
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein corona formation moderates the release kinetics of ion channel antagonists from transferrin-functionalized polymeric nanoparticles†
Priya S. R. Naidu a,
Eleanor Denhamb,
Carole A. Bartlettb,
Terry McGonigleb,
Nicolas L. Taylorac,
Marck Norreta,
Nicole. M. Smitha,
Sarah A. Dunlopd,
K. Swaminathan Iyer*a and
Melinda Fitzgerald*bde
a,
Eleanor Denhamb,
Carole A. Bartlettb,
Terry McGonigleb,
Nicolas L. Taylorac,
Marck Norreta,
Nicole. M. Smitha,
Sarah A. Dunlopd,
K. Swaminathan Iyer*a and
Melinda Fitzgerald*bde
aSchool of Molecular Sciences, The University of Western Australia, 30 Stirling Highway, Crawley, WA 6009, Australia. E-mail: swaminatha.iyer@uwa.edu.au
bCurtin Health Innovation Research Institute, Curtin University, Kent Street, Bentley, WA 6102, Australia. E-mail: lindy.fitzgerald@curtin.edu.au
cAustralian Research Council Centre of Excellence in Plant Energy Biology, The University of Western Australia, 30 Stirling Highway, Crawley, WA 6009, Australia
dSchool of Biological Sciences, The University of Western Australia, 30 Stirling Highway, Crawley, WA 6009, Australia
ePerron Institute for Neurological and Translational Science, Ralph and Sarich Neuroscience Research Institute Building, Verdun Street, Nedlands, WA 6009, Australia
First published on 15th January 2020
Abstract
Transferrin (Tf)-functionalized p(HEMA-ran-GMA) nanoparticles were designed to incorporate and release a water-soluble combination of three ion channel antagonists, namely zonampanel monohydrate (YM872), oxidized adenosine triphosphate (oxATP) and lomerizine hydrochloride (LOM) identified as a promising therapy for secondary degeneration that follows neurotrauma. Coupled with a mean hydrodynamic size of 285 nm and near-neutral surface charge of −5.98 mV, the hydrophilic nature of the functionalized polymeric nanoparticles was pivotal in effectively encapsulating the highly water soluble YM872 and oxATP, as well as lipophilic LOM dissolved in water-based medium, by a back-filling method. Maximum loading efficiencies of 11.8 ± 1.05% (w/w), 13.9 ± 1.50% (w/w) and 22.7 ± 4.00% (w/w) LOM, YM872 and oxATP respectively were reported. To obtain an estimate of drug exposure in vivo, drug release kinetics assessment by HPLC was conducted in representative physiological milieu containing 55% (v/v) human serum at 37 °C. In comparison to serum-free conditions, it was demonstrated that the inevitable adsorption of serum proteins on the Tf-functionalized nanoparticle surface as a protein corona impeded the rate of release of LOM and YM872 at both pH 5 and 7.4 over a period of 1 hour. While the release of oxATP from the nanoparticles was detectable for up to 30 minutes under serum-free conditions at pH 7.4, the presence of serum proteins and a slightly acidic environment impaired the detection of the drug, possibly due to its molecular instability. Nevertheless, under representative physiological conditions, all three drugs were released in combination from Tf-functionalized p(HEMA-ran-GMA) nanoparticles and detected for up to 20 minutes. Taken together, the study provided enhanced insight into potential physiological outcomes in the presence of serum proteins, and suggests that p(HEMA-ran-GMA)-based therapeutic nanoparticles may be promising drug delivery vehicles for CNS therapy.
1 Introduction
Secondary degeneration that follows injuries to the central nervous system (CNS), or neurotrauma, results in progressive and debilitating functional loss.1–4 Defined by a cascade of neurometabolic events that includes glutamate excitotoxicity, cytosolic Ca2+ overload and oxidative stress, the progression of secondary degeneration may be limited by protecting vulnerable yet intact CNS tissue.5–8 It is now widely recognized that the redistribution of Ca2+ within the CNS following injury is a fundamental and viable therapeutic target for secondary degeneration.9,10 Given that the accrual of intracellular Ca2+ can occur via various Ca2+-permeable pathways post-injury,11–13 preclinical studies have identified that the administration of a novel combination of ion channel antagonists at specific concentrations collectively mitigated secondary degeneration and preserved function at both acute (up to 1 week)14 and chronic (3 months) phases of injury.15 This combinatorial therapy that was administered directly to adult rats subjected to partial optic nerve transection,16 or repeated mild traumatic brain injury,17 consisted of lomerizine hydrochloride (LOM), zonampanel monohydrate (YM872) and oxidized adenosine triphosphate (oxATP) that inhibited voltage-gated calcium channels, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate and purinergic P2X7 receptors respectively.The therapeutic efficiency of the combinatorial treatment may be enhanced in vivo by therapeutic nanoparticles that non-invasively enter the CNS from circulation via the protective blood–brain barrier (BBB). While it is known that the BBB is temporarily open upon CNS injury, the extent and duration of this transient breach depends upon the nature and severity of injury.18 Given that polymers are renowned for their ease of chemical modifications,19,20 specifically functionalized polymeric nanoparticles that target the closed BBB may be beneficial in improving drug transport to the CNS. Conjugation of transferrin (Tf), for example, has been a widely utilized functionalization for nanoparticles designed to improve specificity for the CNS.21,22 Tf receptors are over-expressed in brain micro-vascular endothelial cells that form the BBB, making these receptors highly attractive targets for nanoparticles designed for CNS delivery.23 Correspondingly, enhanced brain tissue accumulation by receptor-mediated transcytosis is reported with the use of various Tf-conjugated nanoparticles in in vivo models.24,25
Polymeric nanoparticles may also protect therapeutic agents from degradation within the physiological system prior to reaching their target sites.19,26 Moreover, repeated dosing, which can be inconvenient in clinical settings of neurotrauma, may be overcome by controlled release of therapeutic cargo from the nanoparticles.27 With such potential advantages, a wide range of nanoparticles for CNS therapy have been designed using biocompatible and synthetic polymers such as poly(ethylenimines), poly(alkylcyanoacrylates), poly(methylidene malonates) and polyesters.19 Choice of matrix material for nanoparticle formulations has been typically dependent on the extent of drug solubility, as high drug loading and entrapment efficiency are desirable properties for successful therapeutic nanoparticles.27,28 However, despite the promise of nanoparticle-mediated drug delivery for CNS therapy in pre-clinical in vivo studies, to-date, clinical translation remains a challenge.29 As the neuro-pharmaceutical sector has the highest growth potential within the pharmaceutical field,30 there is an urgent need to identify and overcome the hurdles that hamper the progress of encouraging CNS nanotherapies, such that they may be successfully implemented clinically.
One of the key factors for the lack of clinical success of nanotherapeutics, not just for CNS therapy, but also for biomedical applications in general, includes poor correlations between in vitro assessments and in vivo outcomes. While therapeutic nanoparticles have been extensively explored over the past 40 years,19 it is only in the past decade that it became apparent that the engineered surfaces of nanoparticles can be altered within the physiological environment due to the inevitable adsorption of biomolecules in the circulation.31 Also known as the protein corona, complex biomolecular layers rapidly (<30 seconds) adsorb on nanoparticles within the physiological milieu due to high surface free energy on engineered surfaces.32 The consequential bio-nano interface endows nanoparticles with a biological identity that varies with different nanoparticle formulations, as the composition of the protein corona is dependent on the physico-chemical properties of nanoparticle formulations such as hydrodynamic size, shape, curvature, surface charge and functionalization.33–36 With the adsorption and accumulation of specific biomolecules such as serum proteins with explicit properties, therapeutic fates of nanoparticles such as biodistribution, immune responses as well as drug delivery can be influenced.37–39 Many in vitro nanoparticle assessments are conducted in 10–20% serum and thus do not account for the complexities of the protein corona formed in vivo, which may consist of up to 55% serum content.40 As such, these studies have not accurately predicted nanoparticle behaviour in vivo.41,42 To improve the correlation between preliminary in vitro assessments and the eventual in vivo outcomes to aid clinical translation of nanotherapies, it is therefore critical for any newly developed therapeutic nanoparticle to be assessed on a case-by-case basis for their potential benefits in physiologically relevant environments.
Herein, Tf-functionalized, hydrophilic p(HEMA-ran-GMA) nanoparticles are designed for use as therapeutic carriers for the delivery of a combination of ion channel antagonists for treatment of secondary degeneration following neurotrauma. As the drug release assessments with these newly developed functionalized nanoparticles must be carried out in vitro, this study importantly elucidates the effect of serum protein interactions on drug release kinetics under conditions that closely represent the physiological milieu that the nanoparticles would encounter upon intravenous administration. Cyanine5 (Cy5) (Lumiprobe®) is used for fluorescent labelling of nanoparticle variants developed in this study for bio-imaging purposes, and unfunctionalized nanoparticles are used as controls to assess the effect of Tf functionalization on protein corona formation and uptake into cultured cells. Control nanoparticles are referred to as ‘NP’ and Tf-functionalized nanoparticles as ‘Tf–NP’.
2 Experimental
2.1 Materials
All materials and reagents related to nanoparticle synthesis and assessments were sourced from Sigma-Aldrich unless specified otherwise. All tissue culture materials and reagents were obtained from Gibco™ unless specified otherwise.2.2 Cy5-labelled p(HEMA-ran-GMA) nanoparticle synthesis
p(HEMA-ran-GMA) copolymer (100 mg), synthesized by atom-transfer radical polymerization, was completely dissolved in 10 mL 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES buffer; 50 mM; pH 7) aided by intermittent sonication in a water bath at room temperature. Copolymer synthesis is detailed in the ESI.†The aqueous copolymer solution was added to a mixture of 30 mL n-hexane and 33 g sodium dioctyl sulfosuccinate with moderate and continuous stirring at room temperature. Ethylene diamine (1:100 in MilliQ water; 50 μL) was added to the optically clear and homogenous spontaneous micro-emulsion to cross-link p(HEMA-ran-GMA) chains in the aqueous micelles via epoxide functional groups to form discrete nanoparticles overnight at room temperature.
To retrieve the cross-linked nanoparticles, the oil-based emulsion (∼10 mL) was layered upon HEPES buffer (∼25 mL; 50 mM; pH 7) and centrifuged for 30 minutes at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g at 4 °C. The aqueous layer (bottom) containing cross-linked nanoparticles in suspension was carefully collected and lyophilized immediately. The lyophilized nanoparticles were reconstituted in excess ammonia (25%; ∼50 mL) and allowed to react at 60 °C for 48 hours with moderate stirring for amine-functionalization. The reaction solution was then dialysed (SnakeSkin™ Dialysis Tubing MWCO 10K, ThermoFisher Scientific®) against MilliQ water over 24 hours (4 × 5 L changes), and the purified suspension was lyophilized.
000 × g at 4 °C. The aqueous layer (bottom) containing cross-linked nanoparticles in suspension was carefully collected and lyophilized immediately. The lyophilized nanoparticles were reconstituted in excess ammonia (25%; ∼50 mL) and allowed to react at 60 °C for 48 hours with moderate stirring for amine-functionalization. The reaction solution was then dialysed (SnakeSkin™ Dialysis Tubing MWCO 10K, ThermoFisher Scientific®) against MilliQ water over 24 hours (4 × 5 L changes), and the purified suspension was lyophilized.
Cyanine5®-N-hydroxysuccinimide ester (Cy5-NHS; 0.25 mg; Lumiprobe) was dissolved in dimethyl sulfoxide (DMSO) and added to suspension of amine-functionalized p(HEMA-ran-GMA) nanoparticles that was reconstituted in sodium bicarbonate solution (NaHCO3; 0.1 M), according to the fluorophore manufacturer's protocol. Cy5-labelling of nanoparticles took place over 2 hours at room temperature. Unbound Cy5 dye and other unreacted components were removed by dialysis against MilliQ water overnight (4 × 5 L changes). The resulting suspension was centrifuged (20000 × g; 30 minutes; 3×) to wash and collect the nanoparticles as a pellet. Cy5-p(HEMA-ran-GMA) nanoparticles (NP) were lyophilized and stored in the dark at room temperature in a desiccator. The synthesis of NP had been adapted from the protocol previously described by Naidu, et al.43
2.3 Tf-functionalization of Cy5-labelled p(HEMA-ran-GMA) nanoparticles
The protocol for Tf functionalization was adapted from Hristov, et al.44 succinimidyl-([N-maleidoproprionamido]-dodecylethyleneglycol) ester (SM(PEG)12; 250 mM in DMSO; Thermo Scientific™) diluted in HEPES buffer was added to the NP suspension (10 mg mL−1) at a 1:1 volumetric ratio to make up a final concentration of 1 mM of SM(PEG)12. The mixture was stirred moderately at room temperature for 2 hours. After which, the PEGylated nanoparticles were washed and retrieved by centrifugation and redispersed at a concentration of 10 mg mL−1 in HEPES buffer.Holo-Tf was dissolved in 1× phosphate buffered saline (PBS) (pH 7.2) to make a protein concentration of 5 mg mL−1. N-Succinimidyl S-acetyl(thiotetraethylene glycol) (250 mM SAT(PEG)4 in DMSO; Thermo Scientific™) was added to the Tf solution in a 1:1 molar ratio, and the mixture was left to stir at room temperature for 30 minutes. 1 M glycine was then added at final concentration of 30 mM to quench the reaction for 15 minutes. The resulting Tf solution was purified by centrifugation through pre-packed desalting columns (Thermo Scientific™ Zeba™ Spin Desalting Column). A deacetylation solution (0.5 M hydroxylamine and 25 mM ethylenediaminetetraacetic acid (EDTA) in 1× PBS pH 7.2) was immediately added (0.1 mL per mL of reaction volume) to the purified SAT(PEG)4-modified Tf solution and allowed to stir at room temperature for 2 hours. Following this step, the modified Tf was purified by centrifugation through pre-packed desalting columns and collected in 1× PBS (pH 7.2) with 10 mM EDTA. Tris(2-carboxyethyl)phosphine (TCEP) was added in a 1:1 molar ratio of TCEP to Tf and the mixture was mixed well.
Tf conjugation of NP was performed within 15 minutes of TCEP addition. During which, the suspension was left to stir for 2 hours at room temperature. 2-Mercaptoethanol was added to the dispersion at a final concentration of 1 mM and left for 5 minutes. Tf–NP were then washed and retrieved by centrifugation (20000 × g; 20 minutes; 3×) with HEPES buffer (50 mM, pH 7.4) and stored at 4 °C until required. Results of supporting analyses for Tf conjugation of NP are found in the ESI.† Scheme 1 describes the three-stage process required for Tf–NP synthesis.
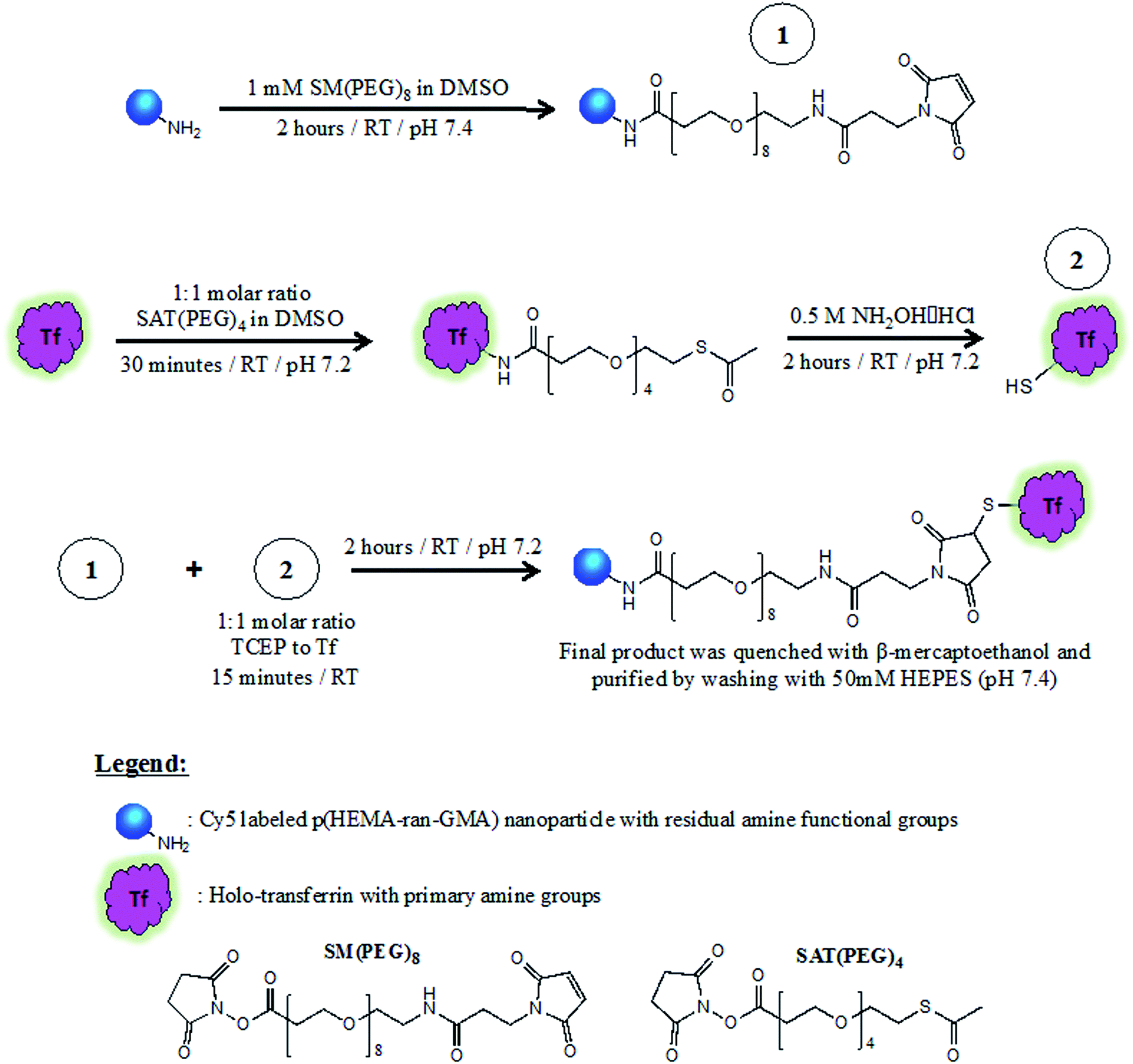 | ||
| Scheme 1 Synthesis of transferrin-functionalized and Cy5-labelled p(HEMA-ran-GMA) nanoparticles (Tf-NP) via a three-stage process that utilizes hetero-bifunctional PEGylated cross-linkers, SM(PEG)8 and SAT(PEG)4. Full description of Tf-NP synthesis is provided in the ESI.† | ||
2.4 Nanoparticle characterization
Characterization of nanoparticle variants was conducted at each stage of the synthesis process using dynamic light scattering (DLS) and zeta potential measurements (Malvern Zetasizer™ Nano ZS, Malvern Panalytical®) using a 4 mW He–Ne laser operating at 633 nm with a scattering angle of 173°.100 μg mL−1 nanoparticle variant samples were prepared in 1× PBS and ∼1 mL volumes were sampled in appropriate cuvettes for hydrodynamic size (12 mm square polystyrene cuvettes) and zeta potential (folded capillary zeta cell) measurements taken in triplicate after an initial equilibrium time of 1 minute. The DLS and zeta potential results were calibrated against measurements recorded for PGMA (refractive index 1.515; viscosity 0.05) in water at 25 °C as dispersant (refractive index 1.33; viscosity 0.887). The scattered light intensity-weighted hydrodynamic radii and zeta potentials of the nanoparticle variants were presented as mean ± standard deviation (S.D.).
Lyophilized NPs were reconstituted in MilliQ water (1 mg mL−1) and washed by centrifugation (20000 × g; 20 minutes; 3×) for further purification for transmission electron microscopy (TEM) imaging. The resulting NP pellet was reconstituted in 250 μL of MilliQ water, and 10 μL of the suspension was deposited on carbon-coated copper grid and dried overnight. All TEM images were obtained at 120 kV using JEOL JEM-2100.
2.5 Protein quantification
Micro BCA™ Protein Assay Kit (Thermo Scientific™) was utilized in the study for (a) confirmation of Tf-conjugation on Cy5-p(HEMA-ran-GMA) nanoparticles and (b) quantification of adsorbed serum proteins on nanoparticle variants. Details of individual analyses have been provided in the ESI.† The quantification assay was performed according to the manufacturer's protocol. In brief, bovine serum albumin standards at known concentrations ranging from 5–50 μg mL−1, and test samples were loaded into individual wells of a clear, flat-bottomed 96-well micro-titre plate. All standard and sample volumes were kept consistent at 150 μL, and tested in triplicate. 150 μL of Micro BCA™ working reagent, as described by the manufacturer, was delivered into each well and the plate was incubated for 2 hours at 37 °C. After cooling to room temperature, the absorbance of all samples was measured at 562 nm on a plate reader (PerkinElmer™ EnSpire® Multimode Plate Reader) to determine protein concentration.2.6 Drug loading and release profile measurements from Tf-NP by high performance liquid chromatography (HPLC)
HPLC coupled with UV/Vis detector was used to determine drug loading and the respective release profiles from Tf-NP. Details of drug loading in nanoparticles by backfilling, and HPLC detection of LOM, YM872 and oxATP have been provided in the ESI.† To assess drug release in the presence of serum proteins, drug-loaded Tf-NP were placed in 55% (v/v) human serum in 1× PBS at 37 °C at a final nanoparticle concentration of 10 mg mL−1. At designated release assessment time points, 150 μL aliquots of the samples from the sinks were rapidly centrifuged (<2 minutes) through filter units (Amicon® Ultra-4 Centrifugal Filter units NMWL 3 kDa) according to the manufacturer's specifications. The filtrate collected was then used for the detection of drug concentration by HPLC at specific time points over 1 hour. HPLC auto-sampler vials were fitted with polypropylene micro-volume inserts with bottom-springs to contain the filtrate samples for analysis.2.7 Culture of primary mixed cortical cells
All experimental procedures were carried out in strict accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes as stipulated by the National Health and Medical Research Council, and approved by The University of Western Australia Animal Ethics Committee (Approval Number RA/3/100/673 and RA/3/100/1485). Brains were dissected from humanely euthanized rat pups using a sterile scalpel (Braun™) and placed into a Petri dish containing Hank's Balanced Salt Solution (HBSS). Cortices were surgically freed from subcortical structures and meninges, using forceps and fine operating scissors, and placed into a new Petri dish containing clean, ice-cold HBSS, where they were dissected into ∼1 mm3 pieces. Digestion solution containing 13.6 mL HBSS, 0.8 mL DNaseI (0.2 mg mL−1) and 0.6 mL trypsin (0.25%), was added for 15 minutes in 5% CO2 at 37 °C. Digested cortices were transferred into 5 mL DMEMS20 (DMEM, 4 mM 100× GlutaMAX, 1 mM sodium pyruvate, 20% (v/v) fetal bovine serum (FBS), 100000 U mL−1 penicillin-streptomycin). Residual tissue in the Petri dish was gently washed with ∼2–3 mL DMEMS20 and added to the digested cortices, which were then centrifuged for 5 minutes at 100 × g, at which point, a clear tissue pellet could be distinguished. 5 mL of DMEMS20 was added to the tissue pellet, which was then triturated using a transfer pipette until almost homogenous and settled on ice for 10 minutes. Avoiding the loose tissue pellet, the cell suspension was passed through a 70 μm nylon strainer (VWR International) and into 5 mL DMEMS20. Cells were counted using trypan blue exclusion assay (0.4% trypan blue; Invitrogen™) with a Neubauer Improved Bright-lined Haemocytometer (ProSciTech®) on an Olympus® CK30 culture microscope at 20× magnification. Cell suspension was added to DMEMS20 (+10 ng mL−1 platelet derived growth factor-AA (PDGF-AA) and 10 ng mL−1 basic fibroblast growth factor (bFGF)) and plated onto poly-DL-ornithine (10 μg mL−1) coated 18 mm2 glass coverslips in clear 12 well tissue culture plates at a density of 1 × 104 cells per cm2. Cultures were incubated at 37 °C in 5% CO2 for 10 days, with half-volume media changes of DMEMS20 + (20 ng mL−1 PDGF-AA and 20 ng mL−1 bFGF) every 2–3 days.
2.8 Treatment application to primary mixed cortical cultures
Stock nanoparticle variant suspensions of known concentrations were obtained by dispersion in 50 mM HEPES buffer (pH 7.4) and sterilized by UV irradiation (45 minutes in 1.5 mL Eppendorf® tubes). After 10 days of culture of the mixed cortical cells, the stock nanoparticle suspensions were added to test wells containing fresh media (DMEMS20; +20 ng mL−1 PDGF-AA and 20 ng mL−1 bFGF) at a volumetric ratio of 1:10 ratio respectively. Final nanoparticle concentrations were applied to test wells as required for the designated assessments: 25 μg mL−1 for immunohistochemistry labelling; 25–1000 μg mL−1 for cell viability assay by Live/Dead™ assay.
2.9 Cytotoxicity assessment
Cultured primary mixed cortical cells were incubated with nanoparticle variants at defined final nanoparticle concentrations, and stained using the Live/Dead™ reagent (4 μM ethidium homodimer-1 (EthD-1) and 2 μM calcein AM) (Molecular Probes™) to identify live versus dead cells on the basis of membrane integrity and esterase activity. After a 45 minute incubation at room temperature, cytotoxicity was quantified by recording fluorescence with a plate reader set to 494/517 nm (excitation/emission) for calcein AM (live cells) and 528/617 nm for EthD-1 (dead cells). Experiments were repeated three times using cultures derived from separate litters of rat pups. Representative wells were imaged using fluorescence microscopy with a 40× objective lens to assess four standardized fields of view (FOV) of 293.703 × 293.703 μm per technical replicate (n = 3). The numbers of appropriately florescent live and dead cells were counted and expressed as mean percentages of the total cell population within the FOV.2.10 Immunohistochemistry and confocal microscopy
Nanoparticle variants at a final concentration of 25 μg mL−1 were applied to well plates in duplicate, to allow for immuno-histochemical identification of multiple cell types and nanoparticle localization therein, and cultures were incubated for a further 24 hours at 37 °C in 5% CO2, with the inclusion of 20% FBS in the culture media as described in Section 2.7. Experiments were repeated three times using cultures derived from separate litters of rat pups. Immunohistochemistry was conducted in accordance with established procedures5 using primary antibodies recognizing markers as follow: microglial activation markers Iba1 (1:500; Abcam, goat Ab5076) and ED1 (1:500; Merck Millipore, mouse MAB1435); oligodendroglial indicator oligodendrocyte transcription factor 2 (Olig2; 1:500; R&D Systems, goat AF2418); astrocyte indicator glial fibrillary acidic protein GFAP (1:500; Abcam, rabbit AB33922); and neuronal indicator β-III tubulin (1:500; Merck Millipore, mouse MAB1637). Antibodies were diluted in PBS containing 0.2% Triton™ X-100 and 5% normal donkey serum. Secondary antibodies were Alexa Fluor 488 or 555 (1:400; Thermo Fisher Scientific™), together with Hoechst 3342 (1:1000; Thermo Fisher Scientific™) diluted in 1× PBS containing 0.2% Triton™ X-100. Finally, the sections were mounted on glass slides with cover slips using Fluoromount-G (Thermo Fisher Scientific™). The slides were viewed using Nikon Eclipse Ti-inverted microscope. Four randomly sampled, non-overlapping Z-stack images were taken per well at 20× magnification. A series of 13 optical images were taken at 0.5 μm increments along the z-axis, and deconvoluted using Nikon Elements NT software. This was followed by the assessment of consistently sized (293.703 × 293.703 μm), non-overlapping regions of interests (ROIs) in the confocal images in order to determine the proportion of distinct cell populations that exhibit Cy5 fluorescence from the uptake of NP and Tf-NP. All image analyses were performed on Fiji image processing software (National Institutes of Health). In conjunction with representative confocal images obtained using all of the above-mentioned markers, total numbers of Iba1+ resident reactive microglia and ED1+ activated microglia/macrophages with and without nanoparticles were counted within a ROI in a 40× image and expressed as the mean number of cells per mm2. Quantification was confined to microglia/macrophages due to the prevalence of nanoparticle variants within these cell populations, and the biological relevance of immune cell engulfment of nanoparticles.2.11 Statistical analysis
All statistical data were processed and analyzed using GraphPad Prism® 6.0.3 Results and discussion
3.1 p(HEMA-ran-GMA) copolymer is a suitable matrix for functionalized nanoparticle formulation designed for the incorporation of water-soluble therapeutic cargo
Of the three ion channel antagonists that collectively exhibited promising therapeutic outcomes for secondary degeneration in preclinical studies,15,45 YM872 and oxATP have high solubility in aqueous medium (YM872, 83 mg mL−1 solubility in Britton–Robinson buffer;46 oxATP, 50 mg mL−1 solubility in H2O47). LOM on the other hand, is lipophilic in nature, and known to be soluble in chloroform, DMSO and methanol, with limited solubility in water.48 However, water-based solutions of LOM may be prepared by dissolution in methanol, followed by dilution in water.49 Poly (HEMA) nanoparticles that swell in aqueous media, due to interactions with the hydroxyl pendants on the polymer chains, have the capacity to load and release water-soluble drugs such as doxorubicin and 5-fluorouracil in a controlled manner.50,51 Thereby, the HEMA component of the polymeric backbone in the hydrophilic p(HEMA-ran-GMA) nanoparticles presents the opportunity to incorporate LOM, YM872 and oxATP within a single polymeric delivery formulation.Highly reactive epoxide functional groups conferred by glycidyl methacrylate (GMA) monomer used in the synthesis of p(HEMA-ran-GMA) are useful in enabling functionalization of targeting ligands and/or fluorescent labels on the nanoparticles via ring-opening reactions.52 Additionally, the epoxide groups in the p(HEMA-ran-GMA) backbone are vital in this study as they facilitate the structural formation of nanoparticles in a spontaneous water-in-oil (W/O) microemulsion by crosslinking chemistry. Taken together, the random copolymer p(HEMA-ran-GMA) is endowed with essential characteristics that suit the development of functionalized, hydrophilic and cross-linked nanoparticles for the incorporation of water-soluble drugs.
Details of the fabrication of cross-linked p(HEMA-ran-GMA) nanoparticles, Cy5-labeling, and Tf-functionalization of the nanoparticles are found in the ESI (Fig. S1 and S2†).
3.2 Physico-chemical properties of Tf-NP suggest suitability for CNS delivery
Dynamic Light Scattering (DLS) intensity histograms (Fig. 1A) of NP and Tf-NP suspensions show that the nanoparticle variants have narrow size distributions, and TEM images (panels (i) and (ii) in Fig. 1A) reveal that the nanoparticles are uniform with spherical conformation. The nanoparticle polydispersity index (PDI) values of both NP (0.107) and Tf-NP (0.045) are small (Fig. 1B), signifying that the nanoparticle variants are suitably mono-dispersed for biological applications.53 Tf-NP (285 nm) has a slightly larger mean hydrodynamic size than NP (220 nm) (Fig. 1B). The lower limit of hydrodynamic size of therapeutic nanoparticles designed for CNS delivery is thought to be ∼20 nm, which allows the avoidance of rapid renal glomerular filtration.54,55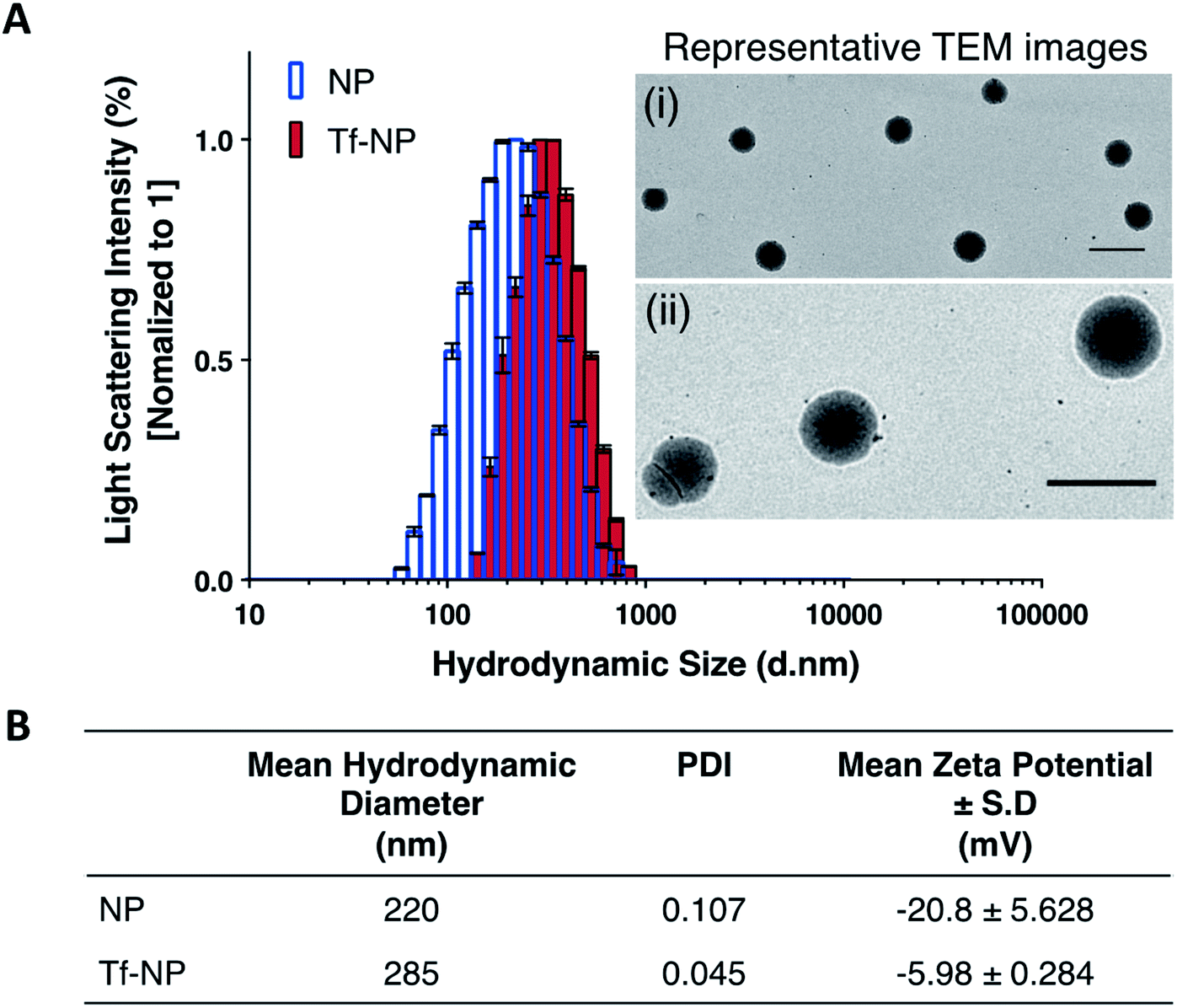 | ||
| Fig. 1 Nanoparticle characterization: (A) Dynamic Light Scattering (DLS) demonstrate the size distribution of Cy5-labelled p(HEMA-ran-GMA) nanoparticles (NP) and transferrin (Tf)-functionalized nanoparticles (Tf-NP). Light scattering intensity percentages for NP and Tf-NP (n = 3) are normalized to 1. Representative transmission electron microscope (TEM) images of NP are depicted in panels (i) 5000× and (ii) 20000× magnifications. Scale bar = 500 nm. (B) Tabulated data summarizes the mean hydrodynamic diameters (±standard deviation (S.D.)), nanoparticle polydispersity (PDI), and nanoparticle surface charges in terms of zeta potential measurements (±S.D.) of NP and Tf-NP (n = 3). | ||
The appeal for this approximate nanoparticle size also stems from the narrow width (∼40–60 nm) of the extracellular space in the healthy human brain.56 However therapeutic nanoparticles up to 300 nm, particularly those that are modified with polyethylene glycol (PEG), have been reported to successfully accumulate in the CNS following systemic delivery.57,58 Thus, the hydrodynamic size range of Tf-NP that are functionalized with the aid of a PEGylated cross-linker, may be suitable for non-invasive CNS delivery. Moreover, it is noteworthy to acknowledge that larger therapeutic nanoparticles (∼200–300 nm) can be advantageous for clinical application due to improved drug-loading efficiency and payload, and can lead to enhanced drug dispersion and extended periods of drug release.59 However, the exact size criterion for CNS-specific nanoparticles remains unclear, as the overall design of the nanoparticles largely controls the mechanisms involved in CNS uptake.
The surface charges of NP and Tf-NP indicated by zeta potential measurements at pH 7.4 are −20.8 and −5.98 mV respectively (Fig. 1B). The general negative charges of the polymeric nanoparticle variants in the aqueous environment can be attributed to the dissociation of hydroxyl groups in p(HEMA-ran-GMA) copolymer chains.60 In addition, the positive shift in the surface charge of Tf-NP with respect to NP may be indicative of the protonation of amine groups on the conjugated Tf.25 Therapeutic nanoparticles with near-neutral, slightly negative zeta potentials have been reported to successfully access brain tissue when administered intravenously.58,61 Accordingly, it is considered advisable for CNS-specific nanoparticles to have zeta potentials that are slightly negative at physiological pH (−1–15 mV at pH 7.4) in order to limit unfavourable biodistribution outcomes as a response to net surface charge.62–64 Taken together, the hydrodynamic size and surface charge of p(HEMA-ran-GMA)-based nanoparticles developed for this study may be suitable for CNS delivery.
3.3 Tf-NP are not cytotoxic to primary mixed cortical cultures, and are less susceptible to uptake by microglial populations than NP
It is fundamental that engineered therapeutic nanoparticles are not cytotoxic such that their proposed beneficial qualities are not negated. In vitro assessments in this study were conducted in primary mixed cortical cultures in 20% serum, as this is the highest concentration of serum in which cells maintain viability in vitro. Live/Dead™ assay confirms that NP and Tf-NP are not cytotoxic to the cultures at concentrations up to 500 μg mL−1 nanoparticle (Fig. S3 in ESI†). The interaction and localization of the Cy5-labelled nanoparticle variants with relevant cell types are further determined by confocal microscopy of immuno-labelled primary cultures.Co-localization of Cy5 is not observed in GFAP+ cells, or in those labelled with βIII-tubulin, suggesting that neither NP nor Tf-NP are taken up by astrocytes or neurons (representative confocal images in Fig. 2A). Likewise, olig2+ cells do not take up the nanoparticle variants regardless of functionalization (representative confocal images in Fig. 2B). Interestingly, both nanoparticle variants predominantly localize within resident microglia (Iba1+ cells) as well as activated microglia/macrophages (ED1+ cells) (representative confocal images in Fig. 2C). Further quantitative analysis from confocal images reveals that Tf-functionalization does not cause a significant difference in nanoparticle uptake by Iba1+ cells. However, a greater percentage of ED1+ cells contain NP than Tf-NP (Fig. 2D).
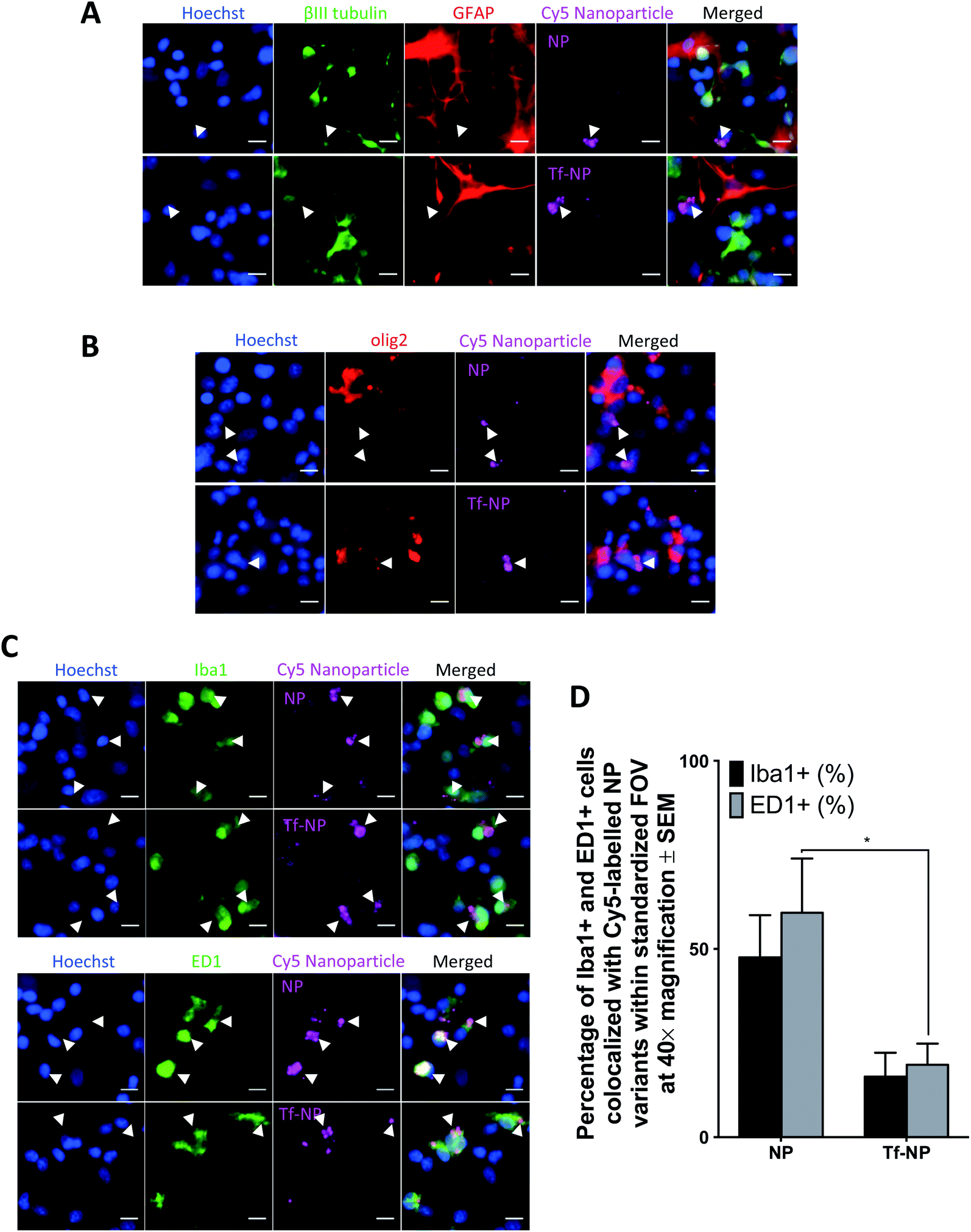 | ||
| Fig. 2 In vitro uptake of Cy5-labelled p(HEMA-ran-GMA) nanoparticles (NP) and transferrin-functionalized Cy5-labelled p(HEMA-ran-GMA) nanoparticles (Tf-NP) in primary mixed cortical cultures. Representative confocal microscopy images (40× magnification) are of NP and Tf-NP treated cultures that are immunohistochemically stained to detect: (A) GFAP+ astrocytes (red) and βIII-tubulin+ neurons (green); (B) olig2+ oligodendroglia (red); (C) top panel: Iba1+ microglia (green); bottom panel: ED1+ microglia/macrophages (green). All cell nuclei are labelled with Hoechst (blue) and Cy5-labelled nanoparticle variants appear magenta in the confocal microscopy images. Scale bar = 10 μm. White arrowheads indicate nanoparticle colocalization in the representative confocal images. (D) Uptake of nanoparticle variants (NP and Tf-NP) by Iba1+ and ED1+ cells in primary mixed cortical cultures expressed as percentages of total count of respective cell types in standardized fields of view at 40× magnification. Mean values are presented with standard error of measurements (±SEM). Statistical analyses were conducted using two-way ANOVA with post-hoc analysis by Sidak's multiple comparison's test (n = 3, *p ≤ 0.05). | ||
The results suggest that Tf-NP are less susceptible to uptake by activated microglia/macrophages than NP. Given that microglial populations in the CNS are highly phagocytic and act as the first line of defence for immunological responses,65 the reduced uptake of Tf-NP reported in this in vitro assessment may be favourable as it suggests that the nanoparticles may not be subjected to rapid systemic clearance under in vivo test conditions. However, it is acknowledged that the elevated presence of serum proteins in the physiological milieu (55%) in comparison to the serum content in the culture environment used in this study (20%) may result in variations in the nanoparticle biodistribution in vivo due to increased serum protein interactions at the nanoparticle surfaces.
3.4 Combinatorial ion channel antagonist treatment can be loaded into Tf-NP by backfilling
A simple backfilling strategy is used to load LOM, YM872 and oxATP into Tf-NP. This drug loading method has been described for mesoporous silica nanoparticles whereby active molecules are allowed to diffuse into the empty pores of the nanoparticles by exposure to either vapour or solution.66 A significant advantage of backfilling is that it does not result in chemical modification of the drugs thereby maximizing drug potency.67 The cross-linked, water-permeable p(HEMA-ran-GMA)-based nanoparticles developed in this study do not lose structural integrity upon lyophilization; a process known to improve long-term colloidal stability.68 Since LOM, YM872 and oxATP can be dissolved to make up an aqueous drug solution, drug loading of Tf-NP is possible by rehydration of the lyophilized nanoparticles. As the nanoparticle structure swells in the aqueous medium, the drugs in solution are absorbed within. The maximum loading efficiencies of LOM, YM872 and oxATP are 11.8 ± 1.05% (w/w), 13.9 ± 1.50% (w/w) and 22.7 ± 4.00% (w/w) respectively as detected by HPLC (Fig. 3A).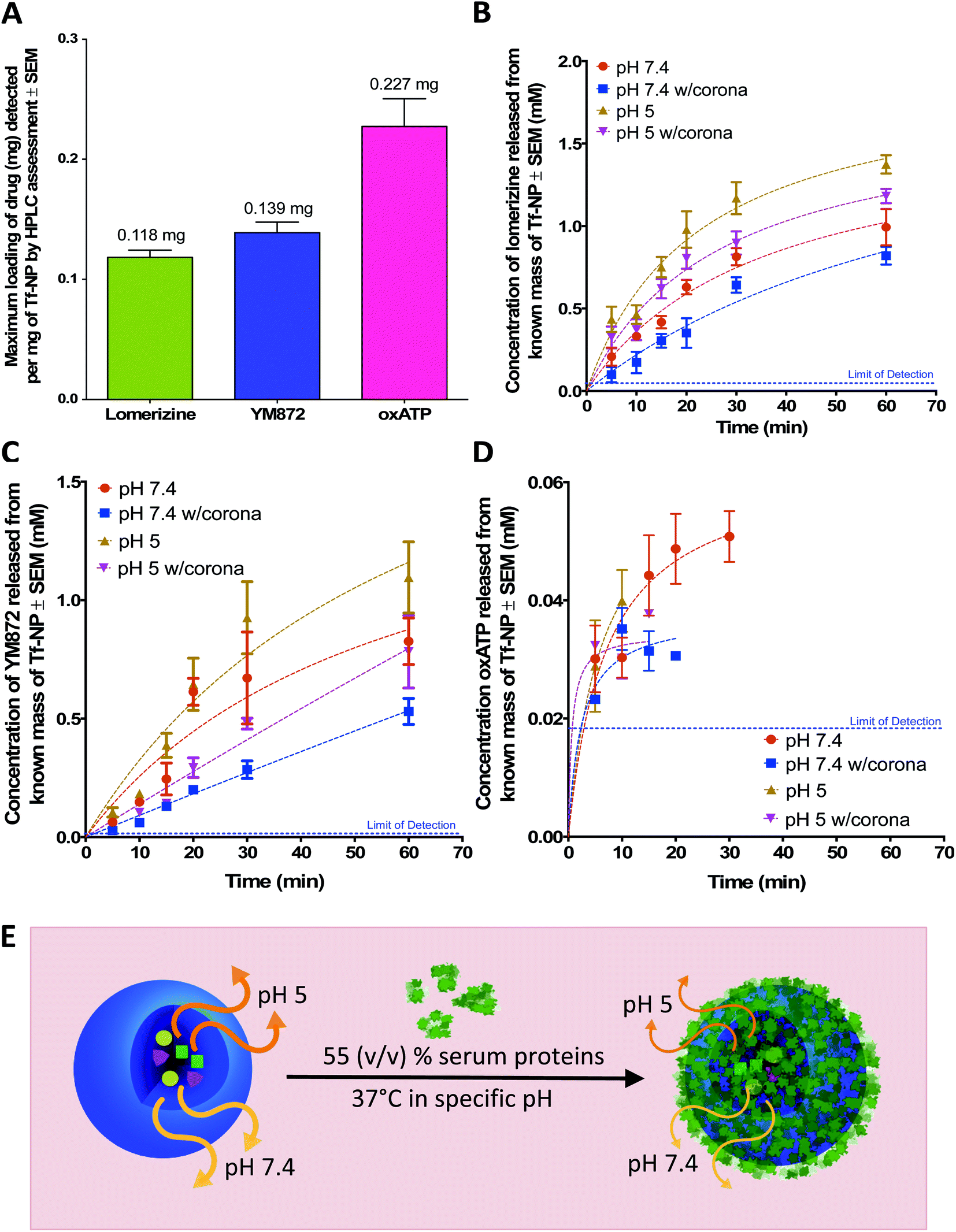 | ||
| Fig. 3 Assessment of drug-loaded, transferrin-functionalized p(HEMA-ran-GMA) nanoparticles (TF-NP) by High Performance Liquid Chromatography (HPLC). (A) Maximum loading of individual ion channel antagonists (mg) per mg Tf-NP. Release profiles of (B) lomerizine, (C) YM872 and (D) oxATP from known masses of drug-loaded Tf-NP incubated at 37 °C in 1× phosphate buffered saline (PBS) at pH 7.4 and 5, with and without the inclusion of 55% (v/v) human serum. Lines of best fit of the data are drawn using the Michaelis–Menten fitting method. Blue dotted lined in plots B-D indicate the limit of detection of individual drugs assessed by respective HPLC methods. Data provided in this figure (A–D) are presented as mean values (n = 3) with standard error of measurements (±SEM). HPLC data stated as ‘w/corona’ refer to those obtained in the presence of serum proteins. (E) Schematic diagram demonstrating the effects of pH and the presence of the protein corona on drug release from drug-loaded Tf-NP. Orange and yellow arrows denote release of ion channel antagonists from Tf-NP under specific pH. Thickness of arrows correlate to concentration of released drugs. Image not drawn to scale. | ||
3.5 Serum protein on drug-loaded Tf-NP impacted drug release kinetics
Commonly utilized in vitro conditions are either serum-free, or supplemented with 10–20% (v/v) serum depending on culture protocols.69 Standard in vivo environments however may consist of up to 55% (v/v) serum.40,42,70 In the presence of higher serum protein concentrations, the effect of the protein corona on the behaviour of therapeutic nanoparticles is likely to be greater, due to increased concentration of adsorbed serum proteins on the surfaces of the nanoparticles.37,71 Using physiologically relevant human serum concentration of 55% (v/v), Micro BCA™ protein quantification assay of the tightly adsorbed serum proteins closest to the nanoparticle surface (hard corona) on NP and Tf-NP reveals that the adsorbed protein concentration on Tf-NP is significantly higher (1.5 mg protein per mg nanoparticle) than that on an equivalent mass of NP (0.4 mg protein/mg nanoparticle) (Fig. S4A in ESI†).Given that the constituency of the protein corona may be modified with variable surface functionalizations,32,72 equal amounts of proteins from the hard corona proteins from NP and Tf-NP are separated for assessment by SDS-PAGE (Fig. S4B†). There are no substantial differences in the types and intensities of the protein bands derived from the hard corona of both nanoparticle variants, suggesting that the Tf functionalization of p(HEMA-ran-GMA)-based nanoparticles likely do not enrich the protein corona with serum proteins of different molecular weights. Indeed, a qualitative assessment of the protein bands by mass spectrometry may offer further insight about the types of adsorbed serum proteins on the nanoparticle variants. However quantitative protein corona assessment is more relevant to the scope of this study.
The release kinetics of ion channel antagonists from Tf-NP are assessed by HPLC over a period of 1 hour at 37 °C, either in 1× PBS in neutral physiological pH of 7.4, or in a slightly acidic environment (pH 5) that correlates to the endosomal environment73 (Fig. 3B–D). NP are not evaluated for drug loading or release in this study as the key objective of the assessment is to elucidate the capacity of nanoparticles that are specifically functionalized for controlled drug release in the CNS.
While the general trends of the plots demonstrate sustained release of the ion channel antagonists over at least 20 minutes, statistical analyses of the data reveal details of the impact of pH and serum on the release of each drug from Tf-NP. Statistically significant differences in the released drug concentrations that are obtained from two-way ANOVA analysis with post-hoc analysis by Tukey's multiple comparisons test are summarized in Table 1.
| LOM: F(5, 48) = 94.81 | 5 min | 10 min | 15 min | 20 min | 30 min | 60 min |
|---|---|---|---|---|---|---|
| a “*” Indicates significant difference in released drug concentration at specific time point, *p ≤ .05, **p ≤ .01, ***p ≤ .001; “ns” = not significant; “ND” = no data (therefore unable to perform statistical analysis); “v.” = versus; “w/corona” = with protein corona; “min” = minutes. | ||||||
| pH 7.4 v. pH 5 | ns | ns | ** | ** | ** | *** |
| pH 7.4 v. pH 7.4 w/corona | ns | ns | ns | * | * | ns |
| pH 5 v. pH 5 w/corona | ns | ns | ns | ns | * | ns |
| pH 7.4 w/corona v. pH 5 w/corona | ns | ns | ** | **** | * | ** |
| YM872: F(5, 47) = 52.34 | 5 min | 10 min | 15 min | 20 min | 30 min | 60 min |
|---|---|---|---|---|---|---|
| pH 7.4 v. pH 5 | ns | ns | ns | ns | ns | ns |
| pH 7.4 v. pH 7.4 w/corona | ns | ns | ns | ** | ** | ns |
| pH 5 v. pH 5 w/corona | ns | ns | ns | * | ** | * |
| pH 7.4 w/corona v. pH 5 w/corona | ns | ns | ns | ns | ns | ns |
| oxATP: F(5, 48) = 7.157 | 5 min | 10 min | 15 min | 20 min | 30 min | 60 min |
|---|---|---|---|---|---|---|
| pH 7.4 v. pH 5 | ns | ns | *** | *** | * | ND |
| pH 7.4 v. pH 7.4 w/corona | ns | ns | ns | ** | ND | ND |
| pH 5 v. pH 5 w/corona | ns | ns | ND | ND | ND | ND |
| pH 7.4 w/corona v. pH 5 w/corona | ns | ns | ND | ND | ND | ND |
Under serum-free conditions, the lower pH of 5 significantly enhances the release of LOM and oxATP beyond the 15 minute time point in comparison to pH 7.4. The pH variation, however, does not significantly affect the concentrations of released YM872 at 1 hour. It is postulated that the enhanced release of LOM and oxATP under acidic conditions is likely a result of structural shrinkage of the nanoparticles, as cross-linked p(HEMA)-based hydrogels have previously demonstrated variable swelling capacity in water in response to pH, which consequently affects drug release from within.74 This assumption is tested in this study using a mathematical model of drug release, which can be useful to establish the mechanism of drug release from a matrix. The Higuchi simulation is a well-known and widely used controlled release model for spherical hydrogel systems that is based on several key hypotheses; one of which assumes that matrix swelling or dissolution is negligible.75,76 By fitting the drug release data of LOM obtained from the tested pH conditions into the Higuchi model, it is found that better linearity is obtained at pH 7.4 (R2 = 0.8868) in comparison to pH 5 (R2 = 0.6980). Data for this simulation is provided in Fig. S5 in the ESI.† While this finding suggests that non-negligible structural changes could have occurred to the Tf-NP under acidic conditions to impact the release kinetics of LOM, the mathematical simulation model could not be applied to the release data obtained with oxATP due to the unavailability of a complete drug release data set. At least initially, oxATP is present at detectable concentrations. However, the release kinetics of oxATP from Tf-NP is not measurable over the entire assessment period and conditions defined in this study (Fig. 3D). Being a small Schiff-base molecule,77 oxATP may be inclined to hydrolytic cleavage of azomethine bonds present in the compound, causing interferences in assessments conducted in the aqueous environment.78 As such, it is possible that the instability of the oxATP released from Tf-NP under the described test conditions may have contributed to the lack of its detection by HPLC beyond ∼15 minutes at pH 5. However, at pH 7.4, oxATP remains detectable by HPLC analysis for up to 30 minutes in serum-free conditions. Given these observed variations in the release kinetics of the ion channel antagonists, it is likely that the differences in the molecular properties of the therapeutic agents played a role in modulating their release rates from Tf-NP in different pH environments.
The presence of serum proteins, or in other words, formation of protein corona, results in a statistically significant difference in the concentrations of released LOM, YM872 and oxATP from Tf-NP at delayed stages (beyond 10 minutes). It is noted however that the analysis of oxATP release is limited due to the aforementioned incomplete data set. Protein corona formation on Tf-NP does not impact the rate of release of all three ion channel antagonists during the first 10 minutes of the assessment at both pH 5 and 7.4. Beyond the 15 minute time point up to the end of the assessment at 1 hour, the protein corona significantly lowers the amount of LOM released from Tf-NP at pH 7.4. Interestingly, at pH 5, the protein corona only impacts on LOM release beyond the 30 minute time point. The protein corona mitigates the release of YM872 after 20 minutes, regardless of pH. However, at pH 7.4, the amount of YM872 released is not affected by the protein corona at the 1 hour time point. It is likely that the YM872 release from Tf-NP was approaching equilibrium concentrations by this stage of the assessment. The highest detected oxATP concentration released from Tf-NP is 0.05 ± 0.006 mM at pH 7.4 at 30 minutes under serum-free conditions. In the presence of serum proteins at pH 7.4, the detectable amount of released oxATP diminishes to approximately 0.03 ± 0.001 mM at the 20 minute time point. In a more physiologically relevant assessment that compares drug release from Tf-NP in the presence of serum proteins at pH 5 to that at 7.4, it is revealed that only the release of LOM is significantly enhanced in the acidic environment, and this is beyond the 15 minute time point up to 1 hour. Taken together, this study demonstrates that the protein corona can induce a shielding effect on drug release.37,79 However, it appears that the impact of the protein corona on drug release from Tf-NP varied temporally with respect to the specific ion channel antagonist.
The results reveal an important consideration for the development of therapeutic nanoparticles, whereby the assessment of drug release kinetics in the presence of serum and variable pH provides a more realistic outcome of therapeutic potentials. In this case, biocompatible Tf-NP loaded with LOM, YM872 and oxATP release their therapeutic cargo at both physiological and endosomal pH. While sustained release is exhibited by both LOM and YM872 from Tf-NP over a period of 1 hour, potential instability of oxATP in the biological environment may limit its therapeutic potential to a shorter time span (<30 minutes). Nevertheless, in the presence of a protein corona induced by exposure to 55% (v/v) serum, LOM, YM872 and oxATP may still be released in combination from Tf-NP for up to 20 minutes.
Partition equilibrium effects may moderate the distribution of released therapeutic agents, thereby affecting measured released drug concentrations.80–82 Released drugs from therapeutic nanoparticles may bind to the serum proteins present in either the protein corona, or to those present in the aqueous media, thereby altering the measured concentration of released drugs. The present work has not distinguished between corona effects and free serum binding effects. Therefore, it is acknowledged that the concentration of released drugs detected in the presence of serum protein may represent the fraction of released drug that was not bound by serum proteins. While this study focuses on the changes in drug release kinetics as a result of serum protein presence, future work may benefit from experimental measurements of partition coefficients for the ion channel antagonists, to determine the relative contributions of unbound serum proteins and protein bound as part of a corona.
Research has revealed that the circulation half-life of systemically administered targeting nanoparticles ranges from a few minutes up to half hour, prior to elimination from the physiological system.83,84 Therefore, the preliminary drug release outcomes from this study obtained by taking typical blood serum concentration into consideration, suggest that systemically delivered Tf-NP may be capable of delivery of ion channel antagonists over clinically relevant durations. Additionally, taking into consideration that transient openings can occur in the BBB upon acute CNS trauma,18 it may also be possible that the Tf-NP can advantageously accumulate within the CNS from circulation to deliver the ion channel antagonist combination. Tf functionalization may thus serve as an auxiliary mechanism to improve CNS biodistribution in the event that the BBB returns to its normal closed state by the time the therapeutic nanoparticles are introduced into physiological circulation.
4 Conclusions
Hydrophilic p(HEMA-ran-GMA) random copolymer is used to develop biocompatible Tf-functionalized nanoparticles in this study to successfully incorporate the largely water-soluble combination of ion channel antagonists (LOM, YM872 and oxATP) identified for secondary degeneration therapy. There is now increased recognition that preliminary in vitro assessments of therapeutic nanoparticles designed for intravenous administration require the inclusion of physiologically relevant serum proteins to account for the effects of the inevitable formation of the protein corona in vivo. In doing so, measured therapeutic outcomes may more closely match prospective in vivo results such that the potential for clinical translation of promising nanotherapies may be accelerated due to enhanced understanding of their biological behaviour. As such, release kinetics of the respective ion channel antagonists from Tf-functionalized p(HEMA-ran-GMA) nanoparticles are assessed in the presence of serum proteins as a critical demonstration of therapeutic capability of the polymeric carriers in biological milieu. Recent studies suggest the inclusion of dynamic flow conditions in in vitro assessments as circulating serum proteins under physiological conditions may affect the protein corona composition, which may then moderate biological outcomes of therapeutic nanoparticles.85–88 Although flow dynamics is not accounted for in this study, it is acknowledged that future work could benefit from incorporating this factor. While controlled release of the therapeutic agents is established from the p(HEMA-ran-GMA)-based nanoparticles, it is importantly revealed that the presence of serum proteins moderated the rate of specific drug release, which may consequently impact the scale of therapeutic success under physiological conditions. Moreover, the inclusion of conditions such as physiological temperature and variable pH in the drug release assessment, suggests that oxATP might not be as stable as LOM or YM872 under biological conditions. Overall, this in vitro study is a pivotal first step to gauge the effectiveness of Tf-functionalized p(HEMA-ran-GMA) nanoparticles to deliver the specific combination of ion channel antagonists for the treatment of secondary degeneration under the influence of the protein corona. The results obtained suggest that future in vivo assessments should factor in the circumstance that the collective exposure of the LOM, YM872 and oxATP might be limited to approximately 20 minutes of nanoparticle circulation within the physiological system.Abbreviations
| BBB | Blood–brain barrier |
| BCA | Bicinchoninic acid |
| CNS | Central nervous system |
| Cy5 | Cyanine5 fluorophore |
| DLS | Dynamic light scattering |
| DMEM | Dulbecco's modified eagle's medium |
| DMSO | Dimethylsulfoxide |
| EDTA | Ethylenediaminetetraacetic acid |
| FBS | Fetal bovine serum |
| FOV | Field of view |
| GMA | Glycidyl methacrylate |
| HEMA | 2-Hydroxyethyl methacrylate |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid |
| HPLC | High performance liquid chromatography |
| LOM | Homerizine hydrochloride |
| MWCO | Molecular weight cut-off |
| NHS | N-Hydroxysuccinimide |
| NMWL | Nominal molecular weight limit |
| NP | Cy5-labeled p(HEMA-ran-GMA) nanoparticle |
| oxATP | Oxidized adenosine triphosphate |
| PBS | Phosphate buffered saline |
| PDI | Polydispersity index |
| PEG | Polyethylene glycol |
| ROI | Region of interest |
| S.D. | Standard deviation |
| SEM | Standard error of measurement |
| TCEP | Tris(2-carboxyethyl)phosphine |
| TEM | Transmission electron microscopy |
| Tf | Transferrin |
| Tf-NP | Transferrin-functionalized Cy5-labeled p(HEMA-ran-GMA) nanoparticle |
| YM872 | Zonampanel monohydrate |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Health and Medical Research Council (NHMRC), Australia (APP1082403). MF was supported by an NHMRC Career Development Fellowship (APP1087114).References
- M. D. Norenberg, J. Smith and A. Marcillo, J. Neurotrauma, 2004, 21, 429–440 CrossRef PubMed.
- O. Farkas and J. T. Povlishock, Prog. Brain Res., 2007, 161, 43–59 CAS.
- V. L. Arvanian, L. Schnell, L. Lou, R. Golshani, A. Hunanyan, A. Ghosh, D. D. Pearse, J. K. Robinson, M. E. Schwab and J. W. Fawcett, Exp. Neurol., 2009, 216, 471–480 CrossRef PubMed.
- H. Levkovitch-Verbin, H. A. Quigley, K. R. Martin, D. J. Zack, M. E. Pease and D. F. Valenta, Invest. Ophthalmol. Visual Sci., 2003, 44, 3388–3393 CrossRef PubMed.
- M. Fitzgerald, C. A. Bartlett, A. R. Harvey and S. A. Dunlop, J. Neurotrauma, 2010, 27, 439–452 CrossRef PubMed.
- C. A. Oyinbo, Acta Neurobiol. Exp., 2011, 71, 281–299 Search PubMed.
- T. I. Peng and M. J. Jou, Ann. N. Y. Acad. Sci., 2010, 1201, 183–188 CrossRef CAS PubMed.
- A. Lau and M. Tymianski, Pflüg. Arch., 2010, 460, 525–542 CrossRef CAS PubMed.
- J. Wells, M. R. Kilburn, J. A. Shaw, C. A. Bartlett, A. R. Harvey, S. A. Dunlop and M. Fitzgerald, J. Neurosci. Res., 2012, 90, 606–618 CrossRef CAS PubMed.
- J. Knöferle, J. C. Koch, T. Ostendorf, U. Michel, V. Planchamp, P. Vutova, L. Tönges, C. Stadelmann, W. Brück and M. Bähr, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6064–6069 CrossRef PubMed.
- D. Pitt, E. Gonzales, A. H. Cross and M. P. Goldberg, Brain Res., 2010, 1309, 146–154 CrossRef CAS PubMed.
- C. Matute, I. Torre, F. Pérez-Cerdá, A. Pérez-Samartín, E. Alberdi, E. Etxebarria, A. M. Arranz, R. Ravid, A. Rodríguez-Antigüedad and M. Sánchez-Gómez, J. Neurosci., 2007, 27, 9525–9533 CrossRef CAS PubMed.
- J. T. Weber, Front. Pharmacol., 2012, 3, 60 Search PubMed.
- R. L. O'Hare Doig, W. Chiha, M. K. Giacci, N. J. Yates, C. A. Bartlett, N. M. Smith, S. I. Hodgetts, A. R. Harvey and M. Fitzgerald, BMC Neurosci., 2017, 18, 62 CrossRef PubMed.
- D. L. Savigni, R. L. H. Doig, C. R. Szymanski, C. A. Bartlett, I. Lozić, N. M. Smith and M. Fitzgerald, Neuropharmacology, 2013, 75, 380–390 CrossRef CAS PubMed.
- M. Fitzgerald, S. C. Payne, C. A. Bartlett, L. Evill, A. R. Harvey and S. A. Dunlop, Invest. Ophthalmol. Visual Sci., 2009, 50, 5456–5462 CrossRef PubMed.
- Y. Mao, A. M. B. Black, H. R. Milbourn, S. Krakonja, M. Nesbit, C. A. Bartlett, B. Fehily, R. Takechi, N. J. Yates and M. Fitzgerald, Int. J. Mol. Sci., 2018, 19, 3408 CrossRef PubMed.
- D. Shlosberg, M. Benifla, D. Kaufer and A. Friedman, Nat. Rev. Neurol., 2010, 6, 393–403 CrossRef CAS PubMed.
- T. Patel, J. Zhou, J. M. Piepmeier and W. M. Saltzman, Adv. Drug Delivery Rev., 2012, 64, 701–705 CrossRef CAS PubMed.
- J. Kreuter, Adv. Drug Delivery Rev., 2014, 71, 2–14 CrossRef CAS PubMed.
- J. Chang, Y. Jallouli, M. Kroubi, X.-b. Yuan, W. Feng, C.-s. Kang, P.-y. Pu and D. Betbeder, Int. J. Pharm., 2009, 379, 285–292 CrossRef CAS PubMed.
- C. W. Gan and S.-S. Feng, Biomaterials, 2010, 31, 7748–7757 CrossRef CAS PubMed.
- C. C. Visser, S. Stevanović, L. Heleen Voorwinden, P. J. Gaillard, D. J. Crommelin, M. Danhof and A. G. de Boer, J. Drug Targeting, 2004, 12, 145–150 CrossRef CAS PubMed.
- J. Chang, A. Paillard, C. Passirani, M. Morille, J.-P. Benoit, D. Betbeder and E. Garcion, Pharm. Res., 2012, 29, 1495–1505 CrossRef CAS PubMed.
- M. Nag, V. Gajbhiye, P. Kesharwani and N. K. Jain, Colloids Surf., B, 2016, 148, 363–370 CrossRef CAS PubMed.
- G. M. Barratt, Pharm. Sci. Technol. Today, 2000, 3, 163–171 CrossRef CAS PubMed.
- R. Singh and J. W. Lillard Jr, Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- J. Panyam, D. Williams, A. Dash, D. Leslie-Pelecky and V. Labhasetwar, J. Pharm. Sci., 2004, 93, 1804–1814 CrossRef CAS PubMed.
- R. F. Service, Science, 2010, 330, 314 CrossRef CAS PubMed.
- W. M. Pardridge, Drug Discovery Today, 2007, 12, 54–61 CrossRef CAS PubMed.
- P. C. Ke, S. Lin, W. J. Parak, T. P. Davis and F. Caruso, ACS Nano, 2017, 11, 11773–11776 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi and C. Reinhardt, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- I. Lynch, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2009, 4, 546–547 CrossRef CAS PubMed.
- M. Mahmoudi, N. Bertrand, H. Zope and O. C. Farokhzad, Nano Today, 2016, 11, 817–832 CrossRef CAS.
- S. Khan, A. Gupta and C. K. Nandi, J. Phys. Chem. Lett., 2013, 4, 3747–3752 CrossRef CAS.
- S. Behzadi, V. Serpooshan, R. Sakhtianchi, B. Müller, K. Landfester, D. Crespy and M. Mahmoudi, Colloids Surf., B, 2014, 123, 143–149 CrossRef CAS PubMed.
- S. Ritz, S. Schöttler, N. Kotman, G. Baier, A. Musyanovych, J. r. Kuharev, K. Landfester, H. r. Schild, O. Jahn and S. Tenzer, Biomacromolecules, 2015, 16, 1311–1321 CrossRef CAS.
- C. Corbo, R. Molinaro, A. Parodi, N. E. ` Toledano Furman, F. Salvatore and E. Tasciotti, Nanomedicine, 2016, 11, 81–100 CrossRef CAS PubMed.
- D. Docter, U. Distler, W. Storck, J. Kuharev, D. Wünsch, A. Hahlbrock, S. K. Knauer, S. Tenzer and R. H. Stauber, Nat. Protoc., 2014, 9, 2030–2044 CrossRef CAS PubMed.
- P. Jain, R. Pawar, R. Pandey, J. Madan, S. Pawar, P. Lakshmi and M. Sudheesh, Biotechnol. Adv., 2017, 35, 889–904 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- P. S. R. Naidu, M. Norret, S. A. Dunlop, M. Fitzgerald, T. D. Clemons and K. S. Iyer, ACS Omega, 2019, 4, 17083–17089 CrossRef CAS PubMed.
- D. R. Hristov, L. Rocks, P. M. Kelly, S. S. Thomas, A. S. Pitek, P. Verderio, E. Mahon and K. A. Dawson, Sci. Rep., 2015, 5, 17040 CrossRef CAS PubMed.
- R. L. H. Doig, C. A. Bartlett, N. M. Smith, S. I. Hodgetts, S. A. Dunlop, L. Hool and M. Fitzgerald, Neuroscience, 2016, 339, 450–462 CrossRef PubMed.
- M. Takahashi, A. Kohara, J. i. Shishikura, S. Kawasaki-Yatsugi, J. W. Ni, S. i. Yatsugi, S. Sakamoto, M. Okada, M. Shimizu-Sasamata and T. Yamaguchi, CNS Drug Rev., 2002, 8, 337–352 CrossRef CAS PubMed.
- Sigma-Aldrich, Adenosine 5′-triphosphate, periodate oxidized sodium salt – Product Specification, USA, 2014 Search PubMed.
- H. Hara, T. Morita, T. Sukamoto and F. M. Cutrer, CNS Drug Rev., 1995, 1, 204–226 CrossRef CAS.
- T. Hibino, M. Yuzurihara, K. Terawaki, H. Kanno, Y. Kase and A. Takeda, J. Pharmacol. Sci., 2008, 108, 89–94 CrossRef CAS PubMed.
- R. Chouhan and A. Bajpai, J. Mater. Sci.: Mater. Med., 2009, 20, 1103–1114 CrossRef CAS PubMed.
- R. Chouhan and A. Bajpai, J. Nanobiotechnol., 2009, 7, 5 CrossRef PubMed.
- H. Chen, S. You, Q. Cai, Y. Zheng, L. Zhang, J. Shen and M. Yin, J. Mater. Chem. B, 2019, 7, 1875–1881 RSC.
- M. Gaumet, A. Vargas, R. Gurny and F. Delie, Eur. J. Pharm. Biopharm., 2008, 69, 1–9 CrossRef CAS PubMed.
- D. H. Jo, J. H. Kim, T. G. Lee and J. H. Kim, Nanomedicine, 2015, 11, 1603–1611 CrossRef CAS PubMed.
- X. Duan and Y. Li, Small, 2013, 9, 1521–1532 CrossRef CAS PubMed.
- R. G. Thorne and C. Nicholson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5567–5572 CrossRef CAS PubMed.
- S. Wohlfart, S. Gelperina and J. Kreuter, J. Controlled Release, 2012, 161, 264–273 CrossRef CAS PubMed.
- E. A. Nance, G. F. Woodworth, K. A. Sailor, T.-Y. Shih, Q. Xu, G. Swaminathan, D. Xiang, C. Eberhart and J. Hanes, Sci. Transl. Med., 2012, 4, 149ra119 Search PubMed.
- R. Langer, Nature, 1998, 392, 5–10 CAS.
- S. Hattori, J. Andrade, J. Hibbs Jr, D. Gregonis and R. King, J. Colloid Interface Sci., 1985, 104, 72–78 CrossRef CAS.
- D. T. Wiley, P. Webster, A. Gale and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8662–8667 CrossRef CAS PubMed.
- M. Bramini, D. Ye, A. Hallerbach, M. Nic Raghnaill, A. Salvati, C. Åberg and K. A. Dawson, ACS Nano, 2014, 8, 4304–4312 CrossRef CAS PubMed.
- C. H. J. Choi, C. A. Alabi, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1235–1240 CrossRef CAS PubMed.
- C. Saraiva, C. Praça, R. Ferreira, T. Santos, L. Ferreira and L. Bernardino, J. Controlled Release, 2016, 235, 34–47 CrossRef CAS PubMed.
- H. Neumann, M. Kotter and R. Franklin, Brain, 2008, 132, 288–295 CrossRef PubMed.
- J. Rytkönen, R. Miettinen, M. Kaasalainen, V.-P. Lehto, J. Salonen and A. Närvänen, J. Nanomater., 2012, 2012, 2 Search PubMed.
- C. Bharti, U. Nagaich, A. K. Pal and N. Gulati, Int. J. Pharm. Invest., 2015, 5, 124 CrossRef CAS PubMed.
- W. Abdelwahed, G. Degobert, S. Stainmesse and H. Fessi, Adv. Drug Delivery Rev., 2006, 58, 1688–1713 CrossRef CAS PubMed.
- S. Fedoroff and A. Richardson, Protocols for neural cell culture, Springer Science & Business Media, 2008 Search PubMed.
- J. Y. Oh, H. S. Kim, L. Palanikumar, E. M. Go, B. Jana, S. A. Park, H. Y. Kim, K. Kim, J. K. Seo and S. K. Kwak, Nat. Commun., 2018, 9, 4548 CrossRef PubMed.
- C. Gräfe, A. Weidner, M. vd Lühe, C. Bergemann, F. H. Schacher, J. H. Clement and S. Dutz, Int. J. Biochem. Cell Biol., 2016, 75, 196–202 CrossRef PubMed.
- P. S. R. Naidu, M. Norret, N. M. Smith, S. A. Dunlop, N. L. Taylor, M. Fitzgerald and K. S. Iyer, Langmuir, 2017, 33, 12926–12933 CrossRef CAS PubMed.
- R. J. Lee, S. Wang and P. S. Low, Biochim. Biophys. Acta, Mol. Cell Res., 1996, 1312, 237–242 CrossRef.
- L. Ferreira, M. Vidal and M. Gil, Int. J. Pharm., 2000, 194, 169–180 CrossRef CAS PubMed.
- J. Siepmann and N. A. Peppas, Int. J. Pharm., 2011, 418, 6–12 CrossRef CAS PubMed.
- J. M. Unagolla and A. C. Jayasuriya, Eur. J. Pharm. Sci., 2018, 114, 199–209 CrossRef CAS PubMed.
- R. Zhao, D. Liang and D. Sun, PLoS One, 2016, 11, e0155953 CrossRef PubMed.
- P. Di Bernardo, P. Zanonato, S. Tamburini, P. Tomasin and P. Vigato, Dalton Trans., 2006, 4711–4721 RSC.
- S. Zanganeh, R. Spitler, M. Erfanzadeh, A. M. Alkilany and M. Mahmoudi, Int. J. Biochem. Cell Biol., 2016, 75, 143–147 CrossRef CAS PubMed.
- A. Hinna, F. Steiniger, S. Hupfeld, M. Brandl and J. Kuntsche, Anal. Bioanal. Chem., 2014, 406, 7827–7839 CrossRef CAS PubMed.
- C. Decker, F. Steiniger and A. Fahr, J. Liposome Res., 2013, 23, 154–165 CrossRef CAS PubMed.
- E. Roese and H. Bunjes, J. Controlled Release, 2017, 256, 92–100 CrossRef CAS PubMed.
- P. Oh, P. Borgström, H. Witkiewicz, Y. Li, B. J. Borgström, A. Chrastina, K. Iwata, K. R. Zinn, R. Baldwin and J. E. Testa, Nat. Biotechnol., 2007, 25, 327–337 CrossRef CAS PubMed.
- S.-D. Li and L. Huang, J. Controlled Release, 2010, 145, 178–181 CrossRef CAS PubMed.
- G. Caracciolo, O. C. Farokhzad and M. Mahmoudi, Trends Biotechnol., 2017, 35, 257–264 CrossRef CAS PubMed.
- S. Y. Khor, M. N. Vu, E. H. Pilkington, A. P. Johnston, M. R. Whittaker, J. F. Quinn, N. P. Truong and T. P. Davis, Small, 2018, 14, 1801702 CrossRef PubMed.
- S. Palchetti, V. Colapicchioni, L. Digiacomo, G. Caracciolo, D. Pozzi, A. L. Capriotti, G. La Barbera and A. Laganà, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 189–196 CrossRef CAS PubMed.
- S. Palchetti, D. Pozzi, A. L. Capriotti, G. La Barbera, R. Z. Chiozzi, L. Digiacomo, G. Peruzzi, G. Caracciolo and A. Laganà, Colloids Surf., B, 2017, 153, 263–271 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09523c |
| This journal is © The Royal Society of Chemistry 2020 |