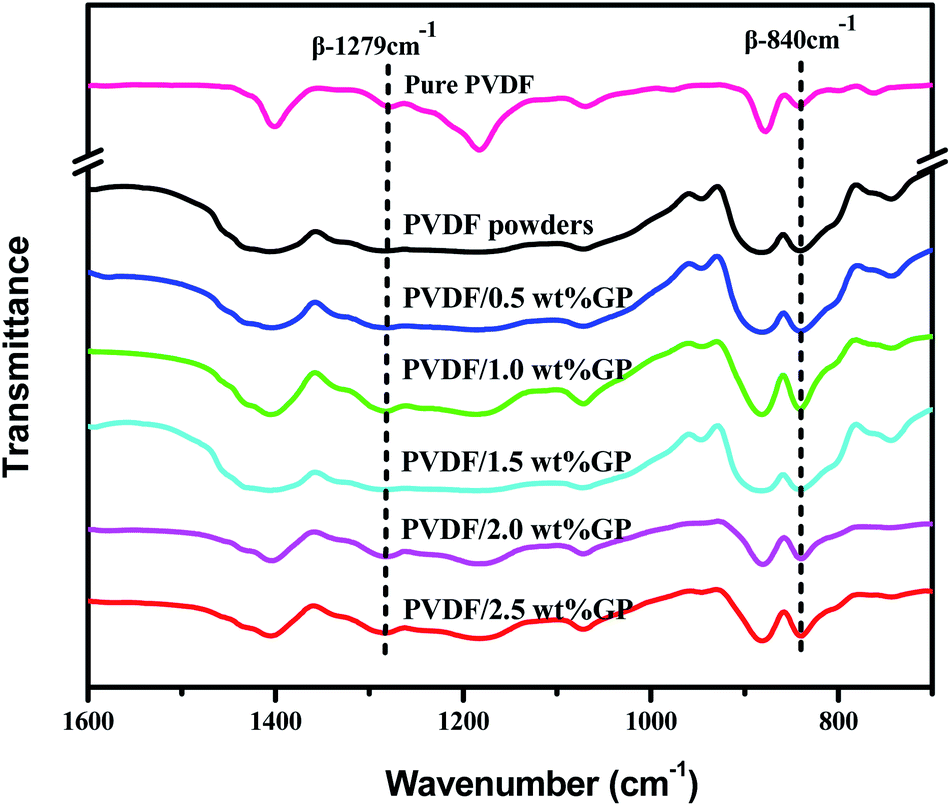 | ||
| Fig. 7 FTIR spectra of pure PVDF pellets and PVDF/GP composite powders. | ||
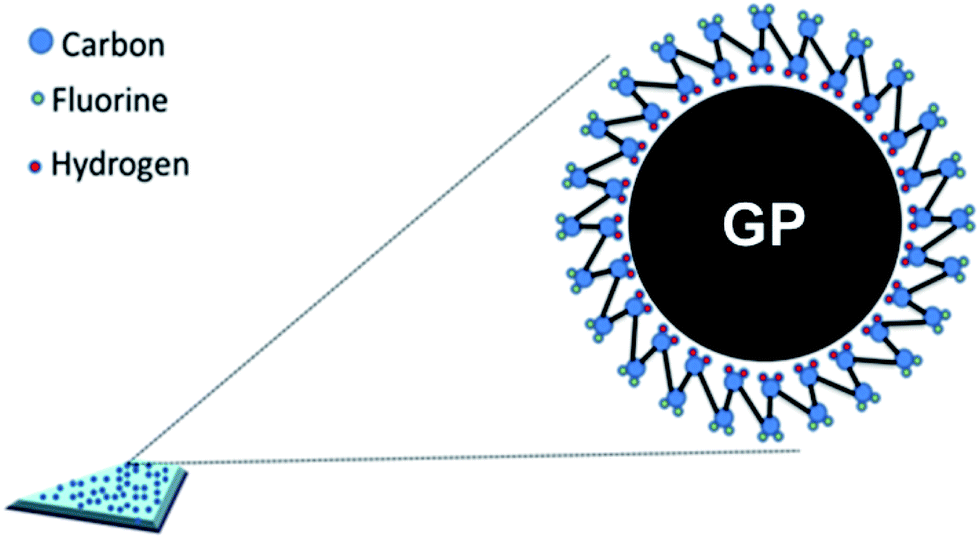 | ||
| Fig. 8 Schematic representation of the formation mechanism of β-phase PVDF on the surface of GP. | ||
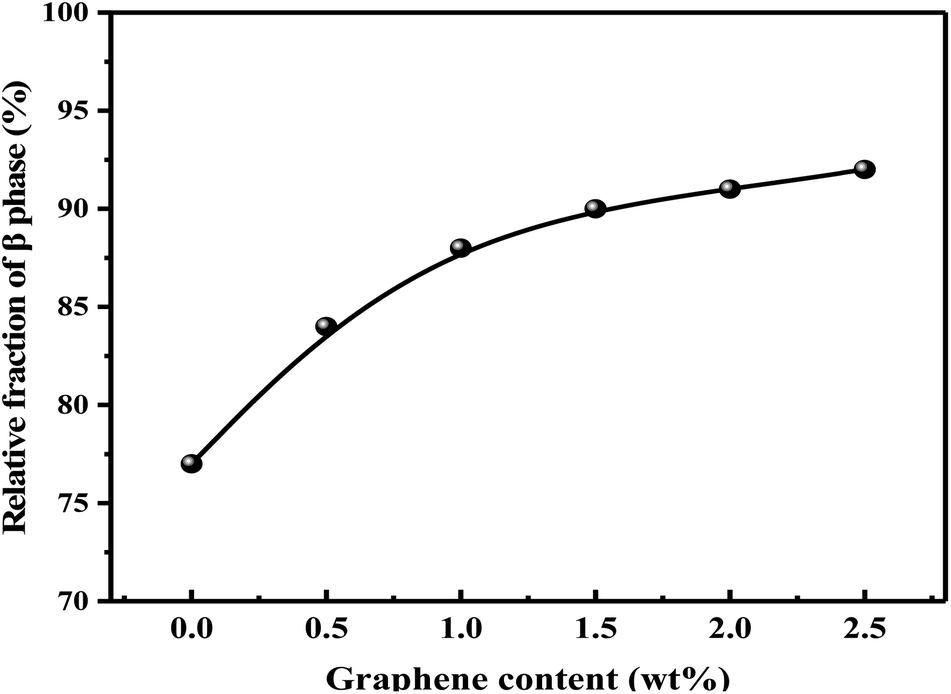 | ||
| Fig. 9 Relative fraction of β phase in PVDF/GP composite powders. | ||
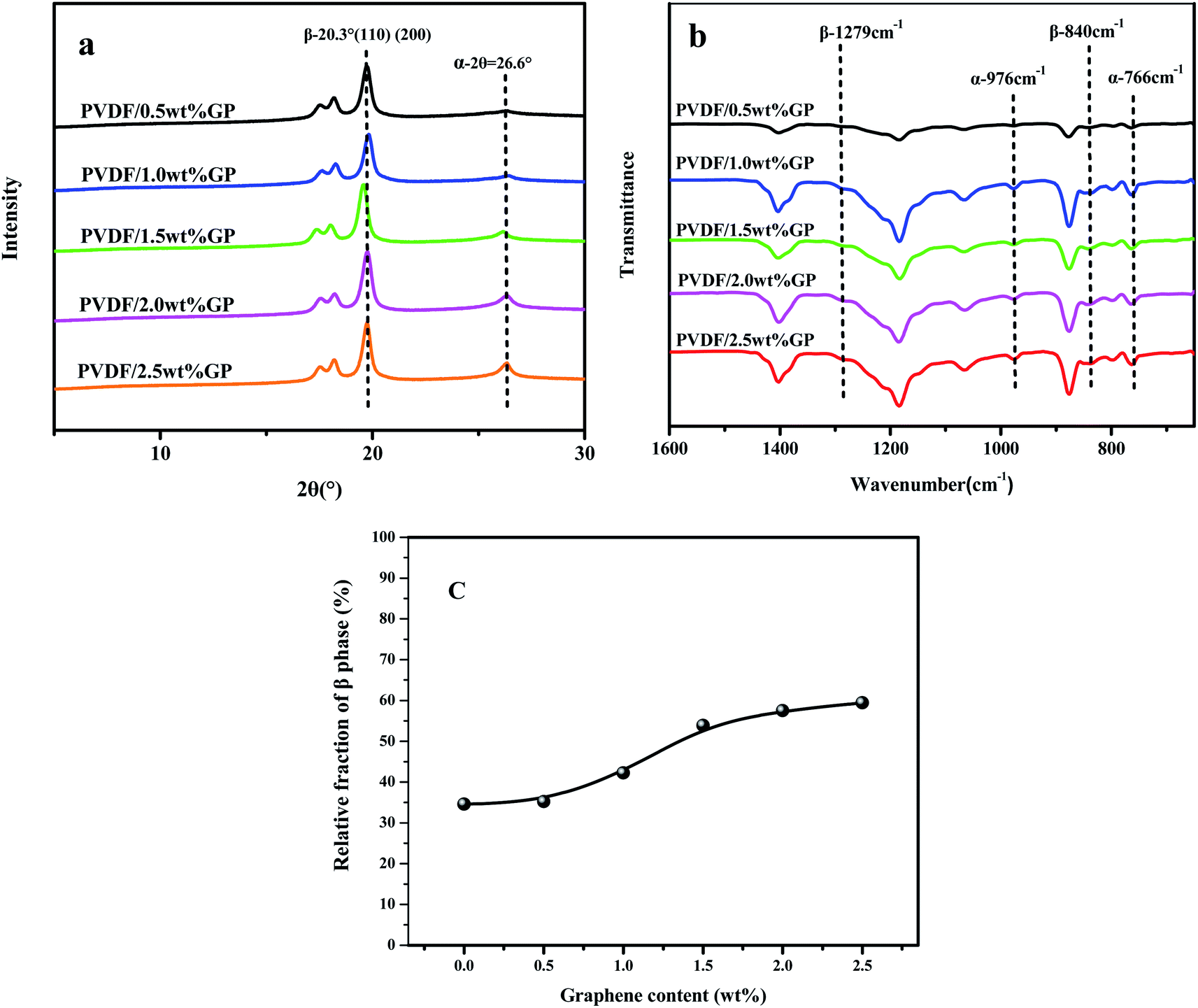 | ||
| Fig. 10 XRD spectra (a), FTIR spectra (b) and relative fraction of β phase of PVDF/GP composites calculated from FTIR (c). | ||
3.4 Thermal properties of PVDF/GP composites
Fig. 11 was the DSC heating (a) and cooling (b) curves of PVDF/GP composites, and the corresponding date including melting temperature, crystallization temperature and crystallinity were listed in Table 1. The crystallinity of PVDF was calculated by using the following eqn (2):43
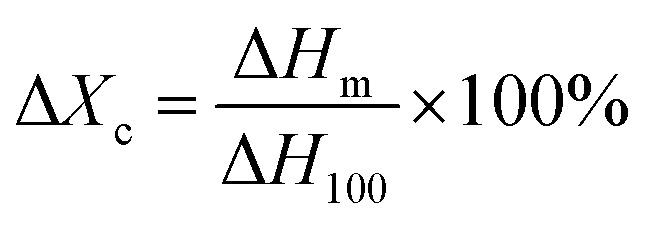 | (2) |
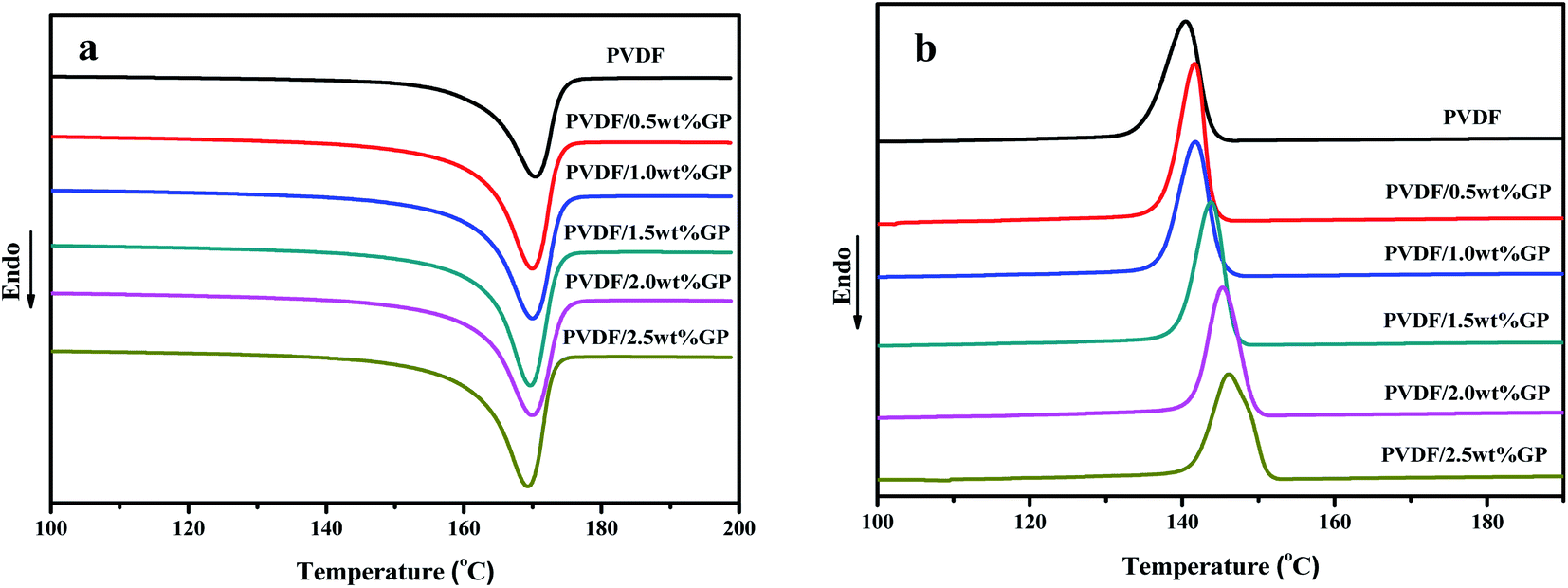 | ||
| Fig. 11 Heating (a) and cooling (b) DSC curves of PVDF and PVDF/GP composites. | ||
| Sample | Tm/°C | Tc/°C | Xc/% |
|---|---|---|---|
| Pure PVDF | 169.7 | 139.6 | 47.5 |
| PVDF/0.5 wt% GP | 170.2 | 141.7 | 51.6 |
| PVDF/1 wt% GP | 170.0 | 142.7 | 52.9 |
| PVDF/1.5 wt% GP | 169.9 | 143.8 | 55.3 |
| PVDF/2 wt% GP | 169.9 | 145.3 | 55.8 |
| PVDF/2.5 wt% GP | 169.2 | 146.1 | 57.6 |
It could be clearly seen from Table 1 that the melting points of PVDF and PVDF/GP composites were all appeared at about 170 °C, indicating that the addition of GP took no effect on the melting point of PVDF, which might be attributed to the similar melting point (167–172 °C) of α phase and β phase (170–175 °C). With the increase of the GP content, both the crystallization temperature and the crystallinity of PVDF gradually raised for the “nucleating effect” of GP on PVDF molecules, thus promoting the heterogeneous nucleation of PVDF and resulting in the increase of the crystallization ability of PVDF molecules.
TGA and DTG curves of PVDF/GP composites under nitrogen atmosphere were shown in Fig. 12. With the addition of GP, the thermal stability of PVDF was improved, which was reflected in TG and DTG curves by that the initial thermal decomposition temperature of PVDF gradually increased (the maximum increase was 15 °C) and the DTG curve shifted to higher temperature. This could be ascribed to the huge specific surface area and good thermal stability of GP, which blocked some PVDF molecules from heat sources to a certain extent, thus effectively improving the thermal stability of PVDF/GP composite. The formation of the three-dimensional network structure of the well dispersed GP was another contribution to the enhanced thermal stability of PVDF by constraining the movement of PVDF molecular chains.
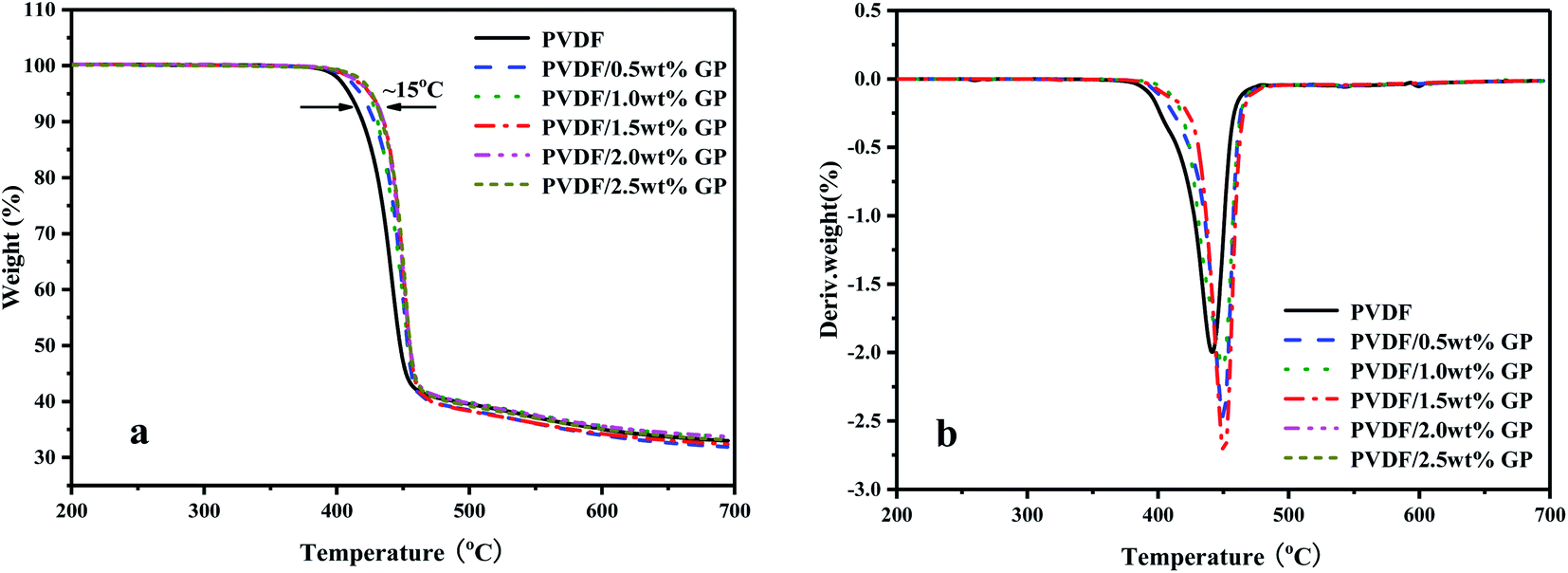 | ||
| Fig. 12 TGA (a) and DTG (b) curves of PVDF and PVDF/GP composites. | ||
3.5 Dynamic mechanical properties of PVDF/GP composites
Dynamic mechanical analysis (DMA) is an important method to study the viscoelasticity, the glass transition temperature (Tg) and the interfacial bonding of material by analyzing the changes of the mechanical internal friction peak. Fig. 13 showed the dynamic viscoelastic spectra of PVDF and PVDF/GP composites. Obviously, both the storage modulus of PVDF and PVDF/GP composites gradually decreased with the increase of temperature due to the accelerated movement of PVDF molecular chains (Fig. 13(a)), which promoted the transformation of PVDF from glass state into high elastic state. At a fixed temperature, PVDF/GP composites showed higher energy storage modulus compared with PVDF, implying that the addition of GP apparently improved the storage modulus of PVDF, i.e. enhancing its rigidity. For example, when temperature was −80 °C, the storage modulus of PVDF was 3639 MPa, but for PVDF/2.5 wt% GP, the storage modulus reached 6548 MPa, almost twice as much as that of PVDF. As well known, the elastic modulus of GP with no defects can reach 1.1 TPa, and still reach 0.25 TPa even some distortion and defects exist.45 Therefore, GP is ideal reinforcement filler for polymer. Its addition would surely remarkably improve the dynamic mechanical properties of PVDF. Furthermore, the hydrogen bonds formed between the residual oxygen functional groups on GP layers and the hydrogen and fluorine atoms on PVDF molecular chains as well as the mechanical interlocking on the folding surface of GP were all beneficial to the improvement of the modulus of the composite materials.30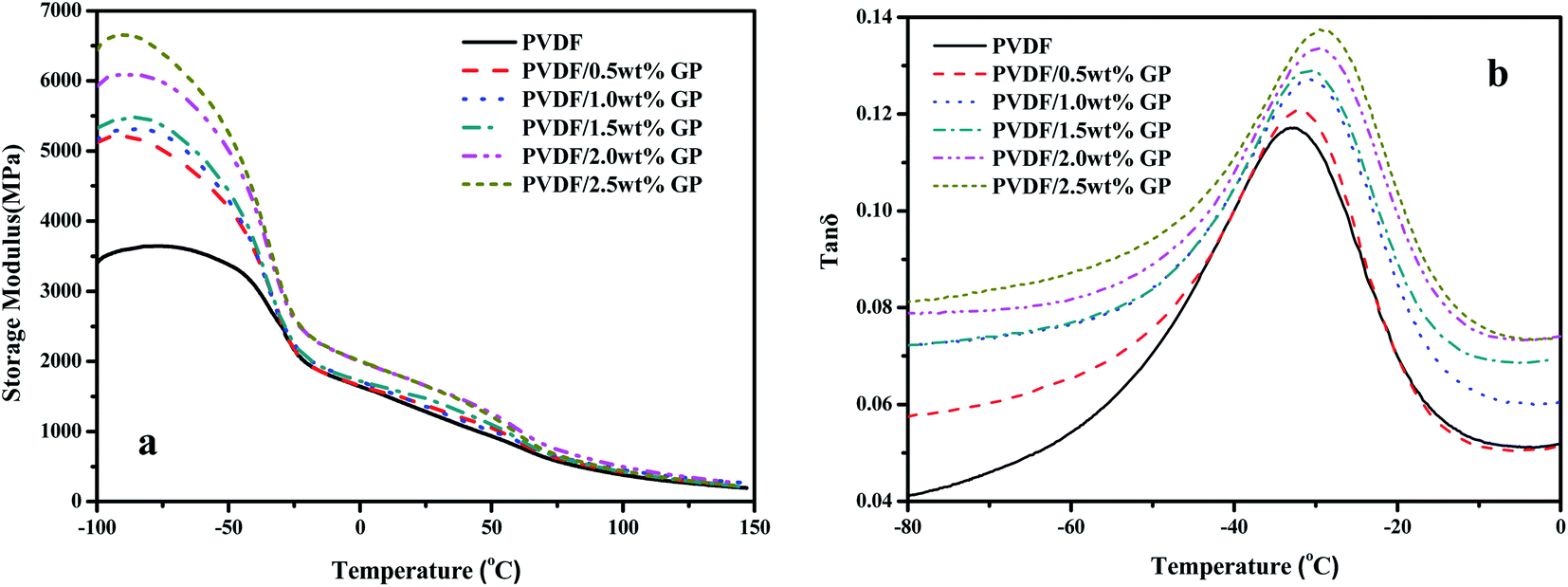 | ||
| Fig. 13 Storage modulus (a) and loss factor (b) of PVDF and PVDF/GP composites. | ||
Fig. 13(b) showed the relationship between mechanical loss factor (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) and the temperature of PVDF/GP composites. The temperature corresponding to the maximum tanδ could be considered as the glass transition temperature (Tg). Obviously, Tg of the composites continuously increased with the increase of GP content, i.e. −33.1 °C, −32.3 °C, −31.3 °C, −30.8 °C, −29.7 °C and −28.9 °C, respectively, confirming again the enhancement of GP on PVDF caused by the restrictive effect of the well dispersed GP layers on PVDF molecular chains and the special network structures constructed between GP and PVDF, which increased the heat needed for the relaxation of PVDF segments.
