
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DMAP-stabilized bis(silyl)silylenes as versatile synthons for organosilicon compounds†
Richard Holzner‡
,
Dominik Reiter‡,
Philipp Frisch and
Shigeyoshi Inoue *
*
Department of Chemistry, WACKER-Institute of Silicon Chemistry and Catalysis Research Center, Lichtenbergstraße 4, 85748 Garching bei München, Germany. E-mail: s.inoue@tum.de
First published on 21st January 2020
Abstract
DMAP-stabilized silylenes 1a–c are obtained from the reductive debromination of the corresponding dibromosilanes in the presence of DMAP. Their distinctly different thermal isomerization reactions via C–H bond activation, dearomative ring expansion and silyl migration are discussed. Furthermore, complexes 1 dissociate at elevated temperatures, providing the corresponding free silylenes in situ, which are even capable of single-site activation of H2. Additionally, a potassium-substituted silicon-centered radical 2 is isolated from overreduction of (tBu3Si)2SiBr2.
Introduction
Silylenes (R2Si:), the heavier congeners of carbenes (R2C:) have attracted much attention in modern main group chemistry in recent years.1 The substituents R can either be monodentate, or cyclic, bidentate ligands, as in the case of the extensively studied class of N-heterocyclic silylenes (NHSis). In general, silylenes possess a lone pair of electrons and an empty 3pz orbital and can therefore display ambiphilic reaction behaviour both as Lewis bases and Lewis acids. This particular reactivity profile even enables the facile activation of small molecules.2 Thus, silylenes are considered to be promising candidates for metal-free catalysis.2 In contrast to carbenes, however, the singlet ground state is energetically favoured for almost all reported silylenes. The two sole exceptions are transient silylenes bearing two bulky and strongly electropositive supersilyl (tBu3Si) substituents, or both supersilyl groups and alkali metal substituents. However, these species were only generated and analyzed in situ at temperatures below 15 K.3 These reports already underline the peculiarity of bis(silyl)silylenes. In fact, no room temperature stable, two-coordinate derivative has been isolated to date. In all synthetic attempts the extremely reactive bis(silyl)silylene was not stable and either silyl migration4 or C–H bond activation occurred, even at low temperatures.3a Very recently, we presented a bis(silyl)silylene that undergoes reversible isomerization to the corresponding tetra(silyl)disilene.5 Although this compound is relatively stable, it eventually decomposes via insertion of the silylene moiety into a C–H bond of a substituent. A convenient method to stabilize silylenes is to control their excessive electrophilicity by coordination of a Lewis base, as already recognized by Tokitoh and co-workers in 1997.6 In fact, electron donation from N-heterocyclic carbenes (NHCs) was the only way so far to isolate bis(silyl)silylenes.7 Sekiguchi et al. successfully employed this approach and obtained the NHC-stabilized silylenes I (Fig. 1).8 Lately, several additional examples of acyclic bis(silyl)silylene NHC complexes were reported by Cowley (II)9 and by our group (III and IV).5 Besides those acyclic representatives, the groups of Scheschkewitz10 and Lips11 synthesized NHC-stabilized silylenes with the low-coordinate silicon center being embedded in a three-membered silicon cycle. Although electron-donation of NHCs to the vacant p-orbital of silylenes is an effective method to allow isolation of these compounds, it brings the downside of a significantly reduced reactivity. Accordingly, none of the examples listed is capable of activating small molecules such as dihydrogen. Therefore, a weaker donor–acceptor interaction is necessary to achieve a balance between reactivity and stability of the respective bis(silyl)silylene compounds. 4-N,N-Dimethylaminopyridine (DMAP) is a much weaker Lewis base, compared to NHCs and was already applied by the groups of Driess12 and So13 to isolate the low-coordinate silicon donor–acceptor complexes V and VI. Thus, we envisioned DMAP to be a suitable Lewis base, strong enough to stabilize elusive bis(silyl)silylenes, yet weak enough to partially maintain their reactivity. Very recently, we reported the first acyclic bis(silyl)silylene–DMAP adduct 1a (cf. Scheme 1).5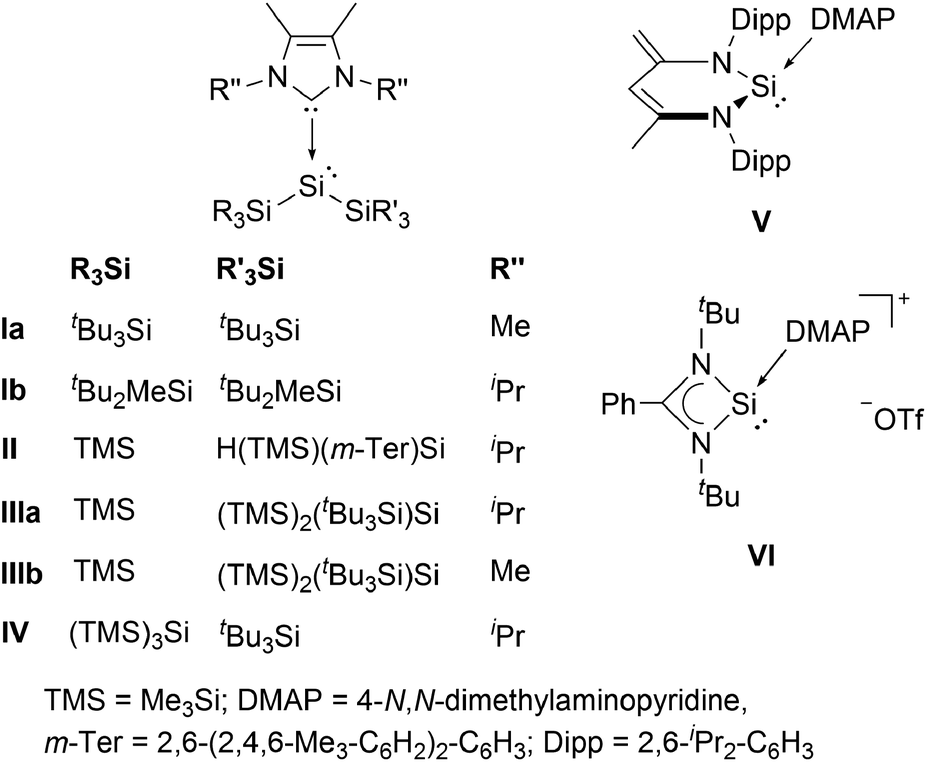 | ||
| Fig. 1 Acyclic NHC-stabilized bis(silyl)silylenes I–IV and low-coordinate silicon DMAP complexes V and VI. | ||
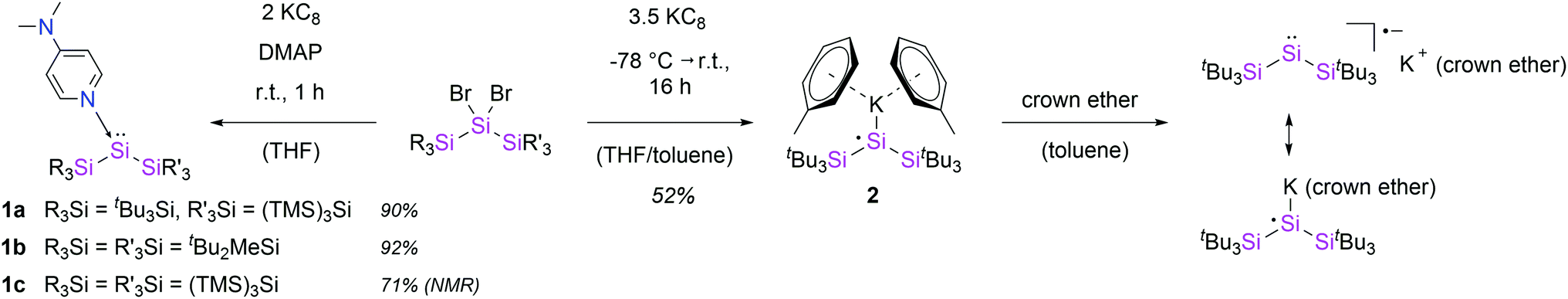 | ||
| Scheme 1 Synthesis of DMAP-stabilized silylenes 1a–c and silyl radical 2. | ||
Herein, we extend this class of donor-stabilized, highly reactive bis(silyl)silylenes. Decomposition pathways and reactivity of these novel silylenes are presented and discussed in detail. Additionally, we report the synthesis and characterization of the potassium-substituted silyl radical 2.
Results and discussion
Synthesis of novel DMAP–silylene complexes 1 and radical 2
In an approach analogue to the synthesis of 1a, we obtained the donor-stabilized bis(silyl)silylene 1b from the reductive debromination of the corresponding dibromosilane with KC8 in presence of DMAP (Scheme 1). Silylene 1b was obtained as red-brown crystals in excellent yield (92%) and fully characterized. Neither the formation of any decomposition products, nor of the disilene (tBu2MeSi)2Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si(SitBu2Me)2
Si(SitBu2Me)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 was observed during the synthesis. Compared to compound 1a, the 29Si NMR signal of the silylene Si atom in 1b is slightly upfield-shifted to 61.5 ppm (68.8 ppm in 1a). Single crystal X-ray diffraction (SC-XRD) analysis revealed a Si:–NDMAP bond length in compound 1b of 1.937(5) Å (Fig. 2). This value is essentially identical to that in 1a (1.942(2) Å)5 and clearly within the range of previously reported low-coordinate silicon–DMAP donor–acceptor complexes (1.84–2.01 Å).9,12,13,15 The high degree of pyramidalization around the silylene center in 1b (sum of bond angles Σθ = 318.1°) results from the stereo-chemically active electron lone pair and also compares very well to 1a (Σθ = 318.7°).5
14 was observed during the synthesis. Compared to compound 1a, the 29Si NMR signal of the silylene Si atom in 1b is slightly upfield-shifted to 61.5 ppm (68.8 ppm in 1a). Single crystal X-ray diffraction (SC-XRD) analysis revealed a Si:–NDMAP bond length in compound 1b of 1.937(5) Å (Fig. 2). This value is essentially identical to that in 1a (1.942(2) Å)5 and clearly within the range of previously reported low-coordinate silicon–DMAP donor–acceptor complexes (1.84–2.01 Å).9,12,13,15 The high degree of pyramidalization around the silylene center in 1b (sum of bond angles Σθ = 318.1°) results from the stereo-chemically active electron lone pair and also compares very well to 1a (Σθ = 318.7°).5
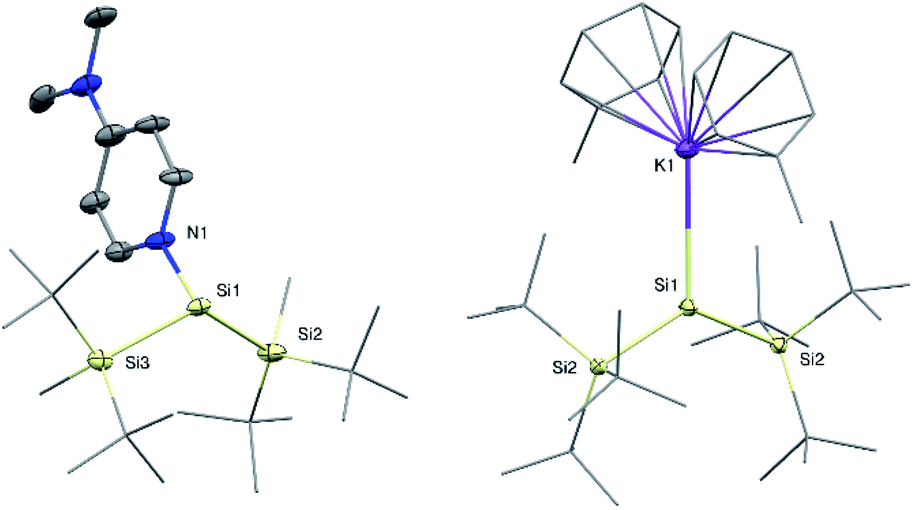 | ||
| Fig. 2 Molecular structures of silylene 1b (left) and silyl radical 2 (right) with thermal ellipsoids drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: 1b: Si1–N1 1.937(5), Si1–Si2 2.390(3), Si1–Si3 2.378(3), Si2–Si1–Si3 123.1(1), Si2–Si1–N1 96.2(2), Si3–Si1–N1 98.8(2); 2: Si1–Si2 2.3936(14), Si1–K1 3.315(2), K1–Si1–Si2 114.91(2), Si2–Si1–Si2* 130.19(3). | ||
Additionally, the steric hindrance of the silylene center was increased by introducing bulky hypersilyl groups ((TMS)3Si), resulting in complex 1c. Compound 1c, which is the first stable bis(hypersilyl)silylene species, was identified by the characteristic 29Si NMR signal of the low-coordinate silicon nucleus (72.5 ppm), similar to the resonances of 1a and 1b.5 Remarkably, in this case, the facile TMS-migrations were prevented by the coordination of DMAP and the silylene could be stabilized successfully. In sharp contrast, we were not able to isolate the bis(hypersilyl)silylene moiety with NHCs. This result underlines the difference in reactivity between NHCs and the weaker Lewis base DMAP. Unfortunately, the reaction was accompanied by the by-product formation of hexakis(trimethylsilyl)trisilirane (4) and Si(TMS)4, reflecting the high propensity of hypersilyl groups towards TMS-group migrations.
Despite several attempts, we were not able to isolate the DMAP-stabilized bis(supersilyl) silylene (tBu3Si)2Si: ← DMAP with the same approach used for the syntheses of 1. Even at low temperatures, the reduction of the corresponding dibromosilane only afforded the decomposition product of the free silylene (disiletane from C–H bond activation).3a With an excess of 3.5 equivalents of KC8, however the potassium-substituted silyl radical 2 was generated, even in the presence of DMAP. The solid state structure of 2 was unambiguously determined by SC-XRD analysis (Fig. 2). Silyl radical 2 exhibits a completely planar geometry (sum of bond angles Σθ = 360.0°) which is typical for alkali metal-substituted silyl radicals.16 The Si–K bond distance (3.315(2) Å) is in the same range as observed in four-coordinate potassium silanides, such as hypersilyl potassium (3.352(4) Å).4a Thus compound 2 is clearly a contact ion pair in the solid state. Unfortunately, 2 is extremely sensitive and decomposes in toluene solution. Therefore, no satisfactory spectroscopic data was obtained. After synthesis in absence of DMAP and stabilization by crown ether (18-C-6) however, we were able to obtain an EPR spectrum which contains a signal with a g value of 2.0056 and a hyperfine coupling a(α-29Si) = 2.92 mT (see ESI, Fig. S7†). Coupling with the β-29Si nuclei was not observable. This g value is in the same range, as it was reported for other alkali metal-substituted silyl radicals.16,17 Furthermore, no signal splitting from coupling of the unpaired electron with the K nucleus was observed. Presumably, in solution, compound 2 in presence of crown ether exists as solvent-separated ion pair. This observation is consistent with reports of a potassium substituted silyl radical.16
Thermally induced isomerization of 1
With the novel silylene complexes 1 in hand, we initially tested their thermal stability. Silylene 1a isomerizes to the respective disiletane VII via DMAP dissociation and subsequent C–H bond activation at elevated temperatures (Scheme 2). The same product, that was observed for the decomposition of the donor-free disilene/silylene equilibrium mixture.5 Surprisingly, upon heating compound 1b to 65 °C, the silylene fragment inserts into the pyridine ring of DMAP, generating azasilepin 3 by dearomative ring expansion in quantitative yield. Silepin formation via insertion of a silylene into an aromatic ring system has previously been reported,18 oftentimes either thermally19 or photochemically20 induced. After transformation from 1b to 3 and thus increase of the coordination number, the 29Si NMR signal of the central silicon atom is strongly upfield-shifted to −28.1 ppm. This value is comparable to that of a similar compound, reported by Tokitoh et al. from the reaction of a transient, in situ generated bis(aryl)silylene with DMAP (−20.8 ppm).21 In comparison to 1b, the Si–N bond distance in 3 is shortened by 10% to 1.750(1) Å, indicating a covalent bonding-type instead of the dative interaction in 1b. This bond length is identical to that in Tokitoh's azasilepin.21 Furthermore, the Si center adopts a tetrahedral coordination sphere within the boat-shaped, seven-membered heterocyclic ring (cf. Fig. 3). In sharp contrast to the related compounds 1a and 1b however, the thermal decomposition of silylene 1c does not proceed via C–H, or C–N bond activation, but in fact by silyl migration. At 65 °C, 1c isomerizes under liberation of DMAP to the cyclic silane 4, which was already observed from rearrangement of ((TMS)3Si)2Si: in the attempted synthesis of the free silylene.4a,d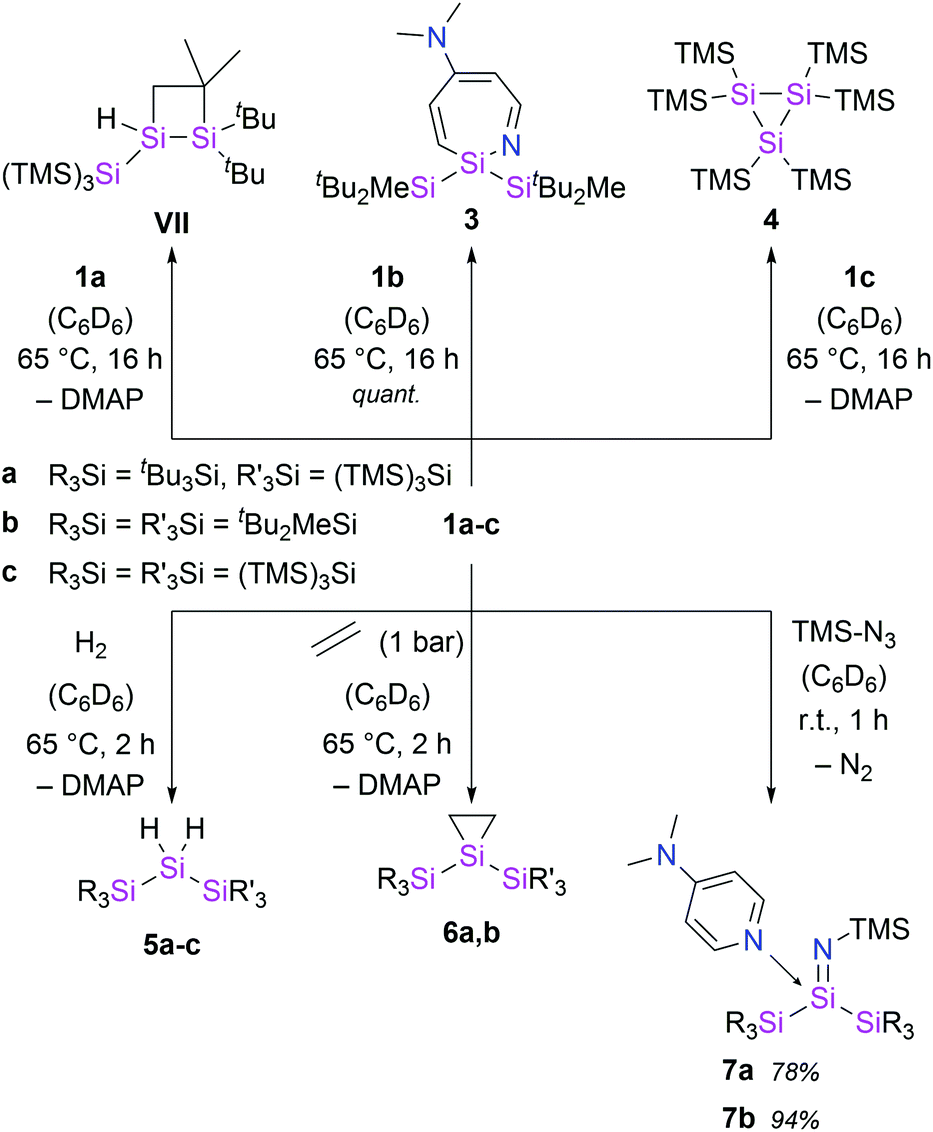 | ||
| Scheme 2 Thermally-induced decomposition of silylenes 1 and synthesis of hydrosilanes 5, siliranes 6 and silaimines 7. | ||
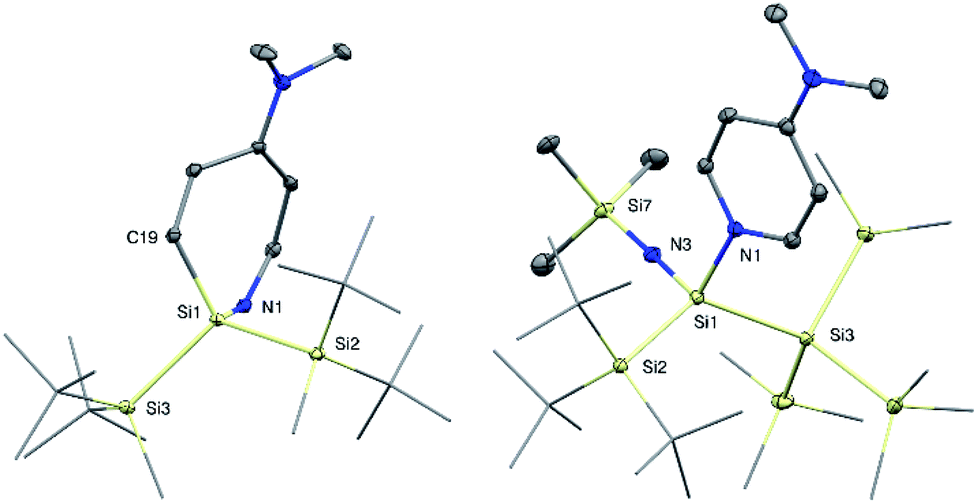 | ||
| Fig. 3 Molecular structures of azasilepin 3 (left) and silaimine 7a (right) with thermal ellipsoids drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: 3: Si1–N1 1.750(1), Si1–C19 1.878(1), Si1–Si2 2.4144(6), Si2–Si1–Si3 113.74(2), Si2–Si1–N1 109.08(4), N1–Si1–C19 104.71(5); 7a: Si1–Si2 2.453(1), Si1–N1 1.928(2), Si1–N3 1.616(2), N3–Si7 1.660(2), Si2–Si1–Si3 125.08(3), N1–Si1–N3 106.08(8), Si1–N3–Si7 177.1(1). | ||
Small molecule activation by silylenes 1
Single-site activation of the enthalpically strong, apolar dihydrogen molecule remains a challenging task for low-coordinate silicon compounds. So far, this was only achieved by few acyclic, donor-free silylenes and a masked iminosilyl silylene.5,18a,22 In fact, to date, there are no reports of H2 activation by a silylene base complex.Although, the thermal decomposition reactions of 1a–c strongly depend on the silyl substituents and proceed via three different mechanisms, they are all based on the extreme reactivity of the respective free silylene. Furthermore, the calculated Gibbs free bond-dissociation energy of 1a (15.3 kcal mol−1),5 which is lower than for the analogous, NHC-coordinated (hypersilyl)(supersilyl)silylene IV5 (16.3 kcal mol−1)5 also suggests a higher reactivity of the DMAP–silylene complexes, compared to the NHC-stabilized bis(silyl)silylenes. Therefore, we conceived compounds 1a–c to be easily accessible synthetic equivalents for these unstable, elusive, donor-free bis(silyl)silylenes and conducted a reactivity study towards activation of small molecules. Indeed, all three DMAP–silylenes underwent dihydrogen addition reactions upon heating to 65 °C, furnishing the reported corresponding dihydrosilanes 5a–c in quantitative yields. (Scheme 2).5,23 Remarkably, the oxidative addition of dihydrogen to the DMAP–silylene complexes proceeds in a selective fashion, without the formation of the respective decomposition products. Free DMAP was simply removed from the product by precipitation with one equivalent of SiBr4 and subsequent filtration. Notably, no reaction was observed upon exposure of NHC-stabilized bis(silyl)silylenes Ia and IIIa to H2, even at elevated temperatures. This result underlines the inherently high reactivity of bis(silyl)silylene–DMAP complexes upon thermal dissociation of the stabilizing donor. Presumably, the H2 addition to the silylene fragments of 1 proceeds via a bimolecular reaction similar to that proposed for the free silylene (tBu3Si)((TMS)3Si)Si:.5
Additional reactivity investigations were carried out with 1a and 1b due to their easier accessibility. Silirane formation – another classical silylene reactivity – was observed after treatment of 1a and 1b with ethylene, yielding compounds 6. The 29Si NMR shift of the central Si-atom in 6b (−174.5 ppm) is similar to that of the earlier reported 6a (−164.3 ppm).5
Since the isolation of the first silaimine by Wiberg et al. in 1985,24 a number of these heavier imine analogues have been published. Besides donor free examples,25 many silaimines need additional stabilization by a coordinating Lewis base, such as NHCs.7 Interestingly, reaction of 1a and 1b with trimethylsilyl azide furnishes the DMAP-coordinated silaimines 7 under liberation of gaseous N2. The 29Si NMR signals of the central Si atoms in 7a and 7b were observed at −25.9 ppm and −25.5 ppm, respectively. Compared to a silaimine–pyridine adduct (δ29Si = −12.6 ppm),25b these resonances are slightly upfield-shifted, presumably due to the electropositive silyl groups. In the solid state, compound 7a displays a tetrahedral coordination sphere around the silicon center. Silaimine 7a contains three unique Si–N bonds, distinguishable by their characteristic lengths: a short SiN bond (1.616(2) Å), a significantly longer Si7–N3 single bond to the TMS group (1.660(2) Å) and an even further elongated, dative Si–NDMAP bond (1.928(2) Å). The central SiN distance is slightly longer, than in the donor-free silaimines, from the groups of Wiberg and Kira (1.57–1.59 Å)25a,25c and essentially identical to Klingebiel's silaimine–pyridine adduct (1.611(2) Å).25b Interestingly, the geometry of the imino group is almost linear (θ = 177.1(1)°). A similar observation was reported by Kira et al. and attributed to the electronic properties of the TMS group.25c Notably, compound 7a slowly decomposes in solution under liberation of DMAP and probably formation of the donor-free silaimine, which decomposes further to a mixture of unidentified species. Complex 7b instead is stable in solution.
Conclusions
In summary, we utilized our recently published method to synthesize two novel DMAP-stabilized silylenes 1b and 1c. Compound 1c is the first stable bis(hypersilyl)silylene complex, which could be synthesized so far. Surprisingly, silyl radical 2 was obtained in a related fashion from the over-reduction of the corresponding dibromosilane. The silylene complexes 1a–c turned out to undergo facile oxidative addition with dihydrogen and ethylene at relatively mild conditions. This remarkable reactivity originates from the respective free silylenes, which are generated in situ from dissociation of complexes 1. Stabilization of transient bis(silyl)silylenes with DMAP is the only method so far to isolate these species and reactivate their extreme reactivity upon dissociation. Therefore, complexes 1 can be considered easily accessible, stable synthetic equivalents of otherwise elusive bis(silyl)silylenes. Additionally, the unprecedented, DMAP-coordinated silaimines 7 were isolated from the reactions of the silylene complexes with trimethylsilyl azide.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are exceptionally grateful to the WACKER Chemie AG and the European Research Council (SILION 637394) for continued financial support. We thank Dr Oksana Storcheva for the EPR measurements.Notes and references
- For selected reviews on silylenes see: (a) M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704–714 CrossRef CAS PubMed; (b) B. Gehrhus and M. F. Lappert, J. Organomet. Chem., 2001, 617–618, 209–223 CrossRef CAS; (c) S. Nagendran and H. W. Roesky, Organometallics, 2008, 27, 457–492 CrossRef CAS; (d) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed; (e) M. Kira, Chem. Commun., 2010, 46, 2893–2903 RSC; (f) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed; (g) S. Yao, Y. Xiong and M. Driess, Organometallics, 2011, 30, 1748–1767 CrossRef CAS; (h) S. S. Sen, S. Khan, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2012, 45, 578–587 CrossRef CAS PubMed; (i) S. S. Sen, S. Khan, P. P. Samuel and H. W. Roesky, Chem. Sci., 2012, 3, 659–682 RSC; (j) B. Blom and M. Driess, in Functional Molecular Silicon Compounds II, ed. D. Scheschkewitz, Springer, Cham, 2013, vol. 156, pp. 85–123 Search PubMed.
- (a) P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed; (b) C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- (a) A. Sekiguchi, T. Tanaka, M. Ichinohe, K. Akiyama and S. Tero-Kubota, J. Am. Chem. Soc., 2003, 125, 4962–4963 CrossRef CAS PubMed; (b) A. Sekiguchi, T. Tanaka, M. Ichinohe, K. Akiyama and P. P. Gaspar, J. Am. Chem. Soc., 2008, 130, 426–427 CrossRef CAS PubMed.
- (a) K. W. Klinkhammer, Chem.–Eur. J., 1997, 3, 1418–1431 CrossRef CAS; (b) M. Ichinohe, R. Kinjo and A. Sekiguchi, Organometallics, 2003, 22, 4621–4623 CrossRef CAS; (c) K. Hassler, A. Dzambaski and J. Baumgartner, Silicon Chem., 2008, 3, 271–288 CrossRef; (d) C. Marschner, Eur. J. Inorg. Chem., 2015, 2015, 3805–3820 CrossRef CAS; (e) S. K. Mueller, A. Dzambaski, N. Altenhuber, A. Torvisco, K. Hassler and M. Flock, J. Mol. Struct., 2015, 1099, 197–203 CrossRef CAS; (f) M. Haas, A. Knoechl, T. Wiesner, A. Torvisco, R. Fischer and C. Jones, Organometallics, 2019, 38, 4158–4170 CrossRef CAS.
- D. Reiter, R. Holzner, A. Porzelt, P. J. Altmann, P. Frisch and S. Inoue, J. Am. Chem. Soc., 2019, 141, 13536–13546 CrossRef CAS PubMed.
- N. Takeda, H. Suzuki, N. Tokitoh, R. Okazaki and S. Nagase, J. Am. Chem. Soc., 1997, 119, 1456–1457 CrossRef CAS.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- H. Tanaka, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2012, 134, 5540–5543 CrossRef CAS PubMed.
- M. W. Stanford, J. I. Schweizer, M. Menche, G. S. Nichol, M. C. Holthausen and M. J. Cowley, Angew. Chem., Int. Ed., 2019, 58, 1329–1333 CrossRef CAS PubMed.
- A. Jana, I. Omlor, V. Huch, H. S. Rzepa and D. Scheschkewitz, Angew. Chem., Int. Ed., 2014, 53, 9953–9956 CrossRef CAS PubMed.
- B. J. Guddorf, A. Hepp and F. Lips, Chem.–Eur. J., 2018, 24, 10334–10338 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, R. Müller, M. Kaupp and M. Driess, J. Am. Chem. Soc., 2010, 132, 6912–6913 CrossRef CAS PubMed.
- H.-X. Yeong, H.-W. Xi, Y. Li, K. H. Lim and C.-W. So, Chem.–Eur. J., 2013, 19, 11786–11790 CrossRef CAS PubMed.
- A. Sekiguchi, S. Inoue, M. Ichinohe and Y. Arai, J. Am. Chem. Soc., 2004, 126, 9626–9629 CrossRef CAS PubMed.
- T. Yamaguchi and A. Sekiguchi, J. Am. Chem. Soc., 2011, 133, 7352–7354 CrossRef CAS PubMed.
- S. Inoue, M. Ichinohe and A. Sekiguchi, Organometallics, 2008, 27, 1358–1360 CrossRef CAS.
- (a) D. Bravo-Zhivotovskii, I. Ruderfer, S. Melamed, M. Botoshansky, B. Tumanskii and Y. Apeloig, Angew. Chem., Int. Ed., 2005, 44, 739–743 CrossRef CAS PubMed; (b) S. Ishida, T. Iwamoto and M. Kira, J. Am. Chem. Soc., 2003, 125, 3212–3213 CrossRef CAS PubMed; (c) G. Molev, D. Bravo-Zhivotovskii, M. Karni, B. Tumanskii, M. Botoshansky and Y. Apeloig, J. Am. Chem. Soc., 2006, 128, 2784–2785 CrossRef CAS PubMed; (d) S. Inoue, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2007, 129, 6096–6097 CrossRef CAS PubMed.
- (a) D. Wendel, A. Porzelt, F. A. D. Herz, D. Sarkar, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 8134–8137 CrossRef CAS PubMed; (b) S. Ishida, T. Tamura and T. Iwamoto, Dalton Trans., 2018, 47, 11317–11321 RSC.
- (a) H. Suzuki, N. Tokitoh and R. Okazaki, J. Am. Chem. Soc., 1994, 116, 11572–11573 CrossRef CAS; (b) H. Suzuki, N. Tokitoh and R. Okazaki, Bull. Chem. Soc. Jpn., 1995, 68, 2471–2481 CrossRef CAS.
- (a) M. Kira, S. Ishida, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 2002, 124, 3830–3831 CrossRef CAS PubMed; (b) M. Kira, S. Ishida, T. Iwamoto, A. de Meijere, M. Fujitsuka and O. Ito, Angew. Chem., Int. Ed., 2004, 43, 4510–4512 CrossRef CAS PubMed; (c) T. Kosai, S. Ishida and T. Iwamoto, Chem. Commun., 2015, 51, 10707–10709 RSC.
- Y. Mizuhata, T. Sato and N. Tokitoh, Heterocycles, 2012, 84, 413–418 CrossRef CAS.
- (a) A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed; (b) A. V. Protchenko, A. D. Schwarz, M. P. Blake, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Angew. Chem., Int. Ed., 2013, 52, 568–571 CrossRef CAS PubMed.
- (a) T. Gross, H. Reinke and H. Oehme, Can. J. Chem., 2000, 78, 1399–1404 CAS; (b) A. Sekiguchi, T. Fukawa, M. Nakamoto, V. Y. Lee and M. Ichinohe, J. Am. Chem. Soc., 2002, 124, 9865–9869 CrossRef CAS PubMed.
- N. Wiberg, K. Schurz and G. Fischer, Angew. Chem., Int. Ed. Engl., 1985, 24, 1053–1054 CrossRef.
- (a) N. Wiberg, K. Schurz, G. Reber and G. Müller, J. Chem. Soc., Chem. Commun., 1986, 591–592 RSC; (b) J. Niesmann, U. Klingebiel, M. Schäfer and R. Boese, Organometallics, 1998, 17, 947–953 CrossRef CAS; (c) T. Iwamoto, N. Ohnishi, Z. Gui, S. Ishida, H. Isobe, S. Maeda, K. Ohno and M. Kira, New J. Chem., 2010, 34, 1637–1645 RSC; (d) S. Inoue and K. Leszczyńska, Angew. Chem., Int. Ed., 2012, 51, 8589–8593 CrossRef CAS PubMed; (e) J. Keuter, A. Hepp, C. Mück-Lichtenfeld and F. Lips, Angew. Chem., Int. Ed., 2019, 58, 4395–4399 CrossRef CAS PubMed; (f) A. V. Protchenko, P. Vasko, D. C. H. Do, J. Hicks, M. Ángeles Fuentes, C. Jones and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 1808–1812 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and crystallographic data. CCDC 1967942–1967945. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra10628f |
| ‡ R. H. and D. R. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |