
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and activation of an iron oxide immobilized drug-mimicking reporter under conventional and pulsed X-ray irradiation conditions†
Anna Barosia,
Petra Dunkel a,
Erwann Guéninbc,
Yoann Lalatonnebd,
Philippe Zeitoune,
Isabelle Fittonf,
Clément Journéb,
Alberto Braving,
Antoine Maruanih,
Hamid Dhimanea,
Laurence Motte*b and
Peter I. Dalko*a
a,
Erwann Guéninbc,
Yoann Lalatonnebd,
Philippe Zeitoune,
Isabelle Fittonf,
Clément Journéb,
Alberto Braving,
Antoine Maruanih,
Hamid Dhimanea,
Laurence Motte*b and
Peter I. Dalko*a
aLaboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, Université Paris Descartes, UMR 8601, 45, Rue des Saints-Pères, 75210 Paris, France. E-mail: peter.dalko@parisdescartes.fr
bLaboratory for Vascular Translational Sciences (LVTS), INSERM, U1148, Université Paris 13, 74 Rue Marcel Cachin, 93017 Bobigny, France. E-mail: laurence.motte@univ-paris13.fr
cSorbonne University – Université de Technologie de Compiègne, Laboratoire TIMR (UTC/ESCOM), EA4297, Centre de recherche de Royallieu, Rue du Docteur Schweitzer, CS 60319, 60203 Compiègne Cedex, France
dServices de Biochimie et de Médecine Nucléaire, Hôpital Avicenne Assistance Publique-Hôpitaux de Paris, 125 Rue de Stalingrad, 93009 Bobigny, France
eLaboratoire d’Optique Appliquée, UMR 7639 ENSTA-ParisTech, CNRS, Institut Polytechnique de Paris, 828 boulevard des Maréchaux, 91120 Palaiseau, France
fService de Radiologie Hôpital Européen Georges Pompidou, Paris, France
gESRF, ID17 Beamline, 71 Avenue des Martyrs, 38043 Grenoble, France
hDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
First published on 21st January 2020
Abstract
An efficient nano-sized delivery system is presented here allowing the immobilized, picolinium-tethered organic ligand to be released by X-ray irradiation. A marked difference was observed in the fragmentation efficiency by using conventional Cs-137 vs. pulsed sources.
Almost a decade ago we reported the first fragmentation of complex organic substrates by X-ray and gamma-ray irradiation providing an opportunity to activate photolabile probes deep within biological tissues where light cannot penetrate.1,2 In this original experiment a tethered gadolinium(III) complex was used as an electron source and also as a contrast agent, allowing the bio-distribution of the probe to be followed in vivo.3,4 The developed system shared common features with “caged substrates” used in molecular biology, physiology and neurosciences allowing the light-gated release of biologically active compounds in high spatial integrity, where the activity of the biomolecule is masked by the photosensitive group and the photolysis restores the biological activity.3,4 We demonstrated that using tissue-penetrating irradiation at 17.5 keV (X-ray), or at 1.17 MeV (γ-ray) the radiolysis produces clean and qualitatively similar fragmentation to near-UV photolysis.1 Although the mechanism of the Gd(III)-sensitized radiolysis is not well understood, our working hypothesis stands on the grounds of converting high-energy photons to electrons by photoelectron and/or Auger/Compton processes in the presence of a metal atom, that converts photons to electrons more efficiently than radiation generates electrons from water. According to this putative mechanism, electrons are transferred in fine to the photodevice, which undergoes ET (electron transfer)-mediated chemical transformation/fragmentation. The role of the metal [i.e. the Gd(III) in the presented model] was double, as it (i) played the role of an efficient sensitizer and (ii) offered an opportunity to follow the distribution of the device in the body in real time under in vivo conditions by using conventional T1 weighted MR imaging modality.
Our preliminary studies established the proof of principle of the selective fragmentation by exposure to X, or, γ-rays, but also showed areas in which improvements are needed to take the approach further. It was observed by MRI that the intravenous (iv) injected Gd-DOTAGA-sensitized devices were eliminated rapidly from the blood stream and cleared to the bladder. The serum residence time of the DOTAGA-sensitized device was in order of minutes. That was essentially attributed to a rapid renal clearance, known also for related DOTAGA analogs. Twenty-four-hour urine samples showed that 60% to 70% of the iv-injected device was excreted. The kinetic of this metabolism was similar to that of the Gd-DOTAGA, and warranted for less polar sensitizers for increased systemic circulation.
Here we report a development with a considerably improved lypophilicity balance and sensitivity of the probe by using ultra small superparamagnetic iron oxide (USPIO) nanoparticles (NPs) as immobilization platforms, which are among the few nanomaterials that are bio-compatible and approved for therapeutic use.5 USPIO NPs were used here (i) for the immobilization/support of the redox probe; (ii) for tracking the biodistribution of the probe by T2 weighted MRI and (iii) for the generation of Auger/Compton electrons upon X or γ-ray irradiation (Scheme 1). In order to increase the ET sensitivity of the probe, the original quinoline scaffold was replaced by our recently developed hydroxyethyl picolinium sensor: the probe undergoes selective fragmentation at E° = −1.14 V (vs. HNE),6 a value low enough for allowing efficient activation by Auger/Compton electrons, while being lower than the limit of biological reducing systems that is approximately −0.65 V, thus may not be activated in vivo without external trigger.5 Our earlier observation was in agreement with the heterolytic (radical ionic) rather than homolytic bond cleavage as the major ET fragmentation path.6
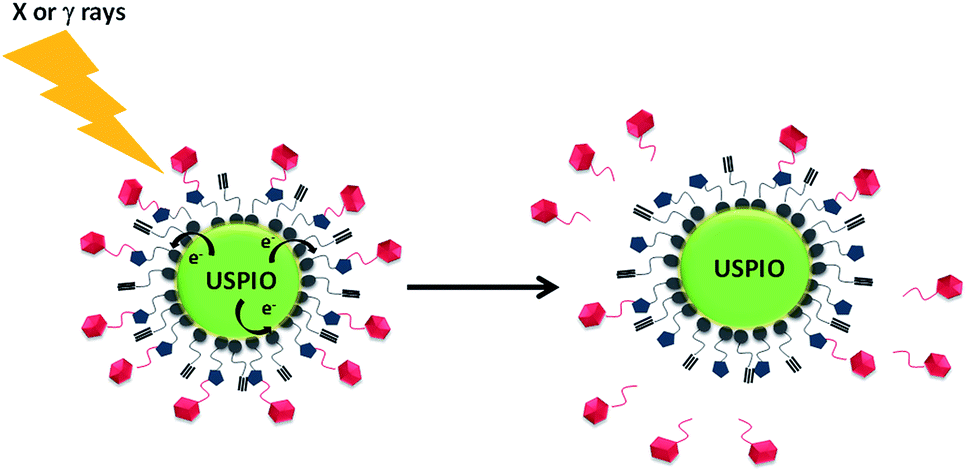 | ||
| Scheme 1 Cartoon of the activation of the USPIO NP immobilized redox probe. Electrons generated by Auger/Compton process by X or γ-ray irradiation results in fragmentation of the covalently linked ligand. | ||
Classical non-aqueous sol–gel synthesis (the ‘benzyl alcohol route’) was used for the preparation of USPIO NPs, opting for microwave activation.7 This protocol allowed reducing considerably the synthesis time (30 min, vs. 2 days in autoclave), and also gave hand on the precise control of the size, resulting in NPs with an average diameter of 9 nm, with high reproducibility.7 The prepared NPs were coated by bisphosphonate anchoring groups. Bisphosphonates have remarkable capacity to chelate divalent cations such as Ca2+, Mg2+ as well as Fe3+, Fe2+ and the binding affinity can be increased by the presence of an extra hydroxyl group (hydroxylmethylene bisphosphonate, HMBP).8 The increased affinity is probably due to the tridentate binding ability of the ligand to the cation. Noteworthy, the HMBP family is orthogonal to a large variety of terminal functions, a fact that was already exploited in a number of elegant coating strategies.9
HMBP-yne ligand, 2, was prepared by using a straightforward 3 steps synthesis (Scheme 2).10 Accordingly, pent-4-ynoyl acid chloride was reacted with tris(trimethylsilyl)phosphite (neat) at −20 °C, then the product was hydrolyzed in the presence of methanol leading to the desired 2, in 70% overall yield. The surface functionalization was achieved by adding 2 to the emulsion of USPIO NPs at pH 2. At this pH, the protonated, thus positively charged NPs, adsorbed rapidly the negatively charged bisphosphonates. NPs were then separated from the supernatant by precipitation followed by magnet-sorting and were washed 3 times. The coated USPIO-yne NPs were re-dispersed in water at pH 7.4, and the amount of the absorbed ligand was quantified by thermogravimetric analysis (TGA) and also, by energy dispersive X-ray analysis (EDX, Fig. SI2†). A value of 440 ± 40 HMBP-yne anchoring group was obtained based on the iron/phosphorus atomic ratio per NPs. The iron concentration was determined by colorimetric assay (ESI†).
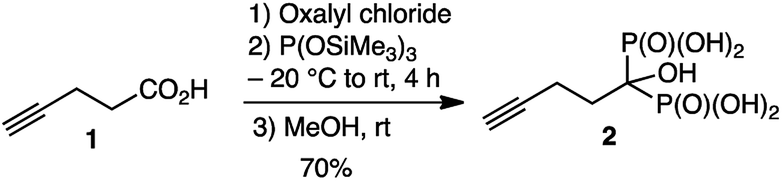 | ||
| Scheme 2 Synthesis of the butyne hydroxylmethylene bisphosphonate (HMBP-yne) linker, 2. | ||
4-Hydroxyethyl picolinium, 4, having pyrene butyrate fluorescent reporter was selected as redox fragmenting element (Scheme 3).6 Pyrene was selected here being the most resistant among all fluorescent tags under redox and radiolytic conditions. Probes were prepared from pyridine hydroxyethylene 3 in a short esterification/quaternarisation sequence by using 2-azidoethyl trifluoromethanesulfonate as alkylating agent.
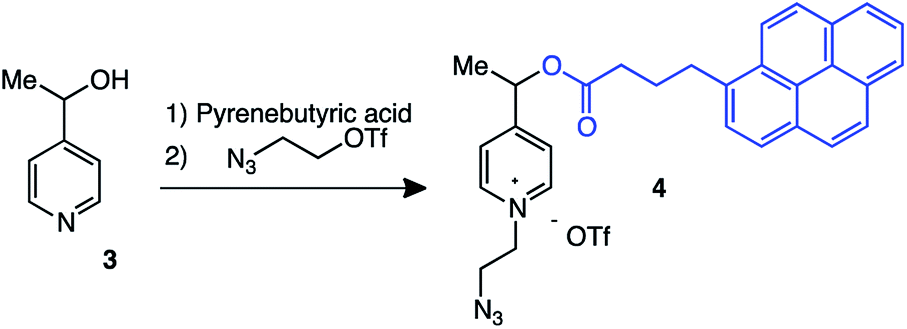 | ||
| Scheme 3 Synthesis of the picolinium redox probe, 4, having pyrene butyrate fluorescent reporter. | ||
Picolinium probes were grafted to the surface-linked bisphosphonate (HMBP-yne) by click chemistry1,6 (Fig. 1a). It was considered, that the polar quaternary picolinium salt may ensure sufficient wetting to stabilize the colloid system in water, thus no other hydrophilic ligands were added. The coupling between HMBP-yne-coated iron oxide and picolinium probes was realized by copper(I)-catalyzed azide/alkyne cycloaddition (CuAAC), in the presence of a catalytic amount of CuI (0.1 equiv.) at rt for 24 h. As the solubility of the pyrene butyric acid-functionalized 4 was very poor in water, the reaction was performed in a mixture of acetonitrile/water (1/1). In the click reaction the picolinium compound, 4, was used in sub-stoichiometric amount (0.2 equiv.) in order to avoid saturation and subsequent particles aggregation that may be driven by inter-NPs strong π–π interaction between the pyrene ligands. The obtained NPs (USPIO-0.2) were washed several times and then re-dispersed in water (pH 7.4).
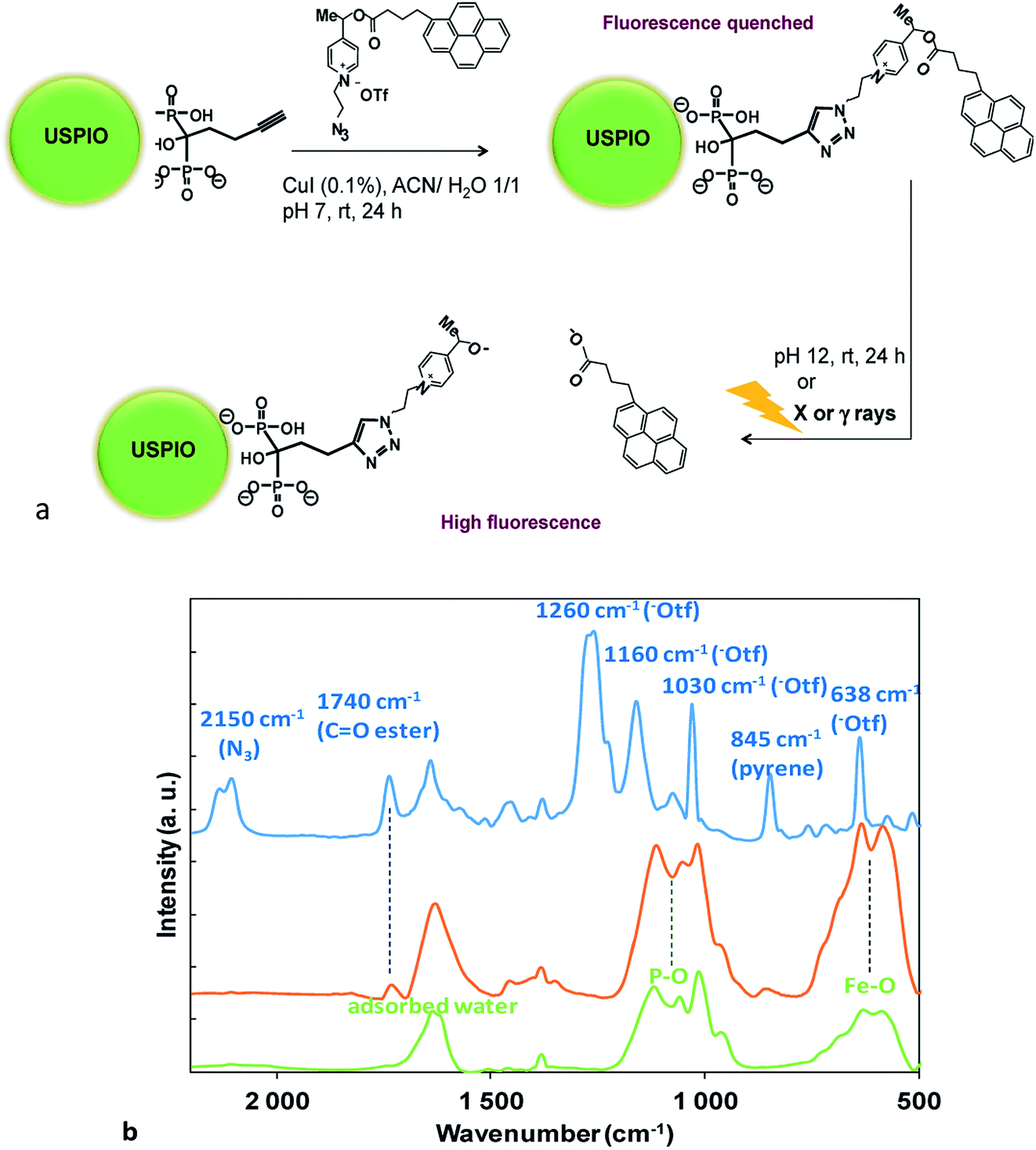 | ||
| Fig. 1 (a) Huisgen 1,3 cycloaddition of picolinium azide on USPIO-yne NPs and schematic representation of the pyrene butyric acid release upon saponification, or, after redox fragmentation under X or γ ray irradiation; (b) FTIR spectra of USPIO-yne NPs (green), USPIO-0.2 (orange) and of picolinium azide (blue) (KBr). (For extended spectra of USPIO-yne NPs see Fig. SI4†). | ||
The hydrodynamic size of the nanoplatforms (USPIO-yne and USPIO-0.2) d = 17 nm (DLS), respectively, which confirmed no aggregation, and also confirmed the colloidal stability of the dispersion at physiological pH (ESI†).§
Nanoplatforms were characterized by FTIR (Fig. 1b). The USPIO-yne NP spectrum (Fig. 1b, green curve and Fig. SI4†) showed a characteristic broad band at 600 cm−1, assigned to the vibration of the iron–oxygen bond at the surface, and strong stretching bands in the region of 1200 to 900 cm−1 that were assigned to P–O stretches of the HMBP-yne ligand.8 The FTIR spectrum of the picolinium azide, 4, exhibited distinctive peaks (Fig. 1b, blue curve), such as the azide stretching (2150 cm−1), ester bond stretching (1750 cm−1), and pyrene aromatics (845 cm−1), respectively. The presence of the triflate counter-ion was confirmed by the presence of strong absorption bands at 1260 cm−1 and at 1160 cm−1 corresponding to the asymmetric stretching of the SO3 and CF3 groups respectively, a strong band at 1030 cm−1 corresponding to symmetric (SO3) stretching mode and a strong band at 638 cm−1 attributed to the bending vibration of the SO3 group.11
The presence of the redox probe on the NP was evidenced by the characteristic IR signals of the functionalized NP (USPIO-0.2 Fig. 1b, orange curve) with those of USPIO-yne and azide 4. The disappearance of the 2100 cm−1 band, characteristic of the azide suggested the transformation of the azide to triazole.‡ Noteworthy is the presence of the ester bond stretching at 1750 cm−1 as well as the pyrene aromatic peak at 845 cm−1. It has to be noticed that the bands previously assigned to the triflate counter-ion disappeared, suggesting their probable displacement during the washing procedure by stronger anions (such as Cl− during the pH adjustment by using HCl (aq)). The optical properties of the various nanoplatforms were also characterized and compared (Fig. SI5†). Noteworthy, the absorption peaks of the picolinium probe were clearly distinguishable in the USPIO-0.2 spectrum (Fig. SI5,† orange curve).
It was observed, that the picolinium group quenched efficiently the fluorescence of the pyrene in aqueous solution, probably by Förster-type quenching/dark complex formation. Moreover, the fluorescence quenching was maintained after immobilisation on the NP surface: here the intra-, or intermolecular quenching and Förster-type energy transfer process between the pyrene and the metal oxide NP may increase the quenching efficiency of the assembled probe. The number of the grafted probes was quantified after saponification of the pyrene fluorophore (NaOH, 1 N) for 24 h. The pH was re-adjusted to neutral with HCl, and the fluorescence of the liberated pyrene butyric acid was quantified (Fig. SI6†). The number of grafted molecules was determined by comparing the fluorescence to a calibration curve; (λex = 341 nm, λem = 376 nm), and an average number of 1.4 probe/NP was obtained.
Since USPIO NPs are negative MRI contrast agents, we used local changes in intensity on T2 weighted MRI to evaluate the USPIO-0.2 NP biodistribution in B6 mice using 7T-MRI. Fig. 2 shows T2 weighted MRI images and the dynamic contrast variation in liver, spleen, and kidneys before injection and 15 min, 45 min, 3 h 30 min and 24 h after NP injection. Shortly after injection (t = 15 min) a large negative contrast enhancement was observed in organs rich in macrophages, such as in liver, kidneys and spleen. Moreover, the contrast in liver and spleen stays constant up to 45 min after injection whereas intensity signal decreases steadily in kidney. This fact suggests NP accumulation in the liver and spleen and also NP clearance from the kidneys. Hence, compared to our previous results obtained with gadolinium(III) complex,1,3 the USPIO-0.2 NP residence time in the body is greatly improved up to 24 h, notably in liver and spleen.
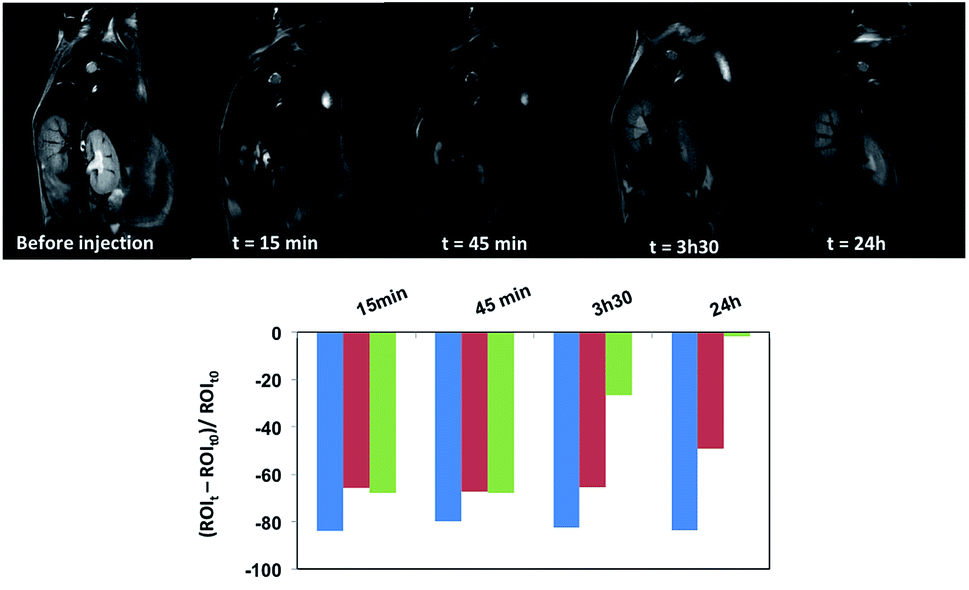 | ||
| Fig. 2 T2 weighted MRI images and contrast variation showing the NP biodistribution after injection in B6 mice. The graphs show the clearance of the contrast signal (in arbitrary units) measured at 15 min, 45 min, 3 h 30 min, and 24 h after injection in the liver (blue), spleen (red) and kidneys (green). | ||
The activation of the probes was validated in vitro at c = 5 μM Fe concentrations§ by using either the ID17 biomedical beamline of the ESRF synchrotron (Grenoble) (conditions: cuvette: quartz, d = 1 mm, beam: 80 keV, bunch length: 48 ps, bunch repetition rate 5.68 MHz, dose rate 1.82 Gy s−1, dose up to 30 Gy), or, a conventional Cs-137 source (BIOBEAM 8000, Gamma-Service Medical GmbH; activity 80.29 TBq ± 20%; dose rate: 5.86 Gy min−1). The fragmentation was quantified by following the appearing fluorescence, and reported in Table 1.¶ While efficient fragmentation was observed under both conditions at low doses (up to 30 Gy) pulsed sources of the ESRF appeared considerably more efficient: 82% of the fluorescence was recovered after 30 Gy irradiation contrasting 27% of fluorescence under conventional conditions. Although the origin of the difference between the efficiency of the two type of redox fragmentation is not well understood, eventually non-linear effects between the use of continuous vs. pulsed sources may be at work.
| 80 keV | 0.6617 MeV | |||||
|---|---|---|---|---|---|---|
| Dose (Gy) | 0.3 | 3 | 30 | 0.3 | 3 | 30 |
| Fragmentation (%) | 25 | 62 | 82 | 6 | 19 | 27 |
Conclusions
Pyrene butyrate fluorescent reporter-armed picolinium probes were immobilized on spherical, homogeneous USPIO NPs. The MRI contrast ability of the prepared probe was evaluated and the biodistribution of the probe was followed in B6 mice by using 7T-MRI modality. A large negative contrast enhancement was observed in liver, kidneys and spleen, whereas T2 signal decreased steadily in kidney. Activation of the probes was validated in vitro at c = 5 μM Fe concentrations by using either the ID17 biomedical beamline facility of the ESRF synchrotron (Grenoble), or, by using conventional Cs-137 source. While efficient fragmentation was observed under both conditions up to 30 Gy, pulsed sources of the ESRF appeared considerably more efficient: 82% of the fluorescence was recovered by 30 Gy irradiation contrasting 27% of fluorescence by using conventional conditions. Results indicate that the coated nanoplatform may be suitable for the use as MRI contrast agent and also as a drug delivery platform, in particular for liver indications. Further studies are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. Barosi acknowledges the NanoK grant of the IdF region. Authors wish to thank to the CNanoMat platform of the University Paris 13 for the use of UV-vis, FTIR and Zetasizer Nano ZS and the FRIM Imaging platform at Université Paris 7 (CHU X Bichat) for the MRI 7T measurements. A. M. would like to acknowledge the Ramsay Memorial Trust for provision of a Ramsay Fellowship. We also thank the ImagoSeine core facility of the Institut Jacques Monod, member of IBiSA and France-Bioimaging (ANR-10-INBS-04) infrastructures, particularly Dr Rémi le Borgne for sample preparation and assistance with TEM imaging.Notes and references
- M. Petit, G. Bort, B.-T. Doan, C. Sicard, D. Ogden, D. Scherman, C. Ferroud and P. I. Dalko, Angew. Chem., Int. Ed., 2011, 50, 9708 CrossRef CAS PubMed; M. Petit, G. Bort, D. Ogden, C. Sicard and P. I. Dalko, Int. Pat. Appl., WO 2011/158189 A1, 2011.
- W. Deng, W. Chen, S. Clement, A. Guller, Z. Zhao, A. Engel and E. M. Goldys, Nat. Commun., 2018, 9, 2713 CrossRef CAS PubMed; F. Liu, Q. Liu, D. Yang, W. B. Bollag, K. Robertson, P. Wu and K. Liu, Cancer Res., 2011, 71, 6807 CrossRef; H. Minato, M. Matsumoto and T. Katayama, J. Chem. Soc., Perkin Trans. 1, 1973, 1819 RSC.
- M. J. Davis, C. H. Kragor, K. G. Reddie, H. C. Wilson, Y. Zhu and T. M. Dore, J. Org. Chem., 2009, 74, 1721 CrossRef CAS PubMed; O. D. Fedoryak and T. M. Dore, Org. Lett., 2002, 4, 3419 CrossRef PubMed; Y. Zhu, C. M. Pavlos, J. P. Toscano and T. M. Dore, J. Am. Chem. Soc., 2006, 128, 4267 CrossRef PubMed; Y. M. Li, J. Shi, R. Cai, X. Y. Chen, Q. X. Guo and L. Liu, Tetrahedron Lett., 2010, 51, 1609 CrossRef; M. C. Pirrung, T. M. Dore, Y. Zhu and V. S. Rana, Chem. Commun., 2010, 46, 5313 RSC.
- G. Bort, T. Gallavardin, D. Ogden and P. I. Dalko, Angew. Chem., Int. Ed., 2013, 52, 4526 CrossRef CAS PubMed; G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900 CrossRef PubMed; A. Specht, F. Bolze, Z. Omran, J.-F. Nicoud and M. Goeldner, HFSP J., 2009, 3, 255 CrossRef PubMed; H.-M. Lee, D. R. Larson and D. S. Lawrence, ACS Chem. Biol., 2009, 4, 409 CrossRef PubMed; G. C. R. Ellis-Davies, Nat. Methods, 2007, 4, 619 CrossRef PubMed.
- A. L. Cortajarena, D. Ortega, S. M. Ocampo, A. Gonzalez-García, P. Couleaud, R. Miranda, C. Belda-Iniesta and A. Ayuso-Sacido, Nanobiomedicine, 2014, 1, 2 CrossRef CAS PubMed; B. Dalzon, M. Guidetti, D. Testemale, S. Reymond, O. Proux, J. Vollaire, V. Collin-Faure, I. Testard, D. Fenel, G. Schoehn, J. Arnaud, M. Carrière, V. Josserand, T. Rabilloud and C. Aude-Garcia, Nanoscale, 2019, 11, 9341 RSC.
- P. Dunkel, A. Barosi, H. Dhimane, F. Maurel and P. I. Dalko, Chem. – Eur. J., 2018, 24, 12920 CrossRef CAS PubMed.
- S. Richard, V. Eder, G. Caputo, C. Journe, P. Ou, J. Bolley, L. Louedec, E. Guenin, L. Motte, N. Pinna and Y. Lalatonne, Nanomedicine, 2016, 11, 2769 Search PubMed; N. Pinna, S. Grancharov, P. Beato, P. Bonville, M. Antonietti and M. Niederberger, Chem. Mater., 2005, 17, 3044 CrossRef CAS.
- Y. Lalatonne, C. Paris, J. M. Serfatty, P. Weinmann, M. Lecouvey and L. Motte, Chem. Commun., 2008, 44, 2553 RSC.
- C. Dubreil, O. S. Catherine, Y. Lalatonne, C. Journé, P. Ou, P. van Endert and L. Motte, Small, 2018, 14, 1802053 CrossRef CAS PubMed; A. Plan Sangnier, R. Aufaure, L. Motte, C. Wilhelm, E. Guenin and Y. Lalatonne, Beilstein J. Nanotechnol., 2018, 9, 2947 CrossRef PubMed; R. Aufaure, R. Buendia, L. Motte, J. Hardouin, Y. Lalatonne and E. Guénin, New J. Chem., 2017, 41, 12153 RSC; R. Aufaure, J. Hardouin, N. Millot, L. Motte, Y. Lalatonne and E. Guénin, Chem. – Eur. J., 2016, 22, 16022 CrossRef PubMed; J. Bolley, E. Guénin, N. Lievre, M. Lecouvey, M. Soussan, Y. Lalatonne and L. Motte, Langmuir, 2013, 29, 14639 CrossRef PubMed; B. Basly, D. Felder-Flesch, P. Perriat, C. Billotey, J. Taleb, G. Pourroy and S. Begin-Colin, Chem. Commun., 2010, 46, 985 RSC.
- E. Guénin, J. Hardouin, Y. Lalatonne and L. Motte, J. Nanopart. Res., 2012, 14, 965 CrossRef.
- D. H. Johnston and D. F. Shriver, Inorg. Chem., 1993, 32, 1045 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: HMBPyne, USPIO and picolinium derivative synthesis; USPIO surface functionalization and physicochemical methods; TEM size distribution; hydrodynamic size and zeta charge. See DOI: 10.1039/c9ra09828c |
| ‡ The triazole characteristic peaks (IR) is usually displayed between 1650 and 1690 cm−1: here are buried under other peaks. |
| § Samples were hydrolytically stables in water at pH 7.4 for (at least) 1 week at rt as was seen from the absence of fluorescent signal of the examined aliquot. NPs were stable in aqueous solution under extended storage at 4 °C. |
| ¶ The bleaching of the pyrene was studied in ethanol at rt by irradiation using the Cs-137 source up to 30 Gy and was seen marginal. |
| This journal is © The Royal Society of Chemistry 2020 |