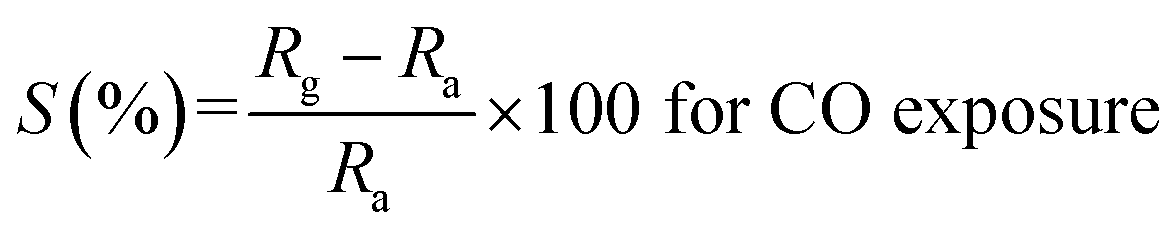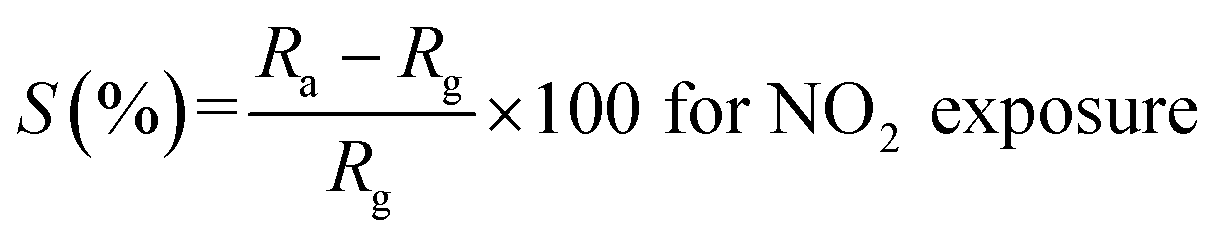DOI:
10.1039/C9RA09460A
(Paper)
RSC Adv., 2020,
10, 1297-1308
Fabrication of LaFeO3 and rGO-LaFeO3 microspheres based gas sensors for detection of NO2 and CO†
Received
13th November 2019
, Accepted 18th December 2019
First published on 7th January 2020
Abstract
In the present report, gas sensing devices based on LaFeO3 and rGO-LaFeO3 were fabricated by a photolithography technique. The X-ray diffraction, Raman spectra and FT-IR results confirm the formation of a perovskite phase and composite. XPS and TEM give the chemical compositions for both products. The higher roughness, greater surface area (62.1 m2 g−1), larger pore size (16.4 nm) and lower band gap (1.94 eV) of rGO-LaFeO3 make it a suitable candidate to obtain high sensitivity. The gas sensing performance of the devices was investigated for various concentrations of NO2 and CO gases at temperatures of 200 and 250 °C. It was observed that the rGO-LaFeO3 based device exhibited a high relative response (183.4%) for a 3 ppm concentration of NO2 at a 250 °C operating temperature. This higher response is attributed to the large surface area, greater surface roughness, and numerous active sites of rGO-LaFeO3. The gas sensing properties investigated show that rGO-LaFeO3 is an excellent candidate for an NO2 sensor.
1. Introduction
Environmental concerns about health hazards due to the presence of toxic gases, such as CO, CO2, NO2, H2, and O3, and consequent safety regulations have necessitated the increased use of sensors in various settings from industrial sites to automobiles, the workplace and even homes. Among the various toxic gases, CO and NO2 are the most hazardous air pollutants and are risky for humans, animals, and plants. According to the Occupational Safety and Health Administration (OSHA), the lowest tolerance limits for CO and NO2 gases over a period of 8 h are ∼20 ppm and ∼5 ppm, respectively. Over-exposure to these gases could be a cause of disease and in extreme cases even loss of human life.1 Therefore, the development of CO and NO2 gas sensors which can detect minute concentrations of these toxic gases at low temperature is in high demand for environment protection. Nano-materials have been shown to be promising materials for gas sensing because their high surface area provides more active sites for gas adsorption. Single and composite metal oxides, like SnO2,2 TiO2,3 ZnO,4 In2O3,5 WO3,6 Fe2O3,7 ZnO–TiO2,8 and Fe doped ZnO,9 are important materials for detecting small concentrations of harmful gases, but they have associated problems, such as issues about long-term stability and high operating temperature. Graphene, a very famous carbonic material, shows excellent properties and the very high surface area which is necessary for the adsorption of minute concentrations of gases and shows high sensitivity towards the detection of dangerous gases. Graphene also displays a significant change in its electrical resistance after the adsorption of target gases, which further confirms graphene as a promising candidate for gas sensing.10 Up to now, graphene-based materials synthesized by diverse methods have been used for the detection of gases at low operating temperatures, such as reduced graphene oxide (rGO) prepared by the thermal reduction of graphene oxide (GO)11,12 and the chemical reduction of (GO),13,14 and graphene obtained by chemical vapor deposition (CVD).15,16 In particular, rGO has attracted much interest due to its great advantages of low cost and production in bulk quantities.17 M. Gautam et al.18 and Nanto et al.19 have used graphene as a gas sensor and observed high sensitivity towards CH4 and NH3 gases. Also perovskite oxides (ABO3) like LaFeO3 and SmFeO3 are well-known materials for gas sensing applications. In these materials the presence of a transition metal ion at a B-site and oxygen deficiencies play an important role in adsorption properties and catalytic behavior. LaFeO3 exhibits outstanding chemical and physical properties for multifunctional applications like chemical sensors,20 catalysts,21 electrolytes,22 fuel cells,23 biosensors24 and gas sensors.25,26 A large number of rare earth sensing materials have been synthesized for the detection of CO and NOx (toxic and combustible) gases.26–29 The use of composites is a favorable scheme to improve sensor performance. Perovskite oxide and reduced graphene oxide are promising candidates as individual sensing materials: rGO is a highly sensitive gas sensing material whereas PO (ABO3) provides a great ability to adjust its formulation by changing the elements on the A and/or B sites and doping. Hence a composite of rGO and LaFeO3 would enhance the gas sensing performance, which is the objective of this work. The authors believe that this is the first work that has used rGO decorated LaFeO3 for a gas sensing application. Gas sensing performance is generally influenced by the morphology and architecture of the materials, which are dependent on the synthesis route taken. There are several methods for the preparation of LaFeO3 and rGO-LaFeO3: viz. sol–gel,30 co-precipitation,31 ball milling32 and hydrothermal33 methods. Among these, the hydrothermal method is an effective tool for the controlled synthesis of perovskite oxide LaFeO3 and rGO decorated LaFeO3. Low-temperature growth, cost-effectiveness and an ability to control the size also make this method an efficient process.
We report the fabrication of gas sensors based on LaFeO3 and its composite with rGO for the detection of CO and NO2 gases. The gas sensing devices were fabricated on an IDE (interdigitated electrode) substrate through photolithography. The sensing materials were deposited on the IDE substrate by the drop cast method. A gas sensing study was undertaken for LaFeO3 and rGO-LaFeO3 devices under different concentrations of NO2 and CO at temperatures of 200 and 250 °C. Synthesized samples of LaFeO3 and rGO-LaFeO3 were characterized by XRD, FTIR, TGA, Raman, XPS, FESEM, AFM, HRTEM, UV-Vis, and BET.
2. Experimental
2.1 Raw materials
Fine graphite powder (99.5%) and potassium permanganate (99%) were purchased from CDH. Sulfuric acid (98%) and hydrogen peroxide (30%) were purchased from RANKEM. Hydrogen chloride (12 N) was purchased from Merck. Lanthanum nitrate hexahydrate (99.9%), iron nitrate nonahydrate (98+%) and citric acid (99+%) were purchased from Alfa Aesar.
