
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-enzymatically crosslinked and injectable hyaluronic acid hydrogels for potential application in tissue engineering†
Luyu Wang,
Jinrui Li,
Dan Zhang ,
Shanshan Ma,
Junni Zhang,
Feng Gao,
Fangxia Guan* and
Minghao Yao*
,
Shanshan Ma,
Junni Zhang,
Feng Gao,
Fangxia Guan* and
Minghao Yao*
School of Life Science, Zhengzhou University, 100 Science Road, Zhengzhou 450001, P. R. China. E-mail: yao453343550@126.com; guanfangxia@126.com
First published on 15th January 2020
Abstract
Recently, in situ formed injectable hydrogels have shown great potential in biomedical applications as therapeutic implants or carriers in tissue repair and regeneration. They can seal or fill the damaged tissue to function as cell/drug delivery vehicle perfectly through a minimally invasive surgical procedure. In this study, hyaluronic acid (HA) is functionalized with tyramine to produce an injectable hydrogel dual-enzymatically crosslinked by horseradish peroxidase (HRP) and galactose oxidase (GalOX). This new tyramine-modified HA (HT) hydrogel exhibited good injectability, favorable cytocompatibility to mice bone marrow mesenchymal stem cells (BMSCs), and low inflammatory response verified by cytotoxicity assay in vitro and an in situ subcutaneous injection study in vivo. In addition, the gelation time, swelling behavior, and degradation rate of the HT hydrogel could be adjusted through varying the concentrations of HT and GalOX in a certain range. These encouraging results suggest that such biocompatible HT hydrogels might have potential application in three-dimensional stem cell culture and tissue engineering.
1. Introduction
Hydrogels, a three-dimensional hydrophilic polymer-based network, have been extensively explored for three dimensional cell culture and tissue engineering due to their remarkable properties including high water content, high porosity, acceptable biocompatibility, and minimized immune response, which are similar to those of living tissues and the extracellular matrix.1–3 Nowadays, injectable hydrogels have gained significant traction in tissue engineering, as they exhibit greater accessibility to sites of injury and minimal invasiveness with patient discomfort significantly reduced after delivery compared with the pre-fabricated scaffold.4–8As an integral component of the ECM, hyaluronic acid (HA) is involved in many cellular processes including motility, signaling and matrix remodeling.4,9 Along with its favourable properties such as inherent biocompatibility, water adsorption ability and biodegradability, mediated by natural hyaluronidases in the body, HA-based hydrogel has been considered as one of the most suitable biomaterials for a variety of tissue engineering applications.10–14 Despite these outstanding features, its further applicability is still hindered by some shortcomings, such as, low mechanical strength and quick degradation kinetics.
Several strategies have been explored to improve the stability and mechanical strength of HA-based hydrogel by chemical modification.15 For example, methacrylated HA16–18 adipic acid dihydrazide modified HA,19 N-hydroxysuccinimide modified HA,20 phenol-rich HA,21–23 and so on. Especially, the phenol-rich HA hydrogel crosslinked by horseradish peroxidase (HRP) and hydrogen peroxide (H2O2) could be well performed under physiological environment, and therefore has been argued as a superior HA-based injectable hydrogel. For this crosslinking system, HRP catalyzes the crosslinking of phenol-rich polymers by consuming hydrogen peroxide (H2O2) as an oxidizing agent, and the direct addition of H2O2 aqueous solution from an external environment is the most common approach. However, the formation of heterogeneous hydrogel and inevitable cytocompatibility due to initially high localization H2O2 concentration and fast gelation rate limit the further application of this kind hydrogel.24–26 To overcome this drawback, the authors' group has proposed a method of HRP-mediated gelation of phenol-gelatin through “indirectly and slowly” addition of H2O2 generated by galactose oxidase (GalOX).27 The control of gelation rate, homogeneity, and cytocompatibility of hydrogel could be obviously enhanced. However, the phenol-rich HA hydrogel enzymatically crosslinked by HRP and GalOX has not been studied, yet.
Herein, the phenol-rich HA was synthesized between the amino of tyramine and the carboxyl of HA through a general carbodiimide/active ester-mediated coupling reaction. Then HT hydrogel was formed by dual-enzymatically crosslinking of HRP and GalOX in the physiological condition. Furthermore, the gelation time, water content, swelling behavior, degradation rate, mechanical strength, cytotoxicity in vitro and immune response in vivo were investigated by a series of tests.
2. Materials and methods
2.1. Materials
Hyaluronic acid sodium salt (HA) from Streptococcus equi, peroxidase from horseradish (HRP, ≥160 units per mg, solid), tyramine hydrochloride, 2-(N-morpholino)ethanesulfonic acid (MES), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), galactose oxidase (GalOX, ≥30 units per mg, solid), and D-galactose were all purchased from Aladdin (Shanghai, China). Calcein-AM/PI (Live/Dead kit) was obtained from Sigma Aldrich (St. Louis, MO, USA). The dialysis membrane (molecular cutoff = 3500 Da) was purchased from Spectrum Laboratories (Rancho Dominguez, CA). Mice bone mesenchymal stem cells (BMSC) were obtained from the Cell Bank, Type Culture Collection, Chinese Academy of Sciences, Shanghai, China. CCK-8 was obtained from US Everbright, China.2.2. Synthesis and characterization of hyaluronic acid–tyramine
Synthesis of hyaluronic acid–tyramine (HT) has been described in many literatures.28,29 Briefly, HA (0.5 g, 1.25 mmol) was first dissolved in 100 mL of MES (1.07 g, 50 mmol) solution, and EDC (0.48 g, 25 mmol) and NHS (0.545 g, 47 mmol) were added to the solution to activate HA for 30 minutes. Tyramine (1 g, 73 mmol) was then added to the activated HA solution and allowed to react at room temperature for 24 hours. The resulting solution was transferred to a dialysis bag, dialyzed against deionized water for 3 days, filtered and lyophilized to give HT conjugation. HT was characterized by 1H NMR spectroscopy (AS400, OXFORD instruments, U.K.), and the phenol content of the conjugate was quantitatively measured at 275 nm using a UV-Vis spectrophotometer (V-750 UV/vis/NIR, Jasco, Japan).2.3. In situ hydrogel formation and gelation time determination
A precursor solution of HT hydrogel was prepared by dissolving a sample of HT conjugate (0.5%, 1%, or 2% wt) in 50 mmol L−1 D-galactose solution, and then different concentrations of GalOX (0.5, 1, 2 U mL−1) and 1 U mL−1 HRP were added to the solution, and the gelation time was determined using an inverted tube test.2.4. Rheological measurements of HT hydrogels
The rheological behavior of the HT hydrogels was evaluated by detecting their modulus of elasticity using a rheometer platform (Leica DHR2, Germany). The dynamic oscillation scanning frequency ranged from 1 to 100 Hz, and the temperature and strain were set as 37 °C and 1%, respectively.2.5. Water content
The water content of the HT hydrogels was calculated with the following formula: D (%) = [(Ww − Wd)/Ww] × 100, where D denotes the water content of the hydrogels, Ww denotes the wet weight of the hydrogel, and Wd denotes the dried weight after freeze-drying.2.6. Morphological study
The surface and internal morphologies of the HT hydrogel was characterized by scanning electron microscopy (SEM, FEI Quanta200, Netherland) after lyophilizing, breakage, and gold spraying.2.7. Swelling and degradation behavior of HT hydrogel in vitro
The equilibrium water content (EWC) of the formed hydrogels was defined as the weight ratio of water content to the swollen hydrogels.30 All the hydrogels were soaked in PBS (pH 7.4) at 37 °C. Then, the hydrogels were removed from the buffer solution at different time points, placed between two pieces of dried filter paper to remove the excess solution, and then weighed (Ws). The swollen hydrogels were weighted, and the swelling ratio (%) was calculated by the following equation:where Ws is the weight of hydrogel after swollen equilibrium, and Wi is the initial weight of hydrogel. The measurements were repeated with triplicate samples.
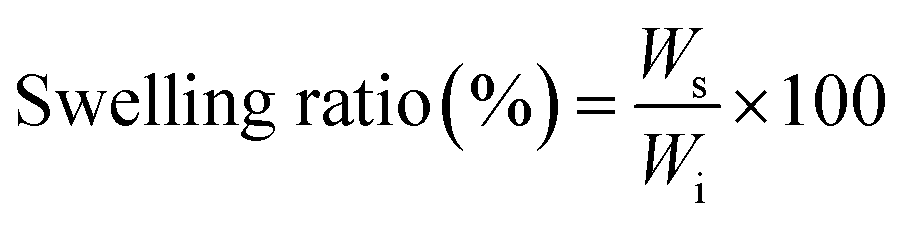
The in vitro degradation behavior of hydrogels was investigated by incubating the preformed hydrogels (100 μL) in PBS (pH 7.4) at 37 °C. At predetermined time intervals, the hydrogels were taken out from the buffer solution, placed between two pieces of dried filter paper to remove the excess solution, and their weights were measured immediately.
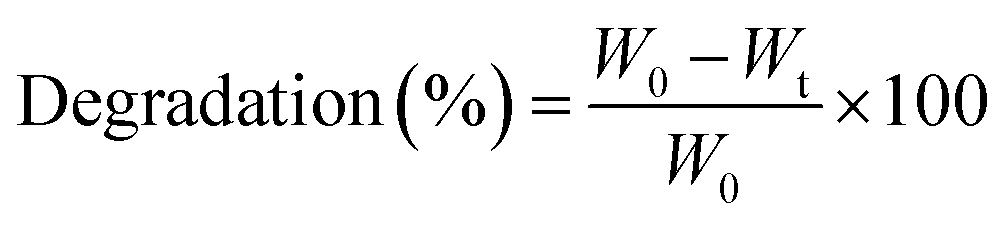
2.8. Three-dimensional culture of BMSCs within HT hydrogel
BMSCs were suspended in the 1% HT solution containing HRP (1 U mL−1) and D-galactose (50 mmol L−1) at a concentration of 1 × 106 cells per mL. Then, GalOX (0.5, 1, and 2 U mL−1) was added to the HT/HRP/D-galactose/BMSCs solution to induce gelation at 37 °C for 15 min. The BMSCs-loaded HT hydrogels were cultured using fresh DMEM/F12 complete medium (with 10% fetal bovine serum) at 37 °C in a humidified atmosphere containing 95% air and 5% CO2.After culturing for 1, 3 and 5 days, the BMSCs-loaded hydrogels were stained with cell Live/Dead kit (Calcein-AM/PI) at 37 °C for 20 min and then observed using fluorescence microscopy (Leica DFC7000T, Germany).
2.9. Biocompatibility and biodegradability in vivo
Animal experiments were performed in strict accordance with the Guide to the Management and Use of Laboratory Animals issued by the National Institutes of Health. The protocol of animal experiments was approved by the Animal Ethics Committee of Zhengzhou University (Zhengzhou, China). C57 mice (male, 6–8 weeks, 22–26 g) were obtained from the Experimental Animal Center of Zhengzhou University.To evaluate the biocompatibility and biodegradability of the HT hydrogel in vivo, 0.1 mL sterilized HT/HRP/GalOX/D-galactose (1% HT, 1 U mL−1 HRP, 2 U mL−1 GalOX, 50 mmol L−1 D-galactose) solution was subcutaneously injected into the dorsum of C57 mice. The solution formed the hydrogel quickly in situ. On day 3, 7, and 14, the surrounding tissues were sectioned and hematoxylin and eosin (HE) staining was used to examine the immune response to the HT hydrogel. In addition, the HT hydrogels were taken out, and the weight and volume of hydrogels were measured.
3. Results and discussion
3.1. Synthesis and characterization of the HT conjugate
The hyaluronic acid–tyramine conjugate (HT) was successfully synthesized in the present of EDC and NHS, and confirmed by 1H NMR and UV-vis spectra analysis (Fig. 1). Synthetic scheme of HT is shown in Fig. 1a. From the 1H NMR spectra data (Fig. 1b), it was clear that the integration value corresponding to phenol groups (6.7–7.1 ppm) of HT was much higher than that of HA (Fig. 1b). In addition, compared to the unmodified HA, the HT conjugate showed UV absorbance in the range with a peak at 275 nm corresponding to the absorbance of tyramine (Fig. 1c). Both results of 1H NMR and UV-vis spectra analysis indicated the successful conjugation of tyramine to HA backbones.23 The standard curve between absorbance of 275 nm and concentration of tyramine was drawn to determine the degree of carboxyl substitution by phenol moiety (Fig. S1†). According to the standard curve and UV-vis spectra analysis of HT, the phenolic contents of these HT derivatives reached 69.29 μmol in 1 g of HT. HT polymers could in situ formed hydrogel enzymatically cross-linked by HRP and H2O2 generated by the oxidase of D-galactose with GalOX. The cross-linking of phenol moieties is through either C–C bonds between ortho-carbons of the aromatic ring or through C–O bonds between ortho-carbons and phenolic oxygen (Fig. 1d).31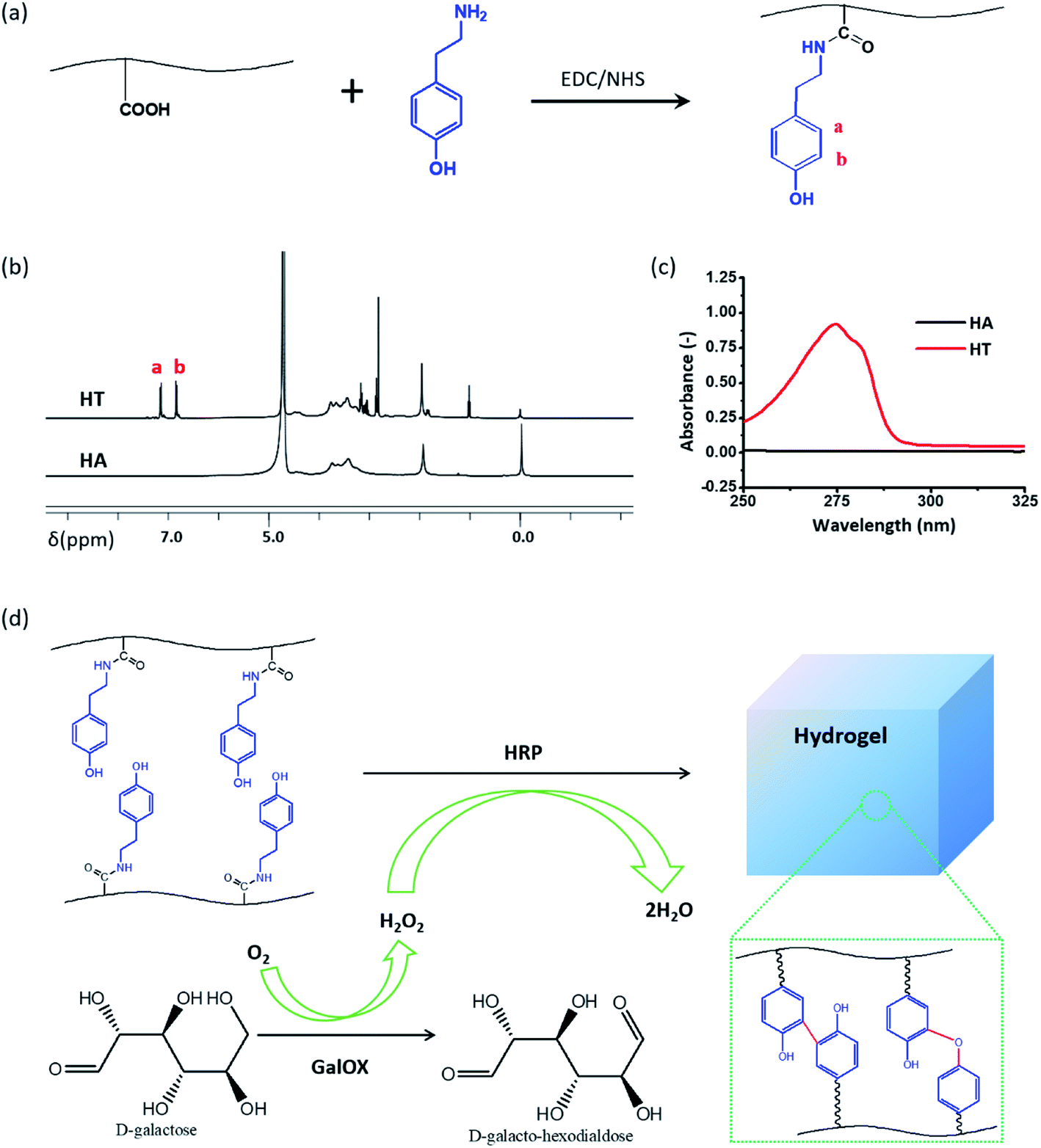 | ||
| Fig. 1 (a) Synthetic scheme of the hyaluronic acid–tyramine (HT) conjugate; (b) 1H NMR spectra of the HT conjugate and unmodified hyaluronic acid; (c) UV-vis spectra of the HT conjugate and unmodified hyaluronic acid; (d) scheme of the HT hydrogel dual-enzymatically cross-linked by HRP and GalOX. | ||
3.2. In situ formation and characterization of GH hydrogel
In situ injectable hydrogel received more and more interests in regenerative medicine and tissue engineering application.32–34 The HT hydrogel dual-enzymatically cross-linked by HRP and GalOX could be in situ fabricated under physiological condition, and therefore possesses a flexible injectability (Fig. 2a). Its shape could be readily controlled by the mold. From the viewpoint of practical, tardy gelation process might cause the encapsulated entity leaking away from injection site as well as initial flowing away of hydrogel components in the injection process. However, immediate gelation might instead lead to the formation of needle blocking, heterogeneous hydrogel and insufficient filling of the tissue defect. Therefore, an applicable gelation time plays an important role in the practical application of hydrogel.35 The effect of HT and GalOX concentrations on gelation rate was studied. The gelation time of 0.5%, 1%, and 2% HT hydrogel crosslinked by 1 U mL−1 HPR and 0.5, 1, or 2 U mL−1 GalOX (aliased as HRP1 UGalOX0.5 U, HRP1 UGalOX1 U, and HRP1 UGalOX2 U hydrogel, respectively) was determined by the inverted tube method. As shown in Fig. 2b, the gelation time of hydrogels ranges from 5 min to 12 min. The content of GalOX has no obvious effect on the gelation rate. The gelation rate of 1% HT hydrogel is the fastest (about 5–6 min), while the gelation rate of 2% HT hydrogel is the slowest (about 10–12 min). Result from Fig. 2c shows that all samples possess high water content exceeding 90%, similar with the natural extracellular matrix. Furthermore, the linear viscoelastic behavior of hydrogels was characterized by oscillatory frequency sweep measurements, and the results are shown in Fig. 2d. The storage modulus of HRP1 UGalOX0.5 U and HRP1 UGalOX1 U hydrogels is about 125 and 200 Pa at the angular frequency of 100 rad s−1. However, when the GalOX content increased to 2 U mL−1, the storage modulus of HRP1 UGalOX2 U hydrogel is still about 200 Pa, close to that of HRP1 UGalOX1 U hydrogel, suggesting that the content of GalOX might be excessive. The micromorphology of HT hydrogel was observed by SEM. This hydrogel possesses a porous three-dimensional network structures, and the internal pore diameter ranges from dozens to hundreds of microns (Fig. 2e). The relatively large and continuous pores may contribute by the high swelling ability of HT hydrogel, which would facilitate the nutrients permeation, oxygen and carbon dioxide exchange, metabolites discharge, and finally ensure the great cellular viability.36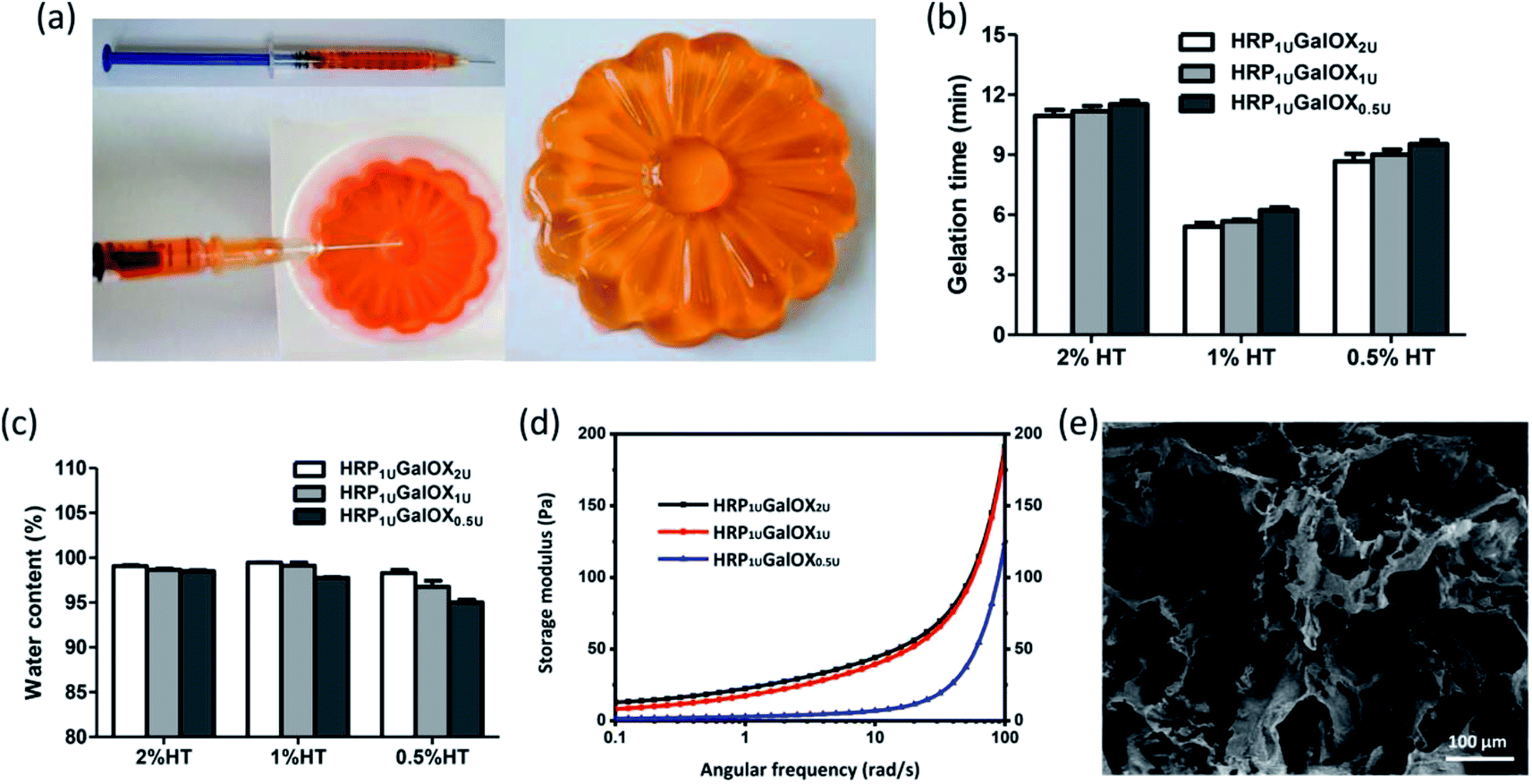 | ||
| Fig. 2 (a) Injectability, (b) gelation time, (c) water content, (d) mechanical property, (e) internal microtopography of HT (0.5, 1, and 2% wt) hydrogel cross-linked by different amount of GalOX (0.5, 1, and 2 U mL−1). 50 mM L−1 D-galactose and 1 U mL−1 HRP. | ||
The good stability and swelling behavior are two important factors for hydrogel applying in three-dimensional cell culture or tissue engineering. Therefore, the degradation and swelling ratio of all sample hydrogels were investigated using the gravimetric method. From Fig. 3a–c, more HT or GalOX concentration contributed to higher stability. And the stability could be controlled from 3 to 28 days by adjusting the HT and GalOX content. The swelling ratio of 1% or 2% HT hydrogel could reach as high as 400% (Fig. 3d), which might benefit the exchange of nutrition and metabolite. In addition, the stability of 1% HT hydrogel of HRP1 UGalOX2 U is the same as that of 2% HT hydrogel of HRP1 UGalOX2 U. The viscosity of 2% HT is so high that it is not suitable for practical operation. Therefore, the authors chose the 1% HT hydrogels of HRP1 UGalOX2 U, HRP1 UGalOX1 U, and HRP1 UGalOX0.5 U for three-dimensional cell culture and 1% HT hydrogel of HRP1 UGalOX2 U for in vivo test subsequently.
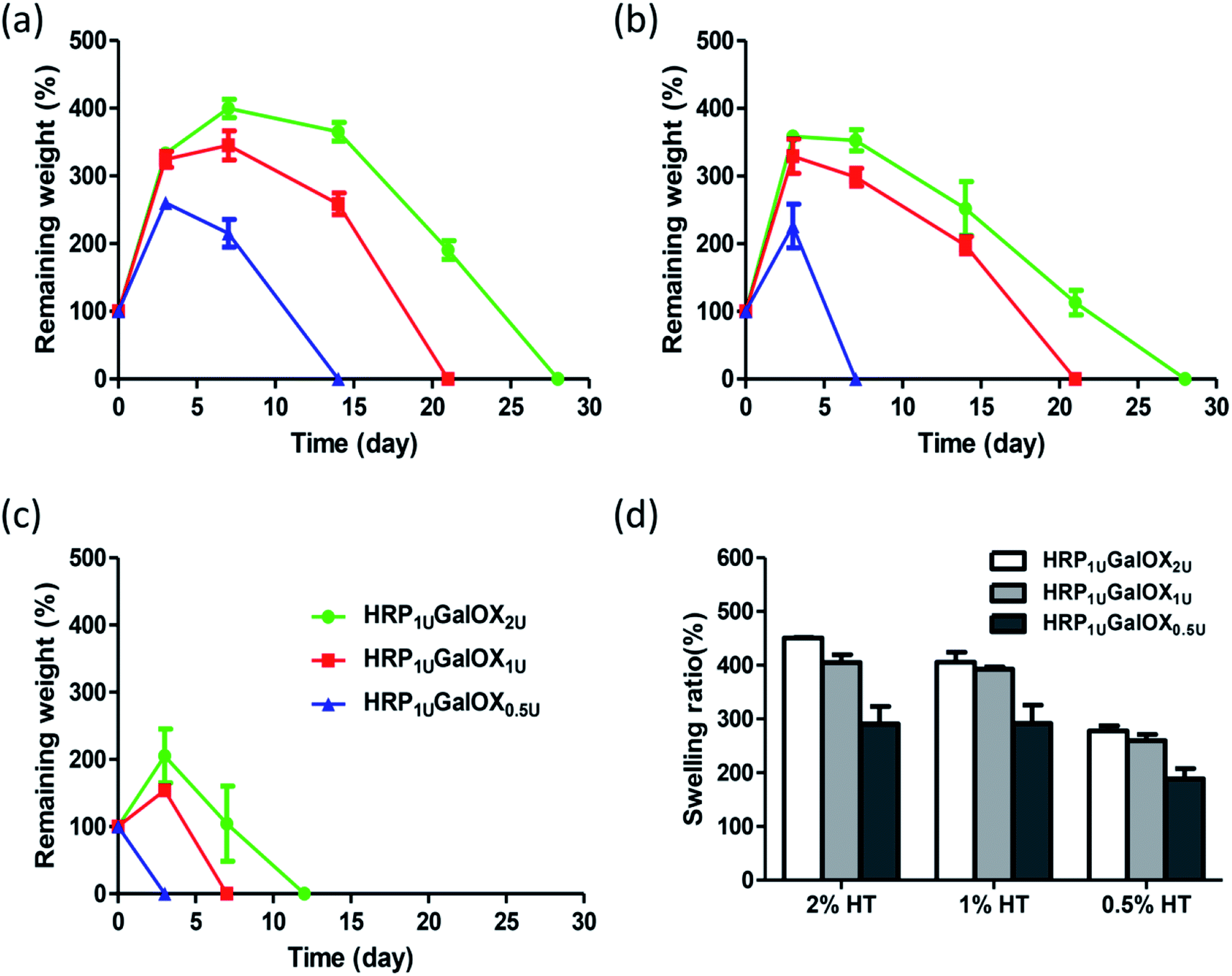 | ||
| Fig. 3 Degradation rate under the conditions of PBS buffer at 37 °C with 2% wt HT (a), 1% HT (b) and 0.5% HT (c) hydrogels; swelling ratio (d). All 2%, 1%, 0.5% hydrogels were crosslinked by 1 U mL−1 HPR and 0.5, 1, or 2 U mL−1 GalOX, and was aliased as HRP1 UGalOX0.5 U, HRP1 UGalOX1 U, and HRP1 UGalOX2 U hydrogel, respectively. | ||
3.3. Cytocompatibility of HT hydrogel in vitro
Biomaterials, application as the scaffold for three-dimensional cell culture or tissue engineering, should possess good biocompatibility.37 The cytocompatibility was firstly assessed using a three-dimensional culture method of BMSCs in vitro for 1, 3, and 5 days. It can be seen clearly from Fig. 4 and S2† that almost all BMSCs encapsulated within HRP1 UGalOX0.5 U, HRP1 UGalOX1 U, and HRP1 UGalOX2 U hydrogels displayed a well viability over 90% within 5 days culture (no significant difference among three kinds of hydrogels). In addition, the magnified fluorescent and bright views of BMSCs culturing within the hydrogels for 1, 3, and 5 days are shown in Fig. S3 and S4.† These data confirm the great cytocompatibility of these hydrogels to BMSCs.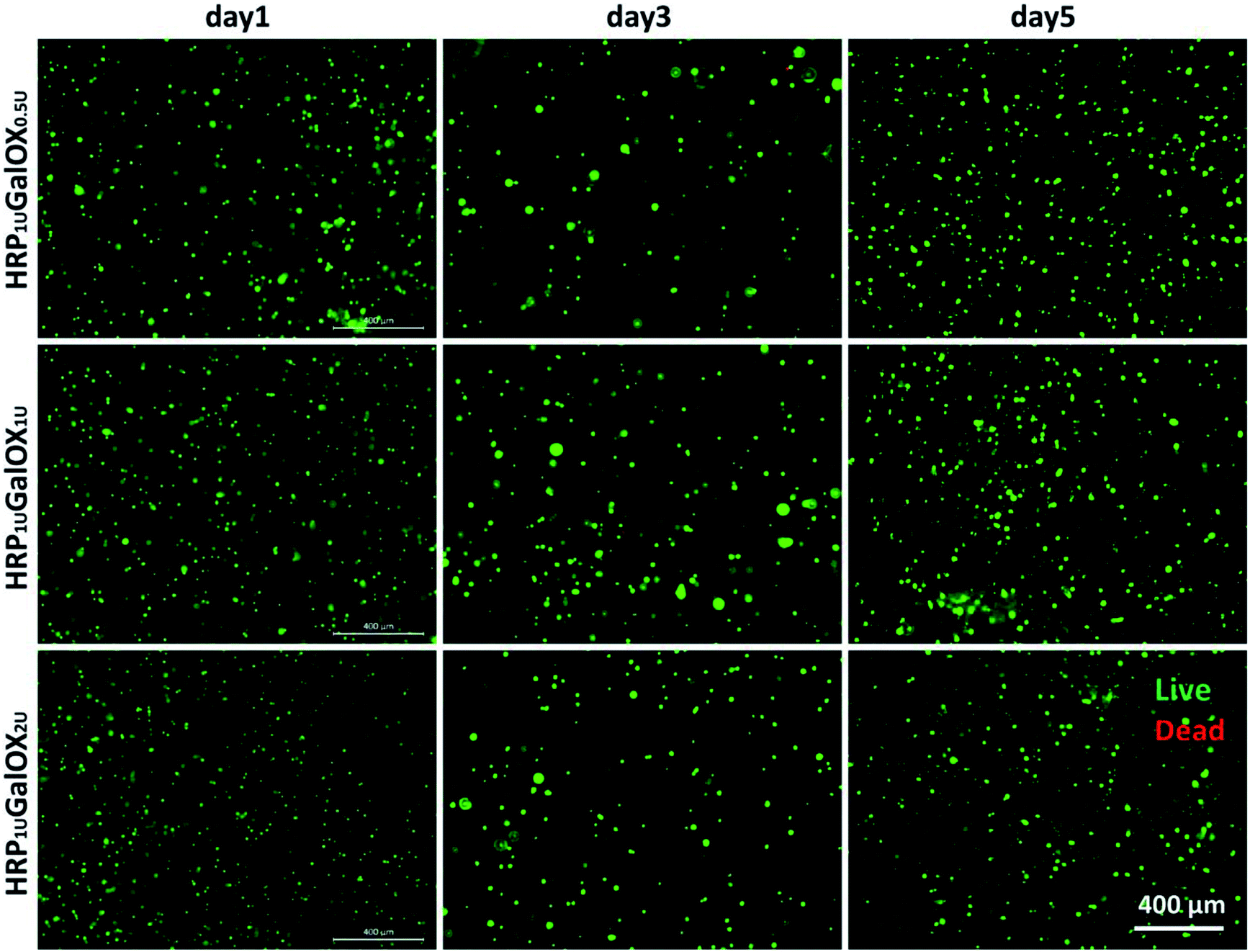 | ||
| Fig. 4 Fluorescent images of BMSCs encapsulated within 1% HT hydrogels of HRP1 UGalOX0.5 U, HRP1 UGalOX1 U and HRP1 UGalOX2 U on day 1, 3, and 5. Green labels the living cells and red labels the dead cells. Scale bar is 400 μm. | ||
3.4. Histocompatibility and biodegradation of HT hydrogel in vivo
Moreover, the biocompatibility of HT hydrogel was evaluated by the HE staining, and the biodegradation was determined via measuring the weight and volume of the remaining hydrogel on day 3, 7, and 14. According to Fig. 5a and b, the weight and volume of remaining hydrogels gradually decreased, and degraded nearly 90% on day 14. From Fig. 5c, barely inflammatory cells were observed in the subcutaneous tissue surrounding hydrogel on day 3, 7, or 14. All results demonstrated that this kind hydrogel possesses a good biocompatibility and biodegradation, which ensures the application in vivo in the future.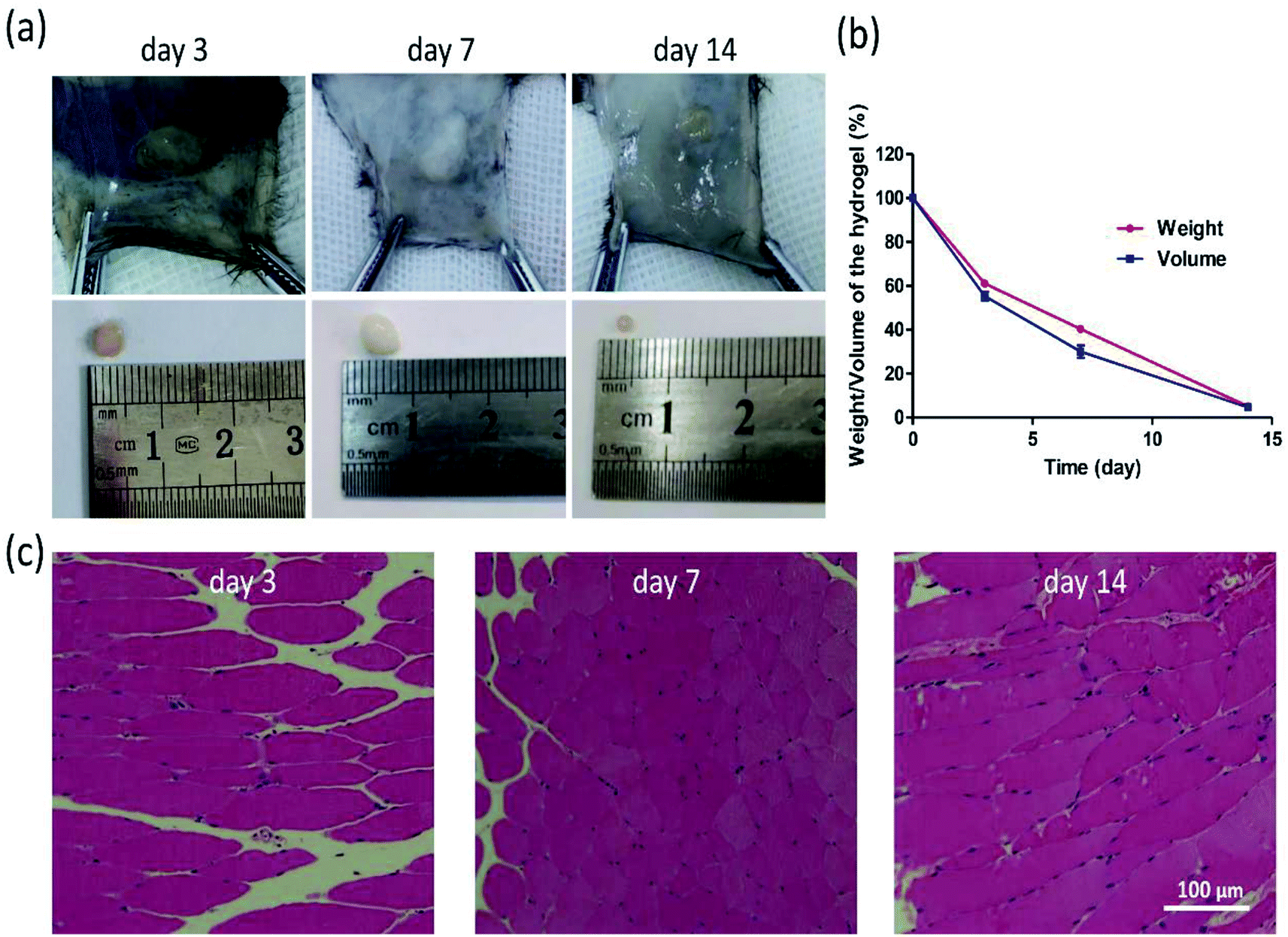 | ||
| Fig. 5 Morphology (a) and degradation rate (b) of hydrogels in C57 mice. HE staining was performed on surrounding tissues on day 3, 7, 14 (c). (Mean ± SD, n = 3). | ||
4. Conclusions
A new HT hydrogel in situ dual-enzymatically cross-linked by HRP and GalOX was developed and characterized. This hydrogel possesses a flexible injectability, quick gelation process within 12 min, adjustable stability, and excellent cytocompatibility to BMSCs and almost no immune response in vivo. Therefore, such injectable HT hydrogel might hold great application potential in three-dimensional stem cell culture and tissue engineering.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the Joint Fund of the National Natural Science Foundation of China and Henan province (U1804198), National Natural Science Foundation of China (31700820), Key Scientific Research Projects of higher education institutions in Henan province (18A180003), the Joint Fund for Fostering Talents of NCIR-MMT & HNKL-MMT (No. MMT2017-04).References
- X. Li, Q. Sun, Q. Li, N. Kawazoe and G. Chen, Front. Chem., 2018, 6, 499 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed.
- K. Liang, K. H. Bae and M. Kurisawa, J. Mater. Chem. B, 2019, 7, 3775–3791 RSC.
- A. Sivashanmugam, R. Arun Kumar, M. Vishnu Priya, S. V. Nair and R. Jayakumar, Eur. Polym. J., 2015, 72, 543–565 CrossRef CAS.
- E. Jooybar, M. J. Abdekhodaie, M. Alvi, A. Mousavi, M. Karperien and P. J. Dijkstra, Acta Biomater., 2019, 83, 233–244 CrossRef CAS PubMed.
- R. Dimatteo, N. J. Darling and T. Segura, Adv. Drug Delivery Rev., 2018, 127, 167–184 CrossRef CAS PubMed.
- L. Teng, Y. Chen, Y.-G. Jia and L. Ren, J. Mater. Chem. B, 2019, 7, 6705–6736 RSC.
- A. Borzacchiello, L. Russo, B. M. Malle, K. Schwach-Abdellaoui and L. Ambrosio, BioMed Res. Int., 2015, 2015 DOI:10.1155/2015/871218.
- J. Shin, J. S. Lee, C. Lee, H. J. Park, K. Yang, Y. Jin, J. H. Ryu, K. S. Hong, S. H. Moon and H. M. Chung, Adv. Funct. Mater., 2015, 25, 3814–3824 CrossRef CAS.
- N. R. Raia, B. P. Partlow, M. McGill, E. P. Kimmerling, C. E. Ghezzi and D. L. Kaplan, Biomaterials, 2017, 131, 58–67 CrossRef CAS PubMed.
- P. Ren, H. Zhang, Z. Dai, F. Ren, Y. Wu, R. Hou, Y. Zhu and J. Fu, J. Mater. Chem. B, 2019, 7, 5490–5501 RSC.
- M. Y. Kwon, C. Wang, J. H. Galarraga, E. Puré, L. Han and J. A. Burdick, Biomaterials, 2019, 222, 119451 CrossRef CAS PubMed.
- S. Tan, A. Yamashita, S. J. Gao and M. Kurisawa, Acta Biomater., 2019, 94, 320–349 CrossRef CAS PubMed.
- W. Hu, Z. Wang, Y. Xiao, S. Zhang and J. Wang, Biomater. Sci., 2019, 7, 843–855 RSC.
- L. Messager, N. Portecop, E. Hachet, V. Lapeyre, I. Pignot-Paintrand, B. Catargi, R. Auzély-Velty and V. Ravaine, J. Mater. Chem. B, 2013, 1, 3369–3379 RSC.
- K. Gwon, E. Kim and G. Tae, Acta Biomater., 2017, 49, 284–295 CrossRef CAS PubMed.
- R. Beninatto, C. Barbera, O. De Lucchi, G. Borsato, E. Serena, C. Guarise, M. Pavan, C. Luni, S. Martewicz and D. Galesso, Mater. Sci. Eng. C, 2019, 96, 625–634 CrossRef CAS PubMed.
- P. Luo, L. Liu, W. Xu, L. Fan and M. Nie, Carbohydr. Polym., 2018, 199, 170–177 CrossRef CAS PubMed.
- A. T. Chan, M. F. Karakas, S. Vakrou, J. Afzal, A. Rittenbach, X. Lin, R. L. Wahl, M. G. Pomper, C. J. Steenbergen and B. M. W. Tsui, Biomaterials, 2015, 73, 1–11 CrossRef CAS PubMed.
- K. Xu, K. Narayanan, F. Lee, K. H. Bae, S. Gao and M. Kurisawa, Acta Biomater., 2015, 24, 159–171 CrossRef CAS PubMed.
- Y. Zhang, H. Chen, T. Zhang, Y. Zan, T. Ni, Y. Cao, J. Wang, M. Liu and R. Pei, Mater. Sci. Eng., C, 2019, 96, 841–849 CrossRef CAS PubMed.
- K. S. Kim, S. J. Park, J. A. Yang, J. H. Jeon, S. H. Bhang, B. S. Kim and S. K. Hahn, Acta Biomater., 2011, 7, 666–674 CrossRef CAS PubMed.
- B. Y. Kim, Y. Lee, J. Y. Son, K. M. Park and K. D. Park, Macromol. Biosci., 2016, 16, 1570–1576 CrossRef CAS PubMed.
- S. Sakai, K. Komatani and M. Taya, RSC Adv., 2012, 2, 1502–1507 RSC.
- M. Yao, F. Gao, R. Xu, J. Zhang, Y. Chen and F. Guan, Biomater. Sci., 2019, 7, 4088–4098 RSC.
- M. Yao, J. Zhang, F. Gao, Y. Chen, S. Ma, K. Zhang, H. Liu and F. Guan, ACS Omega, 2019, 4, 8334–8340 CrossRef CAS PubMed.
- M. Kurisawa, J. E. Chung, Y. Y. Yang, S. J. Gao and H. Uyama, Chem. Commun., 2005, 4312–4314 RSC.
- F. Lee, J. E. Chung and M. Kurisawa, Soft Matter, 2008, 4, 880–887 RSC.
- L. Li, C. Lu, L. Wang, M. Chen, J. White, X. Hao, K. M. McLean, H. Chen and T. C. Hughes, ACS Appl. Mater. Interfaces, 2018, 10, 13283–13292 CrossRef CAS PubMed.
- M. Khanmohammadi, M. B. Dastjerdi, A. Ai, A. Ahmadi, A. Godarzi, A. Rahimi and J. Ai, Biomater. Sci., 2018, 6, 1286–1298 RSC.
- X. Tong and F. Yang, Adv. Healthcare Mater., 2018, 7, 1701065 CrossRef PubMed.
- B.-S. Kim and C.-S. Cho, Tissue Eng. Regener. Med., 2018, 15, 511–512 CrossRef CAS PubMed.
- J.-A. Yang, J. Yeom, B. W. Hwang, A. S. Hoffman and S. K. Hahn, Prog. Polym. Sci., 2014, 39, 1973–1986 CrossRef CAS.
- F. Jiang, Z. Tang, Y. Zhang, Y. Ju, H. Gao, N. Sun, F. Liu, P. Gu and W. Zhang, Biomater. Sci., 2019, 7, 2335–2347 RSC.
- M.-H. Yao, J. Yang, J.-T. Song, D.-H. Zhao, M.-S. Du, Y.-D. Zhao and B. Liu, Chem. Commun., 2014, 50, 9405–9408 RSC.
- S. Naahidi, M. Jafari, M. Logan, Y. Wang, Y. Yuan, H. Bae, B. Dixon and P. Chen, Biotechnol. Adv., 2017, 35, 530–544 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09531d |
| This journal is © The Royal Society of Chemistry 2020 |