
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethanol to aromatics: isolated zinc phosphate groups on HZSM-5 zeolite enhance BTX selectivity and catalytic stability†
Jian Qiao a,
Jianqiang Wanga,
Anatoly I. Frenkelb,
Jiawei Tenga,
Xiqiang Chena,
Jingxian Xiaoa,
Tiezhu Zhanga,
Zhendong Wanga,
Zhiqing Yuana and
Weimin Yang*a
a,
Jianqiang Wanga,
Anatoly I. Frenkelb,
Jiawei Tenga,
Xiqiang Chena,
Jingxian Xiaoa,
Tiezhu Zhanga,
Zhendong Wanga,
Zhiqing Yuana and
Weimin Yang*a
aState Key Laboratory of Green Chemical Engineering and Industrial Catalysis, Sinopec Shanghai Research Institute of Petrochemical Technology, Shanghai 201208, China. E-mail: yangwm.sshy@sinopec.com; Fax: +86-21-68462283; Tel: +86-21-68462197
bDepartment of Materials Science and Chemical Engineering, Stony Brook University, Stony Brook, NY 11794, USA
First published on 5th February 2020
Abstract
HZSM-5 zeolite combined with unique zinc and phosphorus species, yields excellent selectivity (∼85%) to BTX (benzene, toluene, xylenes) in aromatic products. It was found that both zinc and phosphorus species were highly distributed in the pores of the zeolite channel network to form isolated zinc phosphate groups, which directly bond to the surface of zeolite, leading to a strong Lewis acidic center and an optimized surface acidity distribution favorable for BTX formation and the hydrothermal stability of the catalyst.
The conversion of methanol to aromatics (MTA) has been turning into an increasingly important alternative source of light aromatics including benzene (B), toluene (T), xylenes (X), with the development of coal-, shale gas-, or biomass-derived methanol via the syngas route.1–4 By modifying HZSM-5 with zinc ions,1–14 light aromatics could be selectively produced. However, it has many drawbacks by exchanging zinc ions directly to HZSM-5, such as low selectivity, loss of zinc15 and the coke formation on the surface of the catalyst. The catalytic dehydration of methanol produces large amounts of water vapor during MTA and the subsequent regeneration process at temperatures higher than the reaction temperature,16–18 call for catalysts with high hydrothermal stability.19 Therefore, the preservation of good activity and selectivity in the severe reaction and regeneration conditions of MTA processes requires the catalysts to maintain good thermal and hydrothermal stabilities, moreover, understanding the nature of, and controlling over stability of both zinc ions and HZSM-5 framework of the catalyst are among the main challenges in the field.
Modification of HZSM-5 by phosphorus (referred to as P/HZSM-5) is extensively used to improve the hydrothermal stability of HZSM-5 zeolite18,20–24 in converting methanol to light olefins,25 alkylation and/or disproportionation of aromatics.26,27 However, the sole use of P-modified HZSM-5 catalyst is rarely reported in MTA reaction,7,28 to the best of our knowledge. As shown in this work, the BTX formation over P/HZSM-5 catalyst is limited in comparison with Zn/HZSM-5 catalyst. Nevertheless, it is generally accepted that phosphorus inhibits the dealumination of zeolites, which, in turn, increases their hydrothermal stability and catalytic selectivity. Our hypothesis is that the combination of P-modified HZSM-5 catalyst and Zn/HZSM-5 catalyst could enable both the enhancement of hydrothermal stability and aromatics selectivity of MTA catalysts. Several groups have reported that adding phosphorus to Zn/HZSM-5 promotes the activity and stability of the catalysts in light alkane aromatization by decreasing the loss of zinc,29–31 as well as in aromatization of fluid catalytic cracking (FCC) fractions.31–34 There are only a handful of ZnPHZ catalyst types used in MTA reaction,35,36 and most of such reports have been focused on the reaction studies, not the catalyst structure. In the remainder of the Introduction, we will present our motivation of synthesizing a hybrid ZnPHZ catalyst.
Controlling acid properties is among the key factors for achieving selective transformation using HZSM-5 zeolites37,38 In MTA reaction, the Lewis nature of hydroxyl zinc ions can accelerate the recombination of surface hydrogen,39,40 suppress the cracking process to C3 fragments by decreasing stronger Brönsted acid and enhance cyclization and dehydrogenation of C6=–C9= oligomer intermediates to aromatics.40 Hersh et al.41 reported reaction of NH3 with the zinc ions of zinc phosphate glasses to form coordinately bound NH3, showing the Lewis nature of the insoluble zinc phosphate. Yuan et al.42 reported the possibility to synthesize hydroxyl zinc phosphate materials by reacting phosphate groups with zinc nitrate. Hence, in order to reduce zinc loss in exchanged bridging hydroxyl groups during MTA process, an incorporation of stabilized phosphorus groups seems to be an attractive alternative. By anchoring the phosphorus group on P/HZSM-5 catalyst with the zinc species to form the insoluble zinc phosphate groups, it may inherit the hydrothermal stability from P-modified catalysts. In addition, the formation of insoluble zinc phosphate groups might contribute its Lewis nature for altering the acidity distribution, causing better aromatization properties in MTA reaction, as well as stabilizing the hydroxyl zinc ions by the formation of insoluble zinc phosphate groups.
Herein, we report preparation of Zn-P/HZSM-5 catalyst with isolated zinc phosphate groups, verification of its catalyst performance and characterization of the local structure and chemical state of Zn and its nearest neighbors that shed light on the nature of the Zn-P/HZSM-5 complex. The formation of zinc phosphate groups by adding zinc ions to P/HZSM-5 catalyst is studied by NMR. Detection of possible incorporation of zinc into phosphate groups and characterization of its nearest coordination sphere are accomplished by using XANES and EXAFS techniques in combination with FTIR measurements of the change of bridging hydroxyl groups. The Lewis nature of as-prepared zinc phosphate groups on Zn-P /HZSM-5 catalyst are identified by NH3-TPD (temperature programmed desorption of ammonia) and Py-FTIR (FTIR spectroscopy using pyridine as a probe molecule) measurements. Finally, the catalyst performance of ZnPHZ (short for Zn-P /HZSM-5) in MTA reaction is interpreted by correlating catalyst structure and its acidic properties (the distribution of Brönsted acid and Lewis acid), as well as the comparative research with the reference catalysts including the parent HZSM-5, P/HZSM-5, Zn/HZSM-5 and P-Zn/HZSM-5, denoted as HZ, PHZ, ZnHZ and PZnHZ respectively as mentioned above.
The structural and crystalline characteristics of the samples were studied by XRD (ESI, Fig. S1a†). All of the samples were well crystallized as the MFI structure, as deduced from the XRD spectra. Table 1 shown the relative crystallinity of the samples as compared to the parent HZ sample. The relative crystallinity decreased to a certain extent for sample impregnated with orthophosphoric acid, and this might be due to the dealumination of the zeolite sample, surface modification and thermal treatment. After the addition of zinc to PHZ sample, the crystallinity of HZ for ZnPHZ sample decreased significantly, but the HZ support still kept the origin structure. Synchrotron based XRD (ESI, Fig. S1b†) and scanning TEM (ESI, Fig. S2†) were used to further confirm the absence of zinc oxides or zinc phosphate particles formed on the catalyst surface, indicating that the zinc species was highly dispersed, amorphous or singly bonds to the catalyst surface.
| Catalysts | SBET, m2 g−1 | Smicroa, m2 g−1 | Sexta, m2 g−1 | Vmicro, mL g−1 | Cry.b (%) | Xc, % | Xd, % |
|---|---|---|---|---|---|---|---|
| a t-Plot method.b Cry. represents crystallinity based on XRD spectra, estimated by comparing the total XRD peak area of the zeolite sample in range of 2θ from 22° to 25° with that of the parent HZ having the strongest diffraction intensity.c X represents the Si/Al ratio based on NMR data by using dm2002 software.50d X represents the Si/Al ratio based on ICP-AES measurement. | |||||||
| HZ | 361 | 226 | 135 | 0.104 | 100 | 44.2 | 46.4 |
| PHZ | 357 | 210 | 147 | 0.096 | 91 | 51.5 | 53.4 |
| ZnPHZ | 349 | 199 | 150 | 0.091 | 65 | 50.1 | 51.1 |
Surface area and micropore analysis datum were listed in Table 1, including the BET surface area (SBET), microporous surface area (Smicro), external surface (Sext) and microporous volume (Vmicro) of the parent HZ, PHZ and ZnPHZ samples. The results indicated that the BET surface area and microporous surface area were both slightly reduced by impregnation of phosphoric acid to the HZ sample and further reduced by the following introduction of zinc species to PHZ sample meanwhile, the external surface area was gradually increased, which gave the further explanation that both phosphorus acid and zinc ions were impregnated into the pore channels.47
MAS NMR spectra of HZ (no. 1), PHZ (no. 2) ZnPHZ (no. 3), reference material Zn3(PO4)2, (no. 4, denoted as r-Zn3(PO4)2) and PZnHZ (no. 5) samples were shown in Fig. 1 as the captions described respectively. Fig. 1a shown the 29Si spectra of HZ, PHZ and ZnPHZ samples, in which there were four characteristic peaks, had been deconvoluted and identified as Q4 [Si(OSi)4] species (−112.3 ppm), inequivalent Q4 [Si(OSi)4] (−115.9 ppm),24,48 Q4 [Si(OSi)3(OAl)] (−105.9 ppm) and Q3 [Si(OSi)3(OH)] (−100.0 ppm).24,47,48 Based on Loewenstein's rule,49 the framework Si/Al ratio was calculated and list in Table 1.50 It suggested that H3PO4 impregnation to HZ sample followed by a subsequent thermal treatment at 550 °C led to sort of dealumination of the framework, whereas the addition of zinc species did not affect the zeolite framework.
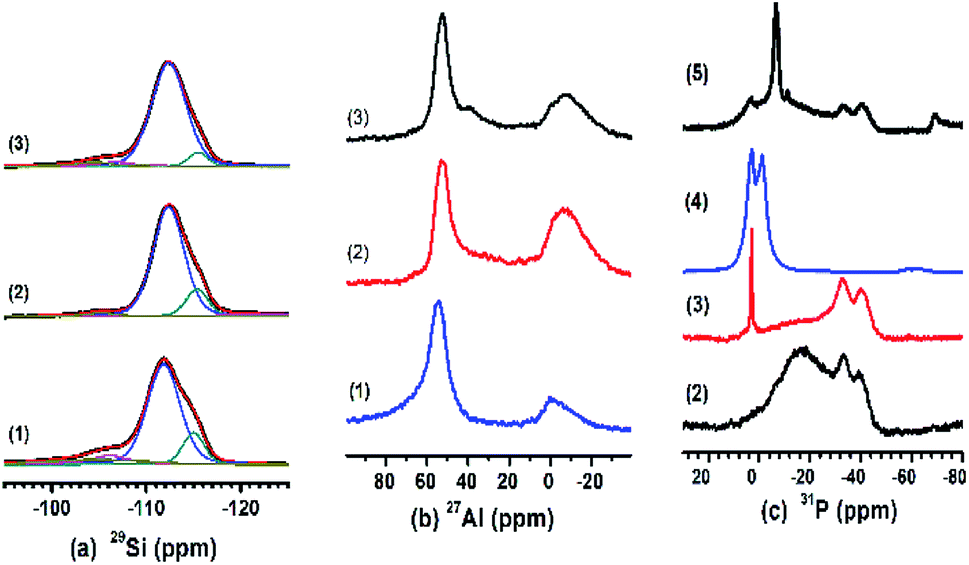 | ||
| Fig. 1 29Si (a), 27Al (b), 31P (c) MAS NMR spectra of catalysts: (1) HZ, (2) PHZ, (3) ZnPHZ, (4) r-Zn3(PO4)2 and (5) PZnHZ. | ||
Fig. 1b shown the 27Al MAS NMR spectra of HZ, PHZ and ZnPHZ samples. Typically, the spectra of the HZ sample consist of tetrahedrally coordinated framework sites (∼54 ppm, Altet-f), distorted tetrahedral coordinated framework sites (30–40 ppm, Altet-dis)51 or tetrahedrally coordinated non-framework sites (Altet-nf),52 octahedrally coordinated non-framework aluminum species (−20 to 8 ppm, Aloct)20 or framework defect sites (−20–8 ppm, Al-(OSi)3(H2O)3, Aloct-f).53 For PHZ sample, the band at −8 ppm could be observed due to the association of the octahedral Al sites with phosphorus (Aloct-o-p).22,47,54 The broad band appeared at the range of 45 to 15 ppm in comparison to the HZ sample. It suggested the increase of Altet-dis and Altet-nf sites, which was correlated with the bonding of Al with phosphorus species. For ZnPHZ sample, the bands at Altet-dis, Altet-nf, and Aloct-o-p sites were decreased, and a sharp band appeared at ∼38 ppm. Obviously, all these phenomena suggested that the local environments of both tetrahedrally coordinated Al phosphoric groups and octahedral Al phosphoric groups redeploy after the addition of zinc species, moreover, the octahedral Al phosphoric groups combined with zinc ions to form a new structure.
Fig. 1c showed the 31P MAS NMR spectra of PHZ, ZnPHZ, PZnHZ samples and r-Zn3(PO4)2. The spectrum of PHZ sample (Fig. 1c(2)) featured two intensive peaks at −39 ppm ((SiO)xAl(OP)4−x species24 with branching P sites in condensed polyphosphate,55 polyphosphoric species,20), −33.5 ppm (Al(H2O)5(PO4)55–58) and a broad peak with a maximum at about −17 ppm from 10 to −40 ppm (the specific Al–O–P structures, which could be short-chain pyrophosphoric acid or pyrophosphates bonding to non-framework Al59 or to framework Al56). The spectrum of ZnPHZ (Fig. 1c(3)) showed a significant decrease for Al–O–P structures and a slight reduction for (SiO)xAl(OP)4−x species and Al(H2O)5(PO4). Comparison of its spectrum with that of r-Zn3(PO4)2, as shown in Fig. 1c(4), indicated that the transformation of short-chain polyphosphoric species (10–−40 ppm) on PHZ sample to zinc phosphate groups (3 ppm) on ZnPHZ occurred. The 31P NMR spectrum of PZnHZ in Fig. 1c(5) featured a sharp contrast in the bands distribution from 20 to −80 ppm, which clearly indicated that only a small amount of zinc phosphate groups showed up, which was totally different in the structure or configuration of zinc and phosphorus species from the ZnPHZ sample. The spectra of PZnHZ sample show that P did see Zn but not too much of it, which meant that zinc ions could find the phosphoric groups to form a new structure – zinc phosphate groups – during the preparation of ZnPHZ sample, whereas such structure was nowhere to be seen from P-perspective in 31P NMR spectrum of PZnHZ sample.
XANES spectra43–45 related to Zn K-edge for ZnPHZ, PZnHZ, and ZnHZ samples and reference materials r-Zn3(PO4)2, and r-ZnO (short for zinc oxide) were shown in Fig. 2a. Absorption edge energies (as calculated for the first inflection point of the edge rise) were 9663.7 eV for all samples except for the r-Zn3(PO4)2 for which the edge energy was 9663.5 eV. The XANES spectra of ZnPHZ showed the similar pattern to the r-Zn3(PO4)2 but quite different from r-ZnO. XANES data (Fig. 2a inset) demonstrated the similarity between Zn mirco-environments in ZnPHZ sample and r-Zn3(PO4)2, whereas the whiteline shape of PZnHZ catalyst was close to that in P-free sample of ZnHZ sample. It should be noted that all the samples were measured at room temperature after in situ water removal at 430 °C to eliminate the effect of water adsorption on XANES spectra. We thus concluded that the similarity of the whiteline shape between ZnPHZ and r-Zn3(PO4)2 accounts for the similar structures of phosphante groups that had different concentrations.
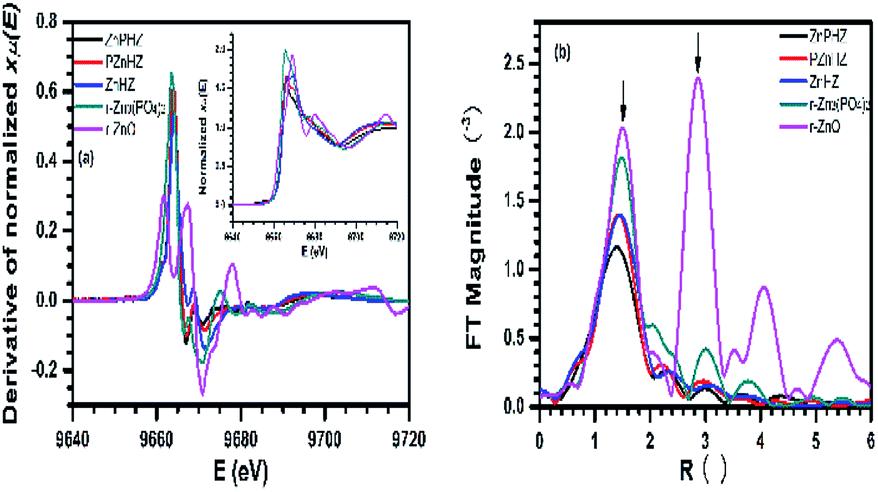 | ||
| Fig. 2 (a) Derivative of normalized XANES region of absorption coefficient of ZnPHZ, PZnHZ, ZnHZ catalysts and r-Zn3(PO4)2 and r-ZnO (inset: normalized XANES region of absorption coefficients), and (b) Fourier transform magnitudes of corresponding k2-weighted EXAFS spectra. | ||
Fourier transforms (FT)-EXAFS data was shown in Fig. 2b. The peak positions were lower than real-space distances between Zn atoms and its nearest neighbors due to the photoelectron phase shift, not corrected for in Fig. 2b. The peak around 1.60 Å for r-Zn3(PO4)2 in the FT magnitude (shown by an arrow in Fig. 2b), which was attributed to the nearest oxygen neighbors with the bond Zn–O–P,60 corresponded to a Zn–O bond length of 2.04 Å. The second main peak around 3.05 Å was associated with the bond of Zn-P with the actual bond length of 3.28 Å. In order to quantitatively analyze the local structure of zinc species, data analysis of EXAFS spectra (ESI, Fig. S6†) was performed by using IFEFFIT package.61–63 Theoretical photoelectron scattering amplitudes and phase shifts of Zn–O and Zn-P contributions were generated by FEFF6 software64 using crystal structure of r-Zn3(PO4)2. The passive electron reduction factor was fixed to 1.0, as found from the fit to the r-ZnO. The results obtained for the ZnPHZ system demonstrated that each Zn ion in the nearest coordination shell was surrounded on average by 3.5 ± 0.6 Å of O atoms with a coordination distance 1.95 ± 0.02 Å and meant squared bond length disorder σ2 = 0.011 ± 0.005 Å2. Relatively large uncertainties prevented us from accurately measuring the number of Zn-P species in the second shell because of the correlation of the coordination numbers and bond length disorder in the fitness. Alternative models were compared, in which we added the Zn–Zn contribution (calculated theoretically using r-ZnO structure) as a possible second neighbor to Zn instead, or in addition to, Zn-P, but no reasonable results were obtained. Notably, we found that a model where Zn–Zn was the only contribution to the second coordination shell, as in the local structure of r-ZnO, was not consistent with the experimental spectra.
For PZnHZ sample, each Zn ion in the nearest coordination shell was surrounded on average by 3.9 ± 0.5 Å of O atoms with a coordination distance 1.96 ± 0.01 Å and σ2 = 0.008 ± 0.002 Å2; for ZnHZ sample, each Zn ion in the nearest coordination shell was surrounded on average by 4.1 ± 0.6 Å of O atoms with a coordination distance 2.01 ± 0.01 Å and σ2 = 0.008 ± 0.002 Å2. The main summary of the above results was that the Zn–O bond length changed significantly from PZnHZ to ZnHZ sample, whereas the coordination number was similar for the two samples. Such coordination number of ca. 4.0 of Zn–O was in agreement with the reported structure of ZnHZ catalyst measured after water removal,65 which suggested Zn species were in a similar coordination environments in the first shell for both PZnHZ and ZnHZ samples. The shorter average Zn–O bond length in the former catalyst was consistent with the contribution of phosphate groups to the nearest environment of zinc ions, because the bond length of Zn–O in Zn–O–P groups in Hopeite structure varied from 1.90 to 2.05 Å (ESI, Fig. S7†), hence, had a fraction of shorter bonds compared to the ZnHZ system.
In brief, the EXAFS fitting results, taking together with other evidences from XANES, NMR and catalytic tests, supported the idea that both zinc species and phosphate groups existed as the form of zinc phosphate groups on ZnPHZ sample, which quite differentiated from those on ZnHZ and PZnHZ samples. The absence of Zn–Zn bond further suggested that the presence of these zinc phosphate groups was isolated.
Fig. 3a displayed FTIR spectra of the HZ, PHZ, and ZnPHZ samples. It could be clearly seen there were four hydroxyl bands, assigned as amorphous silica hydroxyl groups (3740 cm−1),66 terminal framework silanols (Si–OH, 3720 cm−1),67 extra-framework aluminum species or partially hydrolyzed aluminum species (3660–3690 cm−1),68 and bridging hydroxyl groups Si(OH)Al (3610 cm−1).69 Both bands for P–OH groups41 at 3666 cm−1 and bands for Zn–OH groups41 at 3620–3674 cm−1 were not discerned since they were obscured by the bands of Si–OH, Al–OH. It should be noted that the bands at 3610 cm−1 were specific to the bridged hydroxyl groups. The peak height of the band at 3610 cm−1 decreased when phosphorus was impregnated to HZ sample, suggesting that part of the bridging hydroxyl groups (Brönsted acid site) were eliminated by reacting with orthophosphoric acid. On the contrary, the bridging hydroxyl groups kept unchanged or even stronger when zinc was further impregnated to PHZ sample, indicating zinc ions were not exchanged with the bridging hydroxyl groups, properly evidenced that zinc ions reacted with phosphorus groups to form zinc phosphate groups.
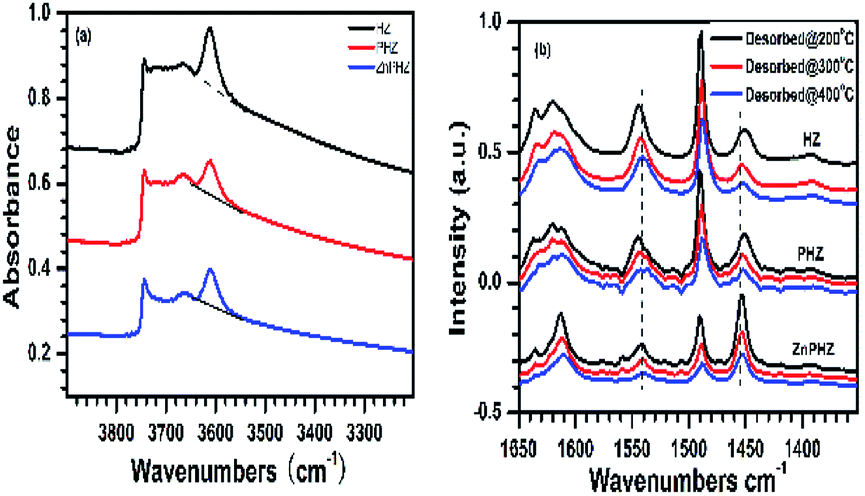 | ||
| Fig. 3 (a) FTIR spectra measured at 40 °C after dehydration at 450 °C for 1 h and (b) Py-FTIR spectra measured at 200 °C, 300 °C and 400 °C after desorption at the corresponding temperatures of catalysts for: (1) HZ, (2) PHZ, (3) ZnPHZ. | ||
Py-FTIR spectra of HZ, PHZ, and ZnPHZ samples after pyridine desorption at different temperatures were shown in Fig. 3b. The characteristic bands67,70 were clearly observed at ∼1450 cm−1 for Lewis acid sites and at 1540 cm−1 for Brönsted acid sites. For both the different conditions of samples at the same temperature and the same catalyst at the different desorption temperature, the bands for Brönsted acid sites shifted to lower wavenumbers, whereas the bands for Lewis acid sites to higher wavenumbers. It was demonstrated that both Lewis acid sites and Brönsted acid sites had different strength of acid sites existed on the zeolite surface. Quantification of weak, medium, and strong acid sites for Brönsted and Lewis acid sites was performed according to the amount of pyridine desorption at different temperature by the same way as everywhere.2,35 To allow semi-quantitative comparisons of the peak intensities, all Py-FTIR spectra were normalized using the area of the overtone lattice vibration bands of the zeolites at 1990 cm−1 and 1873 cm−1, as conducted in the literatures.2,36,71 Fig. 4 showed the acidities of different samples measured by NH3-TPD method.15,40,72–76 It was found that for HZ, PHZ and ZnPHZ samples: (1) the relative total acidity amount was 100%, 99.4% and 78.3%; (2) the weak acid amount was 100%, 128.1% and 118.8%; and (3) strong acid amount was 100%, 69.6%, and 35.4% respectively (ESI, details showed in Fig. S3†).
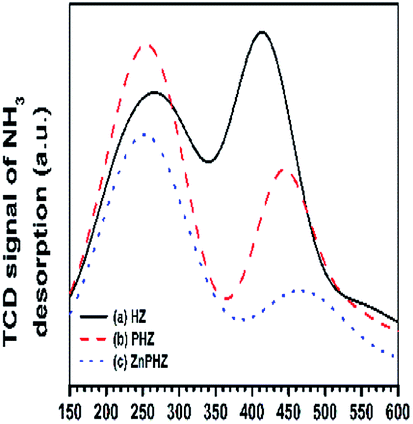 | ||
| Fig. 4 NH3-TPD profiles of catalysts: (a) HZ, (b) PHZ, (c) ZnPHZ. | ||
The effect of the reaction time on methanol conversion over ZnPHZ sample was shown in Fig. 5a. It could be seen that the catalyst exhibited a stable performance during the reaction procedure. All of the yields of C1–C5+ and benzene (C6A) hydrocarbons slightly decreased with time on stream (TOS), while the yields of toluene initially increased and then steadily remained. The C9+ aromatics (C9+A) yield continuously increased as prolong the reaction time. These phenomena implied that BTX and C9+ aromatics might be produced through a common intermediate, such as (C6=–C9=) oligomer intermediates. The yield of BTX (YBTX) was stable, and the selectivity to BTX shown only a slightly decrease due to the yield of total aromatics (YA) slightly increased. For HZ, PHZ and ZnHZ catalyst, all showed the similar fuction in the effect of the reaction time on methanol conversion (ESI, Fig. S8†). Fig. 5b compared the performances of ZnHZ and ZnPHZ catalysts under the same reaction conditions. It clearly showed that ZnPHZ catalyst owned better selectivity to BTX.
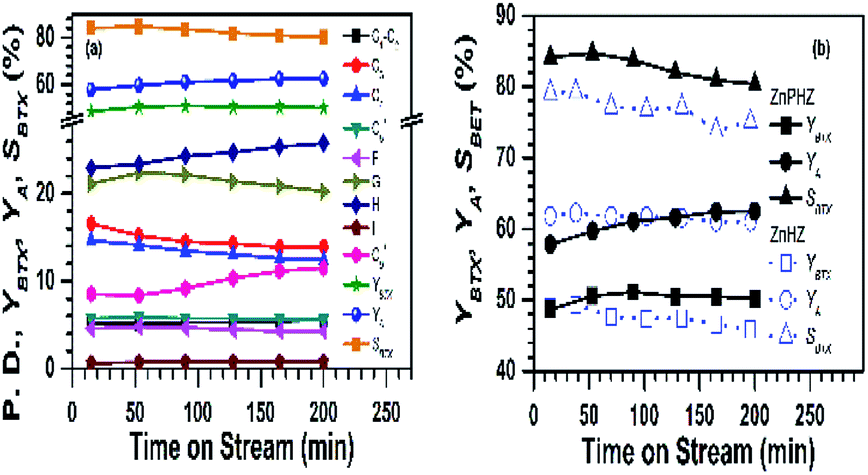 | ||
| Fig. 5 (a) Product distribution (P. D.) of MTA reaction for catalyst ZnPHZ; (b) yield of BTX (YBTX), yield of aromatics (YA), selectivity to BTX (SBTX) of catalyst ZnPHZ in comparison with reference catalyst ZnHZ. Reaction conditions: 0.1 MPa, 430 °C, 3.0 g catalyst, WHSV = 2.0 h−1. | ||
Table 2 and Fig. S8† displayed the catalytic reactivity of ZnPHZ catalyst in the conversion of methanol to light aromatics, including the HZ, PHZ, ZnHZ, and PZnHZ catalysts. Methanol was completely converted on all catalysts under the same reaction conditions. The differences in the reaction performance could be attributed to changes in the nature of the catalytic sites present: (1) the parent HZ catalyst exhibited only 25.61% yield of BTX and 75.58% selectivity to BTX; By addition of Zn to PHZ, the ZnPHZ catalyst showed 50.59% yield of BTX (32.85% for PHZ) and 84.73% selectivity to BTX (81.89% for PHZ); (2) ZnPHZ catalyst showed different product distribution (featured as lower C1–C5+ light hydrocarbons and significantly lower C9+ aromatics) from the ZnHZ catalyst; (3) toluene and xylene were the main products in the MTA reaction for all the catalysts, among which ZnPHZ catalyst had the maximal values of 22.39% and 23.40% for YT and YX, respectively; (4) ZnPHZ catalyst exhibited the superior performance to the reference catalysts, ZnHZ and PZnHZ samples. W. D. Xiao82 et al. reported a MTA catalyst, HZSM-5 zeolite catalyst (SiO2/Al2O3 = 30) modified by 1.0 wt% ZnO and 2.0 wt% P, by using of the special titanium made U-shaped reactor, in the reaction condition of temperature range from 380 to 480 °C, pressure:15–50 kPa, obtained the best aromatic product: YA = 59.61%, with YB = 8.50%, YT = 27.70%, YX = 20.18%, which exhibited nearly the same yield of aromatics, slightly different to the PZnHZ catalyst in the BTX distribution, owing to the different content of Zn and P. Meanwhile, this MTA catalyst, similar to ZnPHZ catalyst, should contain the isolated zinc phosphate groups.
| Catalysts | Yields (wt%) | YBTX (wt%) | YA (wt%) | SBTX (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 + C2 | C3 | C4 | C5+ | B | T | X | EB | C9+ | BTX | A | S | |
| a Data point: TOS@60 min; B = benzene, T = toluene; X = xylene; C5+ = alkanes and olefins with C5 and plus; C9+ = aromatics with C9 and plus; A = total aromatics. | ||||||||||||
| HZ | 5.61 | 22.47 | 24.74 | 13.29 | 2.60 | 9.99 | 13.01 | 0.82 | 7.45 | 25.61 | 33.89 | 75.58 |
| PHZ | 5.15 | 27.51 | 21.09 | 6.14 | 4.07 | 13.78 | 15.00 | 0.64 | 6.62 | 32.85 | 40.12 | 81.89 |
| ZnPHZ | 5.08 | 15.20 | 14.13 | 5.89 | 4.80 | 22.39 | 23.40 | 0.74 | 8.37 | 50.59 | 59.70 | 84.73 |
| ZnHZa | 6.17 | 12.16 | 12.98 | 6.90 | 3.27 | 18.24 | 26.20 | 0.87 | 13.20 | 47.71 | 61.78 | 77.23 |
| PZnHZa | 7.85 | 13.89 | 14.09 | 11.45 | 2.38 | 14.23 | 19.87 | 1.32 | 13.75 | 33.00 | 47.13 | 70.01 |
Discussion
The IZPG (short for isolated zinc phosphate groups), as we mentioned above, were perfectly evidenced by XANES and EXAFS methods, as well as FTIR measurements. From the procedure of catalyst preparation method,47 the local mirco-environment of could be further deduced from the principal phosphorus groups which were confirmed to bond with zinc ions. As from the results of 27Al NMR spectra of ZnPHZ catalyst (ESI, Fig. S4†), the descent of Altet-dis, Altet-nf, and Aloct-o-p and an emerging sharp band at ca. 38 ppm suggested that the transformation of both tetrahedrally Al phosphoric groups and octahedrally Al phosphoric groups to zinc phosphoric groups took place. Meanwhile, from the 31P MAS NMR spectra(ESI, Fig. S5†), different amounts of zinc addition also demonstrates that (SiO)xAl(OP)4−x species (−39 ppm) gradually decreased, the nonreactive Al(H2O)5(PO4) species (−33.5 ppm) remained unchanged, whereas the amount of zinc phosphate groups (3 ppm) increased step by step and the principal peak centred on −17 ppm was related to the specific Al–O–P structures, which could be assigned to short-chain pyrophosphoric acid or pyrophosphates bonding to non-framework Al59 or to framework Al55 and revealed systematic increase occurred at the bands of all distorted tetrahedral Al sites and a systematic decrease in octahedral Al sites. On the other hand, only a slight fluctuation appeared at the peak of tetrahedral Al sites (Brönsted acid sites) as the increase of zinc amount from 0 to 3.0% to PHZ catalyst. It was a compelling proof that preferential reaction between phosphorus groups and zinc ions took place by adding zinc ions to PHZ catalyst.This unique structure of IZPG was also confirmed in comparison with the zinc environment of ZnHZ and PZnHZ samples, from the EXAFS data in r-space (ESI, Fig. S5†), the zinc mirco-environment of ZnHZ and PZnHZ samples, exhibited a similar fluxion, which meant the coordination environments for both catalysts was similar, but quite different from ZnPHZ sample. It was also expected that the lower intensity of the Zn–O peak suggested a lower coordination numbers for Zn-P bonds in the ZnPHZ system compared to the PZnHZ and ZnHZ samples, as well as r-Zn3(PO4)2, consistent with the conclusion made by examination of the XANES data (vide supra) that zinc phosphate groups were generated on ZnPHZ sample during the procedure of adding zinc ions to PHZ catalyst. However, upon adding phosphoric acid to ZnHZ sample, the structure of zinc just slightly changed as evidenced in 31P NMR spectra. It should be noted that the amount of zinc phosphate groups on PZnHZ sample should be very low, if any.
From NH3-TPD and Py-FTIR80,81 results, we could further find the IZPG not only altered the acid strength distribution of the zeolite surface but also affected the total acidic amounts. As shown in Fig. 6 and Table S2,† the contributions of Lewis nature of IZPG could be deduced from the results by the ascent of Lewis acid distribution. Via the addition of zinc ions to PHZ sample, the summit of Lewis acid sites was shifted to a higher frequency, and indicated that zinc phosphate groups act as strong Lewis acid sites. It was reported that the reduction of the bridging hydroxyl groups (Brönsted acid sites) derived from its reaction with the supported othorphosphorus acid,77,78 or as a result of the lattice dealumination by the dislodged aluminum species reacting with H3PO4,47 which interpreted the reduction of the strong acid peak in the curve of NH3-TPD in the temperature range between 300–600 °C (Fig. 4). Meanwhile, the impregnation of HZ with phosphoric acid was accompanied by the creation of Brönsted acid sites with an acid strength lower than that of the bridging hydroxyls.22,66
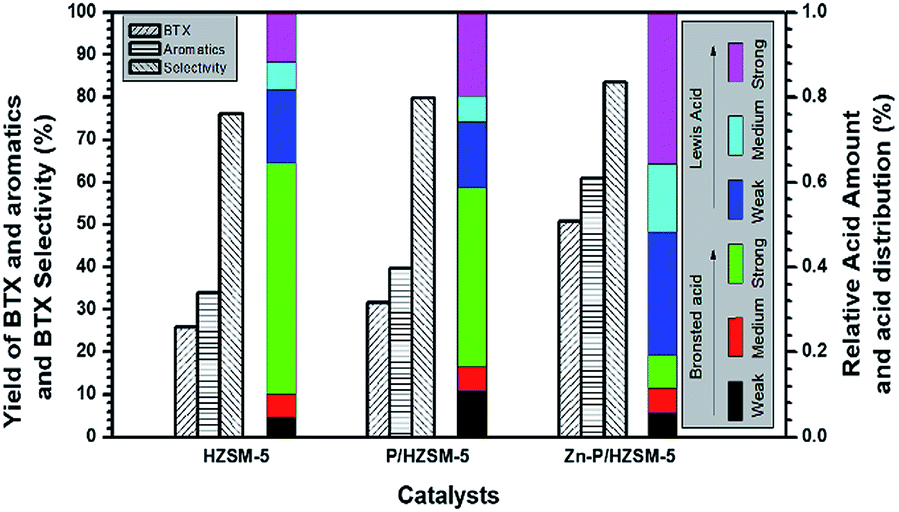 | ||
| Fig. 6 Relationship between yields of BTX, aromatics and BTX selectivity, in MTA reaction, and the distribution of different type of acidic sites in weak, medium and strong acidic strength for HZ, PHZ, and ZnPHZ respectively. Reaction conditions see Fig. 5. | ||
It was thus the acid amount lost by the reduction of Brönsted acid sites or the other acid sites could be compensated by ammonia uptakes from phosphorus hydroxyl groups, which accounted for the total acid amount of PHZ sample was close to the HZ sample, which meant, the phosphoric groups reacted with zinc ions and further transformed to zinc phosphate groups. The loss of acid amount of ZnPHZ sample should be ascribed to the elimination of P–OH groups by the combination of zinc ions and P–OH groups. Therefore, the total acid amount was further reduced. It could be also concluded that the transformation from phosphate groups to zinc phosphate groups increased the high temperature peak of ammonia desorption from 444 °C to 470 °C from Fig. S3 and Table S1.† Therefore, the strength of IZPG could be identified as strong Lewis acid sites in agreement with the result.
Fig. 6 plotted the specific yield of BTX, yield of total aromatics and selectivity to BTX in correlation with the corresponding acidic distribution was plotted by It exhibited that a significant increase was obtained over ZnPHZ catalyst with the change of the acidic distribution. As known to all, the acidic distribution of catalyst played an important role in the catalytic reactivity. The catalyst should have a moderate B/L ratio so as to obtain the high BTX yield, which preferable contribute the formation of BTX and also suppressed the generation of carbon, coke and heavy aromatics.2,36,40 According to the reaction mechanism reported,2,6,40 it could be deduced that the cracking or methylation of C6=–C9= oligomer intermediates was suppressed over ZnPHZ catalyst leading to the decrease of C1–C5 fraction and C9+ aromatics, whereas the dehydrogenation reaction of C6=–C8= oligomer intermediates was enhanced over ZnPHZ catalyst leading to the high BTX selectivity. It was well accepted that exchanged zinc ions promoted the desorption of H atoms to H2, which greatly res trained hydrogen transfer to olefins but facilitate dehydrogenation of cyclic olefins to produce aromatics.40 In this case, IZPG might play a similar role as ZnHZ catalyst but with more suitable acidity distribution and better hydrothermal stability, as well as the MTA catalyst, Zn and P co-modified HZSM-5.35,36,82
As it is known to all, for the MTA process, zinc play the most popular enroller, employed by HZSM-5 zeolite, which associated with the acidic sites of zeolite framework, generated more Lewis acid (ZnOH+, Zn2+-L acid center), make the catalyst more effective to convert the intermediate olefins into aromatics by dehydrogenation, via the expense of the silanol hydroxyl and proton acid sites. And the impregnation of phosphorus to ZSM-5 can also adjust the acidity of zeolite and improve its hydrothermal stability, but excessive introduction of P could lead to the covering of Brönsted acid sites and blockage of pores, resulting in MTA activity decreased.
In the methanol to hydrocarbon (MTH) reaction, both Brönsted acid sites and Lewis acid sites can regulate the conversion of methanol to dimethyl ether and the further formation of DME to low-carbon olefins (C2=–C3=), the polymerization of low-carbon olefins to high-carbon olefins (C4=–C8=). Brönsted acid sites can also catalyse the cyclization of higher olefins and the hydrogen transfer of cycloalkanes. In order to maximize the performance of certain MTA catalyst with IZPG species, there should be kept counterbalance of Brönsted acid sites to Lewis acid sites (B to L) ratio nearly 0.2, as Fig. 6 and Table S2† showed.
Table 3 and Fig. S9† shown the reactivity of the fresh ZnPHZ catalyst and the regenerated catalysts. Technically speaking, fixed bed reactor is not suitable for MTA process due to the deactivation rate.4,16,79 In the present research, the catalyst was operated in the fixed bed reactor for 240 min, then the reaction was terminated to regenerate in situ to simulate the fluidized bed reaction for testing the stability of ZnPHZ catalyst. As indicated in Table 3, ZnPHZ catalyst exhibited excellent recoverable performance during as many as 5 cycles of reaction-regeneration were carried out. Only a slightly decrease could be found in the yield of BTX. It was pronounced that such catalyst has a robust structure with good thermal and hydrothermal stabilities. Table 4 summarized the Zn and P contents for the fresh and regenerated catalysts after reaction-regeneration cycles. The initial P and Zn contents in the fresh ZnPHZ catalyst were 0.97% and 0.75% respectively. After each reaction-regeneration cycle, the content of P remains almost unchanged for all the 6 times running. Meanwhile, the final zinc content decreased slightly from 0.75% to 0.59% after 6 times running. For the ZnHZ catalyst, the zinc content definitely decreased from 0.73% to 0.53% after first run. It is also confirmed that the loss of zinc15,29–31 took place for ZnHZ catalyst, while the ZnPHZ catalyst stabilized the phosphorus and zinc by the form of isolated zinc phosphate groups and further extended the ZnPZ catalyst recycle regeneration times above 20 can be seen in Fig. S10.†
| Run no. | Yields (wt%) | YBTX (wt%) | YA (wt%) | SBTX (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 + C2 | C3 | C4 | C5+ | B | T | X | EB | C9+ | BTX | Aromatics | Selectivity | |
| a Data point: TOS@60 min; B = benzene, T = toluene; X = xylene; C5+ = alkanes and olefins with C5 and plus; C9+ = aromatics with C9 and plus; A = total aromatics. | ||||||||||||
| 0 | 5.08 | 15.20 | 14.13 | 5.89 | 4.80 | 22.39 | 23.40 | 0.74 | 8.37 | 50.59 | 59.70 | 84.73 |
| 1 | 5.69 | 14.82 | 13.78 | 5.96 | 4.52 | 21.55 | 23.95 | 0.74 | 8.99 | 50.01 | 59.74 | 83.71 |
| 2 | 5.97 | 15.02 | 14.18 | 6.37 | 4.26 | 20.66 | 23.83 | 0.75 | 8.98 | 48.74 | 58.47 | 83.37 |
| 3 | 6.21 | 14.88 | 14.33 | 6.71 | 4.14 | 20.41 | 23.56 | 0.78 | 8.97 | 48.12 | 57.87 | 83.15 |
| 4 | 6.60 | 15.10 | 14.70 | 6.71 | 4.19 | 20.51 | 23.15 | 0.74 | 8.29 | 47.85 | 56.88 | 84.11 |
| 5 | 6.77 | 15.09 | 14.87 | 6.98 | 4.11 | 20.36 | 22.82 | 0.78 | 8.22 | 47.30 | 56.30 | 84.01 |
| Components | Fresh (wt%) | Regenerated (wt%) after each cycle | |||||
|---|---|---|---|---|---|---|---|
| 1a | 2a | 3a | 4a | 5a | 6a | ||
| a Regenerated catalysts analyzed using ICP-AES after each reaction-regeneration cycle. Prior to each cycle, the used catalyst were calcined in air at 550 °C for 240 min for coke removal. | |||||||
| P@ZnPHZ | 0.97 | 0.96 | 0.94 | 0.93 | 0.93 | 0.95 | 0.91 |
| Zn@ZnPHZ | 0.75 | 0.76 | 0.75 | 0.73 | 0.70 | 0.62 | 0.59 |
| Zn@ZnHZ | 0.73 | 0.53 | — | — | — | — | — |
Experimental procedures
Catalyst preparation
The starting material, HZSM-5 with silica to alumina mole ratio of 100, was purchased from Shanghai Fuxu Molecular Sieves Co (China). PHZ catalyst, loading with 1.0 wt% phosphorus, was prepared by wetness impregnation as follows: (a) the impregnation solution was prepared by mixing 31.6 g of aqueous solution of orthophosphoric acid (10 wt%, H3PO4) and 118.4 g H2O at room temperature; (b) 100 g of HZSM-5 zeolite was added to the solution (a) with stirring at room temperature until dryness for over 5 hours; (c) the impregnated sample was subsequently dried at 393 K overnight and then calcined at 450 °C for 4 h. The ZnPHZ46 (0.75 wt% zinc), was prepared by adding the 10.0 g P/HZSM-5 catalyst to 15.0 g of Zn(NO3)2 solution (2.30 wt%, Zn(NO3)2·6H2O), followed by the same stirring, drying and calcination procedure as for the PHZ catalyst. ZnHZ catalyst, with the Zn loading of 0.75 wt%, was prepared similar to ZnPHZ catalyst by impregnating HZ with Zn(NO3)2 solution. The reference catalyst PZnHZ was prepared by impregnating ZnHZ with orthophosphoric acid. All the reference catalysts were prepared with the same P and Zn loading weight as ZnPHZ.Characterization
Surface area and micropore analysis were analyzed by nitrogen physisorption data (Micromeritics Tristar 3000, conducted in Sinopec SRIPT). The crystallinity of catalysts was measured by powder X-ray diffraction (Bruker Advance D8, conducted in Sinopec SRIPT; Beamline 14A of the National Synchrotron Light Source at Brookhaven National Laboratory). Scanning Transmission Electron Microscopy is used for confirmation of the absence of zinc oxide particles and the catalyst crystallinity of ZnPHZ (JEOL ARM200F, conducted in Hefei, China). NMR measurements were conducted at Sinopec SRIPT for 29Si, 27Al and 31P MAS NMR spectra (Varian VNMR 400).XAFS measurements were performed to identify the chemical states of zinc species. XAFS measurements were done at beamline X19A of the National Synchrotron Light Source at Brookhaven National Laboratory and Beamline BL2-2 of the Stanford Synchrotron Radiation Light source. The catalyst sample was grounded to fine powder and pressed into a pellet. All spectra were collected at room temperature after water removal at 430 °C in transmission mode.
Hydroxyl surface groups of HZ catalysts were characterized by Fourier Transform Infrared Spectroscopy (Nicolet 5700, conducted in Sinopec SRIPT). NH3-TPD (Set up by Sinopec SRIPT) and Py-FTIR (Nicolet 380, conducted in Sinopec SRIPT) measurements were conducted to measure the catalyst acidity amount and the distribution of acidity type. Chemical analysis of the zeolite samples was performed by Inductively Coupled Plasma-Atomic Emission Spectrometry (ICP-AES, conducted in Sinopec SRIPT). The details are given in ESI.†
Reaction evaluation
The HZ, PHZ, and ZnPHZ catalysts, as well as the reference catalyst ZnHZ, were respectively pressed, crushed and sieved to obtain particles in the range of 12–20 mesh (1.4–1.7 mm) for catalytic evaluation in Sinopec SRIPT. Prior to the reaction, the catalysts were blown out with flowing nitrogen at 430 °C for 2 h. Methanol conversion was performed in a stainless-steel fixed bed reactor (650 mm length and 10 mm i.d.). The reaction conditions were weight hourly space velocity (WHSV) = 2.0 h−1, reaction temperature of 430 °C and atmospheric pressure. Methanol and the products were analyzed by an online gas chromatograph (Agilent GC-7890) with a flame ionization detector (FID). The tubing between outlet of the reactor and GC was kept at 240 °C for prohibiting the reaction products from condensing. The methanol conversion, BTX selectivity and yield of the products were calculated from the number of C atoms in the products.Conclusions
We demonstrated that isolated zinc phosphate groups can be successfully anchored to HZSM-5 zeolite channel network prepared by impregnating zinc ions to P-modified HZSM-5 catalyst, which has a good hydrothermal stability and a suitable Lewis acidity regulating the acidity distribution. The stable zinc phosphate groups on ZnPHZ enhance the hydrogen-transfer pathway to high BTX selectivity and suppress the methylation of BTX to heavy aromatics. This research may provide an alternative way to the effective MTA catalyst design for achieving high BTX selectivity and inhibiting the formation of heavy aromatics (C9+) with good thermal and hydrothermal stabilities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from China Petrochemical Technology Company, Ltd. Use of the NSLS was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. The authors acknowledge the facilities support at the NSLS beamline X19A and SSRL beamline BL2-2 provided by the Synchrotron Catalysis Consortium (U.S. DOE grant no. DE-SC0012335). The authors acknowledge financial support from the National Key R&D Program of China (project number: 2017YFB0702800), China Petrochemical Corporation (Sinopec Group).References
- X. Niu, J. Gao, Q. Miao, M. Dong, G. Wang, W. Fan, Z. Qin and J. Wang, Influence of preparation method on the performance of Zn-containing HZSM-5 catalysts in methanol-to-aromatics, Microporous Mesoporous Mater., 2014, 197, 252–261 CrossRef CAS
.
- G. Zhang, T. Bai, T. Chen, W. Fan and X. Zhang, Conversion of Methanol to Light Aromatics on Zn-Modified Nano-HZSM-5 Zeolite Catalysts, Ind. Eng. Chem. Res., 2014, 53(39), 14932–14940 CrossRef CAS
- F. Wang, W. Xiao and G. Xiao, Atomic layer deposition of zinc oxide on HZSM-5 template and its methanol aromatization performance, Catal. Lett., 2015, 145, 860–867 CrossRef CAS
- T. Wang, X. Tang, X. Huang, W. Qian, Y. Cui, X. Hui, W. Yang and F. Wei, Conversion of methanol to aromatics in fluidized bed reactor, Catal. Today, 2014, 233(15), 8–13 CrossRef CAS
- Y. Ono, H. Adachi and Y. Senoda, Selective conversion of methanol into aromatic hydrocarbons over zinc-exchanged ZSM-5 zeolites, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 1091–1099 RSC
- Y. Xin, P. Qi, X. Duan, H. Lin and Y. Yuan, Enhanced performance of Zn–Sn/HZSM-5 catalyst for the conversion of methanol to aromatics, Catal. Lett., 2013, 143, 798–806 CrossRef CAS
- H. Jing, F. Yang, Y. Xia, J. Feng, J. Liu and Q. Zong, A study on the selectivity of methanol aromatization, Pet. Sci. Technol., 2012, 30(16), 1737–1746 CrossRef CAS
- F. Yang, S. Wang, Y. Xia, W. Bi and W. Wang, Study on Methanol Aromatization over Zeolite Catalyst H/ZSM-5 Modified with Zinc Ions in Different Contents, Pet. Sci. Technol., 2011, 29(16), 1675–1684 CrossRef CAS
- Y. Ni, A. Sun, X. Wu, J. Hu, T. Li and G. Li, Aromatization of Methanol over La/Zn/HZSM-5 Catalysts, Chin. J. Chem. Eng., 2011, 19(3), 439–445 CrossRef CAS
- Y. Ni, A. Sun, X. Wu, G. Hai, J. Hu, T. Li and G. Li, The preparation of nano-sized H [Zn, Al] ZSM-5 zeolite and its application in the aromatization of methanol, Microporous Mesoporous Mater., 2011, 143(2–3), 435–442 CrossRef CAS
- Y. Ni, W. Peng, A. Sun, W. Mo, J. Hu, T. Li and G. Li, High selective and stable performance of catalytic aromatization of alcohols and ethers over La/Zn/HZSM-5 catalysts, J. Ind. Eng. Chem., 2010, 16(4), 503–505 CrossRef CAS
- Y. Inoue, K. Nakashiro and Y. Ono, Selective conversion of methanol into aromatic hydrocarbons over silver-exchanged ZSM-5 zeolites, Microporous Mater., 1995, 4(5), 379–383 CrossRef CAS
- H. Ji, Q. Zhang, L. Chen and H. Shan, Study on the reaction performance of methanol to aromatics over Zn/HZSM-5 catalysts with alumina, J. Fuel Chem. Technol., 2014, 42(11), 1387–1393 CAS
- Y. Bi, Y. Wang, X. Chen, Z. Yu and L. Xu, Methanol aromatization over HZSM-5 catalysts modified with different zinc salts, Chin. J. Catal., 2014, 35(10), 1740–1751 CrossRef CAS
- V. Abdelsayed, D. Shekhawat and M. Smith, Effect of Fe and Zn promoters on Mo/HZSM-5 catalyst for methane dehydroaromatization, Fuel, 2015, 139(1), 401–410 CrossRef CAS
- T. Tian, W. Qian, X. Tang, S. Yun and F. Wei, Deactivation of Ag/ZSM-5 Catalyst in the Aromatization of Methanol, Acta Phys.-Chim. Sin., 2010, 26(12), 3305–3309 CAS
- J. Song, Z. Di, C. Yang and J. &Wu, Preparation of Small Crystalline ZSM-5 and its Application in Conversion of Methanol to Aromatic, Adv. Mater. Res., 2012, 457–458, 245–248 CAS
- H. Van der Bij, F. Meirer, S. Kalirai, J. Wang and B. Weckhuysen, Hexane Cracking over Steamed Phosphated Zeolite H-ZSM-5: Promotional Effect on Catalyst Performance and Stability, Chem.–Eur. J., 2014, 20(51), 16922–16932 CrossRef CAS
- J. Otterstedt, Y. Zhu and J. Sterte, Effects of matrix alumina-silica ratio on the performance of heavy oil cracking catalysts containing zeolite Y in matrices of amorphous silica-alumina, Appl. Catal., 1991, 70(1), 43–52 CrossRef CAS
- T. Blasco, A. Corma and J. Triguero, Hydrothermal stabilization of ZSM-5 catalytic-cracking additives by phosphorus addition, J. Catal., 2006, 237(2), 267–277 CrossRef CAS
- G. Caeiro, P. Magnoux, M. Lopes, F. Ribeiro, S. Menezes, A. Costa and H. Cerqueira, Stabilization effect of phosphorus on steamed H-MFI zeolites, Appl. Catal., A, 2006, 314(2), 160–171 CrossRef CAS
- G. Lischke, R. Eckelt, G. Jerschkewitz, B. Parlitz, E. Schreier, W. Storek, B. Zibrowius and G. Öhlmann, Spectroscopic and physicochemical characterization of P-Modified H-ZSM-5, J. Catal., 1991, 132(1), 229–243 CrossRef CAS
- G. Yang, J. Zhuang, Y. Wang, D. Zhou, M. Yang, X. Liu, X. Han and X. Bao, Enhancement on the hydrothermal stability of ZSM-5 zeolites by the cooperation effect of exchanged lanthanum and phosphoric species, J. Mol. Struct., 2005, 737(1–2), 271–276 CrossRef CAS
- J. Zhuang, D. Ma, G. Yang, Z. Yan, X. Liu, X. Liu, X. Han, X. Bao, P. Xie and Z. Liu, Solid-state MAS NMR studies on the hydrothermal stability of the zeolite catalysts for residual oil selective catalytic cracking, J. Catal., 2004, 228(1), 234–242 CrossRef CAS
- M. Kaarsholm, F. Joensen, J. Nerlov, R. Cenni, J. Chaouki and G. Patience, Phosphorous modified ZSM-5: Deactivation and product distribution for MTO, Chem. Eng. Sci., 2007, 62(18–20), 5527–5532 CrossRef CAS
- W. Kaeding, C. Chu, L. Young and S. Butter, Shape-selective reactions with zeolite catalysts: II. Selective disproportionation of toluene to produce benzene and p-Xylene, J. Catal., 1981, 69(2), 392–398 CrossRef CAS
- W. Kaeding, C. Chu, L. Young, B. Weinstein and S. Butter, Selective alkylation of toluene with methanol to produce para-Xylene, J. Catal., 1981, 67(1), 159–174 CrossRef CAS
- C. Chu, Aromatization reactions with zeolites containing phosphorus oxide. US Patent 4590321, 1986
- H. Long, F. Jin, G. Xiong and X. Wang, Effect of lanthanum and phosphorus on the aromatization activity of Zn/ZSM-5 in FCC gasoline upgrading, Microporous Mesoporous Mater., 2014, 198(1), 29–34 CrossRef CAS
- Y. Li, W. Wu, E. Min and B. Sun, Intrinsic kinetics of ammoximation of cyclohexanone to cyclohexanone oxime over HTS zeolite, Pet. Process. Petrochem., 2003, 34(11), 39–43 CAS
- Y. Zhao, C. Cao, K. Tang and X. Hong, The Aromatization of Different FCC Naphtha Fractions over a P-Zn/HZSM-5 Catalyst, Pet. Sci. Technol., 2011, 29(10), 1009–1015 CrossRef CAS
- L. Zhao, J. Zhu, H. Wang, M. Wei, J. Ma and Y. Bai, Aromatization of FCC Gasoline Over Modified HZSM-5 Catalyst, Pet. Sci. Technol., 2007, 25(5), 577–584 CrossRef CAS
- Y. Zhao, H. Wang, M. Wei and C. Cao, Studies on Aromatization of Different FCC
Naphtha Fraction Over P-Zn/HZSM-5 Catalyst, Acta Pet. Sin., 2007, 23(3), 96–100 CAS
- J. Zhu, L. Zhao, G. Cui, M. Wei, H. Wei, H. Wang and J. &Ma, Influence of ZnO and P2O5 Content on Modified HZSM-5 on Aromatization of FCC Gasoline, Pet. Sci. Technol., 2007, 25(10), 1305–1311 CrossRef CAS
- J. Zhang, W. Qian, C. Kong and F. Wei, Increasing para-Xylene Selectivity in Making Aromatics from Methanol with a Surface-Modified Zn/P/ZSM-5 Catalyst, ACS Catal., 2015, 5(5), 2982–2988 CrossRef CAS
- J. Zhang, W. Qian, X. Tang, K. Shen, T. Wang, X. Huang and F. Wei, Influence of Catalyst Acidity on Dealkylation, Isomerization and Alkylation in MTA Process, Acta Phys.-Chim. Sin., 2013, 29(6), 1281–1288 CrossRef CAS
- C. Costa, I. Dzikh, J. Lopes, F. Lemos and F. Ribeiro, Activity–acidity relationship in zeolite ZSM-5. Application of Brönsted-type equations, J. Mol. Catal. A: Chem., 2000, 154(1–2), 193–201 CrossRef CAS
- R. Weber, J. Fletcher, K. Möller and C. O'Connor, The characterization and elimination of the external acidity of ZSM-5, Microporous Mater., 1996, 7(1), 15–25 CrossRef CAS
- L. Lubango and M. Scurrell, Light alkanes aromatization to BTX over Zn-ZSM-5 catalysts: Enhancements in BTX selectivity by means of a second transition metal ion, Appl. Catal., A, 2002, 235(1–2), 265–272 CrossRef CAS
- L. Yu, S. Huang, S. Zhang, Z. Liu, W. Xin, S. Xie and L. Xu, Transformation of Isobutyl Alcohol to Aromatics over Zeolite-Based Catalysts, ACS Catal., 2012, 2(6), 1203–1210 CrossRef CAS
- L. Hersh, E. Onyiriuka and W. Hertl, Amine-reactive surface chemistry of zinc phosphate glasses, J. Mater. Res., 1995, 10(8), 2120–2127 CrossRef CAS
- X. Yuan, B. Zhu, X. Ma, G. Tong, Y. Su and X. Zhu, Low Temperature and Template-Free Synthesis of Hollow Hydroxy Zinc Phosphate Nanospheres and Their Application in Drug Delivery, Langmuir, 2013, 29(39), 12275–12283 CrossRef CAS
- O. Kleifeld, A. Frenkel, J. Martin and I. Sagi, Active site electronic structure and dynamics during metalloenzyme catalysis, Nat. Struct. Mol. Biol., 2003, 10, 98–103 CrossRef CAS
- O. Kleifeld, A. Frenkel and I. Sagi, Time-dependent XAS studies of trapped enzyme-substrate complexes of alcohol dehydrogenase from Thermoanaerobacter brockii, J. Synchrotron Radiat., 2001, 8, 978–980 CrossRef CAS
- O. Kleifeld, A. Frenkel, O. Bogin, M. Eisenstein, V. Brumfeld, Y. Burstein and I. Sagi, Spectroscopic Studies of Inhibited Alcohol Dehydrogenase from Thermoanaerobacterbrockii: Proposed Structure for the Catalytic Intermediate State, Biochemistry, 2000, 39(26), 7702–7711 CrossRef CAS
- J. Wang, J. Qiao, Z. Jia and Z. Wang, CN Patent CN106607076A, 2015
- J. Caro, M. Bülow, M. Derewinski, J. Haber, M. Hunger, J. Kärger, H. Pfeifer, W. Storek and B. Zibrowius, NMR and IR studies of zeolite H-ZSM-5 modified with orthophosphoric acid, J. Catal., 1990, 124(2), 367–375 CrossRef CAS
- E. Brunner, H. Ernst, D. Freude, T. Fröhlich, M. Hunger and H. Pfeifer, Magic-angle-spinning NMR studies of acid sites in zeolite H-ZSM-5, J. Catal., 1991, 127(1), 34–41 CrossRef CAS
- W. Loewenstein, The distribution of aluminum in the tetrahedra of silicates and aluminates, Am. Mineral., 1954, 39(1–2), 92–96 CAS
- D. Massiot, F. Fayon, M. Capron, I. King, S. Calvé, B. Alonso, J. Dur, B. Bujoli, Z. Gan and G. Hoatson, Modelling one- and two-dimensional solid-state NMR Spectra, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS
- S. Menezes, Y. Lam, K. Damodaran and M. Pruski, Modification of H-ZSM-5 zeolites with phosphorus. 1. Identification of aluminum species by 27Al solid-state NMR and characterization of their catalytic properties, Microporous Mesoporous Mater., 2006, 95(1–3), 286–295 CrossRef
- S. Menezes, V. Camorim, Y. Lam, R. Gil, A. Bailly and J. Amoureux, Characterization of extra-framework species of steamed and acid washed faujasite by MQMAS NMR and IR measurements, Appl. Catal., A, 2001, 207(1–2), 367–377 CrossRef CAS
- G. Woolery, G. Kuehl, H. Timken, A. Chester and J. Vartuli, On the nature of framework Brønsted and Lewis acid sites in ZSM-5, Zeolites, 1997, 19(4), 288–296 CrossRef CAS
- M. Kojima, F. Lefebvre and Y. Taârit, Modification of siliceous zeolites using phosphorus pentachloride, Zeolites, 1992, 12(6), 724–727 CrossRef CAS
- K. Damodaran, J. Wiench, S. Menezes, Y. Lam, J. Trebosc, J. Amoureux and M. Pruski, Modification of H-ZSM-5 zeolites with phosphorus. 2. Interaction between phosphorus and aluminum studied by solid-state NMR spectroscopy, Microporous Mesoporous Mater., 2006, 95(1–3), 296–305 CrossRef CAS
- P. Bautista, J. Campelo, A. Garcia, D. Luna, J. Marinas and A. Romero, A1P04-A12O3 catalysts with low alumina content: I. Structural and textural characterization of catalysts obtained with aqueous ammonia, Appl. Catal., A, 1993, 96(2), 175–199 CrossRef
- L. Montagne, G. Palavit and M. Draoui, Mechanism of polyphosphate gel formation in the Na2O Al2O3 P2O5 system, J. Non-Cryst. Solids, 1993, 155(2), 115–121 CrossRef CAS
- D. Müller, I. Grunze, E. Hallas and G. Ladwig, Hochfeld-27Al-NMR-Untersuchungen zur Aluminiumkoordination in kristallinen Aluminiumphosphaten Anorg, Allg. Chem., 1983, 500, 80–88 CrossRef
- K. Dixon and J. Mason, Multinuclear NMR, Springer US, 1987, p-369 Search PubMed
- R. Hill and J. &Jones, The crystal structure of hopeite, Am. Mineral., 1976, 61, 987–995 CAS
- M. Newville, IFEFFIT : interactive XAFS analysis and FEFF fitting, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS PubMed
- M. Newville, P. Līviņš, Y. Yacoby, J. Rehr and E. Stern, Near-edge x-ray-absorption fine structure of Pb: A comparison of theory and experiment, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 14126–14131 CrossRef CAS PubMed
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
- S. Zabinsky, J. Rehr, A. Ankudinov, R. Albers and M. Eller, Multiple-scattering calculations of x-ray-absorption spectra, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 2995–3009 CrossRef CAS
- J. Biscardi, G. Meitzner and E. Iglesia, Structure and Density of Active Zn Species in Zn/H-ZSM5 Propane Aromatization Catalysts, J. Catal., 1998, 179(1), 192–202 CrossRef CAS
- M. Low and P. Ramamurthy, Infrared study of the surface properties of phosphoric acid impregnated silica, J. Phys. Chem., 1968, 729, 3161–3167 CrossRef
- J. Marques, I. Gener, P. Ayrault, J. Bordado, J. Lopes, F. Ribeiro and M. Guisnet, Dealumination of HBEA zeolite by steaming and acid leaching: distribution of the various aluminic species and identification of the hydroxyl groups, C. R. Chim., 2005, 8(3–4), 399–410 CrossRef CAS
- X. Jiang, In situ FTIR studies of extraframework aluminum bound methoxy species in H-ZSM-5 zeolites, J. Mol. Catal. A: Chem., 1997, 121(1), 63–68 CrossRef CAS
- M. Guisnet, P. Ayrault, C. Coutanceau, M. Alvarez and J. Datka, Acid properties of dealuminated beta zeolites studied by IR spectroscopy, J. Chem. Soc., Faraday Trans., 1997, 93(8), 1661–1665 RSC
- N. Brodu, M. Manero, C. Andriantsiferana, J. Pic and H. Valdés, Role of Lewis acid sites of ZSM-5 zeolite on gaseous ozone abatement, Chem. Eng. J., 2013, 231, 281–286 CrossRef CAS
- S. Zheng, H. Heydenrych, A. Jentys and J. Lercher, Influence of Surface Modification on the Acid Site Distribution of HZSM-5, J. Phys. Chem. B, 2002, 106(37), 9552–9558 CrossRef CAS
- E. El-Malki, R. Santen and W. Sachtler, Introduction of Zn, Ga, and Fe into HZSM-5 Cavities by Sublimation:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Identification of Acid Sites, J. Phys. Chem. B, 1999, 103(22), 4611–4622 CrossRef CAS
Identification of Acid Sites, J. Phys. Chem. B, 1999, 103(22), 4611–4622 CrossRef CAS - P. Jacobs, E. Flanigen, J. Jansen and H. Bekkum, Introduction to zeolite science & practice, Elsevier, 2001 Search PubMed
- B. Hunger, M. Heuchel, L. Clark and R. Snurr, Characterization of Acidic OH Groups in Zeolites of Different Types: An Interpretation of NH3-TPD Results in the Light of Confinement Effects, J. Phys. Chem. B, 2002, 106(15), 3882–3889 CrossRef CAS
- L. Lin, S. Zhao, D. Zhang, H. Fan, Y. Liu and M. He, Acid Strength Controlled Reaction Pathways for the Catalytic Cracking of 1-Pentene to Propene over ZSM-5, ACS Catal., 2015, 5(7), 4048–4059 CrossRef CAS
- Y. Ni, A. Sun, X. Wu, G. Hai, J. Hu, T. Li and G. Li, The preparation of nano-sized H[Zn, Al]ZSM-5 zeolite and its application in the aromatization of methanol, Microporous Mesoporous Mater., 2011, 143(2–3), 435–442 CrossRef CAS
- W. Kaeding and S. Butter, Production of chemicals from methanol: I. Low molecular weight olefins, J. Catal., 1980, 61(1), 155–164 CrossRef CAS
- H. Vinek, G. Rumplmayr and J. Lercher, Catalytic properties of postsynthesis phosphorus-modified H-ZSM5 zeolites, J. Catal., 1989, 115(2), 291–300 CrossRef CAS
- M. Castilla, A. Gayubo, A. Aguayo, J. Ar&es and J. Bilbao, Simulation and Optimization of Methanol Transformation into Hydrocarbons in an Isothermal Fixed-Bed Reactor under Reaction− Regeneration Cycles, Ind. Eng. Chem. Res., 1998, 37(6), 2383–2390 CrossRef CAS
- G. Mekhemer, A. Nohman, N. Fouad and H. Khalaf, Surface to bulk characterization of phosphate modified aluminas, Colloids Surf., A, 2000, 161(3), 439–446 CrossRef CAS
- S. Kouva, J. Kanervo, F. Schüβler, R. Olindo, J. Lercher and O. Krause, Sorption and diffusion parameters from vacuum-TPD of ammonia on H-ZSM-5, Chem. Eng. Sci., 2013, 89(15), 40–48 CrossRef CAS
- H. Li, X. Li and W. Xiao, Deactivation kinetics of individual C6–C9 aromatics' generation from methanol over Zn and P co-modified HZSM-5, RSC Adv., 2019, 9, 22327–22335 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09657d |
| This journal is © The Royal Society of Chemistry 2020 |