2.2 Synthesis of LaFeO3
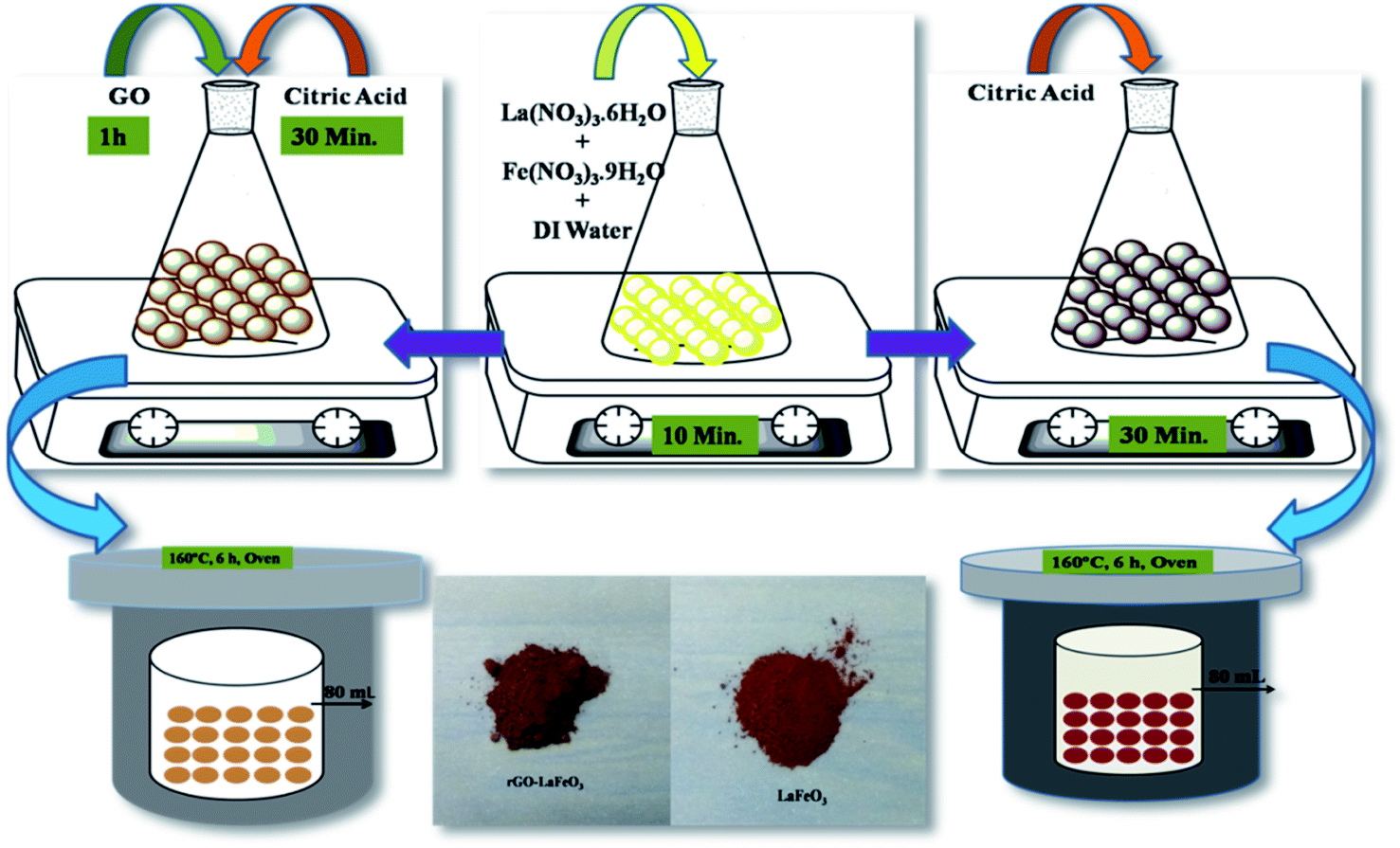
Lanthanum(III) nitrate hexahydrate (La(NO3)3·6H2O) and iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O) were used as precursors and citric acid (C6H8O7·H2O) was used as a chelating agent to prepare LaFeO3 microspheres using the hydrothermal method.34 La(NO3)3·6H2O (0.2 mol) and Fe(NO3)3·9H2O (0.2 mol) were dissolved in 80 mL of DI water under magnetic stirring for 10 minutes. Subsequently, citric acid (0.4 mol) was added to the solution under continuous magnetic stirring for 10 minutes and the reaction mixture was poured into a 100 mL Teflon-lined stainless steel autoclave and kept at 160 °C for 6 h. After cooling down to room temperature (RT), precipitates were collected by centrifugation at 4000 rpm from the autoclave, and washed with DI water and ethanol. The collected precipitates were dried in air at 60 °C, followed by calcination at 800 °C for 4 h to obtain powdered LaFeO3. The reaction between the above salts in the presence of citric acid is assumed to occur in the way shown in eqn (1).34| | |
La(NO3)3·6H2O + Fe(NO3)3·9H2O + C6H8O7·H2O → LaFeO3 + 6CO2 + 3N2 + nH2O
| (1) |
2.3 Synthesis of rGO-LaFeO3
La(NO3)3·6H2O (0.2 mol) and Fe(NO3)3·9H2O (0.2 mol) were dissolved in 80 mL of DI water under magnetic stirring for 10 minutes; then 200 mg of graphene oxide was added to the solution under ultrasonication at 50 °C for 1 h until the GO was well dispersed in the solution.35 GO was prepared by Hummers' method.36 An appropriate amount of citric acid (0.4 mol) was added to the solution under magnetic stirring for 30 minutes with the same follow-up as that for the preparation of LaFeO3 to obtain rGO-LaFeO3 powder. The reaction for the synthesis of the rGO-LaFeO3 composite is assumed to occur as shown in eqn (2). The pathway for the synthesis of LaFeO3 and its composite with rGO is shown in Fig. 1.| | |
La(NO3)3·6H2O + Fe(NO3)3·9H2O + GO + C6H8O7·H2O → rGO-LaFeO3 + 6CO2 + 3N2 + nH2O
| (2) |
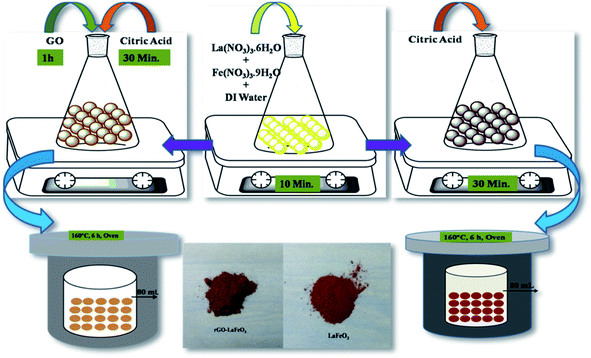 |
| | Fig. 1 Pathway for the formation of LaFeO3 and rGO-LaFeO3 microspheres. | |
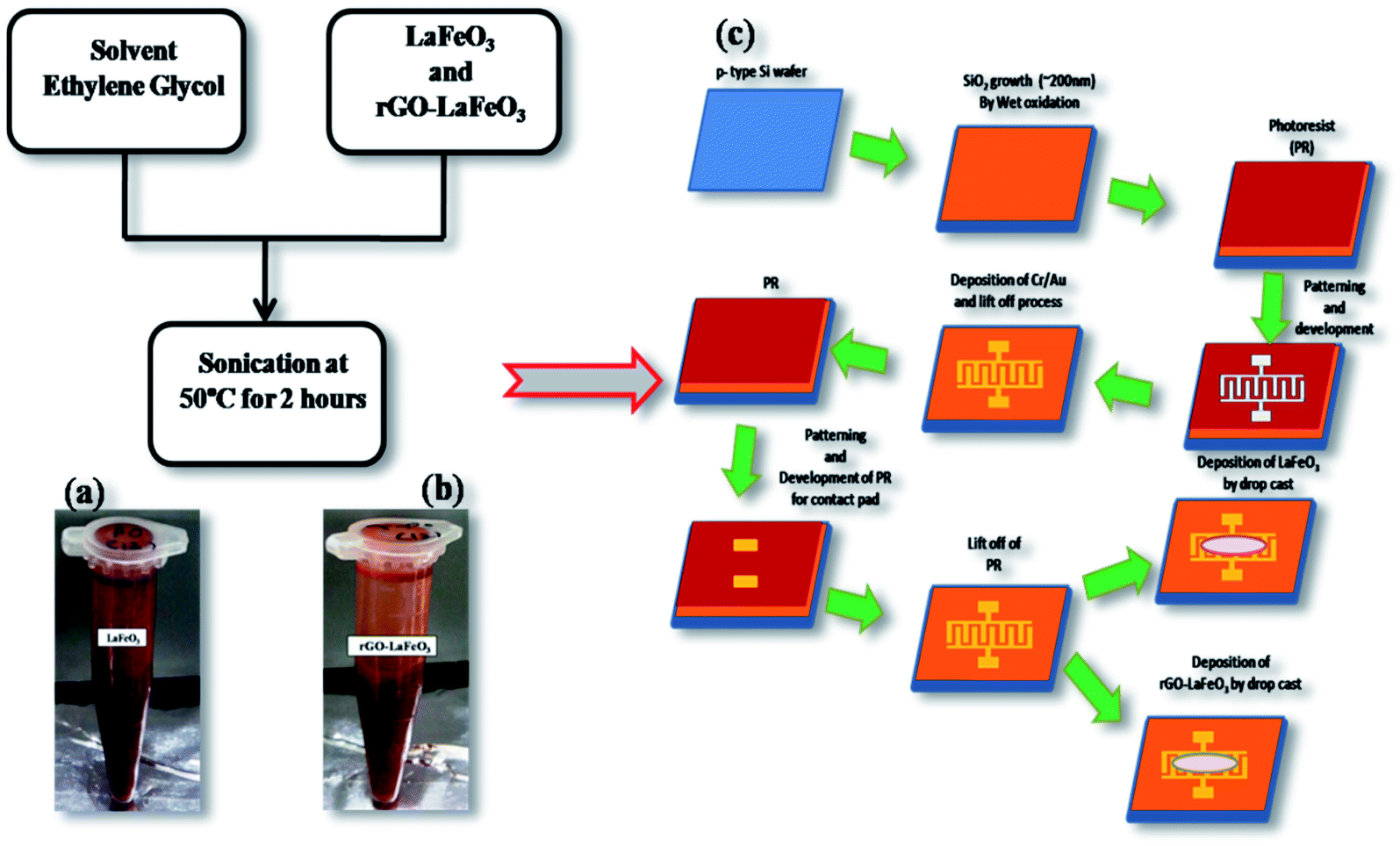
In this work, ethylene glycol (EG) was used as a solvent. For the preparation of the LaFeO3/rGO-LaFeO3 solution, 12 mg of LaFeO3/rGO-LaFeO3 were dissolved in 1 mL of EG. The solution was then sonicated at 50 °C for 2 hours to be used for film preparation, as shown in Fig. 2(a and b).
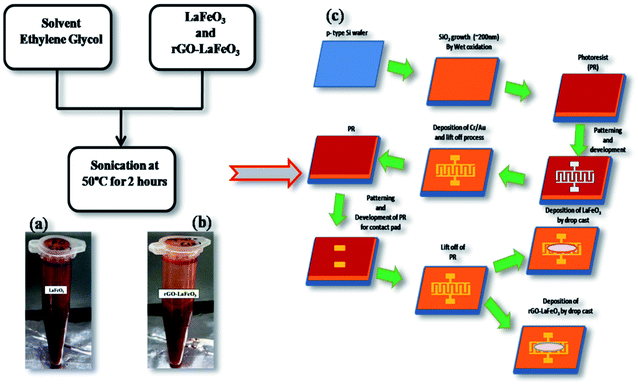 |
| | Fig. 2 Solution formation of LaFeO3 (a) and rGO-LaFeO3 (b) using solvent EG. Fabrication process of IDE deposited LaFeO3/rGO-LaFeO3 (c). Both samples are deposited by drop casting on an IDE pattern. | |
2.4 Fabrication of LaFeO3/rGO-LaFeO3–IDE based device
For CO and NO2 detection, LaFeO3 and rGO-LaFeO3 thin films were deposited on an interdigitated electrode (IDE) substrate. To prepare the IDE substrate, an oxide layer (SiO2 ∼ 200 nm) was grown on a cleaned p-type Si wafer using a pyrogenic wet oxidation process. After that a thick layer of photo resist (AZ5214E PR) was spin coated on the SiO2/p-Si substrate, followed by photolithography, sputtering of 10 nm Cr/120 nm Au and lifting off the photo-resist to produce a pattern of Au heating lines (125 nm thick) on the substrate. After the lift-off process AZ5214E PR was again spin coated above the Cr/Au film followed by photolithography for the gas sensing layer. LaFeO3/rGO-LaFeO3 solutions were then drop caste on the Si-IDE substrate after lifting off the photo resist and dried at 200 °C for 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 hours. The flow process for the fabrication of an IDE deposited LaFeO3/rGO-LaFeO3 gas sensor is shown in Fig. 2(c). The device size was 500 × 500 μm, with 100 fingers. The width of each finger was 5 μm with the same spacing between two fingers.
:30 hours. The flow process for the fabrication of an IDE deposited LaFeO3/rGO-LaFeO3 gas sensor is shown in Fig. 2(c). The device size was 500 × 500 μm, with 100 fingers. The width of each finger was 5 μm with the same spacing between two fingers.
2.5 Characterizations studies
As-synthesized samples of LaFeO3 and its composite with reduced graphene oxide (rGO-LaFeO3) were characterized by X-ray diffraction (XRD; X-pert powder diffractometer with copper rotating anode using incident beam of 1.54 Å wavelength), Fourier transform infrared spectroscopy (FTIR; PerkinElmer system in the KBr mode), Raman spectroscopy (STR 500 Confocal Micro Raman Spectrometer at 532 nm wavelength), thermo-gravimetric analysis (TGA; PerkinElmer STA 6000), a field emission scanning electron microscope (FESEM; 450 FEI, NOVA nano SEM), a transmission electron microscope (TEM-Tecnai G2 XFLESH-6T130 with accelerating voltage 200 kV), an atomic force microscope (AFM using non-contact mode), a UV-Vis spectrometer (UV-Vis, Lambda 750, PerkinElmer), and X-ray photoelectron spectroscopy (XPS, Omicron Nanotechnology, Oxford Instruments Germany). XPS wide scans and C 1s and O 1s spectra were recorded using AlKα radiation (hν = 1486.6 eV). The pass energy for the C 1s and O 1s spectra was kept at 50 eV and 20 eV for full scans. Brunauer–Emmett–Teller (BET; Quantachrome instruments: NOVA touchLX1) was used to examine the surface area, and gas sensing measurements were recorded by Eurotherm 2404, Keithley-2450 at IISc Bangaluru.
3. Result and discussion
3.1 X-ray diffraction studies
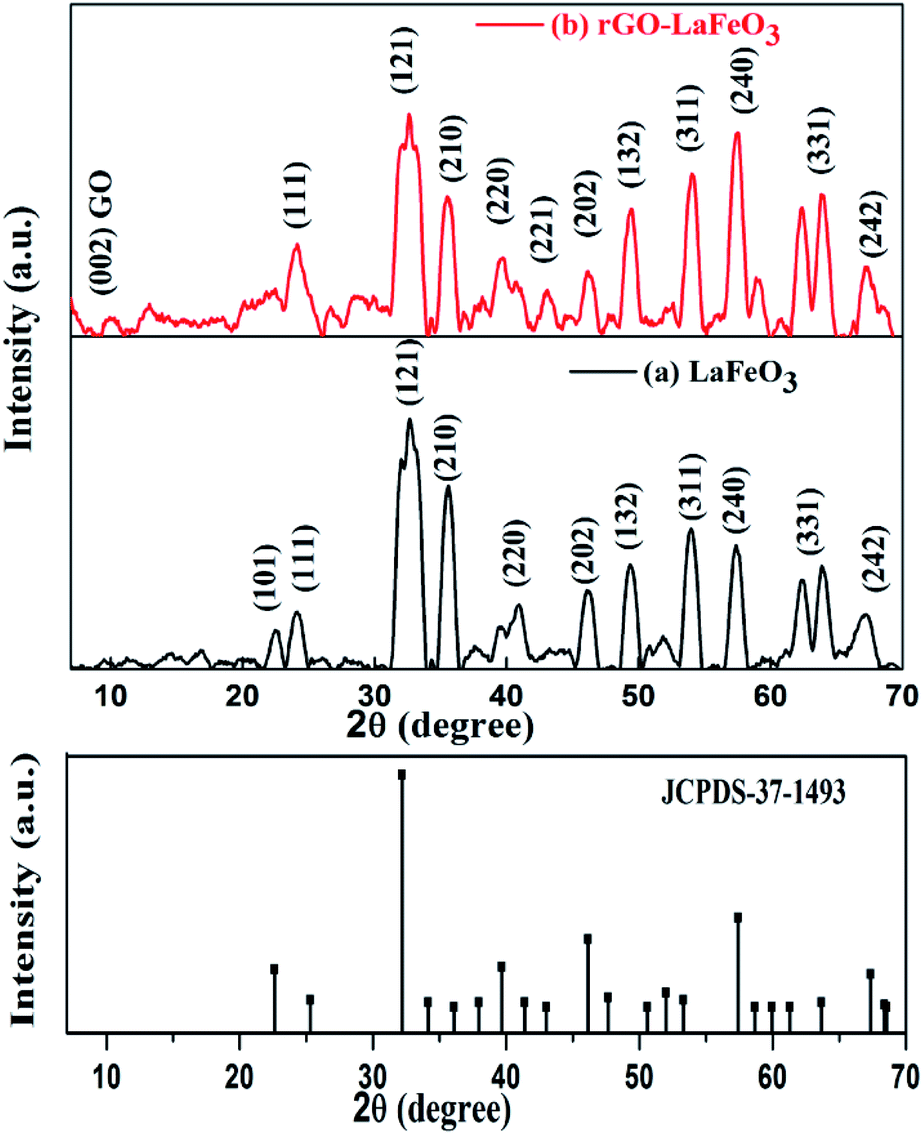
The XRD patterns of LaFeO3 and rGO-LaFeO3 shown in Fig. 3(a) and (b) give information about the phases present and the crystallinity of the samples. Characteristic peaks of obtained LaFeO3 microspheres were indexed using JCPDS-37-1493.24,26,37 Strong and highly intense peaks indicate good crystallinity of synthesized LaFeO3. No impurity peaks were observed in the XRD pattern. A d-spacing of LaFeO3 microspheres corresponding to a highly intense plane (121) was found at 2.7 Å. These results suggest an orthorhombic structure for LaFeO3 with lattice parameters a = 5.565 Å, b = 7.839 Å, and c = 5.577 Å.37 The XRD pattern in Fig. 3(b) for the rGO-LaFeO3 composite comprises peaks assigned to individual LaFeO3, as shown in Fig. 3(a), belonging to the orthorhombic LaFeO3 phase. A small peak at 10.02° was attributed to the (002) plane arising from the presence of rGO in the synthesized composite.38 The d-spacing corresponding to the (121) plane of rGO-LaFeO3 was observed at 2.6 Å. Background noise in XRD spectra may be due to instrumental error, and the splitting of the peaks can be removed after calcining the samples at higher temperature and for a longer duration.37
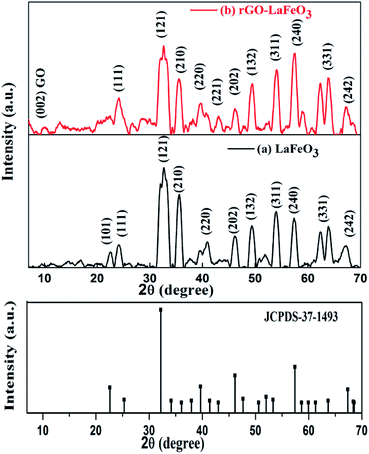 |
| | Fig. 3 XRD spectra for LaFeO3 (a) and rGO-LaFeO3 (b) with JCPDS-37-1493. | |
3.2 FT-IR studies
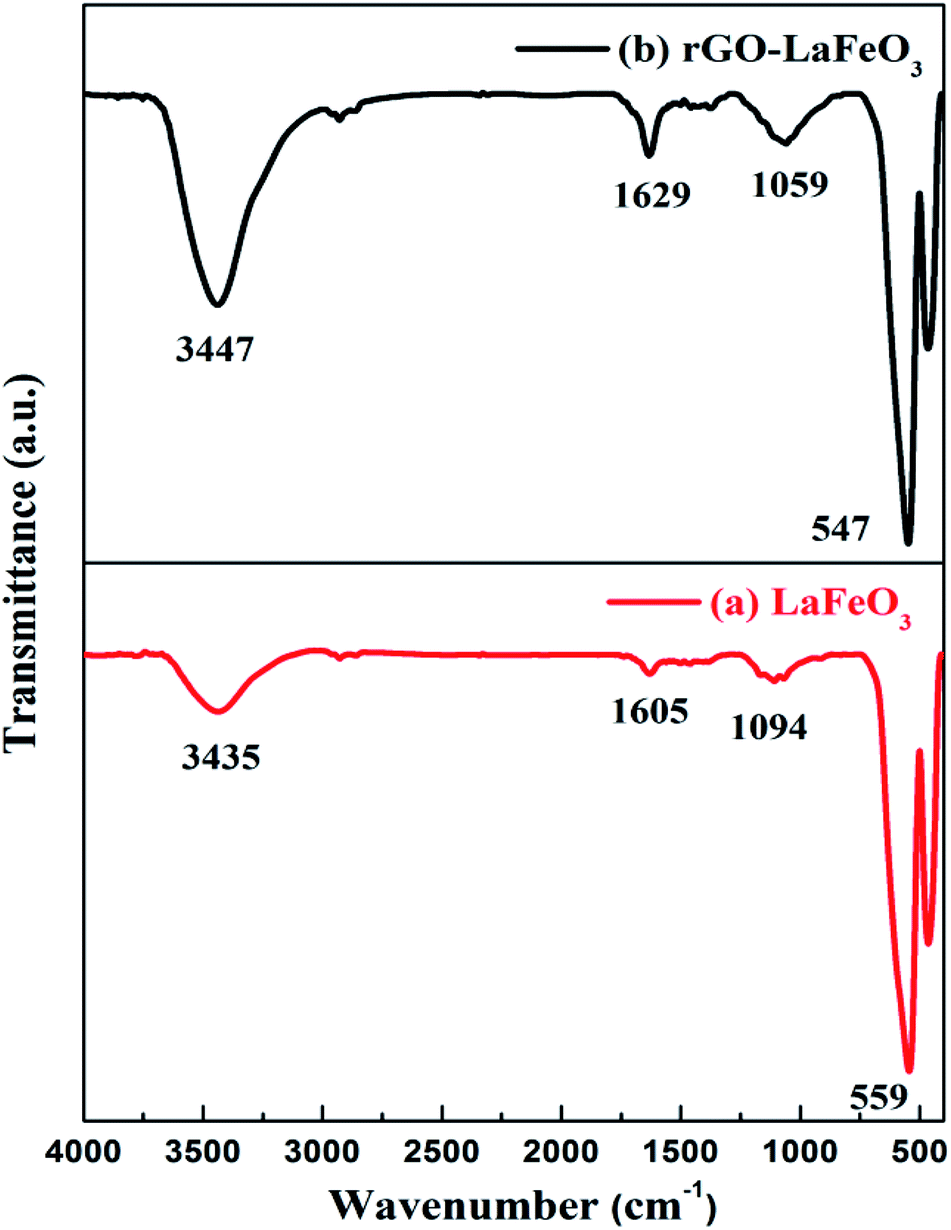
FT-IR spectra for LaFeO3 and rGO-LaFeO3 were recorded in a range of 400–4000 cm−1 wavenumbers to monitor the functional groups present in the materials and are given in Fig. 4(a) and (b), respectively. For LaFeO3, bands were observed at 559, 1094, 1605 and 3435 cm−1. The band at 559 cm−1 is the characteristic band indicating the presence of Fe–O stretching.39,40 This peak confirms the formation of a perovskite phase, and usually appears in the range of 500–700 cm−1. Peaks at 1605 cm−1 and 3435 cm−1 are assigned to asymmetric stretching of the carboxyl group and hydroxyl group.38 These peaks were shifted to 547, 1059, 1629 and 3447 cm−1, when LaFeO3 was decorated with rGO. The peak at 1629 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C stretching of the alkene or aromatic group confirms the sp2 structure of rGO.41 For further confirmation of perovskite formation and the presence of rGO in the composite, Raman spectroscopy was performed and is given in ESI (Fig. S1†). The thermal stability of the synthesized samples was studied by TGA and is given in ESI Fig. S2.†
C/C–C stretching of the alkene or aromatic group confirms the sp2 structure of rGO.41 For further confirmation of perovskite formation and the presence of rGO in the composite, Raman spectroscopy was performed and is given in ESI (Fig. S1†). The thermal stability of the synthesized samples was studied by TGA and is given in ESI Fig. S2.†
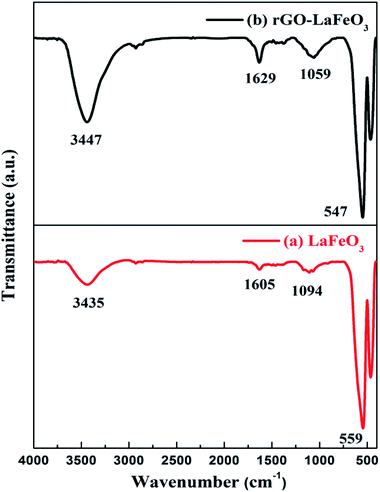 |
| | Fig. 4 FTIR spectra for LaFeO3 (a) and rGO-LaFeO3 (b). | |
3.3 Field emission scanning electron microscopy studies
The surface morphologies of LaFeO3 and rGO-LaFeO3 were observed using FESEM and the micrographs obtained are shown in Fig. 5(a) and (b), respectively. Fig. 5(a) shows the microsphere formation of LaFeO3 with an average size of 1.45 μm. In Fig. 5(b) it can be observed that rGO decorated LaFeO3 microspheres showed an average size of 1.57 μm. The layered structure of rGO between the microspheres of LaFeO3 can clearly be seen in Fig. 5(b). In both samples of LaFeO3 and rGO-LaFeO3 microspheres are formed due to the coagulation of a large number of nanoparticles. The TEM results, as shown in Fig. 6 and 7, provide more information about the microstructure.
 |
| | Fig. 5 FESEM images for LaFeO3 (a) and rGO-LaFeO3 (b). | |
 |
| | Fig. 6 Low (a) and high magnification (b and c) TEM images of porous LaFeO3 microspheres, and the corresponding HRTEM image with labeled lattice spacing (inset: SAED pattern of LaFeO3 microsphere) (d). | |
 |
| | Fig. 7 (a and b) Low and (c) high magnification TEM image of porous rGO-LaFeO3 microspheres, (d) the corresponding HRTEM image with labeled lattice spacing (inset: SAED pattern of rGO-LaFeO3 microsphere). | |
3.4 High-resolution transmission electron microscopy studies
Fig. 6(a)–(c) show low and high magnification TEM images of synthesized LaFeO3 microspheres. It was observed that the prepared LaFeO3 shows a sphere-like structure with high porosity. Fig. 6(b and c) confirm that porous microspheres are arranged in a network form with a large number of particles. The HRTEM image of a porous LaFeO3 microsphere shown in Fig. 6(d) is indicative of the formation of interference fringes with a lattice perpendicular (d-spacing) of about 0.268 nm, in agreement with the plane (121) of LaFeO3 observed from XRD. The selected area electron diffraction (SAED) pattern shown in the inset of Fig. 6(d) clearly indicates the polycrystalline nature of the prepared porous LaFeO3 microspheres. The average diameter of a microsphere was found to be about 1.45 μm, as calculated by Image J software. This size of the microsphere coincides with that obtained from the FESEM study.
Low and high magnification TEM images of prepared rGO-LaFeO3 composites are shown in Fig. 7(a)–(c), respectively. Fig. 7(a) and (b) show that porous microspheres of LaFeO3 are decorated on the surface of the GO layer and the GO sheets show interconnections with the LaFeO3 microspheres. From Fig. 7(c) it is clear that the LaFeO3 microspheres are well distributed on the GO surface, which would be beneficial for making a significant surface area in the composite for sensing gases. The HRTEM image of the rGO-LaFeO3 composite shown in Fig. 7(d) clearly indicates interference fringes with a d-spacing of about 0.279 nm and suggest the growth of LaFeO3 microspheres on the GO surface. The d-spacing for the composite rGO-LaFeO3 (0.279 nm) as compared to that for LaFeO3 (0.268 nm) confirms that no structural change occurred in the composite. The corresponding SAED spectra shown in the inset of Fig. 7(d) show that the microspheres are randomly oriented in the composite. The size of the LaFeO3 microspheres in the composite using Image J software was obtained as 1.57 μm. EDAX spectra and the percentages of the elements in the synthesized samples are shown in ESI Tables S1 and S2,† respectively.
3.5 X-ray photoelectron spectroscopy studies
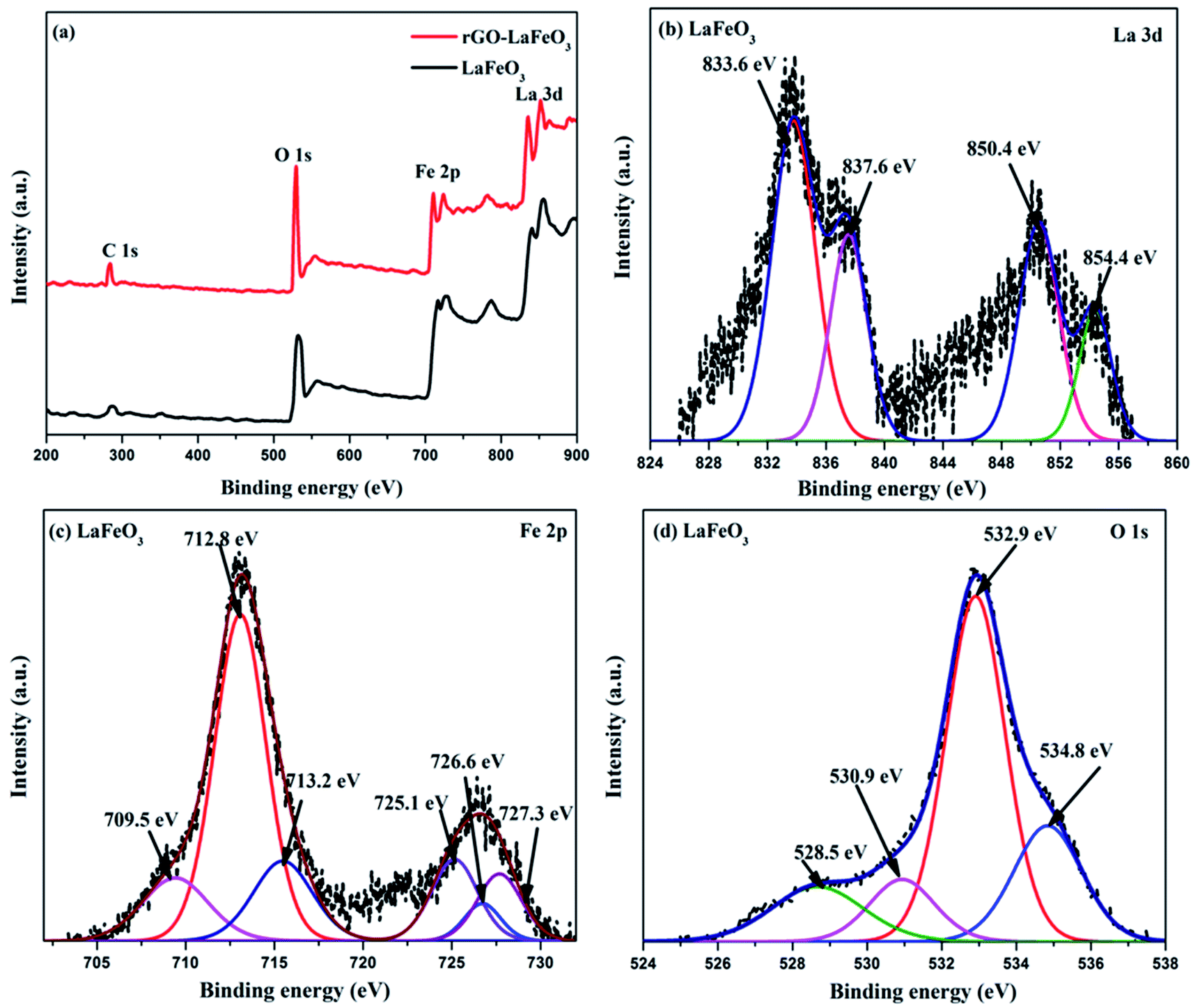
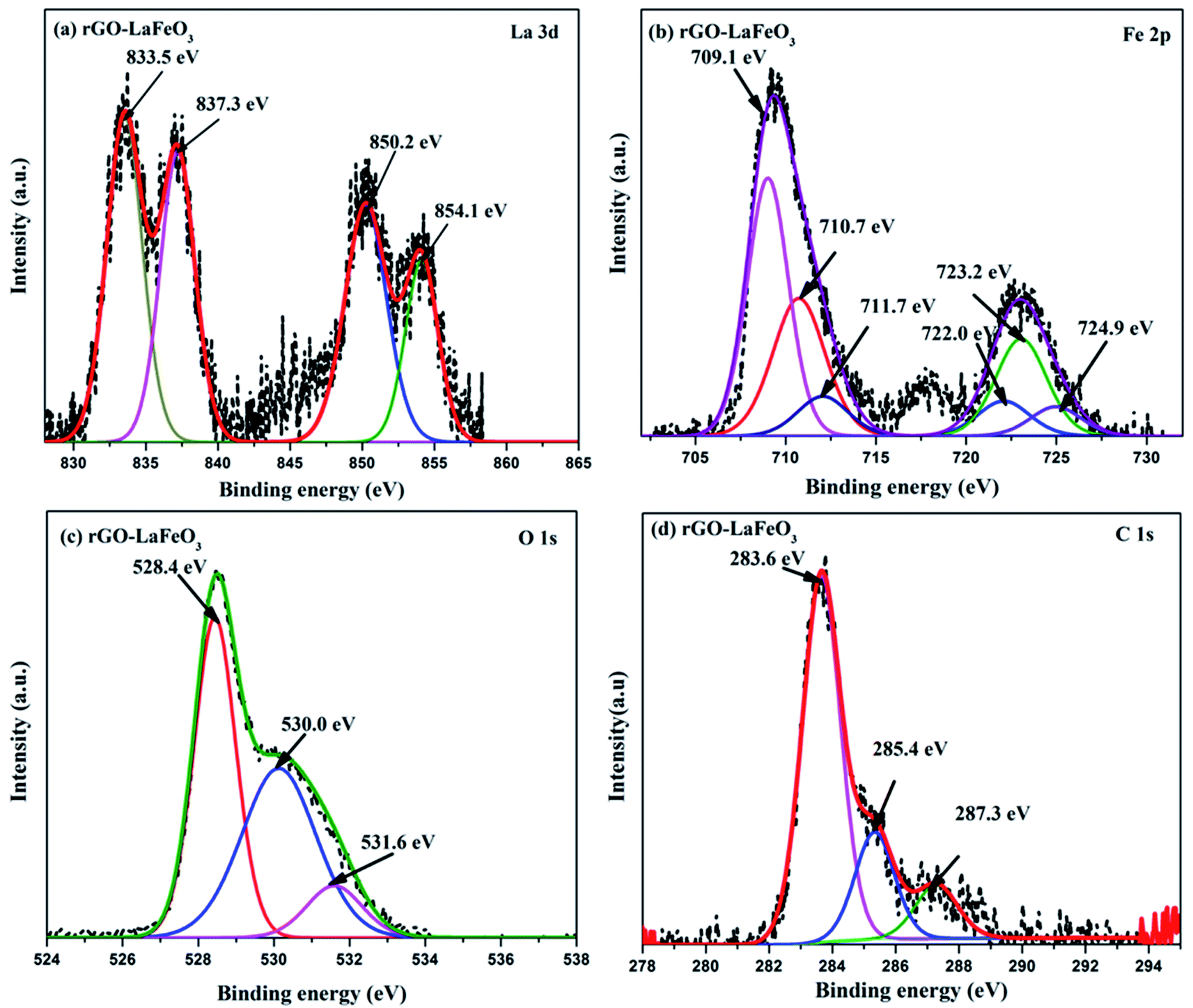
X-ray photoelectron spectroscopy (XPS) has proved to be a useful tool for identifying the valence states of La and Fe in the synthesized samples LaFeO3 and rGO-LaFeO3. Casa XPS software was used for peak fitting of La 3d, Fe 2p, C 1s and O 1s spectra with Shirley background correction. Fig. 8(a) shows wide range survey spectra for LaFeO3 and rGO-LaFeO3 and the observed characteristic peaks attributed to La, Fe, oxygen, and carbon. XPS spectra for LaFeO3 shown in Fig. 8(b)–(d) correspond to La 3d, Fe 2p, and O 1s, respectively. From Fig. 8(b) it can be seen that La deconvoluted into four peaks; among them, peaks corresponding to binding energies (BE) of 833.6 eV and 850.4 eV are assigned to La 3d5/2 and La 3d3/2, respectively.42–44 The other two peaks at binding energies of 837.6 eV and 854.4 eV are satellite peaks corresponding to La 3d5/2 and La 3d3/2, respectively.44 This information about the core level of La indicates that La ions exhibit +3 oxidation states. The BE of the Fe 2p3/2 signal has been found in the energy range 706.5–707.0 eV for metallic iron, 709.5–710.3 eV for Fe2+, and 710.6–711.4 eV for Fe3+.45 In Fig. 8(c), the peaks at binding energies 709.5 eV and 712.8 eV are attributed to the spin–orbit of Fe 2p3/2.45 The binding energy at 725.1 eV is assigned to Fe 2p1/2 for Fe3+.46 The other two peaks at 726.6 eV and 727.3 eV may be due to the presence of some Fe4+ state of Fe 2p1/2, which is not seen in XRD.43 Thus the results indicate that Fe contains mixed valence state of Fe3+ and Fe4+ in the main. This combination of valence states is rare and was observed by Phokha et al. in 2014.43 The XPS peaks of oxygen spectra are deconvoluted into four peaks; among them the peak corresponding to a binding energy of 528.5 eV confirms the existence of crystal lattice oxygen (OL) and the peak located at 530.9 eV binding energy confirms the presence of hydroxyl oxygen (OH).1 The core level of the La 3d region for the composite shown in Fig. 9(a) exhibits peak positions at 833.5 eV and 850.2 eV assigned as La 3d5/2 and La 3d3/2, respectively. This indicates that La ions have mainly an La3+ state. La 3d satellite peaks are assigned at 837.6 eV and 854.5 eV binding energies, respectively.44 Fig. 9(b) for the core level region of Fe 2p for sample rGO-LaFeO3 shows peaks located at 710.7 eV and 711.7 eV binding energies, corresponding to Fe 2p3/2. The peaks at BE 723.2 eV and 724.9 eV are attributed to Fe 2p1/2. This indicates that Fe ions in rGO-LaFeO3 also have a mixed state of Fe3+ and Fe4+.43,45 In addition, the peak at 709.1 eV, corresponding to Fe2p3/2, may be due to the presence of the Fe2+ state of Fe2O3.45 A broad peak at 718.2 eV is a shake-up satellite peak of Fe3+.45,47 For the O 1s spectra of rGO-LaFeO3, similar peak positions at 528.4 eV for OL and at 530.9 eV for OH were observed.24 The XPS spectrum of C ls shown in Fig. 9(d) deconvoluted into three peaks due to hydrocarbons and hydroxyls. The peak at 283.6 eV is associated with (CC), 285.4 eV corresponds to (C–C/C–H) and 287.3 eV is associated with the hydroxyl group (C–OH).48–50
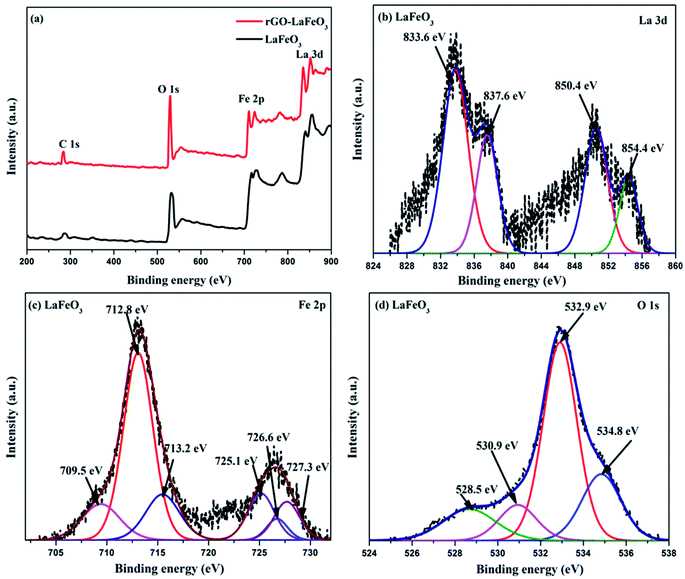 |
| | Fig. 8 Wide range survey spectra (a) of LaFeO3 and rGO-LaFeO3, XPS spectra of La 3d (b), Fe 2p (c), O 1s (d) for LaFeO3. | |
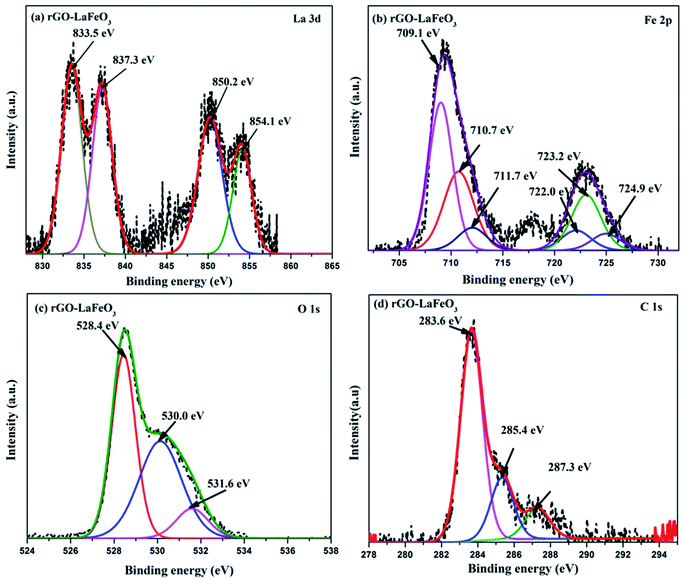 |
| | Fig. 9 XPS spectra of La 3d (a), Fe 2p (b), O 1s (c), and C 1s (d) for rGO-LaFeO3. | |
3.6 UV-visible spectroscopy
Fig. 10(a) shows the UV-Vis absorption spectra of LaFeO3 and its composite with rGO in the wavelength range 200–1300 nm. It was observed from the spectra that both samples show a strong absorption in the ultraviolet (200–400 nm) and visible (400–800 nm) regions. The UV-Vis spectra allow us to calculate the direct band gap Eg by fitting the absorption data to the direct transition using Tauc's formula (αhν = A(hν − Eg)1/2, where α is the absorption coefficient, hν is the photon energy, A is a constant, and Eg is the direct band gap) by plotting (αhν)2 versus hν, as shown in Fig. 10(b). The calculated band gaps are 1.94 eV for rGO-LaFeO3 and 2.01 eV for LaFeO3, indicating a slight narrowing of the band gap on the addition of rGO to LaFeO3. This would be useful for the movement of electrons from the valence band to the conduction band for gas sensing.
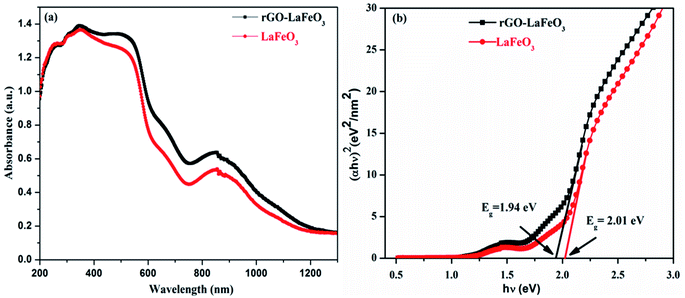 |
| | Fig. 10 Absorption spectra (a) and Tauc plot (b) of LaFeO3 and rGO-LaFeO3. | |
3.7 Adsorption–desorption isotherm, surface area and pore size distribution
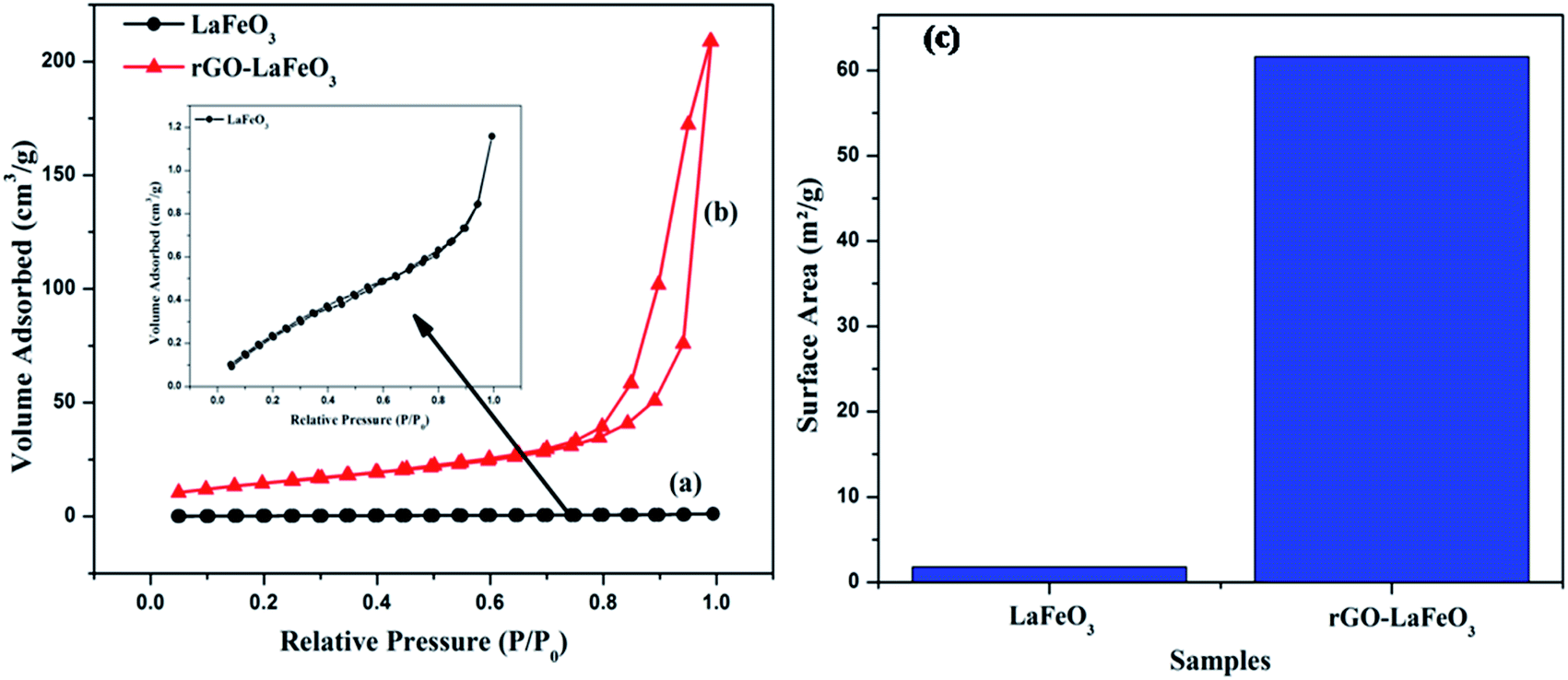
Fig. 11(a–c) display the adsorption–desorption curves (N2) and the obtained surface areas of the synthesized samples. It can clearly be seen that the isotherm changes from type II for LaFeO3 (shown in the inset) to type III for rGO-LaFeO3.51 The pore size for LaFeO3 comes out to be 1.85 nm, and increased to 16.4 nm for rGO-LaFeO3.52 Hence, according to the pore size and type of isotherm, the samples can be categorized as having a micro porous and a mesoporous structure for LaFeO3 and rGO-LaFeO3, respectively. The surface area for composite rGO-LaFeO3 is about forty times ∼62.1 m2 g−1 (Fig. 11(c)) that of LaFeO3 ∼1.41 m2 g−1. This many-fold increase in surface area is due to the addition of rGO to LaFeO3, and is a useful property.
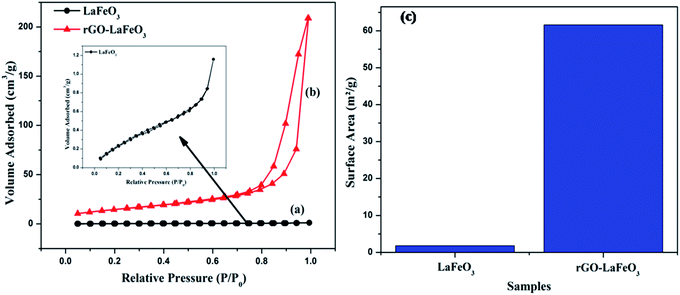 |
| | Fig. 11 N2 adsorption–desorption isotherm spectra of (a) LaFeO3, (b) rGO-LaFeO3 and surface area of (c) LaFeO3, rGO-LaFeO3. | |
3.8 Gas sensing studies
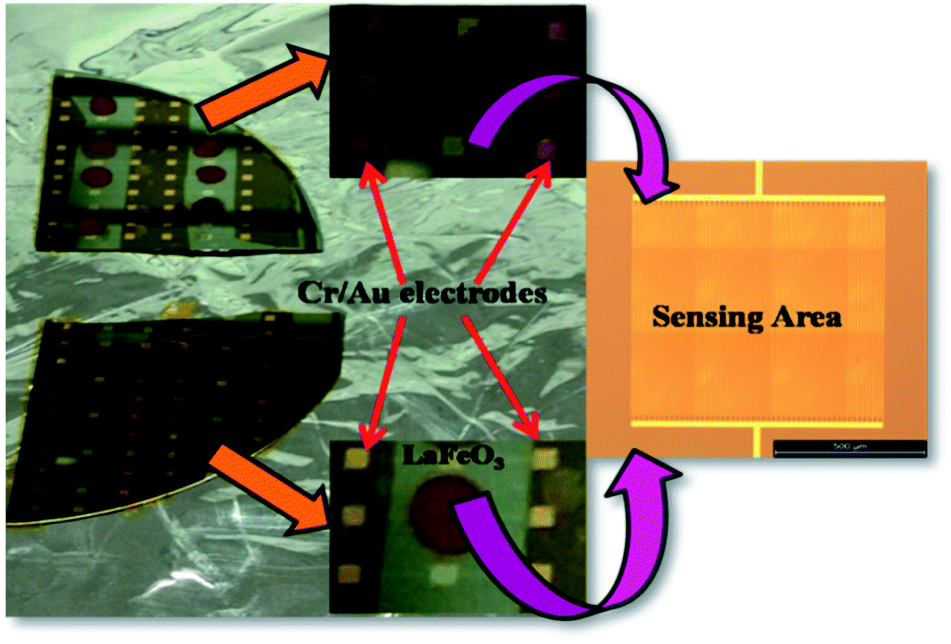
It is evident from the studies carried out that LaFeO3 and rGO-LaFeO3 materials meet the criteria for realization of a gas sensor. These films were then employed for CO and NO2 gas sensing by sourcing a voltage of 6 V and measurement of current/resistance through Cr/Au contact pads resting on an IDE deposited on the films by photolithography. A photograph of the fabricated gas sensor with Cr/Au contact is shown in Fig. 12. Compressed air was used as a carrier gas (N2 + O2 = 80% + 20%) to which 1–3 ppm CO and 1–5 ppm NO2 were added from a second cylinder whilst maintaining a constant flow rate of 30 sccm. The gas sensing characteristics are plotted as change in ratio defined as follows: as change in resistance for CO and NO2 gases in opposite polarity. Here Ra and Rg are the resistance of the specimens in air and gas, respectively.| |
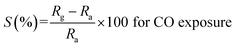 | (3) |
| |
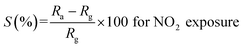 | (4) |
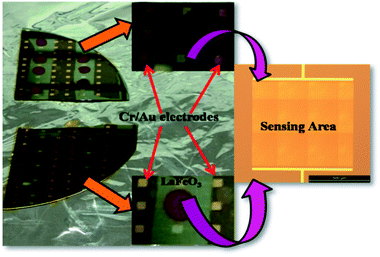 |
| | Fig. 12 Gas sensing device based on LaFeO3 and rGO-LaFeO3. | |
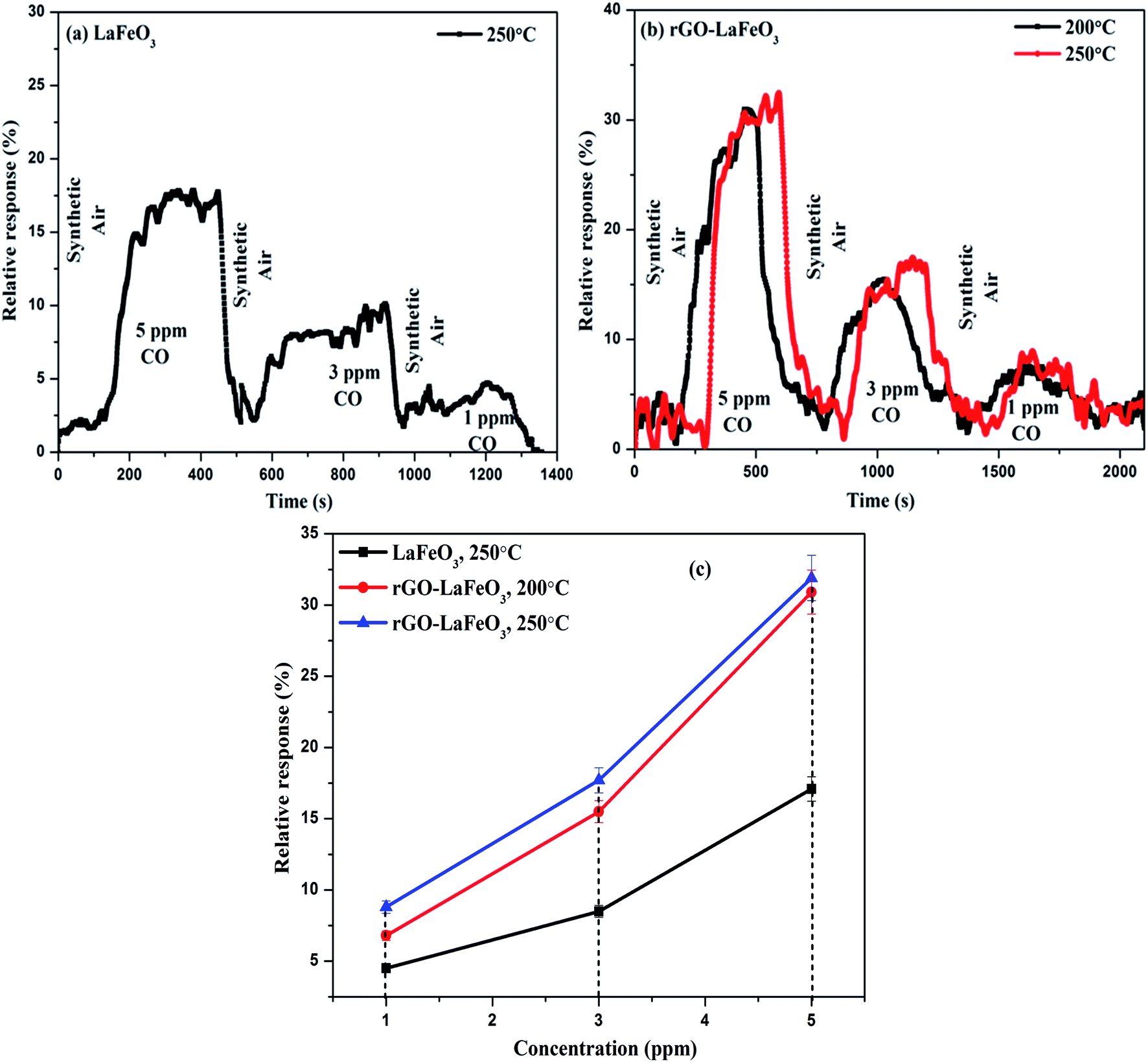
3.8.1 Carbon monoxide gas sensing. Fig. 13(a) and (b) show the plots of relative response(s) for LaFeO3 and rGO-LaFeO3 as a function of time. Both samples were tested in the presence of 5, 3 and 1 ppm CO gas at temperatures of 200 and 250 °C. However, LaFeO3 does not show any response at 200 °C while a good response was found at 250 °C. For the p-type LaFeO3 sensor, exposure to 5 ppm of CO gas at 250 °C leads to an injection of electrons into the valence band and their recombination with holes (generated in an oxygen ambient atmosphere), decreasing the concentration of holes and thereby increasing sensor resistance. After the CO is turned off, the sensing material starts to recover its path towards the initial resistance. The same trend was obtained for 3 and 1 ppm of CO at the same temperature but with a lower response. LaFeO3 microspheres show a response of 17.1% for 5 ppm, 8.5% for 3 ppm and 4.5% for 1 ppm of CO. Fig. 13(b) shows the relative response plot of a sensor based on rGO-LaFeO3 to various concentrations (5, 3 and 1 ppm) of CO at 200 °C where the responses are 30.9%, 15.5%, and 6.8%, respectively, indicating that the sensor thus fabricated can be used for CO detection with a wide range of smaller concentrations. The sensing mechanism is similar for rGO-LaFeO3 to that for an LaFeO3 sensor. As the temperature increases to 250 °C, the response is quite similar to that at 200 °C: viz. 31.9% for 5 ppm of CO, 17.7% for 3 ppm of CO and 8.8% for 1 ppm of CO. Fig. 13(c) represents the change in relative response with respect to concentration of test gases for the fabricated sensors at temperatures of 200 and 250 °C. There is a clear and visible improvement in the sensing performance corresponding to the addition of carbon atoms through doping of reduced graphene oxide (rGO) into the LaFeO3 which leads to an increase in the active sites and surface area for higher adsorption/desorption of gas molecules.
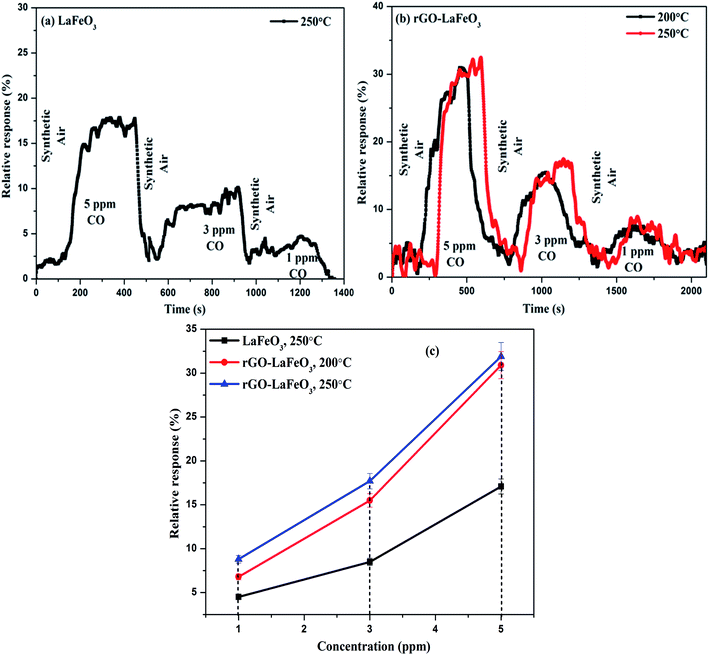 |
| | Fig. 13 Relative response with respect to time for LaFeO3 (a) and rGO-LaFeO3 (b) and relative response with respect to concentration for LaFeO3 and rGO-LaFeO3 (c) towards CO gas at temperatures of 200 and 250 °C. | |
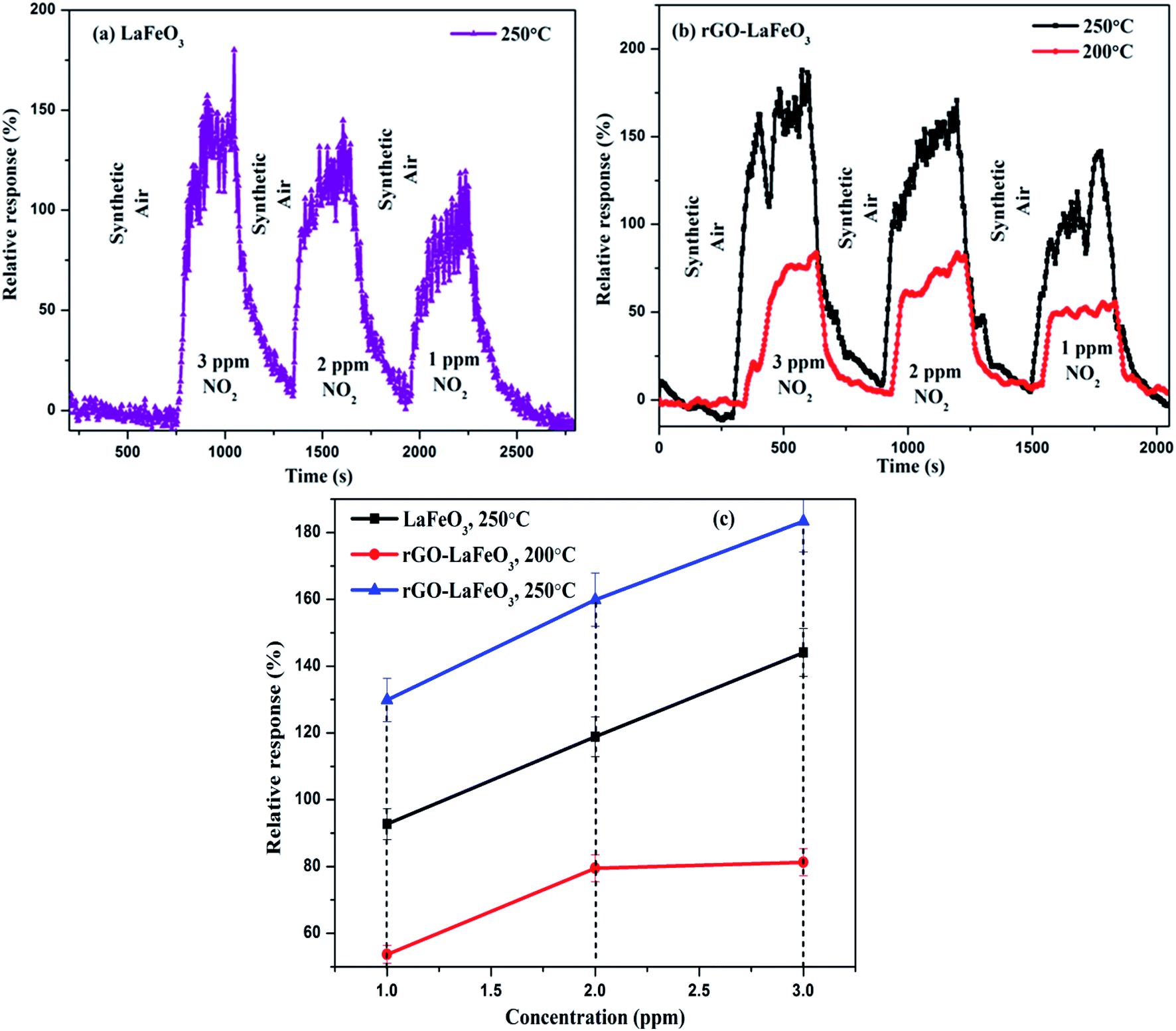
3.8.2 Nitrogen dioxide gas sensing. The gas sensing characteristics of LaFeO3 and rGO-LaFeO3 based sensors for 3, 2 and 1 ppm of NO2 gas are shown in Fig. 14(a) and (b), respectively, at operating temperatures of 200 and 250 °C. Fig. 14(c) shows the relative response with respect to concentration for LaFeO3 and rGO-LaFeO3. LaFeO3 does not show any response at all to concentration of NO2 at 200 °C. However, at 250 °C, LaFeO3 exhibits a high response. When 1 ppm of NO2 is injected onto the surface of p-type LaFeO3, it gives a 92.7% response and it reaches a higher value of 144.1 from 118.9 for an increase in concentration from 2 to 3 ppm. A sensor based on rGO-LaFeO3 gave an even higher response at 200 °C upon exposure to NO2: viz. 53.7%, 79.5% and 81.3% for 1 ppm, 2 ppm and 3 ppm, respectively. This relative response increased further for a temperature of 250 °C: viz. 129.9%, 159.9% and 183.4% for 1 ppm, 2 ppm and 3 ppm, respectively. The increase in response is due to the smaller band gap of rGO-LaFeO3, which makes the flow of electrons/holes easier, to provide conductance. It is evident that the interactions of NO2 with the LaFeO3 surface are more complicated than those of CO. NO2 can be absorbed on the surface of LaFeO3 film in three different states: two nitrosyl types (NO−, NO+) and a nitro type (NO2−).26 The gas sensing mechanisms of LaFeO3 with CO and NO2 are described in ESI.† A comparison Table (S3)† of the gas sensing results for both samples with the already published literature is given in ESI,† and clearly indicates that rGO-LaFeO3 gives a higher sensitivity than LaFeO3 and is a potential material for gas sensing applications.
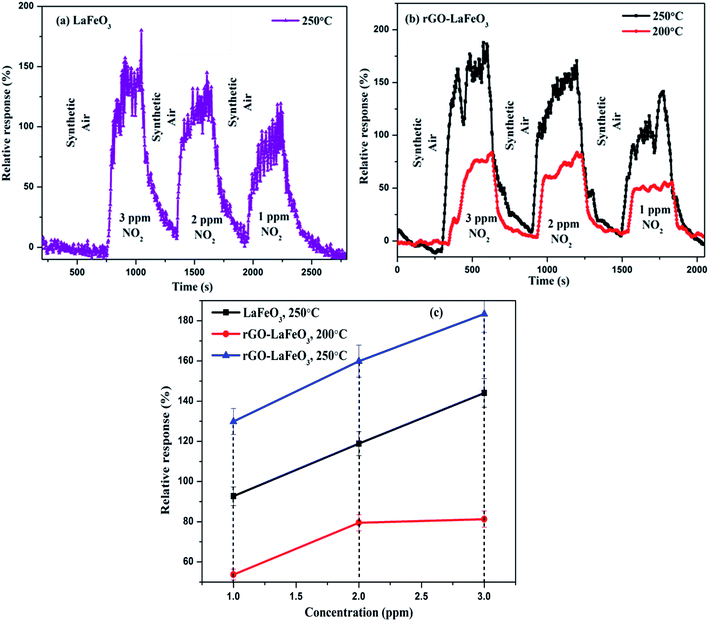 |
| | Fig. 14 Relative response with respect to time for LaFeO3 (a) and rGO-LaFeO3 (b) and relative response with respect to concentration for LaFeO3 and rGO-LaFeO3 (c) towards NO2 gas at temperatures of 200 and 250 °C. | |
4. Conclusion
In summary, microspheres of LaFeO3 and rGO-LaFeO3 were synthesized by a hydrothermal method. Gas sensing devices (dimension 500 × 500 μm) using synthesized nanomaterials were successfully fabricated through photolithography using a drop cast method for film deposition. In terms of structure, morphology, thermal stability, and surface roughness, the samples show potential as gas sensing devices. An orthorhombic structure was observed for both products from X-ray diffraction. The appearance of a D′ band in the Raman spectra and an alkene/organic group in FT-IR confirm the formation of a composite. The synthesized products are thermally stable at a temperature of ∼720 °C. XPS and TEM confirm the presence of La, Fe, O, and C elements. Gas sensing measurements indicate that an rGO-LaFeO3 based device exhibits a higher relative response than LaFeO3 for both CO (5 ppm, 3 ppm, 1 ppm) and NO2 (3 ppm, 2 ppm, 1 ppm) gases. It is also contributed that rGO-LaFeO3 is highly selective for NO2 gas even at 1 ppm concentration at 200 and 250 °C. This is mainly due to the higher surface area and smaller band gap of rGO-LaFeO3. To the best of our knowledge, this is the first study of an rGO-LaFeO3 based gas sensing device with an excellent relative response.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not for profit sector.
Conflicts of interest
On behalf of all authors, the corresponding author states that there are no conflicts to declare.
Acknowledgements
The authors would like to thank Materials Research Center, MNIT Jaipur, India for providing characterization facilities and INUP-CeNSE, Indian Institute of Science, Bengaluru, funded by the Ministry of Electronics and Information Technology (MeitY), Govt. of India for providing sensor fabrication in clean room and gas sensing facilities for the present study. We also thank Dr Vikas Sharma, Dr Satyavir Singh, and Mr Saquib Khan, MNIT Jaipur for their support in carrying out this work.
References
- S. Thirumalairajan, K. Girija, V. R. Mastelaro and N. Ponpandian, ACS Appl. Mater. Interfaces, 2014, 6, 13917–13927 CrossRef CAS PubMed.
- P. Sun, W. Zhao, Y. Cao, Y. Guan, Y. Sun and G. Lu, CrystEngComm, 2011, 13, 3718 RSC.
- H. Wang, Y. Liu, M. Li, H. Huang, H. M. Xu, R. J. Hong and H. Shen, Optoelectron. Adv. Mater., Rapid Commun., 2010, 4, 1166–1169 CAS.
- D. E. Motaung, G. H. Mhlongo, I. Kortidis, S. S. Nkosi, G. F. Malgas, B. W. Mwakikunga, S. S. Ray and G. Kiriakidis, Appl. Surf. Sci., 2013, 279, 142–149 CrossRef CAS.
- L. Gao, F. Ren, Z. Cheng, Y. Zhang, Q. Xiang and J. Xu, CrystEngComm, 2015, 17, 3268–3276 RSC.
- P. Su and S. Peng, Talanta, 2015, 132, 398–405 CrossRef CAS PubMed.
- W. Yan, H. Fan, Y. Zhai, C. Yang, P. Ren and L. Huang, Sens. Actuators, B, 2011, 160, 1372–1379 CrossRef CAS.
- R. Vyas, S. Sharma, P. Gupta, Y. K. Vijay, A. K. Prasad, A. K. Tyagi, K. Sachdev and S. K. Sharma, J. Alloys Compd., 2013, 554, 59–63 CrossRef CAS.
- R. Vyas, S. Sharma, S. Khan, R. Divakar, K. Sachdev and S. K. Sharma, Macromol. Symp., 2015, 357, 99–104 CrossRef CAS.
- R. Ghosh, A. Midya, S. Santra, S. K. Ray and P. K. Guha, ACS Appl. Mater. Interfaces, 2013, 5, 7599–7603 CrossRef CAS PubMed.
- G. Lu, L. E. Ocola and J. Chen, Appl. Phys. Lett., 2009, 94, 8–11 Search PubMed.
- G. Lu, L. E. Ocola and J. Chen, Nanotechnology, 2009, 20, 445502 CrossRef PubMed.
- J. T. Robinson, F. K. Perkins, E. S. Snow, Z. Wei and P. E. Sheehan, Nano Lett., 2008, 8, 3137–3140 CrossRef CAS PubMed.
- J. D. Fowler, M. J. Allen, V. C. Tung, Y. Yang, R. B. Kaner and B. H. Weiller, ACS Nano, 2009, 3, 301–306 CrossRef CAS PubMed.
- F. Yavari, Z. Chen, A. V. Thomas, W. Ren, H. M. Cheng and N. Koratkar, Sci. Rep., 2011, 1, 1–5 CrossRef PubMed.
- A. Salehi-Khojin, D. Estrada, K. Y. Lin, M. H. Bae, F. Xiong, E. Pop and R. I. Masel, Adv. Mater., 2012, 24, 53–57 CrossRef CAS PubMed.
- S. Liu, J. Tian, L. Wang, H. Li, Y. Zhang and X. Sun, Macromolecules, 2010, 43, 10078–10083 CrossRef CAS.
- M. Gautam and A. H. Jayatissa, Mater. Sci. Eng., C, 2011, 31, 1405–1411 CrossRef CAS.
- N. Hu, Z. Yang, Y. Wang, L. Zhang and Y. Wang, Nanotechnology, 2014, 25, 1–10 Search PubMed.
- C. Doroftei, P. D. Popa and F. Iacomi, Sens. Actuators, B, 2012, 161, 977–981 CrossRef CAS.
- C. Doroftei and L. Leontie, RSC Adv., 2017, 7, 27863–27871 RSC.
- E. Abdullah, A. Idris and A. Saparon, ARPN J. Eng. Appl. Sci., 2017, 12, 3218–3221 Search PubMed.
- N. Q. Minh, J. Am. Ceram. Soc., 1993, 76, 563–588 CrossRef CAS.
- S. Thirumalairajan, K. Girija, V. Ganesh, D. Mangalaraj, C. Viswanathan and N. Ponpandian, Cryst. Growth Des., 2013, 13, 291–302 CrossRef CAS.
- A. Benali, S. Azizi, M. Bejar, E. Dhahri and M. F. P. Graça, Ceram. Int., 2014, 40, 14367–14373 CrossRef CAS.
- N. N. Toan, S. Saukko and V. Lantto, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 327, 279–282 CrossRef CAS.
- J. W. Yoon, M. L. Grilli, E. Di Bartolomeo, R. Polini and E. Traversa, Sens. Actuators, B, 2001, 76, 483–488 CrossRef CAS.
- B. A. I. Shouli, S. H. I. Bingjie, M. A. Lijing, Y. Pengcheng, L. I. U. Zhiyong, L. I. Dianqing and C. Aifan, Sci. China, Ser. B: Chem., 2009, 52, 2106–2113 CrossRef.
- M. Cristina, G. Martinelli, Y. Sadaoka, P. Nunziante and E. Traversa, Sens. Actuators, B, 1998, 48, 270–276 CrossRef.
- X. Wang, H. Qin, L. Sun and J. Hu, Sens. Actuators, B, 2013, 188, 965–971 CrossRef CAS.
- S. Nakayama, J. Mater. Sci., 2001, 36, 5643–5648 CrossRef CAS.
- M. Sorescu, T. Xu, J. D. Burnett and J. A. Aitken, J. Mater. Sci., 2011, 6709–6717 CrossRef CAS.
- H. Xiao, C. Xue, P. Song, J. Li and Q. Wang, Appl. Surf. Sci., 2015, 337, 65–71 CrossRef CAS.
- N. Sharma, S. K. Sharma and K. Sachdev, Ceram. Int., 2019, 45, 7217–7225 CrossRef CAS.
- R. D. Kumar, R. Thangappan and R. Jayavel, J. Inorg. Organomet. Polym. Mater., 2017, 27, 892–900 CrossRef.
- N. Sharma, V. Sharma, S. K. Sharma and K. Sachdev, Mater. Lett., 2019, 236, 444–447 CrossRef CAS.
- V. A. Online, S. Thirumalairajan, K. Girija, N. Y. Hebalkar, D. Mangalaraj, C. Viswanathan and N. Ponpandian, RSC Adv., 2013, 3, 7549–7561 RSC.
- X. Ren, H. Yang, S. Gen, J. Zhou, T. Yang, X. Zhang, Z. Cheng and S. Sun, Nanoscale, 2016, 8, 752–756 RSC.
- F. Deganello, M. L. Tummino, C. Calabrese, M. L. Testa, P. Avetta, D. Fabbri, A. B. Prevot, E. Montoneri and G. Magnacca, New J. Chem., 2015, 39, 877–885 RSC.
- J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao and J. Li, ACS Catal., 2014, 4, 2917–2940 CrossRef CAS.
- S. Acharya, D. K. Padhi and K. M. Parida, Catal. Today, 2017, 1–12 Search PubMed.
- K. M. Parida, K. H. Reddy, S. Martha, D. P. Das and N. Biswal, Int. J. Hydrogen Energy, 2010, 35, 12161–12168 CrossRef CAS.
- S. Phokha, S. Pinitsoontorn, S. Maensiri and S. Rujirawat, J. Sol-Gel Sci. Technol., 2014, 71, 333–341 CrossRef CAS.
- B. Ding, C. Han, L. Zheng, J. Zhang, R. Wang and Z. Tang, Sci. Rep., 2015, 5, 1–10 Search PubMed.
- V. Di Castro and S. Ciampi, Surf. Sci., 1995, 333, 294–299 CrossRef.
- Preparation, characterization of perovskite LaFeO3 nanospheres and its visible light, 2013, ch. 4, pp. 65–81 Search PubMed.
- W. Weiss and W. Ranke, Prog. Surf. Sci., 2002, 70, 1–151 CrossRef CAS.
- K. Dave, K. H. Park and M. Dhayal, RSC Adv., 2015, 5, 95657–95665 RSC.
- T. Anh, J. Sik, J. Su and Y. Tae, Colloids Surf., A, 2011, 384, 543–548 CrossRef.
- S. Liu, B. Yu, H. Zhang, T. Fei and T. Zhang, Sens. Actuators, B, 2014, 202, 272–278 CrossRef CAS.
- P. Song, Q. Wang, Z. Zhang and Z. Yang, Sens. Actuators, B, 2010, 147, 248–254 CrossRef CAS.
- J. Čejka and S. Mintova, Cat. Rev., 2007, 4, 457–509 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09460a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab