
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synergistic effect of the PEO–PVA–PESf composite polymer electrolyte for all-solid-state lithium-ion batteries†
Ling Xu,
Kaiyuan Wei,
Yong Cao,
Shiping Ma,
Jian Li,
Yu Zhao *,
Yixiu Cui and
Yanhua Cui*
*,
Yixiu Cui and
Yanhua Cui*
Institute of Electronic Engineering, China Academy of Engineering Physics, Mianyang, Sichuan 621000, P. R. China. E-mail: cuiyanhua@netease.com; yzhao@caep.cn
First published on 3rd February 2020
Abstract
Blending with poly(vinyl alcohol) (PVA) and poly(oxyphenylene sulfone) (PESf) has been investigated to improve the properties of a polymer electrolyte based on a poly(ethylene oxide) (PEO) matrix. The composite electrolyte shows a high ionic conductivity of 0.83 × 10−3 S cm−1 at 60 °C due to the significant inhibition of crystallization caused by the synergistic effects of PVA and PESf. The symmetrical cell Li/CPE/Li is continuously operated for at least 200 hours at a current density of 0.1 mA cm−2 without the enhancement in the polarization potential. In addition, the all-solid-state LiFePO4/CPE/Li cells exhibit small hysteresis potential (about 0.10 V), good cycle stability and excellent reversible capacity (126 mA h g−1 after 100 cycles).
1. Introduction
All-solid-state lithium-ion batteries have aroused researchers' attention in recent years due to the impending demands of high safety and energy density.1–4 One of the arduous challenges is developing an adequate solid electrolyte with high Li+ conductivity, wide electrochemical windows and low interfacial resistance to afford outstanding electrochemical performances. The composite polymer electrolyte (CPE) based on PEO is a promising candidate owing to the low glass transition temperature (−60 °C), high Li+ solvation ability and admirable interfacial stability with lithium.5–9 However, the low room temperature conductivity (10−7 to 10−6 S cm−1), narrow electrochemical windows and inferior mechanical properties of PEO are the main obstacles for further development.4,7,9Blending is one of the valid approaches to ameliorate the properties of PEO, such as with PVP,10 PVDF,11 and PMMA;12 this can decrease crystallization and enhance the fractional movement of polymer chains and the migration amount of Li+.13 Similar to PEO, polyvinyl alcohol (PVA) is another semi-crystalline polymer with the fascinating properties of non-toxic, water solubility and biocompatibility; it has been under the spotlight and applied as an electrolyte in supercapacitors on a large scale.14–16 More recently, PVA-based CPE has been reported and has shown a fairly high room-temperature ionic conductivity of 4.31 × 10−4 S cm−1.14 However, the PEO blend with PVA cannot be used individually as a polymer electrolyte because of unsatisfactory mechanical strength. It is of great importance to introduce a filler into the PEO host. The conventional inorganic fillers, such as TiO2,17 ZrO2,18 Al2O3,8 LiAlO2 (ref. 19) and Li10GeP2S12,20 have been reported for improving the interfacial stability and Li+ transportation in the polymer matrix. However, till date, organic fillers with better phase compatibility and more surface groups than that for inorganic fillers have rarely been used in CPEs based on PEO.
Poly(oxyphenylene sulfone) (PESf) is a promising organic filler with good chemical resistance to organic solvents. As the molecular structure of PESf contains sulfonic acid and benzene ring, there are abundant conjugate electrons, which can bridge with the polymer chains. In this work, the PEO electrolyte blended with PVA and PESf has been successfully prepared with excellent electrochemical properties. PVA and PESf have synergistic effects, thus enhancing the amorphous phase and dissociations of the Li salt in the PEO host. The CPE membranes present an ionic conductivity of 0.83 × 10−3 S cm−1 at 60 °C without polarization degradation for at least 200 hours at a current density of 0.1 mA cm−2. The LiFePO4/CPE/Li cell can run for 100 cycles with 126 mA h g−1 capacity retention.
2. Results and discussion
2.1 Characterization of CPE membranes
Fig. 1a reveals the XRD (D8 Bruker) patterns of the pure PEO, PVA, PESf and CPE membranes with gradient concentrations. Two sharp diffraction peaks at around 19° and 23° are revealed for the PEO membrane, corresponding to its partially characteristic crystallization.13 There are no diffraction peaks of LiTFSI in all the XRD patterns, representing the dissolution of the lithium salt in the polymer matrix by the way of Li+ and TFSI−. There are broad diffraction peaks at around 18° and 19° for PVA and PESf, respectively, due to the semicrystalline character. From the XRD patterns in the middle of Fig. 1a, the peak intensity does not notably decrease with the addition of 30 wt% PVA or 20 wt% PESf, indicating that individual PVA or PESf is not a promising plasticizer. In contrast, the crystallinity of CPE presents a remarkable decrease with the simultaneous addition of the PVA and PESf components. Naturally, PVA is a good solvent of LiTFSI because of the similar monomer relative to PEO, and PESf is a promising organic filler containing sulfonic acid and benzene ring. Thus, the PEO–PVA–PESf system not only enhances the concentration of Li+ in the polymer matrix, but also forces the PEO chain's segmental motion. The synergistic effect can sustain the high ionic conductivity of the CPE membrane. As displayed in Fig. 1b, the crystallinity of the membrane with 30 wt% PVA or 20 wt% PESf is over 32%, while the PEO–PVA–PESf system exhibits lower crystallinity, the value for which is below 28%. The lowest crystallinity is about 24.5%, which is calculated from the XRD pattern for the CPE with 30 wt% PVA and 20 wt% PESf membranes (PEO–LiTFSI–30% PVA–20% PESf). The surface and cross-sectional images of the PEO–LiTFSI–30% PVA–20% PESf membrane are shown in Fig. 1c and d, respectively. From the top view of the CPE electrolyte, the membrane surface is smooth and no aggregation can be observed, which is propitious to form a stable interface between the electrode and CPE; this is good for the transport of lithium ions during cycling. From the cross view of the CPE electrolyte, the membrane is relatively dense with the thickness of 287 μm.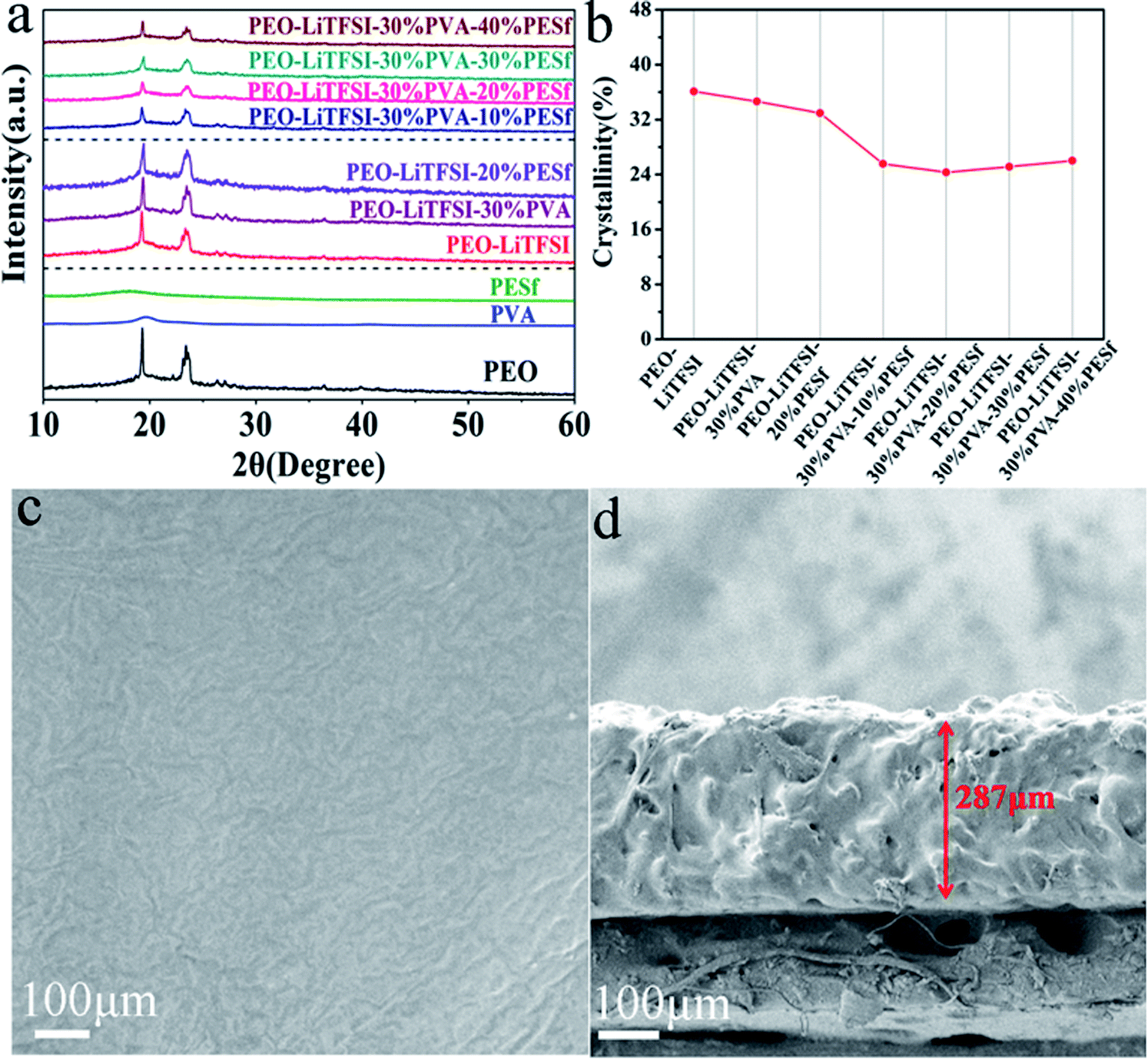 | ||
| Fig. 1 (a) XRD patterns of polymer membranes, (b) the crystallinity calculated from XRD patterns, (c) the surface SEM image and (d) cross-sectional SEM image of PEO–LiTFSI–30% PVA–20% PESf membrane. | ||
The information of the intermolecular interaction between PEO, LiTFSI, PVA and PESf was investigated by FTIR (Nicolet 6700, America) spectroscopy, as shown in Fig. 2. After the introduction of the lithium salt, the intermolecular hydrogen-bonded O–H stretching (3433 cm−1) becomes more evident owing to the interplay with TFSI−, which indicates that more Li+ ions could be freed from the lithium salt.13 The absorption peak of the symmetric CH2 oscillation (1359 cm−1) becomes more acute and intensive, demonstrating the enhancement in the PEO chain's segmental motion by adding the lithium salt.13 Interestingly, with the complex of PVA and PESf, the peak at 3433 cm−1 becomes broader, and the peaks at 787 and 739 cm−1 become more intensive; this demonstrates highly intense interactions between the intermolecular O–H and TFSI− anions, indicating the further dissociation of LiTFSI and more active sites for Li+.21–24 Also, the bands at 1281 and 842 cm−1 become intensive, leading to the stretching of asymmetric C–O–C and the less coordinated structure of the PEO chains and much more dynamic wiggle of the polymer chains.13,25 This phenomenon indicates that PVA and PESf can disorder the coordination of PEO and promote the dissociation of the lithium salt and the movement of Li+.
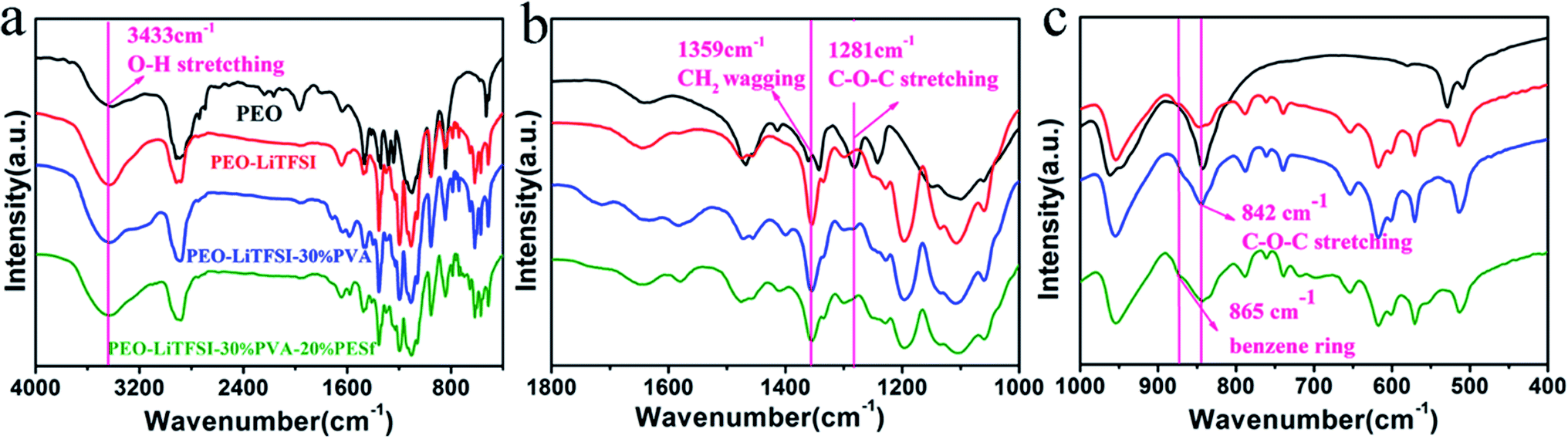 | ||
| Fig. 2 (a) FT-IR spectra of polymer membranes, (b) the magnified plots between 1000 and 1800 cm−1, and (c) the magnified plots between 400 and 1000 cm−1. | ||
2.2 Electrochemical performance analysis
The Nyquist plots of the PEO–LiTFSI, PEO–LiTFSI–30% PVA and PEO–LiTFSI–30% PVA–x% PESf polymer membranes measured at 30 °C are presented in Fig. 3a. The CPE membranes had lower impedance, which illustrated that the modified membranes had higher ionic conductivity compared with the PEO–LiTFSI system. The ionic conductivities of the CPE membranes were calculated based on the equation σ = L/(RS), in which R (Ω), S (cm2) and L (cm) represent the resistance value of the bulk electrolyte, the area of the electrode and the distance of the two electrodes, respectively. The ionic conductivities of the CPE membranes were measured at different temperatures (25–120 °C), as revealed in Fig. 3b. The ionic conductivity of the PEO–LiTFSI–30% PVA–x% PESf polymer membranes increased sharply with the temperature changing from 25 to 60 °C; the PEO–LiTFSI–30% PVA–20% PESf membrane presented a fairly higher ionic conductivity of about 0.83 × 10−3 S cm−1 at 60 °C. Fig. 3c presents the LSV profiles of the CPE membranes to quantify the electrochemical stability window. The PEO–LiTFSI membrane was barely stable under 4.8 V. Furthermore, the CPE with 30 wt% PVA and 20 wt% PESf showed the widest electrochemical window of about 5.38 V, which could meet the need of high-potential cathodic materials. The Li/PEO–LiTFSI/Li cell presented a high polarization potential over 1.5 V and could be merely operated stably for 100 hours at a current density of 0.1 mA cm−2 before short circuit; in contrast, the Li/CPE/Li cell with the polarization potential less than 0.1 V could be stably operated for 200 hours without short circuit and deterioration of polarization at 60 °C. The polarization potential for the Li/CPE/Li cell was about 0.48 V, and it could be cycled stably for about 200 hours at room temperature (25 °C) with the current density of 0.1 mA cm−2 (Fig. 3d), demonstrating that the compatibility between the CPE membranes and the Li-metal electrode was fantastic. As can be seen from Fig. S1,† pristine CPE cannot osculate well with Li before cycling, while CPE and Li metal coalesce to form a stable and indiscerptible interface after 200 hours of cycling, in which lithium dendrites cannot be observed; this indicates that CPE can effectively suppress dendrite formation and growth during lithium plating–stripping.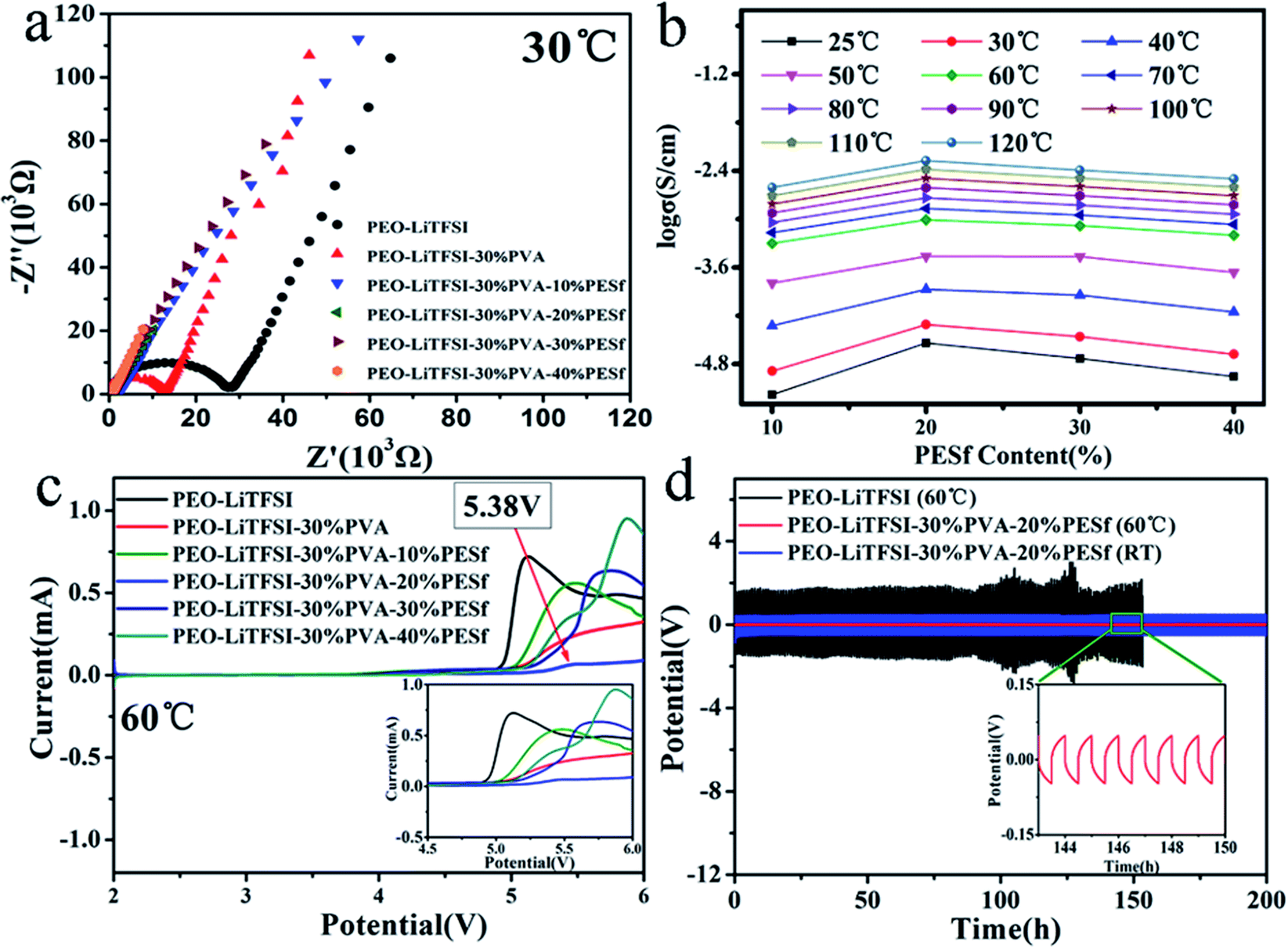 | ||
| Fig. 3 (a) Nyquist plots, (b) ionic conductivity and (c) LSV profiles of polymer membranes. (d) The electrochemical stability of CPE in symmetrical cells of Li/CPE/Li and Li/PEO–LiTFSI/Li. | ||
Fig. 4 shows the performances of the CPE electrolyte in the LiFeO4/CPE/Li half-cell at different operating temperatures. At a temperature of 60 °C, the capacity is 167 mA h g−1 at 0.1C during the initial discharge process (Fig. 4a), which approaches the theoretical value of LiFePO4; this indicates that the interface compatibility between the cathode and CPE is fantastic, and the charge/discharge plateau is only 0.10 V, demonstrating that the interfacial resistance of the CPE/electrode is small enough. The symmetric pair of reduction peaks for Fe3+/2+ can be clearly seen from the dQ/dV–V plot in the inset, indicating that the Li+ intercalation/deintercalation process of LiFePO4 occurs with small transfer resistance. At room temperature, the ionic conductivity of CPE is 3.64 × 10−5 S cm−1, as determined from Fig. 3b, and it is much lower than the value at 60 °C. The LiFePO4/CPE/Li cell can still cycle at 25 °C, at which the capacity is about 100 mA h g−1. The effective Li+ transportation is presented in the charge/discharge profiles in Fig. 4b. The potential hysteresis is about 0.69 V, which is ascribed to the low ionic conductivity and poor interface of CPE and the electrodes. Disappointingly, the LiFePO4/PEO–LiTFSI/Li cell can only run for 10 cycles, while the LiFePO4/CPE/Li cell runs for over 100 cycles continually, with a reversible capacity of 126 mA h g−1 and a coulombic efficiency close to 98% (Fig. 4c) because of the good compatibility between the CPE and the Li-metal electrode and no side reactions. The rate capability differentiation of the LiFeO4/CPE/Li cell is provided in Fig. 4d. The discharge capacity is about 166.0 mA h g−1 with the rate of 0.1C (0.034 mA). When the rate increases, the discharge capacity becomes 160.7, 158.4, 139.2 and 51.2 mA h g−1 with the corresponding rates of 0.2C (0.068 mA), 0.5C (0.17 mA), 1.0C (0.34 mA) and 2.0C (0.68 mA). It is worth noting that a reversible high capacity of 161.0 mA h g−1 is obtained when the cell is charged and discharged at 0.1C again with the coulombic efficiency close to 99%, which may be ascribed to the favourable interfacial stability as well as the mechanical strength of the CPE membranes.
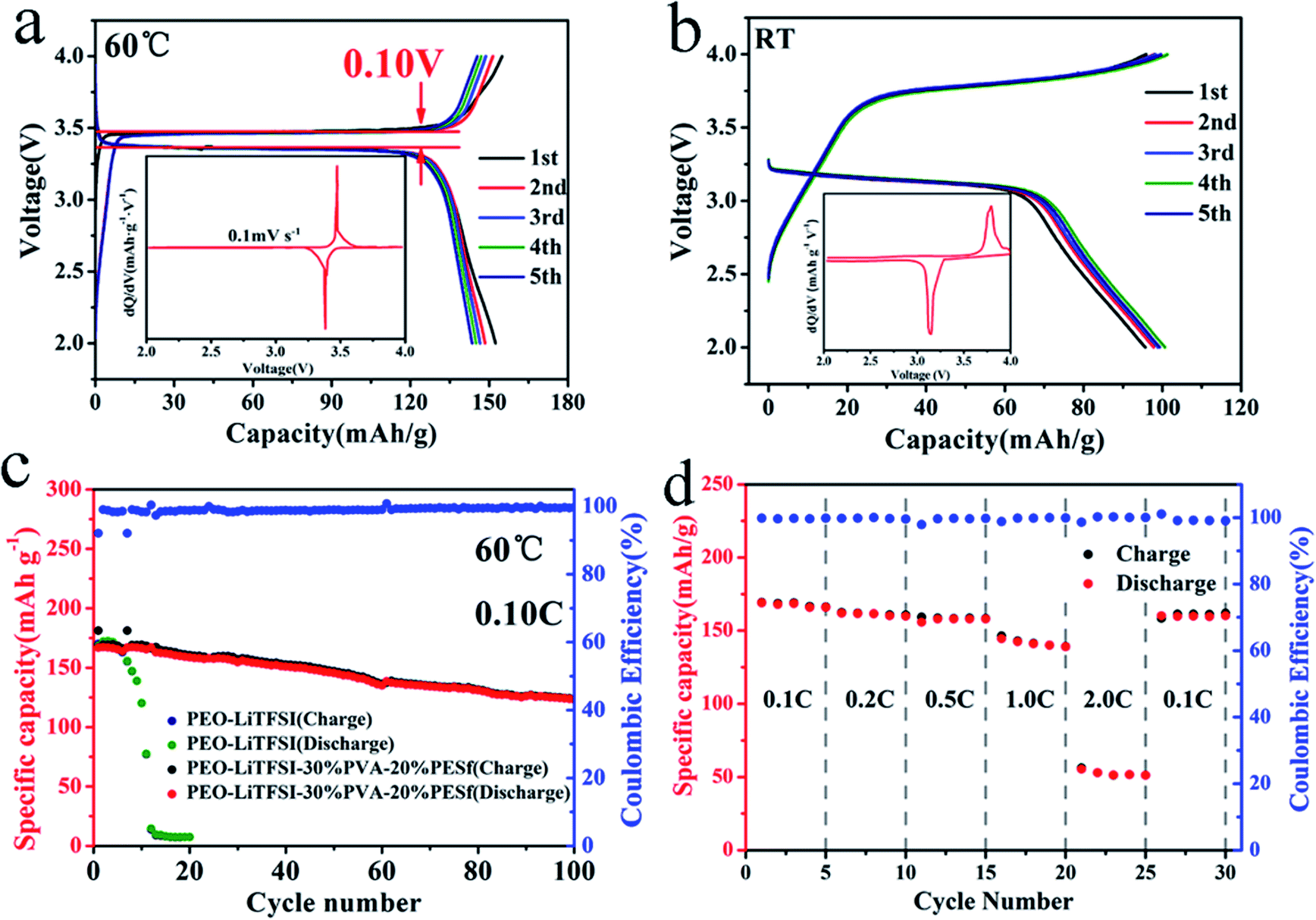 | ||
| Fig. 4 (a) Charge/discharge curves for the first 5 cycles at 60 °C; the inset is differential capacity–voltage curves. (b) Charge/discharge curves for the first 5 cycles at room temperature; the inset is differential capacity–voltage curve. (c) Galvanostatic profiles of LiFeO4/CPE/Li cell and LiFeO4/PEO–LiTFSI/Li cell. (d) The rate capacity of LiFeO4/CPE/Li cell from 0.1C to 2.0C at 60 °C. | ||
3. Conclusion
In summary, the CPE of the PEO–PVA–PESf system has been prepared via the casting method, and it exhibits enhanced properties, such as a wider electrochemical window (about 5.38 V), higher ionic conductivity (0.83 × 10−3 S cm−1 at 60 °C) and better cyclic performance. PVA and PESf have synergistic effects for CPE, resulting in the boosting of the amorphization and dissociation of the lithium salt. Furthermore, the cell with CPE can operate for at least 200 hours at a current density of 0.1 mA cm−2 without short circuit and polarization degradation. In particular, the LiFePO4/CPE/Li cell can run for 100 cycles with 126 mA h g−1 capacity retention.4. Experimental
4.1 Preparation of CPE membranes and composite cathode
Lithium bis(trifluoromethane)sulfonamide (LiTFSI), PEO, PVA, and PESf with the average molecular weights of 287.09, 6.0 × 105, 7.8 × 104 and 6.3 × 104 g mol−1, respectively, were purchased from Aladdin. PESf was ground for 10 hours by planet ball mill. The molding of free-standing CPE membranes has been introduced in a previous publication.13 PEO16–LiTFSI, PVA and PESf were orderly dissolved in anhydrous acetonitrile (ACN) with series of wt%. Then, the precursor solution was poured into the mould and solidified in the glove box filled with argon gas. Eventually, the CPE membrane was transferred to a vacuum oven to eliminate the remaining solvent.The working electrodes were prepared by mixing 70 wt% of the active materials, 10 wt% acetylene black, 20 wt% composite polymer electrolyte (CPE) dissolved in anhydrous acetonitrile (ACN) to form a slurry. The slurry was then pasted onto an aluminum foil and dried at 60 °C for 10 hours in vacuum; then, the composite cathode was obtained for use, and the active mass loading was about 2 mg cm−2. The electrochemical performance of CPE was studied by using CR2032-type coin cells assembled in an argon-filled glove-box. A metallic lithium foil was used as the counter electrode.
4.2 Electrochemical measurement of CPE membranes
The ionic conductivity of the CPE was determined by electrochemical impedance spectroscopy (EIS) on an AutoLab workstation with the frequency range from 10 MHz to 0.01 Hz and the amplitude of 10 mV, in which the cell was assembled by a sandwich structure with a pair of stainless steel as the block electrodes. The liner sweep voltammetry (LSV) of the SS/CPE/Li cell was performed on CHI 660E at a sweep rate of 0.1 mV s−1 within the range from 2.0 to 6.0 V. The interfacial stabilities between the CPE and the Li-metal electrode were performed by galvanostatic cycling of the Li/CPE/Li symmetric cells at the current density of 0.1 mA cm−2. The galvanostatic cycling was performed on LAND CT2001A between 2.0 and 4.0 V with the LiFePO4/CPE/Li cell.Conflicts of interest
We declare that we have no conflict of interest.Acknowledgements
This work is financially supported by NSAF Joint Fund (Grant No. U1930138), National Natural Science Foundation of China (11804312) and Laboratory of Precision Manufacturing Technology, China Academy of Engineering Physics (Grant No. ZD17006, ZM18002).References
- Y. J. Li, C. Y. Fan, J. P. Zhang and X. L. Wu, Dalton Trans., 2018, 47, 14932–14937 RSC.
- L. Zhu, P. Zhu, S. Yao, X. Shen and F. Tu, Int. J. Energy Res., 2019, 43, 4854–4866 CrossRef CAS.
- C. Li, Y. Huang, X. Feng, Z. Zhang and P. Liu, J. Membr. Sci., 2019, 587, 117179–117185 CrossRef CAS.
- J. Lee, T. Howell, M. Rottmayer, J. Boeckl and H. Huang, J. Electrochem. Soc., 2019, 166, A416–A422 CrossRef CAS.
- Y. Zhao, R. Y. Tao and T. Fujinami, Electrochim. Acta, 2006, 51, 6451–6455 CrossRef CAS.
- D. K. Roh, S. J. Kim, H. Jeon and J. H. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 6615–6621 CrossRef CAS PubMed.
- J. Shim, J. W. Lee, K. Y. Bae, H. J. Kim, W. Y. Yoon and J. C. Lee, ChemSusChem, 2017, 10, 2274–2283 CrossRef CAS PubMed.
- B. Jinisha, K. M. Anilkumar, M. Manoj, A. Abhilash, V. S. Pradeep and S. Jayalekshmi, Ionics, 2017, 24, 1675–1683 CrossRef.
- Z. Xiao, B. Zhou, J. Wang, C. Zuo, D. He, X. Xie and Z. Xue, J. Membr. Sci., 2019, 576, 182–189 CrossRef CAS.
- B. Jinisha, K. M. Anilkumar, M. Manoj, V. S. Pradeep and S. Jayalekshmi, Electrochim. Acta, 2017, 235, 210–222 CrossRef CAS.
- S. K. Patla, R. Ray, K. Asokan and S. Karmakar, J. Appl. Phys., 2018, 123, 125102–125112 CrossRef.
- Y. Lim, H.-A. Jung and H. Hwang, Energies, 2018, 11, 2259–2268 CrossRef.
- H. L. Wu, J. L. Wang, Y. Zhao, X. Q. Zhang, L. Xu, H. Liu, Y. X. Cui, Y. H. Cui and C. L. Li, Sustainable Energy Fuels, 2019, 3, 2642–2656 RSC.
- Y. Ma, L. B. Li, G. X. Gao, X. Y. Yang and Y. You, Electrochim. Acta, 2016, 187, 535–542 CrossRef CAS.
- H. N. Fard, G. B. Pour, M. N. Sarvi and P. Esmaili, Ionics, 2019, 25, 2951–2963 CrossRef CAS.
- G. Chen, N. Chen, L. Li, Q. Wang and W. F. Duan, Ind. Eng. Chem. Res., 2018, 57, 5472–5481 CrossRef CAS.
- S. N. Banitaba, D. Semnani, B. Rezaei and A. A. Ensafi, Polym. Adv. Technol., 2019, 30, 1234–1242 CrossRef CAS.
- C. P. Sugumaran and D. E. Selvaraj, J. Nano Res., 2015, 37, 1–12 Search PubMed.
- N. Zhang, J. He, W. Han and Y. Wang, J. Mater. Sci., 2019, 54, 9603–9612 CrossRef CAS.
- Y. Zhao, C. Wu, G. Peng, X. Chen, X. Yao, Y. Bai, F. Wu, S. Chen and X. Xu, J. Power Sources, 2016, 301, 47–53 CrossRef CAS.
- M. T. Caccamo, E. Calabro, S. Coppolino and S. Magazu, Curr. Metabolomics, 2018, 6, 46–48 CAS.
- V. Selvanathan, M. N. A. Halim, A. D. Azzahari, M. Rizwan, N. Shahabudin and R. Yahya, Ionics, 2018, 24, 1955–1964 CrossRef CAS.
- R. Kumar, S. Sharma, D. Pathak, N. Dhiman and N. Arora, Solid State Ionics, 2017, 305, 57–62 CrossRef CAS.
- S. J. Wen, T. J. Richardson, D. I. Ghantous, K. A. Streibel, P. N. Ross and E. J. Cairns, J. Electroanal. Chem., 1996, 415, 197 CrossRef.
- J. F. Zhang, C. Ma, H. Hou, X. F. Li, L. B. Chen, D. G. Ivey and W. F. Wei, J. Membr. Sci., 2018, 552, 107–114 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09645k |
| This journal is © The Royal Society of Chemistry 2020 |