
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization of coal-based fulvic acid and the construction of a fulvic acid molecular model†
Guan-qun Gong‡
 a,
Xin Yuan‡b,
Ying-jie Zhang‡*a,
Ya-jun Lib,
Wei-xin Liub,
Ming Wangb,
Yu-feng Zhaob and
Liang-wei Xub
a,
Xin Yuan‡b,
Ying-jie Zhang‡*a,
Ya-jun Lib,
Wei-xin Liub,
Ming Wangb,
Yu-feng Zhaob and
Liang-wei Xub
aKey Laboratory of Coal Processing and Efficient Utilization of Ministry of Education, Xuzhou 221116, China. E-mail: zhangcumt123@126.com
bSchool of Chemical Engineering & Technology, China University of Mining and Technology, Xuzhou 221116, China
First published on 3rd February 2020
Abstract
Fulvic acid (FA) is important in modern agriculture, ecological restoration, life science, and medicine. The precise characterization of the composition and molecular structure of FA has become a key scientific issue in both basic and applied research. In this study, coal-based FA was separated by microwave-assisted oxygenation from lignite originating from Inner Mongolia in China. Through elemental analysis, infrared spectroscopy, nuclear magnetic resonance spectroscopy, classical quantitative titration experiments, and quantum chemistry combined with software analysis, the representative microscopic molecular structure of FA was determined. The results show that coal-based FA mainly contains three kinds of benzene ring substituents, ether bonds, hydrogen bonds, carbonyl groups, hydroxyl groups, carboxyl groups, phenolic hydroxyl groups, and semiquinonyl groups. The oxygen content is high, the carbon-to-oxygen ratio is less than 1, and the hydrogen-to-carbon ratio is 1.09. The ratio of aromatic carbon to total carbon is approximately 0.6, and benzene rings are connected to each other by an ether–oxygen bridge. The fat chain length of FA is approximately 0.47. FA has a small molecular structure with many acidic groups, primarily carboxyl groups and phenolic hydroxyl groups. The two-dimensional planar molecular structure of FA was established; the chemical formula is C38H32NO24, and the relative molecular mass is 886. The lowest-energy, structurally optimized three-dimensional characteristic ball-and-stick and stick models were also constructed. The calculated infrared spectrum of the molecular structure matches well with the experimental spectrum of FA, and the types and distributions of functional groups agree with the findings of previous studies. The quantum chemical data confirm that the proposed molecular structure is reasonable. The findings provide a scientific reference for applied research on FA in the future.
Introduction
FA is originally derived from low-rank coal and is obtained by acid–base reaction followed by solvent extraction, and it is a mixture of special chemical activities and biological activities composed of a series of molecules whose special complex molecular composition determines the specificity of the action of FA. In recent years, FA has been found to have important applications in many fields such as agriculture, ecological restoration, environmental protection, animal husbandry and aquaculture, veterinary medicine, human medicine,1–6 and life science.7–10 However, the unclear molecular structure of FA constrains the characterization of its properties and mechanism of action. Accurately characterizing the composition and molecular structure of FA has become a key scientific issue in research on FA and relate industries.While the composition of FA has been reported, research on the construction of the FA molecular structure is lacking. Schellekens et al.11 used pyrolysis gas chromatography/mass spectrometry to compare the molecular compositions of humic acid (HA) and FA and found no significant differences among their pyrolysis products. The main chemical groups included carbohydrates, phenols, benzene, and lignin phenols. Murray and Linder12 studied the FA model based on the random assignment of functional groups and aromatic rings and derived the relationship between the percentage of aromatic carbon and the hydrogen-to-carbon molar ratio. Senesi13 found that both FA and HA can be described by a general model structure consisting of a network of differently extended aromatic properties with aliphatic, glucosyl, peptidyl, and lipid-based surface chains along with different chemical functional groups including carboxyl, phenolic hydroxyl and alcoholic hydroxyl, and carbonyl groups with different aromatic, oxygen, nitrogen, or sulfur bridges. Jing et al.14 studied the morphological transformation and structural changes of FA during ozone oxidation and investigated the molecular weight distribution, types of intermediate products, and changes in polar functional groups. Klencsaŕ and Köntös15 completed the EPR analysis of Fe3+ and Mn2+ complex sites in FA, which can be used to track and characterize the structural changes in FA complexes under different conditions and chemical reaction conditions. Chaaban et al.16 showed that a major component of Suwannee River FA is an individual, negatively charged amphiphile with a molecular size similar to that of dodecyl trimethyl ammonium chloride.
Lignite from the northeastern mining area of Inner Mongolia in China was used as a source of FA in this study. The coal-based FA in the lignite was obtained via solvent extraction assisted by the oxygenation of hydrogen peroxide and microwave oxygenation. The purified lignite FA was a dark-brown powder that was easily soluble in water. Preliminary laboratory experiments showed that FA promoted the growth of high-grade flowers such as Clivia, aloe, and vegetables such as cucumber and pepper, typical of FA. In this paper, the physical properties, physicochemical properties, chemical properties, and molecular structure of FA were evaluated using a series of techniques. Through elemental analysis, infrared spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, titration analysis for classical functional groups, and quantum chemistry combined with accurate microstructural characterization, model construction, computational simulation, and experimental detection, the microscopic molecular structure of FA was obtained. The results show that FA contains representative functional groups such as phenolic hydroxyl, carboxyl, keto, and semiquinone groups along with multiple benzene rings connected by bridge bonds. FA is a biologically active class of soluble platform compounds that exhibit acid/base buffering, easy complexation, and reversible dissociation.
Materials and methods
Samples and chemicals
The coal sample used in the experiment was Hulunbuir lignite from Inner Mongolia. First, the lignite was collected using the quarter method. After being broken into smaller particles, the coal sample was crushed and collected with a small universal crusher. All coal samples were passed through a 100-mesh sieve (particle size = 0.147 mm) and then dried at 80 °C for 24 h to remove most of the water in the coal. After drying, the sample was placed into a dry bag followed by sealing and storing.Concentrated sulfuric acid was purchased from Sinopharm Chemical Reagent Co., Ltd, China. Hydrogen peroxide (H2O2) was purchased from Shanghai Zhanyun Chemical Co., Ltd, China. Absolute ethanol was purchased from Damao Chemical Reagent Factory, China.
Experimental
Microwave-assisted H2O2 oxylysis of lignite for extracting FA
A certain amount of the coal sample, H2O2 solution, and deionized water were added to a 250 mL single-neck round-bottom flask. The reaction flask was placed on a polytetrafluoroethylene tray in the center of the bottom of a furnace in a microwave chemical reactor. The mouth of the reaction flask was then connected with a glass connection tube, which was passed through the top of the furnace and connected to a reflux condenser. The reaction parameters of the microwave reactor were set, and the reaction was performed. After the reaction, the reaction flask was removed. The mixture in the flask was cooled to room temperature and then filtered with a circulating water vacuum pump. The filter residue was repeatedly washed with deionized water to reduce the FA residue. Subsequently, the filtrate was concentrated at 60 °C in a rotary evaporator with a vacuum pump. The evaporation dish containing the concentrated liquid was then placed in a blast drying box to evaporate and dry at 60 °C, after which primary FA was obtained.Purification of FA using the sulfuric acid–ethanol method
A certain amount of ethanol, deionized water, and concentrated sulfuric acid were added to a three-necked round-bottom flasks and mixed thoroughly. The three-necked flask was then connected to a reflux condenser and heated in a water bath with magnetic stirring. Subsequently, the filtrate was concentrated at 60 °C. The evaporation dish containing the concentrated liquid was then placed in a drying box to obtain purified FA.Characterization
The infrared spectrum of purified FA was measuring using a Fourier-transform infrared spectrometer (VERTEX 80v, Bruker, Germany). Dried FA and potassium bromide were mixed and fully ground, and a tablet of the sample powder was created using a tablet press. The sample was then scanned in the wavenumber range of 400–4000 cm−1.The solid 13C spectrum of FA was measured by a fully digital NMR spectrometer (AVANCE III HD 600 MHz, Bruker, Germany). The 13C spectrum was measured in the range of 0–220 ppm using a 4 mm dual resonance solid-state probe.
Results and discussion
Experimental determination of elemental content and characteristic acid functional groups
Studies have shown that FA is mainly composed of C, H, and O and sometimes N and S. An elemental analyzer (5E-CHN2000, China Changsha Kaiyuan Instrument Co., Ltd) and CTS7000 automatic sulfur detector (China Xuzhou Tairui company) were used for the elemental analyses of C, H, N, and S; the O content was obtained by subtraction. The results are shown in Table 1.Elemental analysis showed that the FA had a lower carbon content and a higher oxygen content than Suwannee River I FA, the standard FA reported by the International Humic Substances Society (IHSS). This difference is attributed to the use of H2O2 in the extraction of FA.
The FA molecule is composed of a plurality of groups, including methoxy, phenolic hydroxyl, and carboxyl groups.2,17–20 These acidic groups are the structural basis for the unique biological and chemical activities of FA. The acidic groups also contribute the basic characteristics of FA and determine the mechanism of FA action. In this paper, the titration of common barium hydroxide and calcium acetate was used to determine the total acidic functional group and carboxyl functional group contents in FA. The phenolic hydroxyl group content was obtained by subtraction. The results are shown in Table 2.
| Sample | Total acidity (mmol g−1) | Carboxyl (mmol g−1) | Phenolic hydroxyl (mmol g−1) |
|---|---|---|---|
| FA | 11.15 | 8.65 | 2.50 |
The infrared spectrum of the FA in this study is shown in Fig. 1. The spectrum and characteristic peaks are similar to those of Suwannee River I FA, the standard FA published by the IHSS.
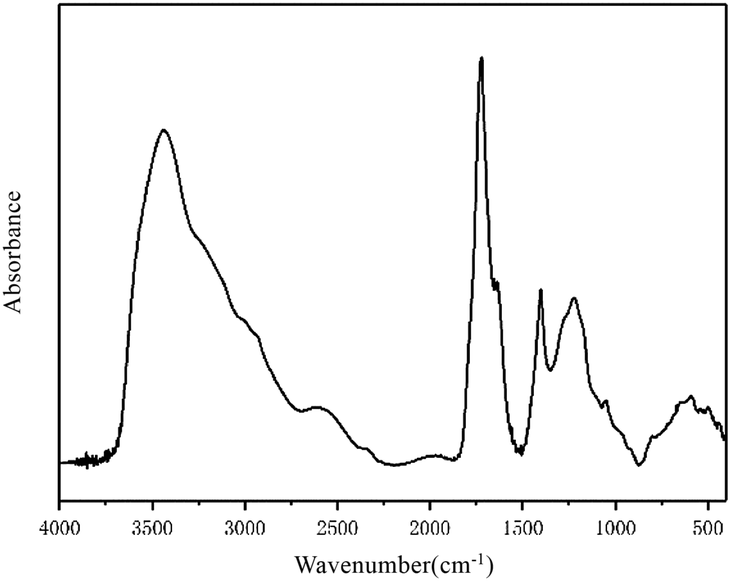 | ||
| Fig. 1 Fourier-transform infrared spectrum of FA. | ||
PeakFit software was used to fit the superimposed characteristic absorption peaks, and the approximate number and position were determined. By continuously adjusting and optimizing the number of characteristic peaks and peak parameters, the fitted spectrum agreed well with the original spectrum.
According to the different functional groups indicated by the FA infrared spectrum shown in Fig. 1, the spectrum can be divided into four main intervals: the hydroxy functional group interval from 3700–3000 cm−1; the aliphatic functional group interval from 3000–2800 cm−1; the oxygen-containing functional group interval from 1800–1000 cm−1; and the aromatic functional group interval from 900–700 cm−1. PeakFit was used to fit the peaks of the four intervals. According to the peak area intensity of each characteristic absorption peak, the attributions of specific characteristic peaks in each interval and the contents of different functional groups were determined quantitatively. The functional group contents were used to characterize the molecular structure of FA and provide partial structural parameters for the molecular model of FA.
The results for each characteristic peak are shown in Fig. 2. According to the characteristic peak information, the corresponding attributions were obtained and are listed in Table 3.
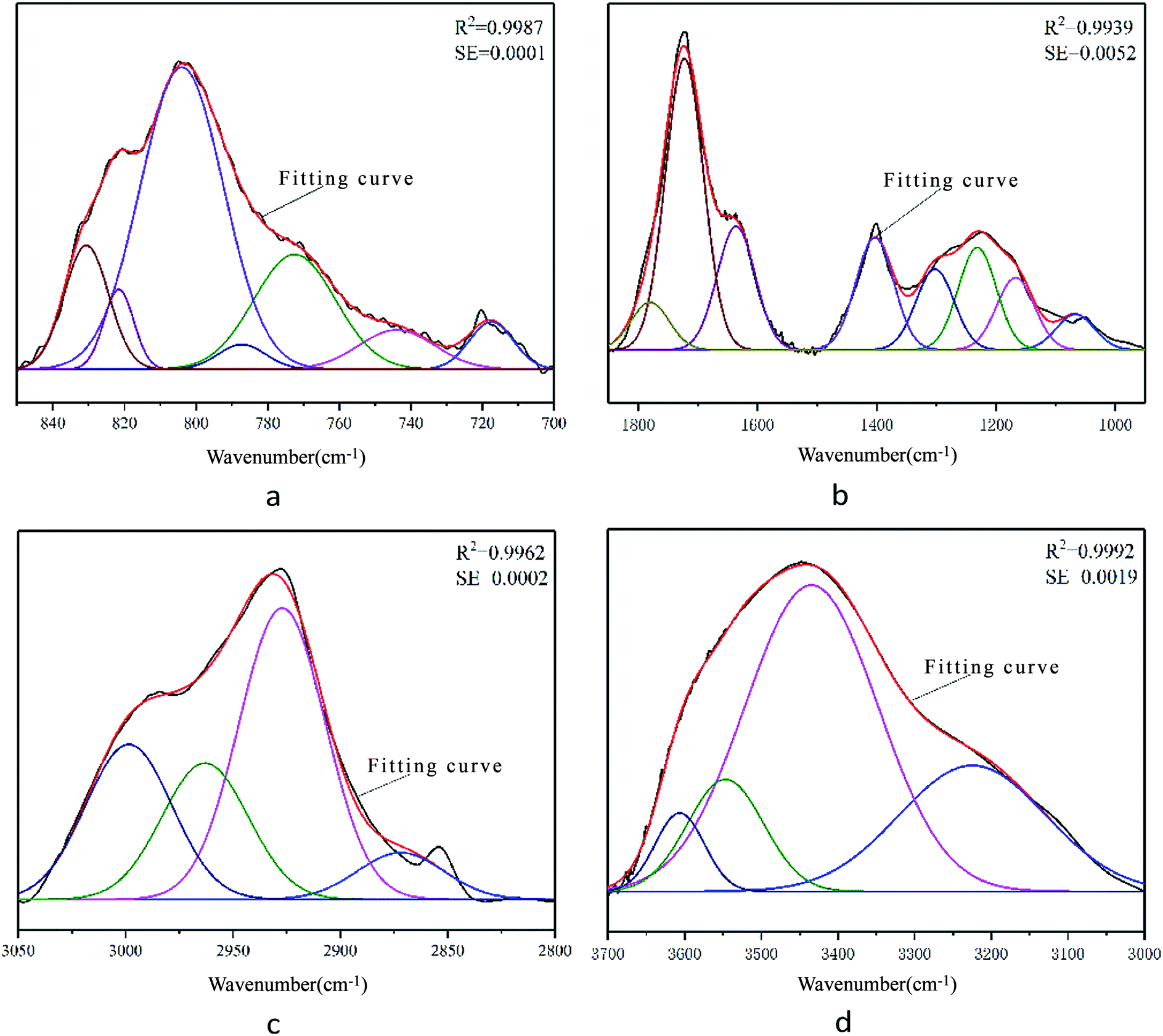 | ||
| Fig. 2 Infrared spectra of FA in different structural intervals showing the peak fitting curves for different functional groups: (a) aromatic functional groups; (b) oxygen-containing functional groups; (c) aliphatic functional groups; and (d) hydroxyl functional groups. | ||
| Number | Peak position (cm−1) | Half width (cm−1) | Peak height | Peak area intensity | Area percentage (%) | Attribution |
|---|---|---|---|---|---|---|
| 1 | 717 | 14.51 | 0.0014 | 0.022 | 4.3 | Di-substituted benzene ring |
| 2 | 744 | 25.05 | 0.0012 | 0.0316 | 6.16 | Tri-substituted benzene ring |
| 3 | 772 | 26.9 | 0.0035 | 0.0989 | 19.29 | Tri-substituted benzene ring |
| 4 | 787 | 15.66 | 0.0007 | 0.0124 | 2.42 | Tri-substituted benzene ring |
| 5 | 810 | 27.35 | 0.0091 | 0.2644 | 51.58 | Tetra-substituted benzene ring |
| 6 | 822 | 9.83 | 0.0024 | 0.0252 | 4.91 | Tetra-substituted benzene ring |
| 7 | 831 | 14.65 | 0.0037 | 0.0581 | 11.33 | Tetra-substituted benzene ring |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| 1 | 1067 | 74.96 | 0.0333 | 2.6535 | 4.1 | Alkyl ether C–O–C stretching vibration |
| 2 | 1167 | 74.96 | 0.0676 | 5.3958 | 8.33 | Aryl ether C–O–C stretching vibration |
| 3 | 1231 | 74.96 | 0.0959 | 7.6523 | 11.82 | Phenolic hydroxyl C–O stretching vibration |
| 4 | 1302 | 74.96 | 0.0757 | 6.0384 | 9.33 | Long carbon chain carboxylic acid C–O–H stretching vibration |
| 5 | 1403 | 74.96 | 0.1052 | 8.3949 | 12.96 | Hydroxyl C–O stretching vibration |
| 6 | 1636 | 74.96 | 0.1158 | 9.2425 | 14.27 | COO− antisymmetric stretching vibration |
| 7 | 1723 | 74.96 | 0.2734 | 21.8138 | 33.69 | Carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration O stretching vibration |
| 8 | 1780 | 74.96 | 0.0446 | 3.5623 | 5.5 | Carboxyl group CO stretching vibration |
|
||||||
| 1 | 2872 | 47.4 | 0.0015 | 0.0768 | 7.42 | RCH3 stretching vibration |
| 2 | 2927 | 47.4 | 0.0095 | 0.4786 | 46.29 | R2CH2 stretching vibration |
| 3 | 2963 | 47.4 | 0.0044 | 0.2239 | 21.66 | R2CH2 stretching vibration |
| 4 | 2999 | 47.4 | 0.005 | 0.2546 | 24.62 | RCH3 stretching vibration |
|
||||||
| 1 | 3225 | 225.34 | 0.0783 | 18.7853 | 25.84 | Alcoholic hydroxyl hydrogen bond |
| 2 | 3435 | 204 | 0.1903 | 41.3142 | 56.83 | Phenolic hydroxyl hydrogen bond |
| 3 | 3547 | 116.91 | 0.0697 | 8.6708 | 11.93 | Phenolic hydroxyl hydrogen bond |
| 4 | 3606 | 75.65 | 0.0488 | 3.9312 | 5.41 | Hydroxyl π hydrogen bond |
Fig. 2 shows that the theoretical fitting curves of the characteristic peaks are in good agreement with the original experimental spectral curves. The correlation coefficients R2 of the peak fittings were high, while the mean-square errors were low, indicating that the data of characteristic peaks obtained by peak fitting are reliable and can be used for the construction of the molecular structure model of FA.
From the peak fitting data of the FA infrared spectrum combined with the results of elemental analysis, important molecular model structural parameters such as aromatic hydrogen rate (fHar), hydrogen-to-carbon atomic ratio (H/C), fat chain length (CH3/CH2), and aromatic-to-carbon ratio (far) were calculated as 0.3314, 1.09, 0.4717, and 0.5951, respectively. The specific calculation process can be found in the ESI.†
13C NMR characterization and related parameter calculations
The FA sample was characterized by NMR spectrometry (AVANCE III HD 600 MHz, Bruker, Germany). The 13C spectrum of the sample in the range of 0–220 ppm is shown in Fig. 3.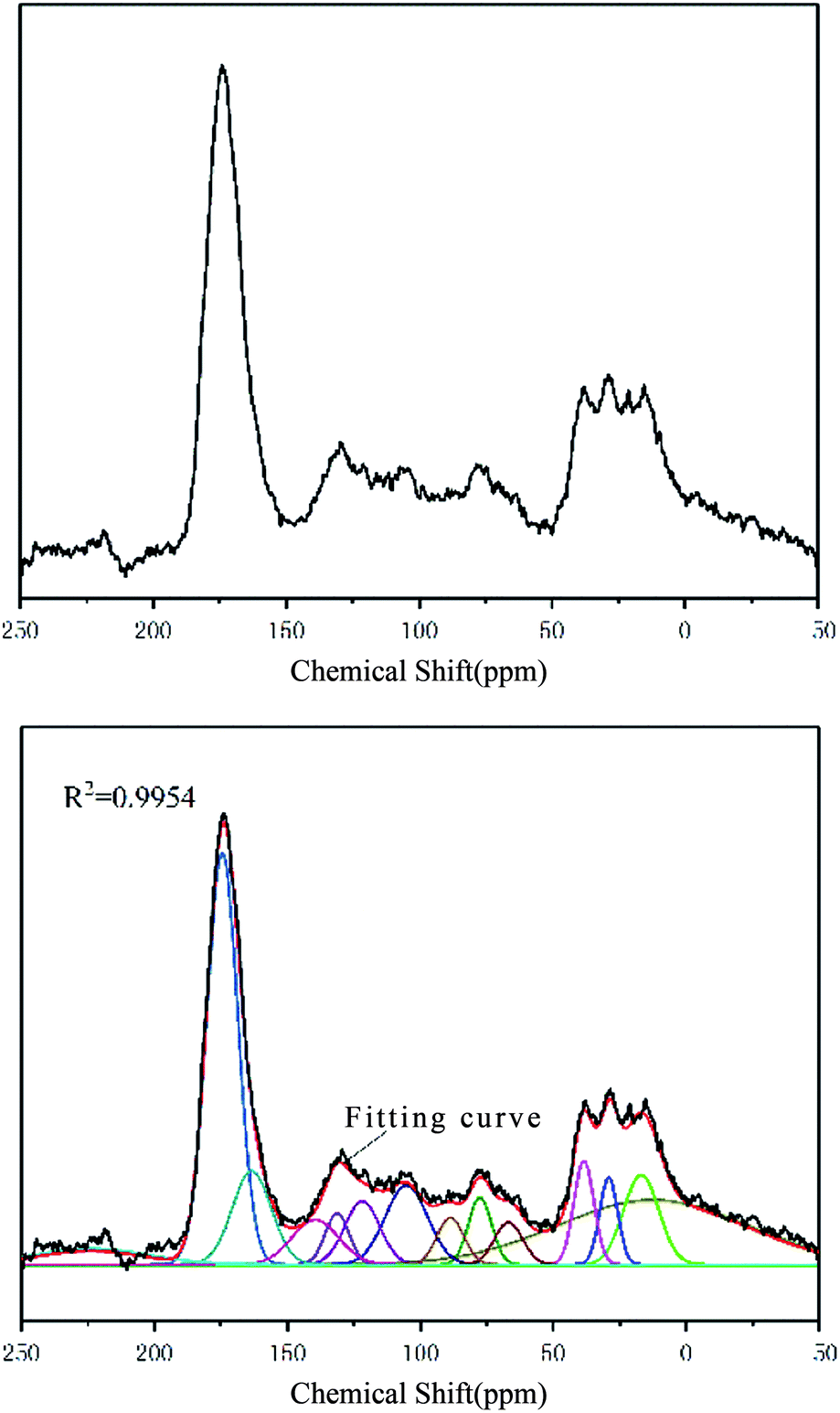 | ||
| Fig. 3 13C NMR spectrum of FA (top) and 13C NMR spectrum curve fitting of FA (bottom). | ||
The 13C NMR spectrum of FA can be obtained by peak fitting to obtain the detailed attributions of various carbon atoms observed at different chemical shifts. The 13C spectrum and chemical shifts of FA were similar to those of Suwannee River I FA. The specific parameters for each NMR peak are shown in Table 4.
| Number | Chemical shift (ppm) | Half width (ppm) | Peak height | Area strength (%) | Carbon atom attribution |
|---|---|---|---|---|---|
| 1 | 12.48 | 84.63 | 1036 | 23.23 | Aliphatic methyl |
| 2 | 16.89 | 15.70 | 1438 | 6.23 | Aromatic methyl |
| 3 | 29.09 | 7.99 | 1390 | 3.06 | Methylene and methine |
| 4 | 38.44 | 8.89 | 1661 | 4.07 | Methylene and methine |
| 5 | 66.75 | 12.56 | 675 | 2.34 | Oxygen connecting methine |
| 6 | 77.50 | 10.29 | 1077 | 3.06 | Oxygen connecting methine |
| 7 | 88.63 | 11.97 | 750 | 2.48 | Oxygen connecting aliphatic carbon in the ring |
| 8 | 105.22 | 18.74 | 1269 | 6.56 | Protonated aromatic carbon |
| 9 | 121.82 | 15.41 | 1019 | 4.33 | Protonated aromatic carbon |
| 10 | 131.28 | 10.59 | 818 | 2.39 | Aromatic bridge carbon |
| 11 | 139.48 | 20.71 | 720 | 4.11 | Lateral aromatic carbon |
| 12 | 163.37 | 17.28 | 1498 | 7.14 | Oxygen substituted aromatic carbon |
| 13 | 174.47 | 13.81 | 6582 | 25.07 | Carboxy carbon |
| 14 | 215.86 | 51.37 | 256 | 3.23 | Carbonyl carbon |
According to the intensity and position of the NMR peaks corresponding to different carbon atoms in FA (Table 4), the relative contents of various carbon atoms were calculated. According to the calculation method23–25 of carbon skeleton parameters, the 12 parameters of the FA carbon skeleton structure and the derived parameter information were obtained as follows:
fa: aromatic carbon, the ratio of sp2 hybrid carbon atoms to the total number of carbon atoms, δ = 90–220 ppm;
fCa: the ratio of carboxyl carbon or carbonyl carbon to the total number of carbon atoms, δ = 165–220 ppm;
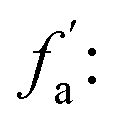 ratio of sp2-hybridized carbon atoms in the aromatic ring to the total number of carbon atoms, δ = 90–165 ppm;
ratio of sp2-hybridized carbon atoms in the aromatic ring to the total number of carbon atoms, δ = 90–165 ppm;
fHa: the ratio of the number of protonated carbon atoms in the aromatic ring to the total number of carbon atoms, δ = 100–129 ppm;
fNa: the ratio of the number of non-protonated carbon atoms in the aromatic ring to the total number of carbon atoms;
fPa: the ratio of the number of carbon atoms to which the phenolic hydroxyl or ether linkage is attached to the total number of carbon atoms, δ = 150–165 ppm;
fSa: the ratio of alkyl-substituted aromatic carbon atoms in the aromatic ring to the total number of carbon atoms, δ = 135–150 ppm;
fBa: the ratio of the carbon atoms of the bridgehead in the aromatic ring to the total number of carbon atoms, δ = 129–135 ppm;
fal: aliphatic carbon, the ratio of sp3-hybridized carbon atoms to the total number of carbon atoms;
fHal: the ratio of the number of carbon atoms in the methine and methylene groups to the total number of carbon atoms, δ = 22–50 ppm;
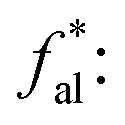 the ratio of methyl carbon and quaternary carbon atoms to the total number of carbon atoms;
the ratio of methyl carbon and quaternary carbon atoms to the total number of carbon atoms;
fOal: the ratio of the number of aliphatic carbon atoms attached to oxygen to the total number of carbon atoms, δ = 50–90 ppm;
fOa: the ratio of the number of carbon atoms in a ketone to an aldehyde to the total number of carbon atoms, δ = 185–220 ppm; and
fOOa: the ratio of amide, lipid, and carboxyl carbon atoms to the total number of carbon atoms, δ = 165–185 ppm.
Using the NMR spectrum processing software MestReNova, the resonance peak areas (Ix, where x is the wavenumber) of different types of carbon atoms in the spectrum were integrated. The total peak area (Itotal) in the range of 0–220 ppm was normalized to calculate the relative contents of various carbon atoms in the FA carbon skeleton structure (Table 5).
| Parameter | Proportion (%) | Parameter | Proportion (%) |
|---|---|---|---|
| fa | 61 | fal | 39 |
| fCa | 39 | fHal | 20 |
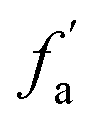 |
22 | 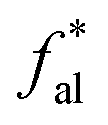 |
28 |
| fPa | 6 | fOa | 10 |
| fSa | 4 | fOOa | 24 |
| fHa | 11 | 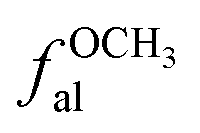 |
2 |
| fBa | 3 | 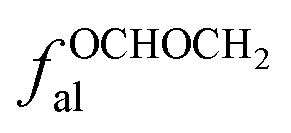 |
10 |
| fNa | 13 |
Establishment of the molecular structure model and its theoretical basis
Currently, when constructing the molecular structure model of coal-based materials, there are two methods for determining the number of carbon atoms in the model. The first way is to directly determine the number of carbon atoms in the model based on relevant research results and experimental data. For example, according to coal quality data from North China, East China, South China, and Southwest China, Wenkuan26 constructed the molecular structure model of lignite; the molecular formula was C102H96O36, and the relative molecular mass was 1736. Kuangzong et al.27 used this method to construct the molecular structure model of brown coal in Huangxian County in China; the relative molecular mass was 1746. The second way is based on relevant experience; the relative molecular weight of the molecular model is first determined, and the number of carbon atoms in the model is then calculated based on experimental detection data to establish the molecular model. For example, based on previous research results, Sanyue et al.28 assumed the relative molecular weight of the lignite molecular structure model to be approximately 3000 and then determined the molecular formula of the Yima brown coal molecular model to be C197H152O32N2S2. The relative molecular mass was 3120. Diallo et al.29 generated a three-dimensional structural model of HA by combining experimental characterization, computer-aided structural analysis, and atomic simulation. These models are consistent with the analytical data and the overall thermodynamic/structural properties of HA. According to the relationship between the structural characteristics of lignite yellow and brown HA and lignite, the second method was used to determine the number of carbon atoms in the model.The systems calculated based on quantum chemistry generally have uniform and fixed molecular structures; however, FA is a relatively complex mixture, and its molecular structure and specific molecular weight are difficult to determine. The structure of FA can be considered to be composed of many molecular structural units with similar characteristic structures. Therefore, based on a combination of molecular dynamics, quantum chemistry, software simulation, chemical bond theory, and experimental data, the characteristic molecular structure was determined, and the structural model was constructed. Because FA is derived from the catalytic oxygenation extraction of lignite, the molecular weight should be significantly smaller than that of lignite. Combined with existing research results and the author's previous experimental research, we can reasonably assume the molecular weight of the constructed FA molecular structure model to be approximately 1000. Combined with the relative molecular weight limitation of FA and the data shown in Table 1, sulfur was neglected when constructing the model due to its low content. Therefore, the molecular model of FA calculated based on elemental analysis data is similar to the chemical formula, which can be C33H36NO33, C34H37NO35, or C35H38NO36. Considering the relative ratio of N to other elements, we chose C33H36NO33, and the relative molecular mass was 974. From the infrared spectrum of FA and the peak-matching data of the 13C solid-state NMR spectrum, the molecular structural parameters of FA were obtained to calculate the compositions of different carbon atoms in the carbon skeleton of the FA molecular model. The calculation process was as follows:
Number of carbon atoms in the benzene ring: 33 × 0.61 = 20.13, approximately 20;
Number of carbon atoms attached to aryl ethers and phenols: 33 × 0.06 = 1.98, approximately 2;
Number of carbon atoms on the carboxyl or carbonyl group: 33 × 0.39 = 12.87, approximately 13;
Number of carbon atoms in aliphatic carbon: 33 × 0.39 = 12.87, approximately 13;
Number of carbon atoms in –OCH and –OCH: 33 × 0.1 = 3.3, about 3;
Number of carbon atoms in methylene and methine groups: 33 × 0.20 = 6.6, approximately 7; and
Number of carbon atoms in the alkyl-substituted aromatic carbon on the benzene ring: 33 × 0.04 = 1.32, approximately 2.
The number of carbon atoms on the benzene ring was approximately 20; thus, the number of benzene rings in the molecular model was tentatively set to three. According to the IR spectral data of FA in the range of 700–900 cm−1, the substitution pattern of the benzene ring in the aromatic structure includes three primary forms: di-substituted, tri-substituted, and tetra-substituted. Among these three substitution patterns, the content tetra-substituted benzene ring was the highest (67.82%) followed by tri-substituted (27.87%) and di-substituted (4.30%). Thus, di-substituted benzene ring was ignored when constructing the molecular model, and the ratio of tetra-substituted to tri-substituted benzene ring was 2.43:1 (approximately 2:1 according to the simulation calculations and the optimal value principle). Thus, the tentative model contained two tetra-substituted benzene rings and one tri-substituted benzene ring. The results of titration experiments with acidic functional groups showed that the carboxyl group content in FA was approximately three times the phenolic hydroxyl group content. Combined with the spectral data for oxygen-containing functional groups in the range of 1000–1800 cm−1 and the relative content of oxygen, the molecular model contained two ether bonds, one alcoholic hydroxyl group, two phenolic hydroxyl groups, and seven carboxyl groups. The remaining oxygen content corresponds primarily to the carbonyl group. Combining the test data3,21,23–27,29–31 with the findings of past research on HA and FA, the numbers and positions of functional groups were continuously adjusted to approximate the experimental test parameters. The resulting two-dimensional planar molecular structure model of FA is shown in Fig. 4.
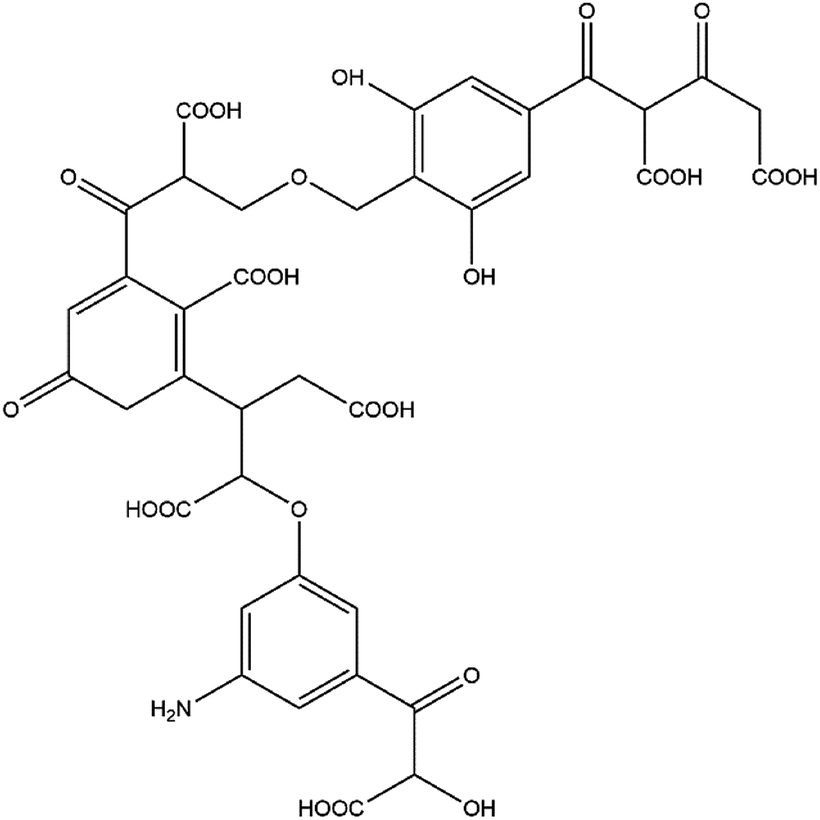 | ||
| Fig. 4 Average molecular structure model of FA. | ||
The molecular formula is C38H32NO24, and the relative molecular mass is 886. In the molecular structure model of FA, the benzene ring is the main component of the aromatic structure, including the tetra-substituted and tri-substituted benzene rings. The model contains many oxygen-containing functional groups, including carboxyl, carbonyl, hydroxyl, ether, and semiquinone groups, consistent with the high oxygen content obtained by elemental analysis. Most of the carboxyl groups are present at the end of the aliphatic structure, and each benzene ring structure is connected by an ether oxygen bridge. The relative content of each element in the molecular model is close to the experimental test data. The parameters of the constructed FA molecular structure model and the experimental test values are in good agreement (within the range of instrumental test error and calculation error). The molecular model can be considered to be close to the true molecular structure of FA.
Quantum chemistry optimization and testing of molecular structure models
In this study, the molecular model was optimized using quantum chemical calculations to minimize the theoretical energy, thereby obtaining the theoretical steady state. A two-dimensional average molecular structure model of FA was constructed using ChemDraw. The model was transformed into a three-dimensional molecular structure using ChemBio3D. The molecular structure was then optimized using molecular mechanics, and the structure data were imported into GaussView software. Quantum chemical calculations were then performed on the molecular model using Gaussian 09W software. According to Hartree–Fock (HF) theory, the molecular structure was optimized via energy minimization at the HF/3-21G level of theory. As the molecular structure was iteratively calculated, the iterative result was gradually reduced. When the result of multiple iterations was below a certain fixed value, the molecular model was considered to be in a convergent state, and the average molecular structure was constructed. At this time, the model reached the theoretical steady state with an optimal structure. Finally, the theoretical frequency spectrum of the determined optimal structure was calculated and compared with the actual experimental spectrum. The optimized molecular model was then adjusted until the calculated and experimental spectra matched closely.We used the same HF/3-21G basis set in the energy optimization process for frequency calculation. The two three-dimensional structural models (ball-and-stick and stick models) corresponding to the optimal average molecular structure of FA were constructed by calculation, as shown in Fig. 5.
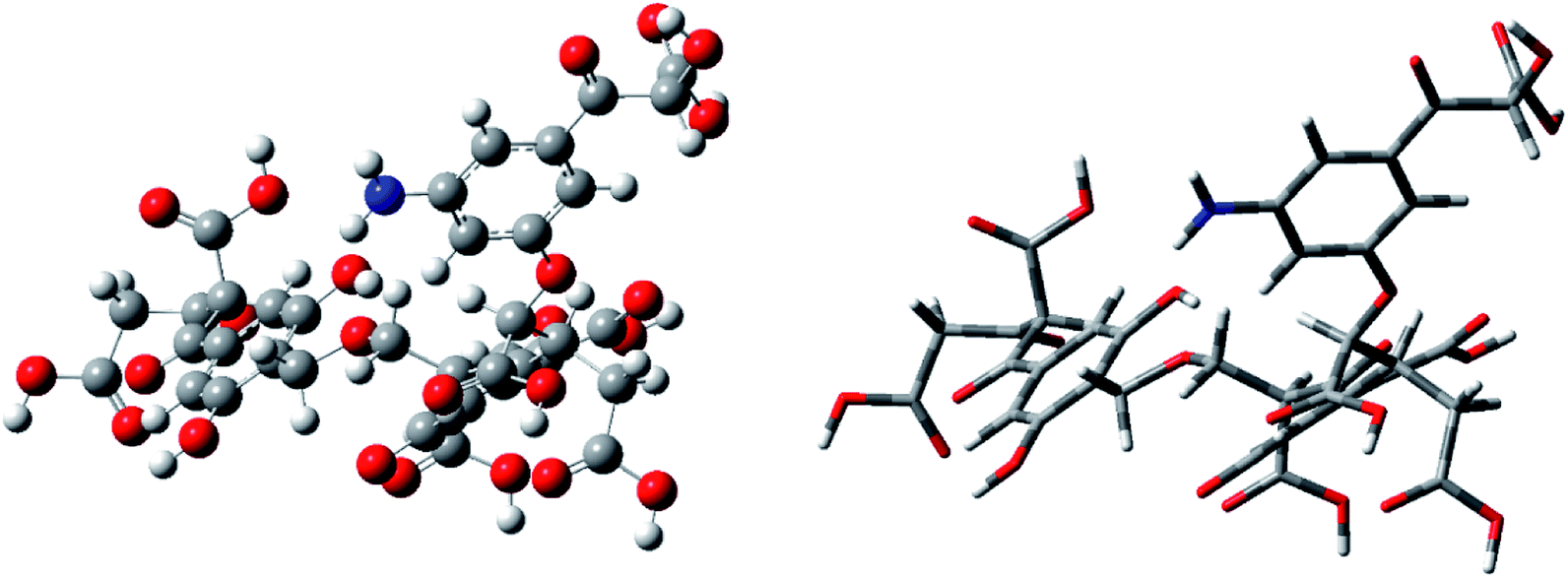 | ||
| Fig. 5 Optimized geometries of FA depicted by ball-and-stick and stick models. In the ball-and-stick model, gray balls represent carbon atoms, white balls represent hydrogen atoms, red balls represent oxygen atoms, and blue balls represent nitrogen atoms. In the stick model diagram, the gray sticks represent carbon bonds, the white sticks represent hydrogen bonds, the red sticks represent oxygen bonds, and the blue sticks represent nitrogen bonds. | ||
Fig. 5 shows that after the optimization using the HF/3-21G basis set, the molecular structure model of FA became very compact, and the three-dimensional structural features became obvious. At this time, the molecular energy of the model is the smallest, and the molecular configuration achieves theoretical stability. In the steady state, the constructed molecular model is closer to the real FA molecular structure, indicating that the model data are highly reliable.
Frequency calculations are usually based on the second-order derivation of the atomic position of the energy. Using Gaussian software and HF theory, the theoretical infrared spectrum of the FA molecular model was calculated at the 3-21G level of theory. It is worth noting that HF calculation does not consider the electron correlation effect, which results in a small amount of systematic error in the infrared spectrum and causes the overall peak position of the characteristic absorption to be too high. To reduce the systematic error, the calculated infrared spectrum was corrected using the HF-related frequency correction factor of 0.9085.32 Fig. 6 compares the theoretical spectrum after error correction with the experimentally measured spectrum. The two are similar in peak position and peak shape, showing a high degree of matching. There is no virtual frequency in the calculation results, indicating that the optimized molecular configuration is located at a small point on the potential energy surface. However, due to the idealization of the Gaussian calculation process, the anharmonic effect of the molecules in the real state is neglected, resulting in a smaller portion of the absorption peak in the calculated spectrum that is not completely consistent with the experimental spectrum.
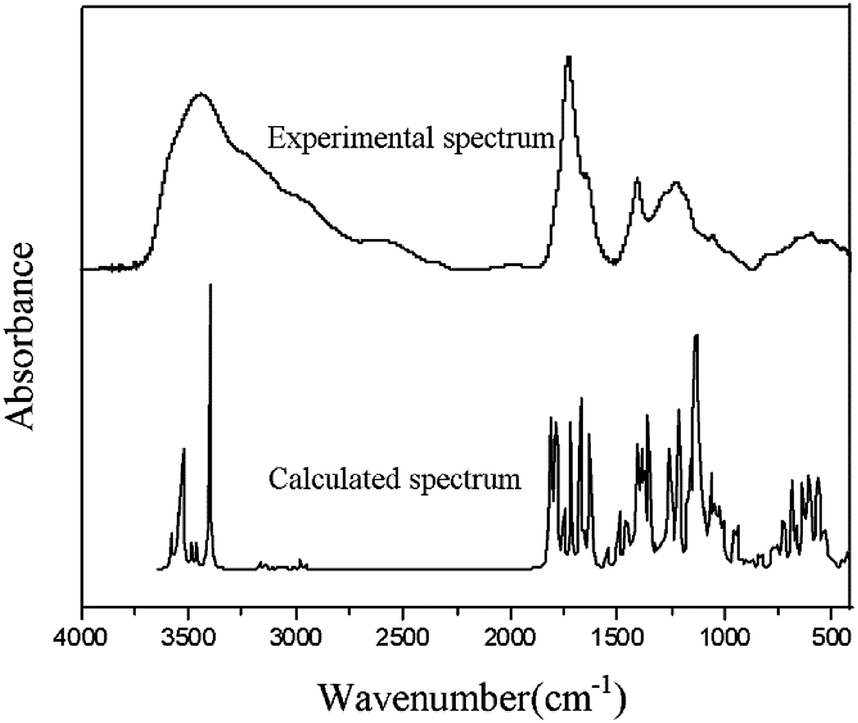 | ||
| Fig. 6 Comparison of the calculated and experimental spectra of FA. | ||
In addition, as shown in Fig. 6, the experimental spectrum includes four wavenumber intervals (3000–3800, 1500–1900, 1000–1500, and 500–900 cm−1), and the calculated spectrum shows obvious characteristic absorption peaks corresponding to those intervals. The positions of most of the characteristic absorption peaks are quite consistent; however, some differences in absorption peak intensity are observed. For example, the experimental spectrum shows a broad and large absorption peak in the range of 3000–3800 cm−1, which is mainly attributed to the vibrations of different types of hydroxyl hydrogen bonds in FA. Several obvious peaks are found in the calculated spectrum in this range (e.g., two strong absorption peaks at 3385 and 3507 cm−1, which are attributed to the stretching vibrations of the alcohol hydroxyl group and the phenolic hydroxyl group). In the calculated spectrum, more characteristic absorption peaks are found in the range of 400–2000 cm−1 than from 3000–3800 cm−1. The coincidence degree of the vibration frequency for the low-wavenumber interval in the two spectra is higher than that of the high-frequency band. Calculating the infrared spectrum reveals the characteristic peaks of each functional group in the molecular structure; therefore, the calculated spectrum contains many narrow and small characteristic peaks. Because of the superposition of the peaks and the absorption of various functional groups in the molecule, the experimental spectrum shows less broad and large superimposed absorption peaks.
Conclusions
In this study, based on classical chemical titration experiments, modern chemical detection techniques, and theoretical molecular modeling using Gaussian 09W, ChemDraw, ChemBio3D, and PeakFit software, the elemental components and molecular structure of FA were revealed. The results indicated a chemical formula of C38H32NO24 and relative molecular mass of 886. Both a two-dimensional planar molecular structure and three-dimensional characteristic model were constructed for FA. Using quantum chemistry and HF theory, the molecular configuration was optimized based on energy minimization, and the three-dimensional structure and characteristic functional group distribution of FA were revealed. The molecular structure model of FA matches well with the experimental data. The model effectively explains the molecular structure of FA and provides a theoretical basis for fundamental and applied FA research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 21576281 and 21776299), the China Coal Industry Association Scientific and Technological Guidance Project (Grants MTKJ2015-220 and MTKJ2012-288), and the China University of Mining and Technology teaching reform project for postgraduate education (YJSJG-2018-01, 2019YJSJG043).Notes and references
- R. Che, L. Huang, J. Xu, P. Zhao, T. Li, H. Ma and X. Yu, Effect of fulvic acid induction on the physiology, metabolism, and lipid biosynthesis-related gene transcription of Monoraphidium sp FXY-10, Bioresour. Technol., 2017, 227, 324–334 CrossRef CAS PubMed.
- J. A. Leenheer, R. L. Wershaw and M. M. Reddy, Strong-acid, carboxyl-group structures in fulvic acid from the suwannee river, georgia. 2. Major structures, Environ. Sci. Technol., 1995, 29(2), 399–405 CrossRef CAS PubMed.
- Y. Qin, H. Zhu, M. Zhang, H. Zhang, C. Xiang and B. Li, GC-MS Analysis of Membrane-Graded Fulvic Acid and Its Activity on Promoting Wheat Seed Germination, Molecules, 2016, 21(10), 1363 CrossRef PubMed.
- Z. Wang, Y. Xu and A. Peng, Influences of fulvic acid on bioavailability and toxicity of selenite for wheat seedling and growth, Biol. Trace Elem. Res., 1996, 55(1), 147 CrossRef CAS PubMed.
- Y. R. Abdel-Baky, H. F. Abouziena, A. A. Amin, M. R. El-Sh and A. M. A. El-Sttar, Improve quality and productivity of some faba bean cultivars with foliar application of fulvic acid, Bull. Natl. Res. Cent., 2019, 43(1), 2 CrossRef.
- M. Wu, M. Song, M. Liu, C. Jiang and Z. Li, Fungicidal activities of soil humic/fulvic acids as related to their chemical structures in greenhouse vegetable fields with cultivation chronosequence, Sci. Rep., 2016, 6, 32858 CrossRef CAS PubMed.
- H. Haan, Effect of a fulvic acid fraction on the growth of a Pseudomonas from Tjeukemeer (The Netherlands), Freshwater Biol., 2010, 4(3), 301–310 CrossRef.
- A. M. Beer, J. Lukanov and P. Sagorchev, The influence of fulvic and ulmic acids from peat, on the spontaneous contractile activity of smooth muscles, Phytomedicine, 2000, 7(5), 407–415 CrossRef CAS PubMed.
- C. P. Mcmurphy, G. C. Duff, M. A. Harris, S. R. Sanders, N. K. Chirase, C. R. Bailey and R. M. Ibrahim, Effect of humic/fulvic acid in beef cattle finishing diets on animal performance, ruminal ammonia and serum urea nitrogen concentration, J. Appl. Anim. Res., 2009, 35(2), 97–100 CrossRef CAS.
- R. G. P. T. Jayasooriya, M. G. Dilshara, C. H. Kang, S. Lee, Y. H. Choi, K. J. Yong and G. Y. Kim, Fulvic acid promotes extracellular anti-cancer mediators from RAW 264.7 cells, causing to cancer cell death in vitro, Int. Immunopharmacol., 2016, 36, 241–248 CrossRef CAS PubMed.
- J. Schellekens, P. Buurman, K. Kalbitz, A. V. Zomeren and R. N. J. Comans, Molecular Features of Humic Acids and Fulvic Acids from Contrasting Environments, Environ. Sci. Technol., 2017, 51(3), 1330–1339 CrossRef CAS PubMed.
- K. Murray and P. W. Linder, Fulvic Acids: Structure and Binding I. A Random Molecular Model, Eur. J. Soil Sci., 1983, 34(3), 511–523 CrossRef CAS.
- N. Senesi, Molecular and quantitative aspects of the chemistry of fulvic acid and its interactions with metal ions and organic chemicals : Part I. The electron spin resonance approach, Anal. Chim. Acta, 1990, 232(3), 51–75 CrossRef CAS.
- L. Jing, J. H. Qu, H. J. Liu, R. P. Liu, Z. Xu and Y. N. Hou, Species transformation and structure variation of fulvic acid during ozonation, Sci. China: Chem., 2008, 51(4), 373–378 CrossRef.
- Z. Klencsár and Z. Köntös, EPR Analysis of Fe3+ and Mn2+ Complexation Sites in Fulvic Acid Extracted from Lignite, J. Phys. Chem. A, 2018, 122(12), 3790–3203 CrossRef PubMed.
- A. A. Chaaban, B. Lartiges, V. Kazpard, C. Plisson-Chastang, L. Michot, I. Bihannic, C. Caillet and B. Prelot, Probing the organization of fulvic acid using a cationic surfactant, Colloids Surf., A, 2016, 504, 252–259 CrossRef CAS.
- V. Romarís-Hortas, A. Moreda-Piñeiro and P. Bermejo-Barrera, Application of microwave energy to speed up the alkaline extraction of humic and fulvic acids from marine sediments, Anal. Chim. Acta, 2007, 602(2), 202–210 CrossRef PubMed.
- A. Fattahi and T. Solouki, Using solution equilibria to determine average molecular weight of the Suwannee River fulvic acids, Anal. Chim. Acta, 2003, 496(1–2), 325–337 CrossRef CAS.
- X. Ma and S. A. Green, Fractionation and spectroscopic properties of fulvic acid and its extract, Chemosphere, 2008, 72(10), 1425–1434 CrossRef CAS PubMed.
- L. M. Y. Iii and R. V. Wandruszka, Functional group analysis of Suwannee River fulvic acid with reactive fluorescent probes, Fresenius' J. Anal. Chem., 1999, 364(8), 746–748 CrossRef.
- K. Shi, X. Tao, Z. Li and D. Kong, Construction of Fushun Coal Macromolecular Structure Model by Infrared Spectroscopy, Chin. Polym. Bull., 2013,(3), 61–66 Search PubMed.
- H. Z. Liang, C. G. Wang, F. G. Zeng, L. I. Mei-Fen and J. H. Xiang, Effect of demineralization on lignite structure from Yinmin coalfield by FT-IR investigation, J. Fuel Chem. Technol., 2014, 42(02), 129–137 CAS.
- L. Yao, J. Zhang and S. Dou, Three-dimensional visualization and characterization of several molecular structures of humic acid and fulvic acid, Chin. J. Soil Sci., 2008, 39(01), 57–61 CAS.
- M. S. Solum, R. J. Pugmire, M. Jagtoyen and F. Derbyshire, Evolution of carbon structure in chemically activated wood, Carbon, 1995, 91, 247–1254 Search PubMed.
- H. Yang, Y. Li and Y. Zhang, Characterization of molecular structure of humic acid by infrared spectroscopy and nuclear magnetic resonance, Meitan Zhuanhua, 2013, 36(4), 72–76 CAS.
- W. Yang, Thermal degradation mechanism and hydrocarbon generation rate of humic coal, Oil Gas Geol., 1987, 8(01), 26–37 Search PubMed.
- K. Qin and P. Zhao, Study on the chemical structure of lignite in Huangxian County by solid 13C NMR technique, J. Fuel Chem. Technol., 1990, 18(01), 3–9 Search PubMed.
- S. Wang, F. Zeng, C. Tian and T. Zhang, Application and Progress of Molecular Simulation in the Study of Coal Macromolecular Structure Evolution, J. Taiyuan Univ. Technol., 2004, 35(05), 541–544 CAS.
- M. S. Diallo, S. Andre, G. Paul, F. Jean Loup, J. H. Johnson, W. A. Goddard and P. G. Hatcher, 3-D structural modeling of humic acids through experimental characterization, computer assisted structure elucidation and atomistic simulations. 1. Chelsea soil humic acid, Environ. Sci. Technol., 2003, 37(9), 1783–1793 CrossRef CAS PubMed.
- Y. I. Tarasevich, M. Y. Tryfonova, S. A. Dolenko and E. V. Aksenenko, Adsorption-based approach to determine the size and mass of humic acids molecules, Adsorpt. Sci. Technol., 2016, 34(2–3), 125–133 CrossRef CAS.
- M. Klučáková, Size and Charge Evaluation of Standard Humic and Fulvic Acids as Crucial Factors to Determine Their Environmental Behavior and Impact, Front. Chem., 2018, 6, 235 CrossRef PubMed.
- A. P. Scott and L. Radom, Harmonic Vibrational Frequencies: An Evaluation of Hartree–Fock, Møller–Plesset, Quadratic Configuration Interaction, Density Functional Theory, and Semiempirical Scale Factors, J. Phys. Chem., 1996, 100(41), 16502–16513 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09907g |
| ‡ Guan-qun Gong, Xin Yuan and Yingjie Zhang contributed equally to this work and all of them are the co-first author. |
| This journal is © The Royal Society of Chemistry 2020 |