
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater structure in solution and crystal molecular dynamics simulations compared to protein crystal structures†
Octav Caldararu a,
Majda Misini Ignjatovića,
Esko Oksanenb and
Ulf Ryde*a
a,
Majda Misini Ignjatovića,
Esko Oksanenb and
Ulf Ryde*a
aDepartment of Theoretical Chemistry, Lund University, Chemical Centre, P. O. Box 124, SE-221 00 Lund, Sweden. E-mail: Ulf.Ryde@teokem.lu.se; Fax: +46-46-2228648; Tel: +46-46-2224502
bInstruments Division, European Spallation Source Consortium ESS ERIC, P. O. Box 176, SE-221 00 Lund, Sweden
First published on 26th February 2020
Abstract
The function of proteins is influenced not only by the atomic structure but also by the detailed structure of the solvent surrounding it. Computational studies of protein structure also critically depend on the water structure around the protein. Herein we compare the water structure obtained from molecular dynamics (MD) simulations of galectin-3 in complex with two ligands to crystallographic water molecules observed in the corresponding crystal structures. We computed MD trajectories both in a water box, which mimics a protein in solution, and in a crystallographic unit cell, which mimics a protein in a crystal. The calculations were compared to crystal structures obtained at both cryogenic and room temperature. Two types of analyses of the MD simulations were performed. First, the positions of the crystallographic water molecules were compared to peaks in the MD density after alignment of the protein in each snapshot. The results of this analysis indicate that all simulations reproduce the crystallographic water structure rather poorly. However, if we define the crystallographic water sites based on their distances to nearby protein atoms and follow these sites throughout the simulations, the MD simulations reproduce the crystallographic water sites much better. This shows that the failure of MD simulations to reproduce the water structure around proteins in crystal structures observed both in this and previous studies is caused by the problem of identifying water sites for a flexible and dynamic protein (traditionally done by overlaying the structures). Our local clustering approach solves the problem and shows that the MD simulations reasonably reproduce the water structure observed in crystals. Furthermore, analysis of the crystal MD simulations indicates a few water molecules that are close to unmodeled electron density peaks in the crystal structures, suggesting that crystal MD could be used as a complementary tool for identifying and modelling water in protein crystallography.
Introduction
Protein structural information is essential for understanding the function of proteins, for designing new enzymes with improved catalytic activity and for developing potent new drug molecules that act on specific target proteins. Such information is currently obtained mainly with X-ray diffraction. However, the structure and function are not determined by the protein alone, but also by the surrounding water molecules, through solvation and the hydrophobic effect, as well as by forming specific hydrogen bonds.1 The solvent also greatly affects the dynamics of the protein.2 Therefore, the positions of the water molecules surrounding the protein are of major interest.3Hydrogen atoms are not visible in X-ray crystallography, so only the positions of the oxygen atoms of water molecules can be determined. Furthermore, only ordered water molecules can be modelled in an electron density map, i.e. those water molecules that interact strongly enough with the protein that they occupy the same position in every molecule of the crystal. The remaining solvent molecules in a protein crystal are modelled as a bulk-solvent contribution.4 However, there is no established consensus on the criteria for modelling an ordered water molecule, so it is a somewhat subjective decision of each crystallographer how many explicit water molecules are modelled in each protein structure. This decision is based on manual inspection of difference density peaks in the electron density map.
Biomolecular simulations also critically depend on the correctness of the solvent structure surrounding the protein. Free energies of ligand binding calculated from simulations are especially sensitive to the position of water molecules in the vicinity of the ligand-binding site.5–7 Thus, the ability of molecular dynamics (MD) simulations to recover positions of water molecules determined experimentally, for example from X-ray crystal structures, is an useful indicator of the reliability of the solvent structure in the simulations. MD simulations also indicate how ordered each water molecule is and should therefore be able to identify ordered water molecules that have been missed in the initial model building of crystal structures. Thus, comparing water structures in MD simulations and in crystal structures has two goals: evaluation of the quality of the water structure from MD and prediction of new ordered water molecules in the X-ray crystal structure that fit into the electron density.
Several studies comparing the water structure in MD simulations to crystal structures have been published and most of them show only partial agreement between the simulations and the experimental data. In an early study of pancreatic trypsin inhibitor, only 19% of the crystallographic waters were found within 1 Å of water molecules in a MD simulation of a crystallographic unit cell.8 On the other hand, Higo and Nakasako compared a 1 ns solution MD simulation of lysozyme, finding 60% of the crystallographic waters to be within 1.4 Å of an MD water.9 Another MD study in crystal was carried out by Altan et al. for a mannose-binding protein and 70% of the crystallographic waters were found to be within 1.4 Å of an MD water.10 Rudling et al. managed to reproduce >70% of water molecules in the crystal structures of 12 different proteins within 1 Å.11 However, only water molecules in the binding site of the proteins were studied, which are usually well-ordered. Finally, a recent study by Wall et al.12 showed the best results so far. The authors performed crystal MD simulations of endoglucanase and compared it to its room temperature crystal structure. Their results show that 80% of the crystallographic waters were within 0.5 Å and 98% were within 1.4 Å of a water molecule in a MD simulation with protein atoms restrained to their positions in the crystal structure. However, in unrestrained simulations, the corresponding percentages were reduced to only 25% and 62%. To our best knowledge, no studies have so far used MD simulations to insert new water molecules in the protein crystal structure.
Thus, there is clearly a discrepancy between the solvent structure in MD simulations and X-ray crystal structures. This is not fully unexpected, as traditional MD simulations in a water box do not model the same kind of system as in a crystal structure. The solvent content in a crystal structure is much lower than that employed in a solution MD simulation and crystal contacts between protein units may influence biomolecular solvation. MD simulations in a crystallographic unit cell can be performed to model these crystal effects effectively. Previous studies have demonstrated that crystal MD simulations reproduce the diffraction data better.13–15 Additionally, MD simulations are usually performed at room temperature, whereas most protein crystal structures are collected at cryogenic temperature (100 K), which reduces the dynamics of the atoms. Ideally, the best comparison would be between MD simulations and room temperature crystal structures, but few high-resolution data sets have been collected at room temperature.
In this paper, we compare the water structure of galectin-3C in complex with two ligands in MD simulations performed both in solution and in a crystallographic unit cell with crystal structures obtained both at 100 K and at 298 K. We show that the results depend strongly on how the water molecules are clustered in the MD simulations. Moreover, we show the MD simulations can be used to identify unmodeled peaks in the electron-density map.
Methods
Crystal structures
Galectin-3 is a mammalian β-galactoside binding protein involved in glycoprotein trafficking, signalling, cell adhesion, angiogenesis, macrophage activation and apoptosis.16–20 It has been implicated in inflammation, immunity, cancer development, metastasis and the pathology of Alzheimer's disease.21,22 The C-terminal domain is easily crystallisable with various ligands.23–25 We studied the binding of two diastereomeric ligands, (2R)- and (2S)-2-hydroxy-3-(4-(3-fluorophenyl)-1H-1,2,3-triazol-1-yl)-propyl-2,4,6-tri-O-acetyl-3-deoxy-3-(4-(3-fluorophenyl)-1H-1,2,3-triazol-1-yl)-1-thio-β-D-galactopyranoside, to the C-terminal domain of galectin-3 (galectin-3C). The two ligands will simply be denoted R and S in this article. Coordinates, B-factors, occupancies and reflection data of the complexes collected at 100 K and 298 K, were obtained from the protein data bank (PDB entries 6QGF and 6QGE for the cryo-structures and entries 6RHL and 6RHM for the room-temperature structures of R and S, respectively).25,26 The resolutions of these structures were 1.34 Å and 1.16 Å for the cryo-structures and 1.30 Å and 1.60 Å for the room-temperature structures.The 2mFo–DFc density maps were generated from the existing reflection data using the phenix.maps module.27 The maps were sigma-normalised and shifted to have a mean of zero.
Molecular dynamics simulations
All MD simulations were run with the Amber 14 software suite.28 Two different types of MD simulations were run: normal MD simulation in a periodic octahedral water box and simulations of a single unit cell of the protein crystal. All simulations were started from the X-ray crystal structures of R- and S-galectin-3C determined at 100 K.For the normal MD simulations, each galectin-3C complex was solvated in an octahedral box of water molecules extending at least 10 Å from the protein using the tleap module, so that 4965–5593 water molecules were included in the simulations. All crystal water molecules were kept. The simulations were set up in the same way as in our previous studies of galectin-3C.24,25,29,30 In agreement with neutron structures,31 all Glu and Asp residues were negatively charged and all Lys and Arg residues positively charged, whereas the other residues were assumed to be neutral. The His158 residue was protonated on the ND1 atom, whereas the other three His residues were protonated on the NE2 atom, in accordance with neutron crystal structures, NMR measurements and previous extensive test calculations with MD.31,32 This resulted in a net charge of +4 for the protein. No counter ions were used in the simulations.
The proteins were described by the Amber ff14SB force field33 and water molecules with the TIP4P-Ewald model.34 The ligands were treated with the general Amber force field with restrained electrostatic potential charges,35 which have been presented before.25 For each complex, the structures were minimised for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps, followed by 20 ps constant-volume equilibration and 20 ps constant-pressure equilibration, all performed with heavy non-water atoms restrained towards the starting structure with a force constant of 209 kJ mol−1 Å−2 (50 kcal mol−1 Å−2). Finally, the system was equilibrated for 2 ns without any restraints and with constant pressure, followed by 10 ns of production simulation, during which coordinates were saved every 5 or 10 ps. For each protein–ligand complex, 10 independent simulations were run, employing different solvation boxes and starting velocities.36 Consequently, the total simulation time for each complex was 100 ns.
000 steps, followed by 20 ps constant-volume equilibration and 20 ps constant-pressure equilibration, all performed with heavy non-water atoms restrained towards the starting structure with a force constant of 209 kJ mol−1 Å−2 (50 kcal mol−1 Å−2). Finally, the system was equilibrated for 2 ns without any restraints and with constant pressure, followed by 10 ns of production simulation, during which coordinates were saved every 5 or 10 ps. For each protein–ligand complex, 10 independent simulations were run, employing different solvation boxes and starting velocities.36 Consequently, the total simulation time for each complex was 100 ns.
All bonds involving hydrogen atoms were constrained to the equilibrium value using the SHAKE algorithm,42 allowing for a time step of 2 fs. The temperature was kept constant at 300 K using Langevin dynamic,43 with a collision frequency of 2 ps−1. The pressure was kept constant at 1 atm using a weak-coupling isotropic algorithm44 with a relaxation time of 1 ps. Long-range electrostatics were handled by particle-mesh Ewald summation45 with a fourth-order B spline interpolation and a tolerance of 10−5. The cut-off radius for Lennard-Jones interactions between atoms of neighbouring boxes was set to 8 Å.
The MD simulations in crystal unit cells were set up using the Amber XtalUtilities package, with the unit cell size extracted from the CRYST1 record in the PDB files. One unit cell contained four protein monomers, resulting in four protein monomers simulated for the simulations. All crystal water molecules were kept in the simulations. Seven Na+ and eleven Cl− counter ions were added to match the 0.4 M ionic strength used in the crystallographic experiments. Water molecules were added successively to the existing crystallographic water molecules until all empty space in the unit cell was filled. As this is difficult to evaluate visually, multiple starting structures with 350, 400, 450 and 500 added water molecules per unit cell were tested in the equilibration step. The simulation containing 500 water molecules kept the volume of the system closest to the unit cell volume and was used for the production runs. Fig. S1† shows that the size of the box was stable during the simulation. The same protocol as in the normal MD simulation was used, resulting in 100 ns (10 × 10) of simulation time for each galectin-3C–ligand complex.
It has been repeatedly shown that MD simulations tend to stay close to the starting structure and that it is more effective to run many short simulations rather than a single long simulation.37–41 However, we have also performed one 100 ns simulation for each simulated system. 100 ns is long compared to the average residence time of the water molecules in the crystallographic water sites, 52–84 ps (Table S1†) and to the water relaxation time-scales estimated in ref. 10, 15 and 180 ps. Fig. S2† shows that the RMSD of the protein atoms are reasonably stable in the various simulations. The RMSD shows a slight increase in the 100 ns solution MD simulation of R-galectin-3C and in the crystal MD simulation of S-galectin-3C. However, the water structure in the simulations is similar, as shown by the results in Tables 1 and S2.†
| MD | Crystal | 1.0 | 1.5 | 2.0 | 2.5 | 3.0 |
|---|---|---|---|---|---|---|
| R-Galectin-3C | ||||||
| Crystal | Cryo | 40 (19%) | 119 (57%) | 180 (86%) | 196 (93%) | 204 (97%) |
| Solution | Cryo | 34 (16%) | 94 (45%) | 163 (78%) | 186 (89%) | 192 (92%) |
| Crystal | RT | 30 (31%) | 64 (66%) | 90 (93%) | 97 (100%) | 97 (100%) |
| Solution | RT | 18 (19%) | 61 (63%) | 81 (83%) | 89 (92%) | 89 (92%) |
|
||||||
| S-Galectin-3C | ||||||
| Crystal | Cryo | 65 (31%) | 120 (57%) | 170 (81%) | 200 (95%) | 202 (96%) |
| Solution | Cryo | 50 (24%) | 114 (54%) | 163 (78%) | 192 (91%) | 201 (96%) |
| Crystal | RT | 27 (35%) | 55 (71%) | 65 (83%) | 71 (91%) | 72 (92%) |
| Solution | RT | 17 (22%) | 48 (62%) | 54 (69%) | 65 (83%) | 69 (88%) |
Comparison of MD water to crystallographic water
The aim of the present investigation is to study how well the MD simulations reproduce water molecules in crystal structures. As in most of the previous studies,8,9,11,12 the comparison is restricted to the well-ordered water molecules reported in the crystal structures. The crystallographic water sites were defined in two separate ways. First, a water site was defined by its coordinates in Cartesian space. To compare the MD water molecules to these water sites, a grid-based analysis was employed: For each 10 ns MD simulation, the water molecule density (only the O atoms) was calculated using the grid command in the cpptraj module of AmberTools.46 For the crystal MD simulations, the whole unit cell was used as grid dimensions with the centre in the centre of the unit cell. For the solution MD simulations, a grid with the centre in the centre of mass of the protein and extending 5 Å away from the protein on each side was used. The default spacing of 0.5 Å was used.The resulting grid files from the 10 simulations were added using the GistPP program47 resulting in a consensus density for the whole 100 ns simulation. Peaks were found in the grid density files using a local script, ensuring a minimum distance of 1.5 Å between peaks. The minimum density for a water peak was considered as one standard deviation (1σ) from the mean in each density grid (calculated considering the solvent density only) and the voxels with the maximum density within 1.5 Å in all direction were chosen as peaks. The minimum density of 1σ corresponds to 5 e− Å−3 for the crystal MD simulations and 1 e− Å−3 for the solution MD simulations, because the water density in the crystal MD simulations is much more well-defined than in the solution MD simulations (discussed further below).
The minimum distance between each crystallographic water in the four crystal structures and the corresponding MD water was calculated after alignment of all heavy protein atoms in the MD structure to the crystal structure, using a PyMol48 script. For crystal MD simulations, this included symmetry-related waters in other protein units. We studied the percentage of crystal water molecules having a close MD water peak at a defined distance threshold of 1.0, 1.5, 2.0, 2.5 or 3.0 Å. Following previous studies, this will be termed the water recall statistics.8–12 We also studied the fraction of MD water peaks that have a corresponding crystal water at the same distance thresholds. As in previous studies, this is defined as the water prediction statistic.
MD water peaks that had no corresponding crystal water within 3.0 Å were identified and the density at their positions was calculated in both cryo- and room-temperature 2mFo–DFc density maps. Water molecules that had an electron density ≥ 1.0 e− Å−3 (equivalent to 1.0σ) were kept and visually inspected to ensure that no clashes occur with the protein.
Second, we also used a local approach to identify water clusters in the MD simulations. In this, each crystal-water site was defined by the distances between the O atom of a crystallographic water molecule and its three closest heavy atoms in the protein or ligand (if any of the three Xi–O–Xj angles was <10° or >170°, instead the fourth or fifth closest atoms were used, to avoid linear dependences). The position in space defined by these three distances was then followed in the MD simulation. The closest MD water molecule to the water site was recorded in each of the 1000 snapshots of an independent 10 ns simulation, and the resulting 1000 water molecules were then clustered based on the three distances with a hierarchical agglomerative algorithm, using the average distance within each cluster and stopping the clustering when the distance between the closest clusters was above 1.5 Å. The distance between the crystal-water site and the centre of the largest cluster was recorded and also if there was a cluster with a shorter distance and at least 16% of the water molecules (i.e. significantly larger than the bulk density). This distance was averaged among the 10 simulations for each water molecule. The recall statistic was defined as for the grid-based analysis. No prediction statistic was computed for this method as only clusters of waters close to the crystallographic waters were considered.
To ensure that the differences observed for the two approaches is not caused by the fact that the first is based electron-density grids, whereas the second is based on clustering of nearest-neighbour distances, we also implemented a global clustering approach, which is identical to the second approach. The only difference is that it is based on the aligned Cartesian coordinates of the water molecules, rather than the nearest-neighbour distances. This is called non-grid-based global clustering in the following.
Result and discussion
Crystal and solution MD simulations of R- and S-galectin-3C were performed to evaluate MD water structures against both cryo- and room-temperature crystal structures. 100 ns (10 × 10) of MD simulations were run for each of the four cases. We first describe the results based on a standard global clustering of the MD water molecules, based on Cartesian coordinates after alignment of the snapshots based on the heavy atoms of the protein. Then, we suggest a novel approach to cluster water molecules, based on the local geometry around each crystal water molecule. Finally, we test if it is possible to identify new water molecules in the crystal structures based on the MD simulations.Recall of crystallographic water molecules in the MD simulations based on global clustering
Water peaks in the MD simulations were determined with grid analysis using AmberTools and the positions were compared to the crystallographic water molecules based on the Cartesian coordinates after alignment of the heavy atoms in the protein. In the crystal MD simulations, the density of the water molecules was pronounced and well-defined. In addition, fewer water peaks were found than in the solution MD simulations: 582 for R-galectin-3C and 446 for S-galectin-3C, compared to 701 and 688 water peaks in the solution MD simulations. These results are not surprising, as the atomic motions of the protein and the solvent are expected to be larger in the solution MD, owing to the absence of crystal contacts.The recall of crystallographic water molecules was rather poor for all MD simulations (Table 1). Comparing the crystal MD simulation of R-galectin-3C to the cryo-crystal structure showed that 19% of crystallographic waters had a MD water cluster within 1.0 Å, 57% within 1.5 Å and 93% within 2.5 Å. The results from the single long 100 ns simulation were similar as can be seen in Table S2.† Furthermore, when comparing with the room-temperature structure, the percentages rise to 31% within 1.0 Å, 66% within 1.5 Å and 100% at 2.5 Å. However, it should be noted that the room-temperature structure had only 97 ordered water molecules compared to the 210 water molecules of the cryo-structure. This means that the absolute number of recalled waters is higher for the cryo structure, even though the percentage is lower. The simulations were started from the cryo structure, so the fact that they also reproduce the room-temperature water structure suggests that these water positions are well conserved.
The solution MD simulations of R-galectin-3C gave slightly worse results than the crystal MD simulations: 16% of the water molecules in the cryo-crystal structure were recalled within 1.0 Å, 45% within 1.5 Å and 89% within 2.5 Å. Similarly, for the room-temperature structure, 19% of the water molecules were recalled within 1.0 Å, 63% within 1.5 Å and 92% within 2.5 Å. These results are clearly worse than for the crystal MD simulations but the difference is not very large.
Another difference between the solution MD and the crystal MD is observed when studying clashes between the MD water molecules and the protein atoms from crystal structure. Ten water molecules in the normal MD simulation were within 2.0 Å of a protein atom in the cryo-crystal structure and 12 water molecules clash with protein atoms in the room-temperature crystal structure. In contrast, no water molecules clashed with any of the crystallographic protein atoms in the crystal MD simulation. This highlights once again the higher amount of dynamics of both the solute and the solvent in the solution MD simulations, which can also be observed in the maximum RMSD of the protein atoms in the simulations compared to the starting structure. The solution MD simulations showed a maximum RMSD of 1.58 Å for R-galectin-3C and 1.54 Å for S-galectin-3C, whereas the crystal MD simulations maximum RMSD was only 0.51 Å and 0.52 Å, respectively.
The simulations for S-galectin-3C gave slightly better results: 31% of crystallographic waters were recalled in the crystal MD simulations within 1 Å, 57% within 1.5 Å and 95% within 2.5 Å. As for R-galectin-3C, better results were obtained when comparing the room-temperature structure of S-galectin-3C: 35% of crystallographic waters were recalled in the within 1 Å, 71% within 1.5 Å and 91% within 2.5 Å. Again, the number of ordered water molecules in the room-temperature structure is lower than in the cryo-temperature structure, 78 compared to 224.
As for R-galectin-3C, the solution MD recall of crystallographic waters for S-galectin-3C is worse, but the difference is quite small. 24% of water molecules from the cryo-structure were reproduced within 1 Å, 54% within 1.5 Å and 91% within 2.5 Å. Interestingly, the solution MD simulation of S-galectin-3C reproduces the cryo-structure better than the room-temperature structure, with only 22%, 62% and 83% of water molecules in the room-temperature structure reproduced within 1 Å, 1.5 Å and 2.5 Å. This is the only case among the four simulations where the recall percentage is higher for the cryo-structures.
The ability of MD simulations to reproduce water molecules in the room-temperature structures even though they were started from the cryo-structures is not wholly surprising. The water structure in the deposited room-temperature structure is not too different from the one found in the deposited cryo-structure. For R-galectin-3C, 75% of water molecules in the room-temperature structure have a corresponding crystallographic water molecule within 1.5 Å in the cryo-structure. The percentage is even higher for S-galectin-3C, 83%. In particular, all the room-temperature crystallographic water molecules reproduced by the four MD simulations have a corresponding water molecule in the cryo-structure.
We also checked whether there is any correlation between the height of each peak in the MD simulations and the minimum distance to a crystallographic water. It could be expected that water molecules with higher peaks would stay closer to their original position in the simulations. However, there was essentially no correlation between these values (R2 < 0.02). Likewise, we studied if lower B-factors of a crystallographic water molecule corresponds to a lower distance to an MD water, as a lower B-factor would indicate a smaller amount of dynamics of that water molecule and stronger interactions with the environment. However, the B-factors of the crystallographic water molecules also did not show any correlation to the minimum distance to an MD water (R2 < 0.01).
The prediction scores are shown in Table 2. The number of predicted water molecules is generally larger than the recall scores, showing that in some cases, multiple MD peaks correspond to one crystallographic water. This is especially true for the solution MD peaks at larger distance thresholds, whereas the prediction to recall ratio is closer to unity for crystal MD water peaks. This shows again that in the crystal MD simulations, the water molecules move less and thus give rise to more well-defined peaks. The percentage of MD peaks close to a crystallographic water is rather low in all cases, between 7 and 21% at 1.5 Å. They are naturally lower when comparing to the room-temperature crystallographic water molecules, as there are fewer of them. Increasing the density threshold improves the prediction percentages, but decreases the recall percentages (Fig. S3†), as was also observed in previous studies.10,12
| MD | Crystal | 1.0 | 1.5 | 2.0 | 2.5 | 3.0 |
|---|---|---|---|---|---|---|
| R-Galectin-3C | ||||||
| Crystal | Cryo | 40 (7%) | 123 (21%) | 185 (32%) | 208 (36%) | 233 (40%) |
| Solution | Cryo | 36 (5%) | 104 (15%) | 205 (29%) | 278 (40%) | 357 (51%) |
| Crystal | RT | 30 (5%) | 66 (11%) | 92 (16%) | 102 (18%) | 108 (19%) |
| Solution | RT | 18 (3%) | 64 (9%) | 104 (15%) | 134 (19%) | 157 (22%) |
|
||||||
| S-Galectin-3C | ||||||
| Crystal | Cryo | 65 (15%) | 124 (28%) | 173 (39%) | 210 (47%) | 240 (54%) |
| Solution | Cryo | 52 (8%) | 122 (18%) | 175 (25%) | 214 (31%) | 259 (38%) |
| Crystal | RT | 27 (6%) | 58 (13%) | 69 (15%) | 98 (22%) | 104 (23%) |
| Solution | RT | 17 (2%) | 50 (7%) | 62 (9%) | 93 (13%) | 133 (19%) |
These results show that the MD simulations reproduce the positions of the crystal water molecules rather poorly. These results agree with most previous investigations.8–11 The recall of 43–70% at 1.4 Å obtained here is comparable to the recall scores obtained by Higo and Nakasako,9 60%, or Altan et al.,10 70% at this distance. These values were obtained at a density threshold of ∼0.6 e− Å−3, a lower threshold than used here, which explains why our lower-bound results are worse than in the previous studies.
The poor agreement between MD and crystallography may indicate that there are major differences between the setup of the simulations and the experiment or that the MM force fields employed in the MD simulations are not accurately enough. However, a recent study showed that much better results (98% recall within 1.4 Å) can be obtained if the protein is restrained to stay close to the crystal structure during the MD simulation.12 This may still be because the force field is not accurate enough to give a reliable protein dynamics. However, an alternative interpretation is that it is not a problem of the simulation but rather of the clustering of the water molecules: the positions of the water molecules are typically defined by interactions with the protein (and the ligand), but if the protein is moving significantly, the water molecules will also move, meaning that a clustering based on positions in Cartesian space (even after alignment) may fail to recognize that the moving water molecules belong to the same cluster. Therefore, we decided to redo the investigation using also local clustering approach.
Recall of crystallographic water molecules in the MD simulations based on local clustering
Consequently, we defined the crystallographic water sites by their distances to the three closest heavy atoms in the protein or ligand (rather than to the Cartesian position). Thereby, the sites move with the protein atoms in the MD simulations. This should cancel errors derived from the alignment of the snapshots and the independent movement of individual side chains throughout the simulations. Furthermore, it should show if MD simulations can reproduce the hydrogen bonding network shown in the crystal structure, which also depends on the positions of the protein atoms. Next, we performed a clustering of the closest water molecule in each snapshot of the MD simulation, based on these three distances and studied how close the largest cluster was to the crystal water molecule (still in terms of the three distances) and also if there was another cluster with a significant size (>16% of the snapshots) with a smaller distance.The results of this local clustering are presented in Table 3. It can be seen that the results are much better than for the traditional global clustering. 57% and 48% of crystallographic water sites in the R- and S-galectin-3C cryo-structure are reproduced by the crystal MD simulations within 0.5 Å. In comparison, for the grid-based analysis, less than 10% of water molecules are reproduced at this distance and this recall level is only found at 1.5 Å. The recall statistic increases to 80% at 1 Å and 86% at 1.5 Å for the R-galectin-3C crystal MD simulation and to 66% and 77% for S-galectin-3C. Interestingly, the solution MD simulations are not consistently worse than the crystal MD simulations. For R-galectin-3C, the recall at 0.5 Å is lower in the solution MD compared to the crystal MD, 51%, but it is higher at all other distances. Conversely, for S-galectin-3C, the recall is higher in the solution MD at 0.5 Å (53% compared to 48%), but lower than in the crystal MD at the larger distances. This suggests that the differences between crystal MD and solution MD observed from the global clustering analysis derive from the larger movement of the protein atoms present in the solution MD. Defining water sites by their distance to protein atoms reduces the effect of the protein dynamics and therefore the differences between crystal MD and solution MD are smaller.
| MD | Crystal | 0.5 | 1.0 | 1.5 | 2.0 | 2.5 | 3.0 |
|---|---|---|---|---|---|---|---|
| R-Galectin-3C | |||||||
| Crystal | Cryo | 120 (57%) | 168 (80%) | 180 (86%) | 186 (89%) | 191 (91%) | 192 (91%) |
| Solution | Cryo | 107 (51%) | 172 (82%) | 191 (91%) | 196 (93%) | 200 (95%) | 201 (96%) |
| Crystal | RT | 72 (74%) | 85 (88%) | 92 (95%) | 90 (93%) | 97 (100%) | 97 (100%) |
| Solution | RT | 70 (72%) | 81 (83%) | 85 (90%) | 94 (88%) | 94 (97%) | 97 (100%) |
|
|||||||
| S-Galectin-3C | |||||||
| Crystal | Cryo | 108 (48%) | 148 (66%) | 172 (77%) | 175 (78%) | 177 (79%) | 179 (80%) |
| Solution | Cryo | 118 (53%) | 142 (63%) | 152 (68%) | 157 (70%) | 168 (75%) | 176 (79%) |
| Crystal | RT | 64 (82%) | 73 (94%) | 78 (100%) | 78 (100%) | 78 (100%) | 78 (100%) |
| Solution | RT | 63 (81%) | 72 (92%) | 75 (96%) | 76 (97%) | 76 (97%) | 76 (97%) |
The crystallographic water sites in the room-temperature structures shows a higher recall than for the cryo-structures. 74% and 82% of room-temperature water sites are reproduced within 0.5 Å in the R- and S-galectin-3C crystal MD simulations, respectively. Furthermore, the R-galectin-3C crystal MD shows full recall (100%) at 2.5 Å, whereas the S-galectin-3C crystal MD shows full recall already at 1.5 Å. The solution MD simulations for both R- and S-galectin-3C show very similar recall statistics, but slightly worse. However, it should be remembered that, as for the global clustering, the absolute number of water sites reproduced is lower than in the cryo-structures.
Clearly, crystallographic water sites defined by their distance to protein heavy atoms are much better reproduced by MD simulations than positions of the water molecules. Up to ten times better recall statistics are found at short distances. It is conceivable that the difference in the recall statistics between the global and local approaches is caused by the fact that the first is based on peaks in the molecule density maps, whereas the other is based on clustering of nearest-neighbour distances. To exclude this possibility, we also calculated recall statistics with the same clustering as in the local approach, but employing instead the aligned Cartesian coordinates of the water molecules. The results in Table S3† are similar to those obtained with the grid-based global approach (Tables 1 and S2†) and much worse than those obtained with the local clustering approach in Table 3. Thus, we can exclude that the improved results are caused by differences in the way the recall statistics is calculated.
These results indicate that MD simulations can actually quite well reproduce the water structure found in protein crystal structures, but the positions of water molecules are highly sensitive to the positions of the surrounding protein atoms and a simple alignment of the snapshots in the trajectory is not sufficient to capture the correct distribution of the water molecules around the protein. This is a very important result, showing that the poor recall statistics found in previous comparisons between MD and crystal water molecules8–11 is not caused by poor force fields, but rather by technical problems with the alignment (i.e. the identification of water sites from MD). Our results also show that these problems can be solved by local clustering algorithms, meaning that it is not necessary to restrain the protein in the MD simulations,12 which of course does not necessarily give fully realistic results.
Identifying new crystallographic water molecules from MD simulations from MD densities
Thus, there are no indications that the MD simulations give unrealistic dynamics of the protein or incorrect water structures. Therefore, it is of interest to check whether it is possible to use the MD simulations to predict preferred positions of the water molecules and use this information to interpret the electron-density maps from crystallography. Therefore, we calculated the electron density in the 2mFo–DFc maps at the positions of MD water peaks that do not have any corresponding crystal water in their vicinity. Any MD water molecule that had a density higher than 1.0σ was visually evaluated in Coot to check if it could be added to the crystal structure. If so, the identified water molecules were added to the model and the resulting structures were refined in phenix.refine with default settings and the electron density maps were evaluated again to check if the added water molecules give rise to any negative difference density.In the case of the solution MD simulations, no water molecules could be added to any of the crystal structures and the same applied to the crystal MD simulations of S-galectin-3C. On the other hand, the crystal MD simulations found three water molecules that were positioned close to unmodeled electron density blobs in the cryo-structure of R-galectin-3C and another two water molecules that could be added to the room-temperature structure of R-galectin-3C. None of these give rise to negative difference density as shown in Fig. 1. The water molecules move by 0.5–0.7 Å from their positions in the MD simulations during the refinement procedure, in order to better fit in the electron density, in agreement with the recall statistics in Table 1. This shows that crystal MD simulations can identify ordered water molecules in the protein crystal structures.
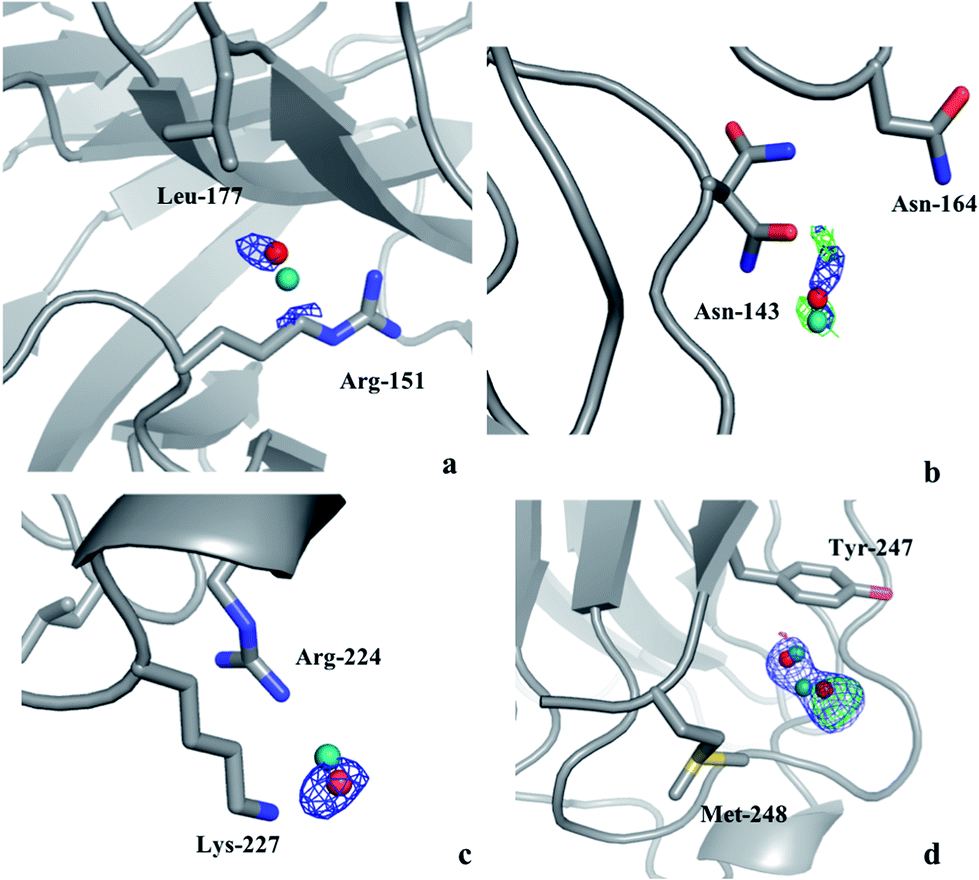 | ||
| Fig. 1 Water molecules that were suggested by the crystal MD simulation of R-galectin-3C after addition to the (a)–(c) cryo- or (d) room-temperature crystal structures of R-galectin-3C (PDB entries 6QGE and 6RGH), followed by refinement, shown in red. The original position of the water molecules in the MD simulation is shown in teal. The 2mFo–DFc maps are contoured at 1.0σ (blue), whereas the mFo–DFc difference maps are contoured at +3.0σ (green) and −3.0σ (red). | ||
Conclusions
In this study, we have compared water structures obtained from crystal and solution MD simulations with water positions in crystal structures of galectin-3C in complex with two ligands, collected at both cryo and room temperature. The comparison was performed in two ways. First, we compared the water molecule positions in the crystal structures to peaks in the water density obtained from a standard global clustering analysis of the MD simulations, after alignment of the protein. Second, we instead defined the crystallographic water sites by their distances to the three closest heavy atoms in the protein (or ligand) and these sites were then compared to MD water molecules in each snapshot of the simulations, using a local clustering approach.The results show that for the traditional, global clustering analysis, both crystal MD and solution MD simulations reproduce the positions of water molecules in the crystal structures rather poorly. The best simulation could reproduce only 35% of crystallographic waters within a distance of 1.0 Å. Crystal MD simulations consistently yielded better results than MD simulations in solution, but the difference was rather small. These differences can be attributed to the smaller dynamics of protein atoms in the crystal MD simulations. Recall percentages were better for the room-temperature crystal structures, showing that MD simulations reproduce better the dynamics at room-temperature, even though they were started from the cryo-temperature structure.
In contrast, the analysis of water sites defined by their relative position to protein atoms gave much better results. 48–82% of the crystallographic water sites were reproduced already within 0.5 Å and 63–94% were reproduced within 1 Å. Similarly, room-temperature crystal structures showed better recall percentages than cryo-temperature structures. However, solution MD simulations were no longer consistently worse than crystal MD simulations. These results suggest that the main problem in comparing MD water structures to crystal water structures arises from the alignment of the system in the MD snapshots. Owing to the dynamics of the protein side chains, the alignment of the water positions is poor and a correct hydrogen-bonding network cannot be defined.
Indeed, the results of the global clustering analysis agree with the studies of van Gunsteren et al.,8 Higo and Nakasako9 or Altan et al.10 On the other hand, the recall statistics from distance-based water site analysis are more similar to the results in the recent article of Wall et al.12 In that article, the authors performed restrained MD simulations, which show 95% recall of crystallographic water molecules at 0.5 Å. Our results suggest that the improved results of Wall et al. are mostly due to the restraints on the protein atoms, eliminating the movements of protein atoms and therefore making the definition of water sites by their positions unambiguous. Our results show that defining the water sites in relation to their distance to the protein gives similar results. Our approach has the advantage of not introducing unphysical restraints, thereby making the simulations more reliable. On the other hand, our recall statistics are slightly worse than that of Wall et al., indicating that we still have some problem with the movement of the protein atoms (the three closest atoms do not necessarily form the interactions that in practice define the binding site). Therefore, there is still room for improvement of the local clustering approach.
We have also used the MD simulations to identify and insert new water molecules in the crystal structures at locations where there is experimental support in the electron-density maps. The results suggest that crystal MD can be used to that end. Five new ordered water molecules were found in the MD simulations that were close to unmodeled density. Thus, MD simulations be a tool for validating positions of less-ordered water molecules and correcting errors in the modelling of the crystal water molecules on a case-by-case basis. The computational effort to produce an MD simulation has nowadays been considerably reduced and a 100 ns simulation can be produced in one day, especially considering the quite small size of the crystallographic unit cell (compared to fully solvated simulations). It should be noted that this water identification is currently based on the global clustering approach and therefore may miss many water sites. It is not fully clear how a local clustering approach can be designed with this aim.
In conclusion, this study indicates that MD simulations can reproduce the crystallographic water structure, but a correct definition of water sites and subsequent analysis needs to be performed. Using water densities from global clustering analysis of the MD simulations based on alignment of the protein gives poor results. However, eliminating the problem of alignment by defining water sites based on their relation to protein atoms gives much better results and is not dependent on the amount of protein dynamics in the MD simulation. In particular, our results show that there is currently no indication that today's force fields are not accurate enough to give proper protein and water dynamics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This investigation has been supported by grants from the Swedish research council (project 2018-05003), from Knut and Alice Wallenberg Foundation (KAW 2013.0022), from eSSENCE: the e-science collaboration and from the Royal Physiographic Society in Lund. The computations were performed on computer resources provided by the Swedish National Infrastructure for Computing (SNIC) at Lunarc at Lund University.References
- Y. Levy and J. N. Onuchic, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 389–415 CrossRef CAS PubMed.
- C. Mattos, Trends Biochem. Sci., 2002, 27, 203–208 CrossRef CAS PubMed.
- E. Nittinger, N. Schneider, G. Lange and M. Rarey, J. Chem. Inf. Model., 2015, 55, 771–783 CrossRef CAS PubMed.
- P. V. Afonine, R. W. Grosse-Kunstleve, P. D. Adams, A. Urzhumtsev and IUCr, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 625–634 CrossRef CAS PubMed.
- P. Setny, R. Baron and J. A. McCammon, J. Chem. Theory Comput., 2010, 6, 2866–2871 CrossRef CAS PubMed.
- R. Baron, P. Setny and J. A. McCammon, J. Am. Chem. Soc., 2010, 132, 12091–12097 CrossRef CAS PubMed.
- M. Misini Ignjatović, O. Caldararu, G. Dong, C. Muñoz-Gutierrez, F. Adasme-Carreño and U. Ryde, J. Comput. Aided Mol. Des., 2016, 30, 707–730 CrossRef PubMed.
- W. F. van Gunsteren, H. J. Berendsen, J. Hermans, W. G. Hol and J. P. Postma, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 4315–4319 CrossRef CAS PubMed.
- J. Higo and M. Nakasako, J. Comput. Chem., 2002, 23, 1323–1336 CrossRef CAS PubMed.
- I. Altan, D. Fusco, P. V. Afonine and P. Charbonneau, J. Phys. Chem. B, 2018, 122, 2475–2486 CrossRef CAS PubMed.
- A. Rudling, A. Orro and J. Carlsson, J. Chem. Inf. Model., 2018, 58, 350–361 CrossRef CAS PubMed.
- M. E. Wall, G. Calabró, C. I. Bayly, D. L. Mobley and G. L. Warren, J. Am. Chem. Soc., 2019, 141, 4711–4720 CrossRef CAS PubMed.
- P. A. Janowski, C. Liu, J. Deckman and D. A. Case, Protein Sci., 2016, 25, 87–102 CrossRef CAS PubMed.
- L. Meinhold and J. C. Smith, Biophys. J., 2005, 88, 2554–2563 CrossRef CAS PubMed.
- L. Meinhold, F. Merzel and J. C. Smith, Phys. Rev. Lett., 2007, 99, 138101 CrossRef PubMed.
- H. Leffler, S. Carlsson, M. Hedlund, Y. Qian and F. Poirier, Glycoconj. J., 2002, 19, 433–440 CrossRef CAS PubMed.
- A. C. MacKinnon, S. L. Farnworth, P. S. Hodkinson, N. C. Henderson, K. M. Atkinson, H. Leffler, U. J. Nilsson, C. Haslett, S. J. Forbes and T. Sethi, J. Immunol., 2008, 180, 2650–2658 CrossRef CAS PubMed.
- D. Delacour, A. Koch and R. Jacob, Traffic, 2009, 10, 1405–1413 CrossRef CAS PubMed.
- F. T. Liu and G. A. Rabinovich, Ann. N. Y. Acad. Sci., 2010, 1183, 158–182 CrossRef CAS PubMed.
- A. Grigorian and M. Demetriou, in Glycobiology, ed. M. Fukuda, Academic Press, 2010, vol. 480, pp. 245–266 Search PubMed.
- G. A. Rabinovich, F.-T. Liu, M. Hirashima and A. Anderson, Scand. J. Immunol., 2007, 66, 143–158 CrossRef CAS PubMed.
- A. Boza-Serrano, R. Ruiz, R. Sanchez-Varo, J. García-Revilla, Y. Yang, I. Jimenez-Ferrer, A. Paulus, M. Wennström, A. Vilalta, D. Allendorf, J. C. Davila, J. Stegmayr, S. Jiménez, M. A. Roca-Ceballos, V. Navarro-Garrido, M. Swanberg, C. L. Hsieh, L. M. Real, E. Englund, S. Linse, H. Leffler, U. J. Nilsson, G. C. Brown, A. Gutierrez, J. Vitorica, J. L. Venero and T. Deierborg, Acta Neuropathol., 2019, 138, 251–273 CrossRef CAS PubMed.
- P. Sörme, P. Arnoux, B. Kahl-Knutsson, H. Leffler, J. M. Rini and U. J. Nilsson, J. Am. Chem. Soc., 2005, 127, 1737–1743 CrossRef PubMed.
- K. Saraboji, M. Håkansson, S. Genheden, C. Diehl, J. Qvist, U. Weininger, U. J. Nilsson, H. Leffler, U. Ryde, M. Akke and D. T. Logan, Biochemistry, 2012, 51, 296–306 CrossRef CAS PubMed.
- M. L. Verteramo, O. Stenström, M. Misini Ignjatović, O. Caldararu, M. A. Olsson, F. Manzoni, H. Leffler, E. Oksanen, D. T. Logan, U. J. Nilsson, U. Ryde and M. Akke, J. Am. Chem. Soc., 2019, 141, 2012–2026 CrossRef CAS PubMed.
- O. Caldararu, R. Kumar, E. Oksanen, D. T. Logan and U. Ryde, Phys. Chem. Chem. Phys., 2019, 21, 18149–18160 RSC.
- P. V. Afonine, R. W. Grosse-Kunstleve, N. Echols, J. J. Headd, N. W. Moriarty, M. Mustyakimov, T. C. Terwilliger, A. Urzhumtsev, P. H. Zwart, P. D. Adams, W. Ralf, J. J. Headd and C. Thomas, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 352–367 CrossRef CAS PubMed.
- D. A. Case, J. T. Berryman, R. M. Betz, D. S. Cerutti, T. E. Cheatham, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, T. Luchko, R. Luo, B. Madej, K. M. Merz, G. Monard, P. Needham, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. E. Roitberg, R. Salomon-Ferrer, C. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, D. M. York and P. A. Kollman, AMBER 14, University of California, San Francisco, 2014 Search PubMed.
- C. Diehl, S. Genheden, K. Modig, U. Ryde and M. Akke, J. Biomol. NMR, 2009, 45, 157–169 CrossRef CAS PubMed.
- S. Genheden and U. Ryde, Phys. Chem. Chem. Phys., 2012, 14, 8662–8677 RSC.
- F. Manzoni, J. Wallerstein, T. E. Schrader, A. Ostermann, L. Coates, M. Akke, M. P. Blakeley, E. Oksanen and D. T. Logan, J. Med. Chem., 2018, 61, 4412–4420 CrossRef CAS PubMed.
- J. Uranga, P. Mikulskis, S. Genheden and U. Ryde, Comput. Theor. Chem., 2012, 1000, 75–84 CrossRef CAS.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- H. W. Horn, W. C. Swope, J. W. Pitera, J. D. Madura, T. J. Dick, G. L. Hura and T. Head-Gordon, J. Chem. Phys., 2004, 120, 9665–9678 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- S. Genheden and U. Ryde, J. Comput. Chem., 2011, 32, 187–195 CrossRef CAS PubMed.
- P. E. Smith, B. M. Pettitt and M. Karplus, J. Phys. Chem., 1993, 97, 6907–6913 CrossRef CAS.
- L. Monticelli, E. J. Sorin, D. P. Tieleman, V. S. Pande and G. Colombo, J. Comput. Chem., 2008, 29, 1740–1752 CrossRef CAS PubMed.
- S. Genheden and U. Ryde, J. Comput. Chem., 2010, 31, 837–846 CAS.
- S. K. Sadiq, D. W. Wright, O. A. Kenway and P. V Coveney, J. Chem. Inf. Model., 2010, 50, 890–905 CrossRef CAS PubMed.
- R. Harada, Y. Takano, T. Baba and Y. Shigeta, Phys. Chem. Chem. Phys., 2015, 17, 6155–6173 RSC.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- X. Wu and B. R. Brooks, Chem. Phys. Lett., 2003, 381, 512–518 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theor. Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- S. Ramsey, C. Nguyen, R. Salomon-Ferrer, R. C. Walker, M. K. Gilson and T. Kurtzman, J. Comput. Chem., 2016, 37, 2029–2037 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC, https://pymol.org Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09601a |
| This journal is © The Royal Society of Chemistry 2020 |