δ) and the temperature of PVDF/GP composites. The temperature corresponding to the maximum tanδ could be considered as the glass transition temperature (Tg). Obviously, Tg of the composites continuously increased with the increase of GP content, i.e. −33.1 °C, −32.3 °C, −31.3 °C, −30.8 °C, −29.7 °C and −28.9 °C, respectively, confirming again the enhancement of GP on PVDF caused by the restrictive effect of the well dispersed GP layers on PVDF molecular chains and the special network structures constructed between GP and PVDF, which increased the heat needed for the relaxation of PVDF segments.
3.6 Mechanical properties of PVDF/GP composites
The effects of GP on the static mechanical properties of PVDF were shown in Fig. 14. Apparently, the Young's modulus of PVDF increased with the increase of GP content, while the tensile strength first increased, then decreased. The maximum tensile strength obtained when GP was 1.0 wt%. As studied in previous section, under the strong squeezing and shearing force of our pan mill, GP was partly exfoliated and GP layers were uniformly dispersed in PVDF matrix, so could effectively bear extra-loads transferred from PVDF and increase the mechanical properties of PVDF.46 However, when GP content increased above 1.0 wt%, the exfoliation of GP layers became more and more difficult, making some small aggregations formed and the tensile strength of the composites slightly decreased.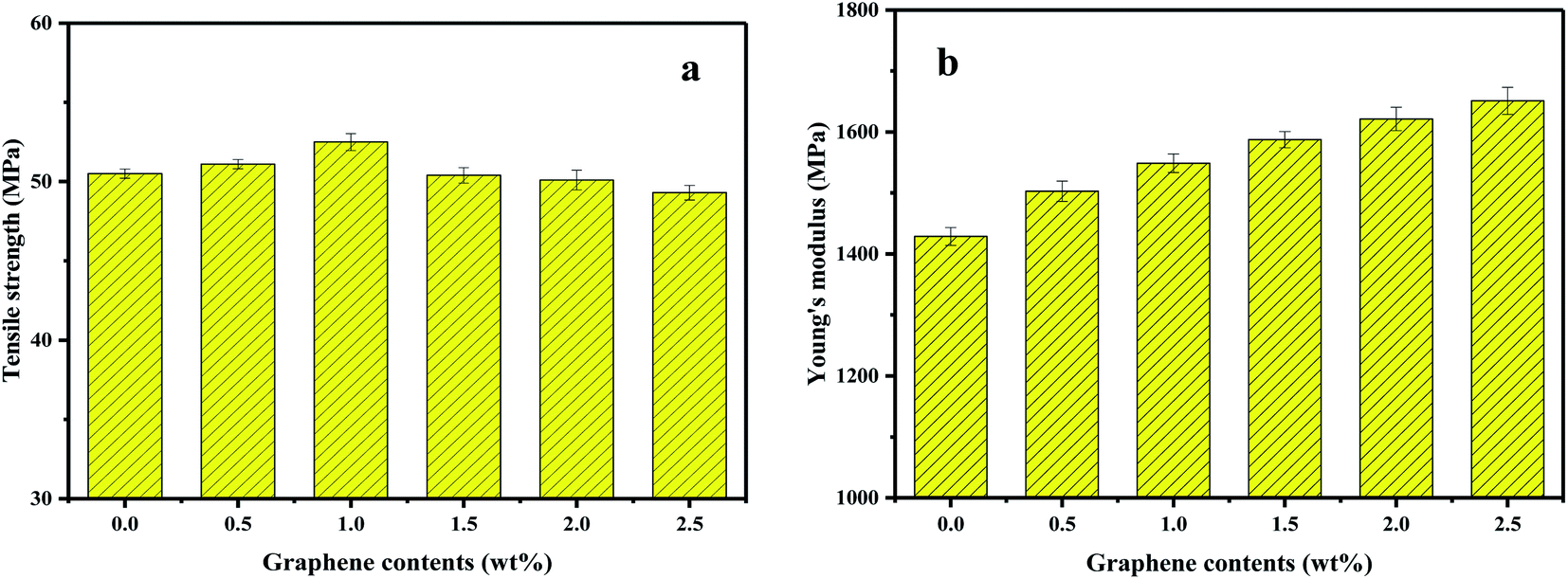 | ||
| Fig. 14 Tensile strength (a) and Young's modulus (b) of PVDF and PVDF/GP composites. | ||
3.7 Dielectric properties of PVDF/GP composites
The dielectric constant of pure PVDF and PVDF/GP composites vs. frequency at room temperature was shown in Fig. 15(a). At the same frequency, the dielectric constant of PVDF significantly increased with the increase of GP content. Ascribing to the effect of our S3M technology, GP with high conductivity and large specific surface area was evenly dispersed in PVDF matrix, enhancing the interface polarization between GP and PVDF and making free charge bounded in the interface between insulators and conductors, so endowed PVDF/GP composites with high dielectric constant.47 The dependence of PVDF composites on fillers could also be explained by the theory of micro-capacitance. The special layered structure of GP is conducive to the formation of “micro-capacitance”, i.e. a layer of dielectric PVDF is sandwiched into two parallel GP layers, similar to parallel plate capacitor.48 With different GP content, the dependence of PVDF composites on frequency was different, and the turning point was 1.0 wt%. When GP content was less than 1.0 wt%, the dielectric constant of PVDF composites almost unchanged, while when GP content was higher than 1.0 wt%, the dielectric constant obviously decreased with frequency. For pure PVDF, only electron displacement polarization and dipole orientation polarization existed in this frequency range, so showed less dependence on frequency. But for the composites, the increased GP led to the enhancement of the interfacial polarization, so with the increase of frequency, the polarization velocity gradually fell behind the change speed of frequency, making the dielectric constant of the composites decreased. According to the variation of dielectric constant, the percolation threshold could be predicted from the percolation effect, i.e. 1.0 wt%. When GP content exceeded this percolation threshold, the conductive pathways would be formed among GP particles due to their mutual contact, and the dielectric constant of the composite increased suddenly (465 at 1000 Hz), almost 42 times than that of pure PVDF (11 at 1000 Hz).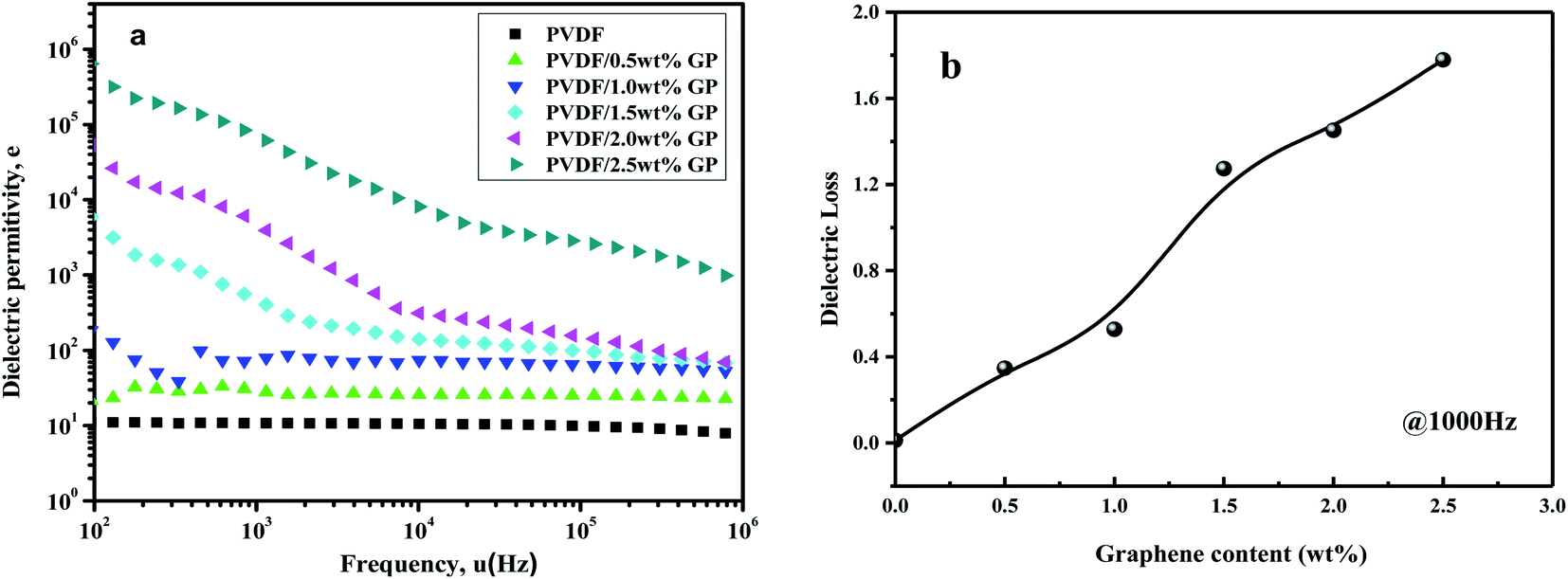 | ||
| Fig. 15 Dependence of dielectric permittivity on frequency (a) and dielectric loss ((b), 1000 Hz) of PVDF and PVDF/GP composites. | ||
Fig. 15(b) showed the curve of the dielectric loss of PVDF/GP composite vs. GP content at 1000 Hz. The dielectric loss values of pure PVDF, PVDF/1.5 wt% GP and PVDF/2.5 wt% GP were 0.012, 1.274 and 1.779 respectively, which were higher than those with other fillers.49,50 This was the results of the super-specific surface area of GP, which made its layers easy to be contacted with each other and the composite material changed into conductor, causing leakage current, and therefore leading to higher dielectric loss.
4 Conclusion
Based on S3M technology, PVDF composites with uniformly dispersed GP and perfect comprehensive properties were successfully obtained. Under the strong squeezed and three-dimensional shear effects of S3M, the exfoliation and solid grafting of GP in PVDF matrix were realized, and with the synergistic effects of GP and S3M on PVDF molecules, α phase in PVDF gradually transformed to β phase, making the co-milled PVDF/GP powders mainly presented β-phase crystal, e.g. the relative content of β phase in PVDF/1.0 wt% GP composite powders was 88%. In this way, PVDF still had relatively high β phase (42.2%) after the melt extrusion and injection. GP could also act as the “nucleating agent” to promote the heterogeneous nucleation of PVDF, thus increasing the crystallization temperature, crystallinity, thermal stability and mechanical properties of PVDF. Most important of all, the composites presented high dielectric constants, and the percolation threshold was 1.0 wt%. When GP content was above this percolation threshold, the dielectric constant of the composite was 465 at 1000 Hz, about 42 times as much as pure PVDF. The approach presented in this paper provides a new idea for preparing PVDF based materials with good comprehensive properties.Conflicts of interest
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.Acknowledgements
The authors are gratefully for the financial support from the National Natural Science Foundation of China (51720105012 and 51721091) and Sichuan Province (2019ZDZX0018).References
- Y. Li, Y. Shi, F. Cai, J. Xue, F. Chen and Q. Fu, Composites, Part A, 2015, 78, 318–326 CrossRef CAS.
- Y.-J. Wan, P.-L. Zhu, S.-H. Yu, W.-H. Yang, R. Sun, C.-P. Wong and W.-H. Liao, Compos. Sci. Technol., 2017, 141, 48–55 CrossRef CAS.
- L. Jia-long, Y. Jing-hua, J. Tianyi, F. Yu, L. Yuan-yuan, Z. He, L. Yan-peng, Z. Cong-cong, Y. Dong and S. Bo, Mater. Lett., 2019, 234, 74–78 CrossRef.
- Z. M. Dang, J. K. Yuan, S. H. Yao and R. J. Liao, Adv. Mater., 2013, 25, 6334–6365 CrossRef CAS PubMed.
- L. Yao, D. Wang, P. Hu, B.-Z. Han and Z.-M. Dang, Adv. Mater. Interfaces, 2016, 3, 1600016 CrossRef.
- Y. Liu, J. Yin, X. Liu, X. Zhao, M. Chen, J. Li, H. Zhao, C. Zhu and B. Su, Composites, Part B, 2019, 156, 252–258 CrossRef CAS.
- P. Martins, A. C. Lopes and S. Lanceros-Mendez, Prog. Polym. Sci., 2014, 39, 683–706 CrossRef CAS.
- H. H. Singh, S. Singh and N. Khare, Polym. Adv. Technol., 2018, 29, 143–150 CrossRef CAS.
- J. Yu, P. Jiang, W. Chao, L. Wang and X. Wu, Polym. Compos., 2011, 32, 1483–1491 CrossRef CAS.
- Z. Chen, H. Huang, S. Yan, Z. Zheng, S. Liu, Y. Yuan, J. Zhao and F. Yi, Ind. Eng. Chem. Res., 2017, 56, 9926–9932 CrossRef CAS.
- M. V. Silibin, V. S. Bystrov, D. V. Karpinsky, N. Nasani, G. Goncalves, I. M. Gavrilin, A. V. Solnyshkin, P. A. A. P. Marques, B. Singh and I. K. Bdikin, Appl. Surf. Sci., 2017, 421, 42–51 CrossRef CAS.
- W. Li, Z. Song, J. Qian, Z. Tan and X. Ran, Carbon, 2018, 141, 728–738 CrossRef.
- S. Song, S. Xia, S. Jiang, X. Lv, S. Sun and Q. Li, Materials, 2018, 11, 347 CrossRef PubMed.
- W. C. Yu, T. Wang, G. Q. Zhang, Z. G. Wang and Z. M. Li, Compos. Sci. Technol., 2018, 167, 260–267 CrossRef CAS.
- T. Men, X. Liu, B. Jiang, X. Long and H. Guo, Thin Solid Films, 2019, 669, 579–587 CrossRef CAS.
- Z. M. Dang, S. H. Yao, J. K. Yuan and J. Bai, J. Phys. Chem. C, 2010, 114, 13204–13209 CrossRef CAS.
- B. Luo, X. Wang, Y. Wang and L. Li, J. Mater. Chem. A, 2013, 2, 510–519 RSC.
- A. Toor, H. So and A. P. Pisano, ACS Appl. Mater. Interfaces, 2017, 9, 6369 CrossRef CAS PubMed.
- P. Saha, S. Das and S. Sutradhar, J. Appl. Phys., 2018, 124, 045303 CrossRef.
- W. L. Deng, T. Yang, L. Jin, C. Yan, H. C. Huang, X. Chu, Z. X. Wang, D. Xiong, G. Tian, Y. Y. Gao, H. T. Zhang and W. Q. Yang, Nano Energy, 2019, 55, 516–525 CrossRef CAS.
- D. Ponnamma, V. Sivakumar, A. Popelka, Y. H. A. Hussein and M. A. Al-Maadeed, Optik, 2019, 176, 372–383 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308 CrossRef CAS PubMed.
- B. Lin, Z. T. Li, Y. Yang, Y. Li, J. C. Lin, X. M. Zheng, F. A. He and K. H. Lam, Compos. Sci. Technol., 2019, 172, 58–65 CrossRef CAS.
- J. Lee and S. Lim, J. Ind. Eng. Chem., 2018, 67, 478–485 CrossRef CAS.
- W. Zhang, M. Liang and C. Lu, Cellulose, 2007, 14, 447–456 CrossRef CAS.
- H. Xu, X. Zhang, Z. Wei, T. Dong and C. Lu, J. Vinyl Addit. Technol., 2014, 20, 177–184 CrossRef.
- D. M. Esterly and B. J. Love, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 91–97 CrossRef CAS.
- Q. Wang, J. Cao, J. Huang and X. Xu, Polym. Eng. Sci., 1997, 37, 1091–1101 CrossRef CAS.
- J. Guan, C. Xing, Y. Wang, Y. Li and J. Li, Compos. Sci. Technol., 2016, 138, 98–105 CrossRef.
- J. H. Yu, X. Y. Huang, C. Wu and P. K. Jiang, IEEE Trans. Dielectr. Electr. Insul., 2011, 18, 478–484 CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov and S. Roth, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- J. Yang, Y. Hu, C. Jin, L. Zhuge and X. Wu, Appl. Surf. Sci., 2020, 503, 144334 CrossRef CAS.
- T. Boccaccio, A. Bottino, G. Capannelli and P. Piaggio, J. Membr. Sci., 2002, 210, 315–329 CrossRef CAS.
- J. Maiz, J. Sacristan and C. Mijangos, Chem. Phys. Lett., 2010, 484, 290–294 CrossRef CAS.
- Q. Wang, H. Q. Chen and Y. Liu, Polym.-Plast. Technol. Eng., 2002, 41, 215–228 CrossRef CAS.
- R. Gregorio, J. Appl. Polym. Sci., 2006, 100, 3272–3279 CrossRef CAS.
- R. Imamura, A. B. Silva and R. Gregorio Jr, J. Appl. Polym. Sci., 2008, 110, 3242–3246 CrossRef CAS.
- S. Yu, W. Zheng, W. Yu, Y. Zhang, Q. Jiang and Z. Zhao, Macromolecules, 2009, 42, 8870–8874 CrossRef CAS.
- P. Martins, C. M. Costa, M. Benelmekki, G. Botelho and S. Lanceros-Mendez, CrystEngComm, 2012, 14, 2807–2811 RSC.
- R. Gregorio Jr and M. Cestari, J. Polym. Sci., Part B: Polym. Phys., 1994, 32, 859–870 CrossRef.
- G. Chen, M. Nie and Q. Wang, J. Vinyl Addit. Technol., 2016, 24, 103–108 CrossRef.
- I. T. S. Garcia and D. Samios, Polymer, 1998, 39, 2563–2569 CrossRef CAS.
- H. Sasaki, P. K. Bala, H. Yoshida and E. Ito, Polymer, 1995, 36, 4805–4810 CrossRef CAS.
- L. Changgu, W. Xiaoding, J. W. Kysar and H. James, Science, 2008, 321, 385–388 CrossRef PubMed.
- X. Liu, T. Ji, N. Li, Y. Liu, J. Yin, B. Su, J. Zhao, Y. Li, G. Mo and Z. Wu, Mater. Des., 2019, 180, 107963 CrossRef CAS.
- N. Maity, A. Mandal and A. K. Nandi, Polymer, 2016, 103, 83–97 CrossRef CAS.
- Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191–3195 CrossRef CAS.
- H. Cheng, Q. M. Zhang and S. Ji, Appl. Phys. Lett., 2003, 82, 3502–3504 CrossRef.
- Y. Jin, X. Ning and R. A. Gerhardt, Nano Energy, 2016, 30, 407–416 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |