
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient one-pot synthesis of functionalised imidazo[1,2-a]pyridines and unexpected synthesis of novel tetracyclic derivatives by nucleophilic aromatic substitution†
Charles R. K. Changunda ,
B. C. Venkatesh,
William K. Mokone,
Amanda L. Rousseau,
Dean Brady,
Manuel A. Fernandes and
Moira L. Bode*
,
B. C. Venkatesh,
William K. Mokone,
Amanda L. Rousseau,
Dean Brady,
Manuel A. Fernandes and
Moira L. Bode*
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Private Bag 3, PO WITS 2050, Johannesburg, South Africa. E-mail: Moira.Bode@wits.ac.za
First published on 25th February 2020
Abstract
Novel tetracyclic imidazo[1,2-a]pyridine derivatives have been prepared by intramolecular nucleophilic aromatic substitution of 5-fluoroimidazo[1,2-a]pyridines under basic conditions. Use of the non-nucleophilic alcoholic solvent tert-butanol, rather than methanol, increased the yield of the tetracycles by reducing the competing intermolecular reaction observed for methanol. In addition, a modified protocol for the dehydration of formamides to isocyanides has been found to be tolerant of an unprotected hydroxyl functional group and one-pot conversion to imidazo[1,2-a]pyridyl-aminocyclohexanol analogues is reported.
Introduction
The imidazopyridine skeleton possesses unique electronic and chemical properties that make it an attractive starting point in the preparation of a broad spectrum of therapeutic agents1 ranging from sedative drugs such as zolpidem 1, antiviral agents 2, anticancer compounds 3, immunomodulators 4 (ref. 2) and antitubercular agents 5 (ref. 3) (Fig. 1), to mention just a few. Other reports indicate activity as gastric proton pump inhibitors4 and as antifungal,5 antibacterial6 and anxiolytic7 agents.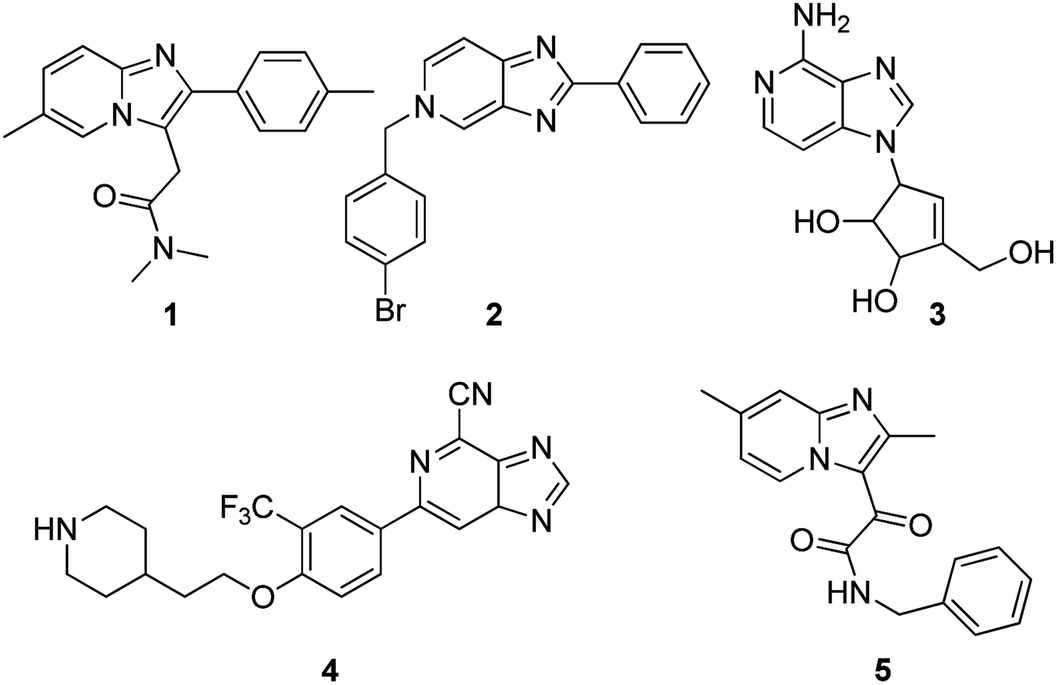 | ||
| Fig. 1 Biologically active imidazopyridines. | ||
Largely as a result of their biological importance, the development of safe synthetic methodologies that efficiently access imidazopyridines and their associated derivatives continues to generate much research interest in synthetic chemistry.8
The utility of the multi-component Groebke–Blackburn–Bienaymé reaction9 for preparation of imidazo[1,2-a]pyridines by reaction of an aldehyde, 2-aminopyridine and an isocyanide is well documented in synthetic chemistry literature.1a,10a,b Given that 2-aminopyridines and aldehydes are generally affordable, the versatility and robustness of this protocol is primarily disadvantaged by the limited variety and high procurement cost of commercially available isocyanides. This mandates researchers to prepare most of the isocyanides that are required to fulfil their research requirements.
More than one and a half centuries ago, Gautier and Hofmann11 first described the preparation of isocyanides. Their apt description of isocyanides as possessing ‘almost overpowering, horrible and extremely distressing odours’ typifies the challenges associated with the preparation and handling of isocyanides even to this present day. Almost a hundred years later, the first generally applicable routes for accessing isocyanides were described, via the dehydration of N-formamides using acyl oxides of group IV–VI elements in the presence of bases.12 Due to the high toxicity and handling difficulties associated with using phosgene,13 phosphorus oxychloride (used together with Et3N base), a method originally described by Ugi and Meyr,14 has become one of the most commonly employed N-formamide dehydrating agents for the preparation of isocyanides in synthetic chemistry today.
Nevertheless, the increasing enactment of tightened environmental, health and safety management laws continues to drive the search for safer synthetic routes for accessing isocyanides. Thus, more research and development is still needed to develop safer methodologies that provide ease of access to a large variety of these key substrates. In an interesting development, Wang and co-workers15 reported the identification of triphenylphosphine and iodine as mild and efficient N-formamide dehydrating agents for generating aromatic isocyanides. Guchhait and colleagues16 reported the development of a one-pot reaction which employed para-toluenesulfonyl chloride (pTsCl) and DABCO for the dehydration of N-formamide substrates to generate isocyanides in situ for subsequent use in multicomponent reactions.
We have previously reported the identification of novel imidazo[1,2-a]pyridine derivatives as non-nucleoside inhibitors of HIV-1 reverse transcriptase.17 The novel lead compound, 2-(2-chlorophenyl)-3-(cyclohexylamino)imidazo[1,2-a]pyridine-5-carbonitrile 6 (Fig. 2) exhibited good antiviral activity (whole cell anti-HIV IC50 = 0.18 μM) and displayed excellent selectivity (SI = 868) when screened against the wild-type HI virus. Molecular modelling results indicated that introduction of groups capable of hydrogen-bonding to amino acids in the allosteric site would potentially lead to compounds with increased potency.
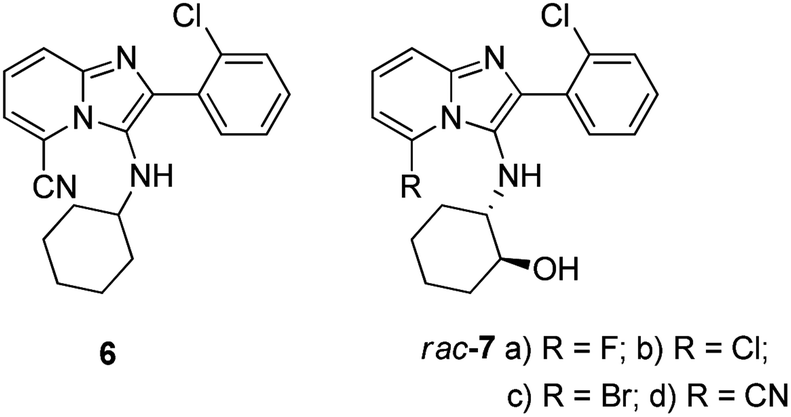 | ||
| Fig. 2 Imidazopyridine lead compound 6; modified target products 7. | ||
Therefore, as part of our ongoing efforts to discover compounds with better antiviral activity profiles against both wild-type and mutant viral strains we planned to expand our imidazo[1,2-a]pyridine library using compound 6 as a starting point. Thus compounds of general structure 7 (Fig. 2) were conceived for synthesis as promising targets for subsequent screening against the HI virus. In this paper, we report a highly efficient modified pTsCl/DABCO protocol16 as a safe and OH functional group tolerant catalyst methodology for accessing novel imidazo[1,2-a]pyridine heterocyclic targets. In addition, we also report the unexpected ring-closure of 5-fluoro-imidazo[1,2-a]pyridine derivatives, giving rise to novel tetracyclic compounds.
Results and discussion
To access our initial small library of target compounds of general structure 7, 2-trans-hydroxyammonium hydrochloride (rac-8) was neutralised and formylated quantitatively to generate formamide rac-9 using a previously reported method18 (Scheme 1). After acetylation, compound rac-10 was then dehydrated using POCl3/Et3N to produce the isocyanide rac-11 for subsequent multi-component Groebke–Blackburn–Bienaymé reaction. Acetylation was used as a means of protecting the OH group during the dehydration step, as free OH groups are incompatible with the POCl3/Et3N methodology. Coupling of rac-11 with 2-chlorobenzaldehyde 12a and 6-substituted-2-aminopyridine 13 (Scheme 1) produced the target acetates 14a–d in excellent yields, specifically over the multicomponent coupling step (v, Scheme 1). Nonetheless, it is noteworthy to point out that a respectable 85% yield of the isocyanide (rac-11) was only achieved at small scale (100 mg of rac-10) whilst efforts to scale up the reaction progressively gave poorer conversions. One of the difficulties of this reaction appeared to be a propensity of rac-11 to undergo a rehydration reaction to regenerate the N-formamide rac-10 during the aqueous work up, given the non-stoichiometric imbalance between the acid and base associated with this protocol.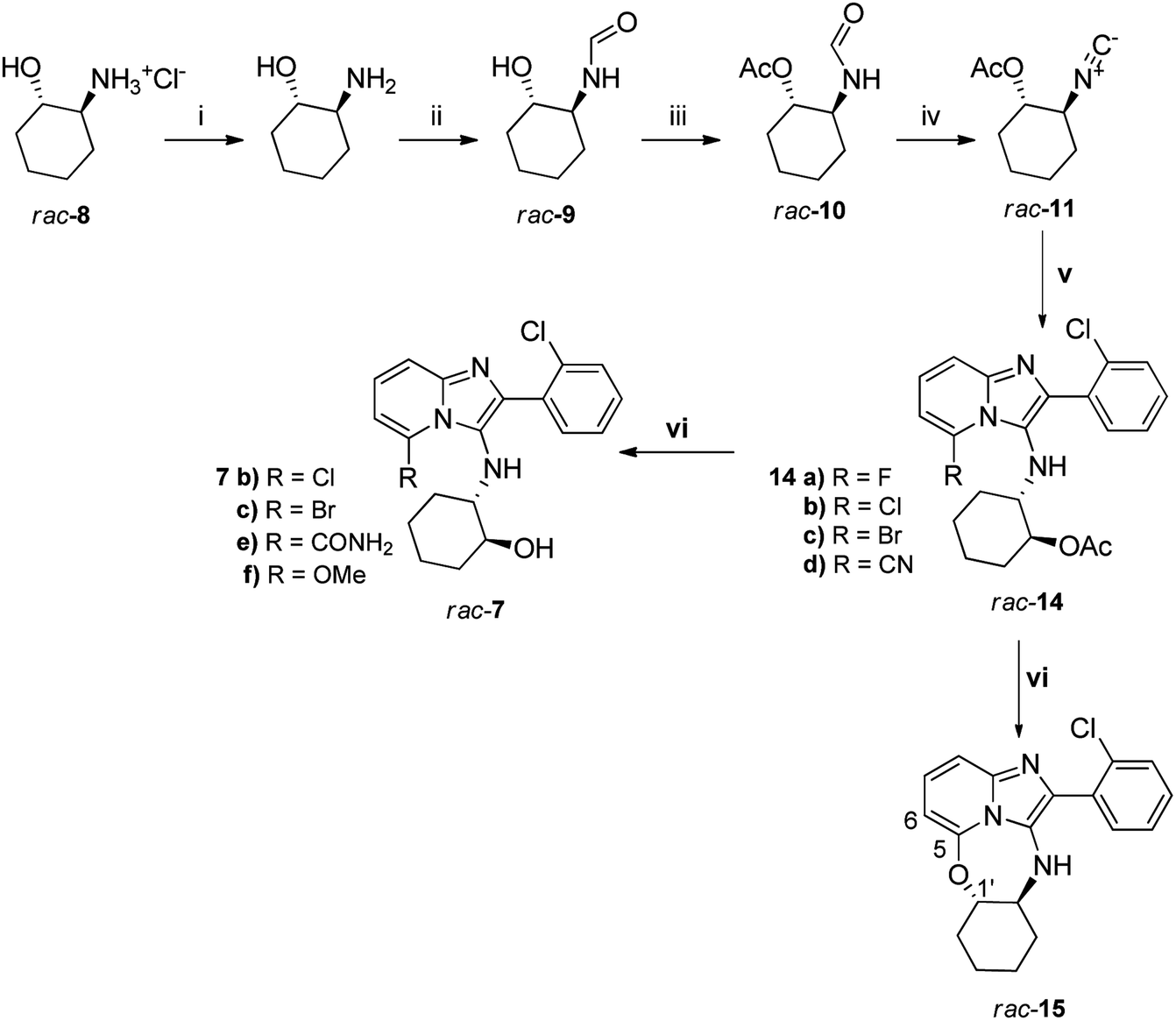 | ||
| Scheme 1 Reagents: (i) NaOMe, MeOH, RT; (ii) methyl formate, RT, 12 h; (iii) Ac2O, pyridine, 4 h; (iv) POCl3/Et3N, DCM; (v) 2-chlorobenzaldehyde 12a, 6-substituted-2-aminopyridine 13a–d, K-10 clay, MW 150W, 100 °C, 30 min; (vi) KOH, MeOH, RT, 4 h. | ||
The final step in the synthesis was KOH-catalysed hydrolysis of the respective acetates in MeOH to obtain alcohols 7. Target compounds 7b and 7c were obtained in excellent yields from 14b and 14c, respectively, while unexpected hydrolysis of the nitrile group of 14d under the basic conditions of the deprotection reaction gave rise to carboxamide 7e. Attempted deprotection of compound 14a also did not lead to the expected deprotected compound; instead a roughly equal mixture of two compounds was obtained. On initial inspection of the 1H NMR spectrum of the first product 15 it was immediately evident that the acetyl group had been removed. The first clear indication that the expected product had not been obtained was the appearance of the signal at 6.17 ppm for H-6 as a doublet showing one ortho-coupling (J = 7.2 Hz). In the starting material 14a, this proton appears at 6.38 ppm as a triplet as a result of ortho-coupling to both H-7 and F with very similar coupling constants (J = 7.1 Hz). The disappearance of F was further confirmed in the 13C NMR spectrum of 15 where C-6 appeared as a singlet at 96.6 ppm, rather than the doublet (2JC–F = 18 Hz) observed at 93.1 ppm for 14a. The signal for C-1′ appeared at 88.4 ppm, far more deshielded than for compound 14a, where this signal appeared at 76.4 ppm. Thus, it appeared that ring-closure of the newly-deprotected hydroxyl group onto the carbon atom originally carrying F had taken place, giving rise to 15. The second product was identified as 7f, where the acetate group had been removed, but where F had been replaced by OMe (Scheme 1). Only these two unexpected products were obtained from the basic deprotection reaction of 14a in MeOH, with none of the expected deprotected hydroxyl product being observed at all. Repeating the KOH hydrolysis reaction of 14a in the non-nucleophilic solvent tert-butanol instead of methanol gave 15 as the sole product in 60% yield.
Excited by the unexpected formation of novel ring-closed heterocyclic product 15, we explored the general applicability of this phenomenon using various fluorine-containing imidazopyridine analogues derived from four randomly selected aldehydes. Given the challenges encountered using the POCl3/Et3N reagents as highlighted above, we explored the utility of an alternative para-toluenesulfonyl chloride-based protocol reported in literature16 that employs equimolar quantities of acid and base as dehydrating agents (Scheme 2).
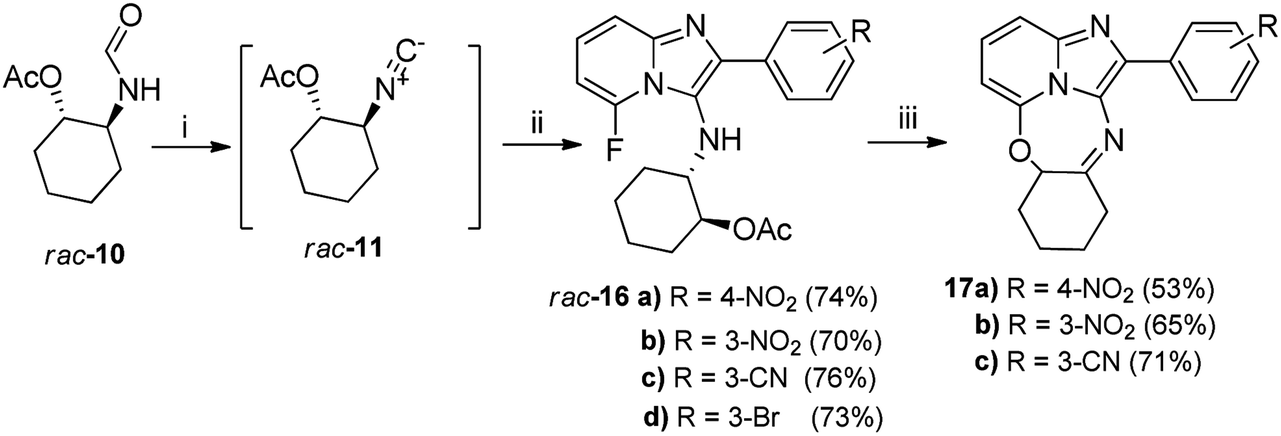 | ||
| Scheme 2 Reagents: (i) DCM, anhydr. Na2SO4 (1 eq.), pTsCl/DABCO, 0 °C–RT, 6 h, (ii) 0–5 °C, aldehyde 12, 2-amino-6-fluoropyridine 13a, anhydr. Na2SO4 (1 eq.); then 50 °C, 12 h; (iii) KOH, MeOH or t-BuOH, RT, 4 h. Yields of 16 are quoted over two steps. | ||
In our case, we observed that the efficiency of the dehydration protocol was highly dependent on the purity of the pTsCl. Thus, the literature reported purification procedure developed by Whitaker19 in 2001 was utilized to purify the pTsCl, which was subsequently stored sealed to reduce moisture ingress. The pTsCl/DABCO protocol proved to be highly convenient and efficient with its main attractive feature being the in situ generation of the desired isocyanide rac-11 which obviated the often tedious aqueous workups encountered during isocyanide purification which are commonplace when employing the POCl3/Et3N protocol. In addition, it removed the unpleasant odour usually associated with isocyanide isolation. The in situ generated isocyanide rac-11 was then coupled with the requisite aldehydes 12b–e and 2-amino-6-fluoropyridine 13a under sealed conditions at moderate temperatures (50 °C) to obtain the respective fluorine-containing imidazopyridine acetates 16a–d, with the pTsCl/DABCO adducts formed during the dehydration reaction subsequently catalysing the multicomponent coupling reaction (Scheme 2). As a slight deviation from the Guchhait protocol,16 we observed that the addition of anhydrous Na2SO4 during the dehydration stage as well as the multicomponent coupling step, not only obviated the need for N2 purging, but further simplified the overall preparation of the desired target compounds. These transformations translated to a minimum 70% yield across both the isocyanide generation as well as the multicomponent coupling phases. The pTsCl/DABCO-catalysed reactions were easily scalable without any discernible drop in target product yields, unlike the problems experienced using POCl3-mediated dehydration. Secondly, the non-stoichiometric addition of dehydrating agents that oftentimes characterises most POCl3/Et3N protocols makes it mandatory for preliminary aqueous workups to remove salts and excess reagents and purify the isocyanide. Such reactive salts and excess reagents could also be responsible for catalysing the hydration of the isocyanide to regenerate the formamide, thereby lowering yields.
Subsequent base-catalysed hydrolysis of the acetate 16a in MeOH did indeed give rise to a ring-closed tetracycle 17a in 53% yield (Scheme 2), together with compound rac-18 (Fig. 3), in 47% yield. Close examination of the 1H NMR spectrum for 17a showed clearly that the fluorine atom had been displaced, as the proton at position 6 appeared as a dd, with one ortho and one meta coupling.
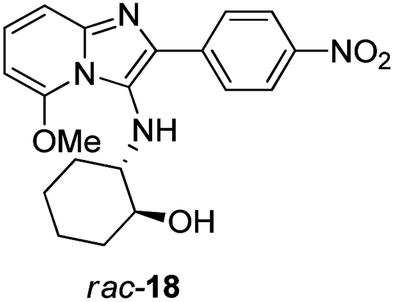 | ||
| Fig. 3 Compound 18 formed from 16a. | ||
However, the signal for the proton on the cyclohexyl ring carbon atom carrying nitrogen had disappeared, together with the NH proton signal, showing that in fact 17a was an imine, representing the oxidised form of compound 15. Similarly, base-catalysed hydrolysis of acetates 16b–c, this time in t-BuOH, gave rise to oxidised tetracycles 17b–c in good yield (step iii, Scheme 2). Hydrolysis of the bromine containing acetate 16d gave rise to an irresolvable mixture. The identities of the ring-closed products 17a and 17c were confirmed by single crystal X-ray crystallographic analysis (Fig. 4). The formation of the oxidised imine-containing products may possibly be attributed to the stronger electron withdrawing effect on the imidazopyridine skeleton by the nitro and the cyano groups as compared to that exerted by the chlorine atom in the case of unoxidised ring-closed product 15.
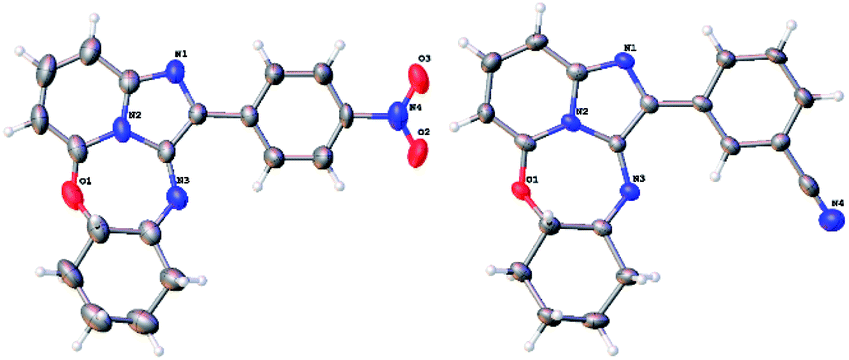 | ||
| Fig. 4 ORTEP diagrams (50% probability level) of 17a (left) and 17c (right). | ||
Given our failure to obtain the originally intended fluorine-containing imidazopyridine targets of general structure 7 via base-catalysed hydrolysis of their respective acetates 14 as explained above, an exploratory attempt was made to directly dehydrate the unprotected 2-trans-hydroxyformamide rac-9 using the modified pTsCl/DABCO protocol and generate the isocyanide rac-19 in situ for the subsequent multicomponent reaction with 2-amino-6-fluoropyridine 13a and selected aldehydes 12 (Scheme 3). To our delight, the expected novel fluorine-containing targets 20a–g were obtained in excellent overall yield, which also demonstrated the excellent OH functional group tolerance of the pTsCl/DABCO protocol. To the best of our knowledge, no similar successful attempts have previously been reported. Guchhait et al.16 did not test their method on functionalised isocyanides. Although rac-19 has not been prepared previously by dehydration of rac-9, it has been prepared by ring-opening of cyclohexene epoxide using TMSCN and ZnI2, to give the TMS-protected alcohol that was subsequently deprotected.20 One example of dehydration of a formamide containing a hydroxyl group to the corresponding isocyanide was reported by McCarthy et al. using Burgess reagent, but the reaction was low-yielding (<50%) and took 2 days.21 Thus, the method reported here is superior in terms of ease of reaction and yield. The small library of novel imidazo[1,2-a]pyridines produced during the course of this research will be screened for activity against the HI virus and the findings will be reported in due course.
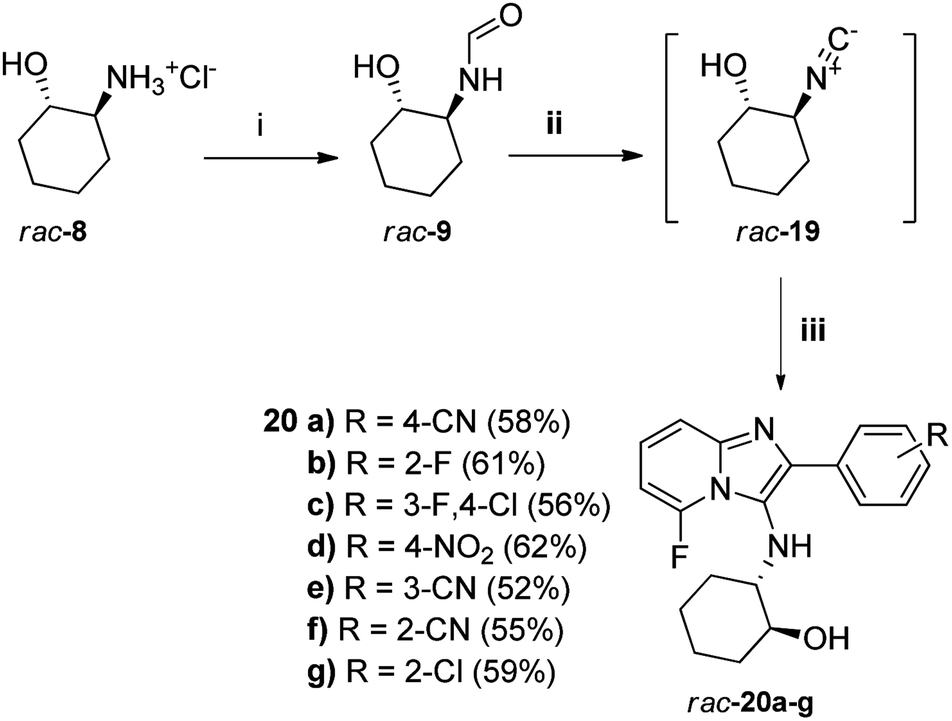 | ||
| Scheme 3 Reagents: (i) NaOMe, methyl formate, MeOH, RT, 4 h; (ii) DCM, anhydr. Na2SO4 (1 eq.), pTsCl/DABCO, 0 °C–RT, 6 h, (iii) 0–5 °C, aldehyde 12, 2-amino-6-fluoropyridine 13a, anhydr. Na2SO4 (1 eq.), then 50 °C, 12 h. Yields for 20 are quoted over all three steps. | ||
Experimental
General
All solvents were freshly distilled prior to use. Other reagents were used as purchased from Sigma-Aldrich. All infrared spectra were recorded neat using a Bruker TENSOR 27 single channel infrared spectrometer. All melting points are uncorrected and were performed using open capillary tubes on a Stuart SMP 10 melting point apparatus. 1H and 13C NMR spectra were recorded using either a Bruker AVANCE 111 300, 400 or 500 MHz spectrometer in deuterated chloroform (CDCl3) with trimethylsilane (TMS) as internal standard (δ = 0) for 1H NMR, and CDCl3 (δ = 77.0 ppm) for 13C NMR. The chemical shift (δ) is reported in ppm and the coupling constants (J) in Hz. High resolution mass spectral data was collected on a Waters Synapt G2 using an ESI positive source and a cone voltage of 15 V. TLC was performed on aluminium-backed Merck silica gel 60 F254 plates. The purification of compounds by column chromatography was performed using gravity (particle size 0.063–0.200 mm) or flash (particle size 0.040–0.063 mm) silica gel 60 purchased from Merck.Synthetic procedures
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (300 MHz, CDCl3) δ: 8.26 (s, 0.7H), 8.06 (d, J = 11.2 Hz, 0.3H), 6.61 (br s, 0.3H), 6.28 (br s, 0.7H), 3.74–3.63 (m, 1H), 3.42–3.20 (m, 1.5H), 3.07–2.98 (m, 0.5H), 2.06–1.97 (m, 2H), 1.90–1.74 (m, 2H), 1.31–1.25 (m, 4H); 13C NMR (75 MHz, CDCl3) δ: 165.14, 162.54, 74.41, 73.23, 58.69, 54.60, 34.38, 33.69, 32.31, 31.57, 24.79, 24.16, 24.44, 24.04; HRMS (ES)+: calculated for C7H14NO2 [M + H]+: 144.1019, found: 144.1019.O ester), 1658 (CO aldehyde); 1H NMR (300 MHz, CDCl3) δ: 8.15–8.00 (m, 1H), 6.56–6.25 (m, N–H), 4.75–4.52 (m, 1H), 4.03–3.88 (m, 0.8H), 3.35–3.20 (m, 0.2H), 2.12–1.92 (m, 5H), 1.83–1.69 (m, 2H), 1.54–1.19 (m, 4H); 13C NMR (75 MHz, CDCl3) δ: 171.5, 170.3, 164.2, 160.9, 74.8, 74.3, 54.8, 51.1, 32.0, 31.7, 30.8, 30.6, 24.0, 23.9, 23.8, 23.5, 21.0, 20.9; HRMS (ES)+: calculated for C9H16NO3 [M + H]+: 186.1125, found: 186.1127.
O); 1H NMR (300 MHz, CDCl3) δ: 8.26 (s, 0.7H), 8.06 (d, J = 11.2 Hz, 0.3H), 6.61 (br s, 0.3H), 6.28 (br s, 0.7H), 3.74–3.63 (m, 1H), 3.42–3.20 (m, 1.5H), 3.07–2.98 (m, 0.5H), 2.06–1.97 (m, 2H), 1.90–1.74 (m, 2H), 1.31–1.25 (m, 4H); 13C NMR (75 MHz, CDCl3) δ: 165.14, 162.54, 74.41, 73.23, 58.69, 54.60, 34.38, 33.69, 32.31, 31.57, 24.79, 24.16, 24.44, 24.04; HRMS (ES)+: calculated for C7H14NO2 [M + H]+: 144.1019, found: 144.1019.O ester), 1658 (CO aldehyde); 1H NMR (300 MHz, CDCl3) δ: 8.15–8.00 (m, 1H), 6.56–6.25 (m, N–H), 4.75–4.52 (m, 1H), 4.03–3.88 (m, 0.8H), 3.35–3.20 (m, 0.2H), 2.12–1.92 (m, 5H), 1.83–1.69 (m, 2H), 1.54–1.19 (m, 4H); 13C NMR (75 MHz, CDCl3) δ: 171.5, 170.3, 164.2, 160.9, 74.8, 74.3, 54.8, 51.1, 32.0, 31.7, 30.8, 30.6, 24.0, 23.9, 23.8, 23.5, 21.0, 20.9; HRMS (ES)+: calculated for C9H16NO3 [M + H]+: 186.1125, found: 186.1127.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 EtOAc/Hex) to give the desired product rac-11 as a light yellow oil (0.077 g, 85%). IR (cm−1): 1736 (CO), 2141 (+N
:1 EtOAc/Hex) to give the desired product rac-11 as a light yellow oil (0.077 g, 85%). IR (cm−1): 1736 (CO), 2141 (+N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C−); 1H NMR (300 MHz, CDCl3) δ: 4.83 (td, J = 9.2, 4.2 Hz, 1H), 3.59–3.47 (m, 1H), 2.25–2.01 (m, 5H), 1.84–1.58 (m, 3H), 1.52–1.20 (m, 3H); 13C NMR (75 MHz, CDCl3) δ: 170.0, 156.5 (t, JC–N = 9.5 Hz), 73.4, 55.1 (t, JC–N = 13.5 Hz), 31.3, 29.4, 22.9, 22.7, 20.9 ppm; HRMS (ES)+: calculated for C9H14NO2 [M + H] +: 168.1019, found: 168.1017.
C−); 1H NMR (300 MHz, CDCl3) δ: 4.83 (td, J = 9.2, 4.2 Hz, 1H), 3.59–3.47 (m, 1H), 2.25–2.01 (m, 5H), 1.84–1.58 (m, 3H), 1.52–1.20 (m, 3H); 13C NMR (75 MHz, CDCl3) δ: 170.0, 156.5 (t, JC–N = 9.5 Hz), 73.4, 55.1 (t, JC–N = 13.5 Hz), 31.3, 29.4, 22.9, 22.7, 20.9 ppm; HRMS (ES)+: calculated for C9H14NO2 [M + H] +: 168.1019, found: 168.1017.General procedure for microwave-assisted synthesis of novel imidazo[1,2-a]pyridine derivatives 14a–d
A mixture of a 6-substituted-2-aminopyridine 13a–d (1 mmol), 2-chlorobenzaldehyde 12a (1 mmol), isocyanide rac-11 (170 mg, 1.02 mmol) and montmorillonite K-10 clay (100 mg) in 1,4-dioxane (2.0 ml) was irradiated in a sealed tube for 30 min (150 W, 100 °C). After cooling to room temperature, the K-10 clay was filtered off through Celite which was later washed with EtOAc and the combined organic solvents were removed in vacuo to give a crude residue that was purified using flash silica by eluting with 20–60% EtOAc/Hex.O str.), 1651 (CN). 1H NMR (500 MHz, CDCl3) δ 7.61–7.58 (m, 1H), 7.50–7.47 (m, 1H), 7.37–7.34 (m, 3H), 7.12–7.08 (m, 1H), 6.38 (t, J = 7.4 Hz, 1H), 4.66–4.55 (m, 1H), 3.55–3.51 (m, 1H), 2.88–2.81 (m, 1H), 1.93–1.87 (m, 4H), 1.71–1.66 (m, 1H), 1.62–1.56 (m, 1H), 1.52–1.47 (m, 1H), 1.24–1.15 (m, 2H), 1.02–0.91 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 170.8, δ 150.6 (d, 1JC–F = 268.1 Hz), 143.6 (d, 4JC–F = 3.4 Hz), 136.2, 133.4, 133.2, 132.5, 129.6, 129.5, 126.8, 126.1 (d, 4JC–F = 2.8 Hz), 124.3 (d, 3JC–F = 6.4 Hz), 113.8 (d, 3JC–F = 5.0 Hz), 93.1 (d, 2JC–F = 17.4 Hz), 76.4, 60.9, 30.9, 30.2, 23.7, 23.7, 21.1 ppm; HRMS (ES+) calculated for C21H22ClFN3O2 [M + H]+: 402.1379, found: 402.1400.O str.), 1653 (CN); 1H NMR (500 MHz, CDCl3) δ 7.61–7.59 (m, 1H), 7.50–7.48 (m, 2H), 7.37–7.34 (m, 2H), 7.06–7.02 (m, 1H), 6.78–6.76 (m, 1H), 4.57–4.52 (m, 1H), 3.67–3.64 (m, 1H), 2.89–2.84 (m, 1H), 1.91–1.84 (m, 4H), 1.61–1.53 (m, 2H), 1.48–1.43 (m, 1H), 1.22–1.07 (m, 2H), 1.02–0.92 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 170.6, 143.8, 138.3, 133.5, 133.4, 132.5, 129.6, 129.5, 127.6, 126.8, 126.3, 123.7, 116.8, 114.2, 76.9, 60.8, 30.3, 30.0, 23.6, 23.6, 21.1 ppm; HRMS (ES+) calculated for C21H22N3O2Cl2 [M + H]+: 418.1089, found: 418.1080.O str.), 1652 (CN); 1H NMR (500 MHz, CDCl3) δ 7.62–7.60 (m, 1H), 7.55–7.52 (m, 1H), 7.49–7.47 (m, 1H), 7.38–7.34 (m, 2H), 7.00–6.94 (m, 2H), 4.58–4.53 (m, 1H), 3.64–3.60 (m, 1H), 2.88–2.82 (m, 1H), 1.92–1.85 (m, 4H), 1.58–1.52 (m, 2H), 1.49–1.43 (m, 1H), 1.22–1.14 (m, 1H), 1.11–1.03 (m, 2H), 0.98–0.90 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 170.5, 143.9, 138.8, 133.6, 133.3, 132.4, 129.6, 129.5, 127.9, 126.8, 124.0, 118.8, 117.3, 112.5, 60.4, 30.1, 29.9, 23.7, 23.6, 21.1 ppm; HRMS (ES+) calculated for C21H22BrClN3O2 [M + H]+: 462.0578, found: 462.0555.O str.), 1653 (CN); 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.9 Hz, 1H), 7.68–7.64 (m, 1H), 7.55–7.51 (m, 1H), 7.45–7.40 (m, 3H), 7.20 (dd, J = 9.0, 7.1 Hz, 1H), 4.59 (td, J = 9.7, 4.4 Hz, 1H), 3.73–3.68 (m, 1H), 2.93–2.86 (m, 1H), 2.00–1.92 (m, 1H), 1.84 (s, 3H), 1.65–1.51 (m, 3H), 1.34–1.21 (m, 3H), 1.12–0.98 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 170.3, 141.3, 139.3, 133.2, 133.0, 132.4, 130.2, 129.9, 127.8, 127.3, 124.3, 123.0, 122.3, 113.8, 108.4, 77.1, 60.0, 30.4, 29.9, 23.8, 23.7, 21.1 ppm; HRMS (ES+) calculated for C22H22ClN4O2 [M + H]+: 409.1426, found: 409.1413.Typical procedure for the preparation of compounds 16a–d
A mixture of 2-trans-hydroxycyclohexylammonium chloride rac-8 (2.00 g, 17.12 mmol), sodium methoxide (20 ml, 6.58 mmol) and methyl formate (2 ml) were stirred at room temperature for 2 h. Thereafter, MeOH (20 ml) was added to the reaction and the mixture was stirred at 40 °C for 2 h, and then at room temperature for a further 24 h. After removing the organic solvent in vacuo, acetone (25 ml) was added to the off-white residue and the precipitated salts were removed by filtration. The solvent was evaporated in vacuo to afford an off-white oily product, 2-trans-hydroxycyclohexyl formamide rac-9 (1.83 g. 99% yield). To a mixture of the formamide rac-9 (1.70 g, 16.18 mmol) and a catalytic amount of dimethylaminopyridine (171 mg) in acetonitrile (20 ml) was added acetic anhydride (15 ml) and the reaction mixture stirred at room temperature for 12 h. After evaporating the solvent, the pale-yellow oil was diluted with acetone (20 ml) and after adding sodium bicarbonate (1.2 eq.), the reaction mixture was filtered to afford 2-formamidocyclohexyl acetate rac-10 as a pale yellow oil in quantitative yield.An appropriate amount of 2-formamidocyclohexyl acetate rac-10 (1.0 mmol (185 mg)–2 mmol (371 mg)), anhydrous Na2SO4 (284 mg, 2 mmol) and a magnetic stirrer were added to freshly distilled dichloromethane (10–15 ml) and chilled in an ice bath (10 min). p-Toluenesulfonyl chloride (pTsCl) (1.0 mmol (191 mg)–2.0 mmol (382 mg)) and DABCO (1.0 mmol (112 mg)–2.0 mmol (225 mg)) were added in succession and the closed reaction mixture was stirred under ice-chilled conditions for 1 h. Thereafter, the reaction mixture was allowed to gradually warm to room temperature with stirring for a further 2 h. To this chilled in situ generated isocyanide rac-11 crude mixture was added anhydrous Na2SO4 (284 mg, 2 mmol), an appropriate aldehyde 12 (1.0–2.0 mmol) and 2-amino-6-fluoropyridine 13a (1.0–2.0 mmol) and the sealed reaction mixture was heated at 50–60 °C in an oil bath for 10–12 h. Thereafter, the reaction was cooled to room temperature, diluted with DCM (20 ml) and filtered. The filtrate was washed successively with distilled water (2 × 10 ml) and saturated brine solution (10 ml). After drying over Na2SO4, the solvent was removed in vacuo and the crude mixture was purified by silica gel flash column chromatography, eluting the title compounds 16a–d with 25–50% EtOAc/hexane.
O str.), 1659 (CN), 1566 (NH bend), 1451 (CC); 1H NMR (500 MHz, CDCl3) δ 8.53–8.48 (m, 2H), 8.30–8.26 (m, 2H), 7.36 (d, J = 9.0, Hz, 1H), 7.17–7.12 (m, 1H), 6.41 (t, J = 7.3 Hz, 1H), 4.79 (td, J = 10.0, 4.5 Hz, 1H), 3.63–3.61 (m, 1H), 3.11–3.06 (m, 1H), 2.09–2.05 (m, 1H), 1.94 (s, 3H), 1.78–1.74 (m, 1H), 1.71–1.68 (m, 1H), 1.62–1.58 (m, 1H), 1.33–1.22 (m, 3H), 1.12–1.06 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 170.7, 150.5 (d, 1JC–F = 262.5 Hz), 146.7, 144.1 (d, 4JCF = 3.7 Hz), 140.5, 134.7, 127.7, 126.0 (d, 4JC–F = 1.7 Hz), 125.4 (d, 3JC–F = 6.8 Hz) 123.7, 114.2 (d, 3JC–F = 4.9 Hz), 93.4, (d, 2JCF = 17.6 Hz), 77.4, 61.4 (d, 5JC–F = 2.1 Hz), 31.2, 30.7, 24.1, 24.0, 21.1; HRMS (ES+) calculated for C21H22FN4O4 [M + H]+: 413.1620, found: 413.1615.O str.), 1654 (CN), 1568 (NH bend), 1434 (CC);.1H NMR (500 MHz, CDCl3) δ 9.20 (t, J = 2.0 Hz, 1H), 8.66 (dt, J = 7.9, 1.3 Hz, 1H), 8.17–8.15 (m, 1H), 7.60 (t, J = 8.0 Hz, 1H), 7.37 (d, J = 9.0 Hz, 1H), 7.16–7.12 (m, 1H), 6.40 (t, J = 7.2 Hz, 1H), 4.77 (td, J = 10.1, 4.5 Hz, 1H), 3.63–3.58 (m, 1H), 3.14–3.08 (m, 1H), 2.09–2.03 (m, 1H), 1.85 (s, 3H), 1.70–1.61 (m, 2H), 1.34–1.22 (m, 4H), 1.16–1.08 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 170.1, 150.5 (d, 1JC–F = 265.7 Hz), 148.6, 143.9 (d, 4JC–F = 3.7 Hz), 135.8, 134.8, 133.1, 129.2, 125.1 (d, 3JC–F = 6.3 Hz), 122.1 (d, 3JC–F = 9.5 Hz), 114.0 (d, 4JC–F = 4.9 Hz), 93.4 (d, 2JC–F = 17.6 Hz), 77.7, 61.1 (d, 5JC–F = 2.2 Hz), 31.2, 30.7, 24.1, 24.0, 21.0; HRMS (ES+) calculated for C21H22FN4O4 [M + H]+: 413.1620, found: 413.1617.O str.), 1657 (CN), 1583 (NH bend), 1443 (CC); 1H NMR (500 MHz, CDCl3) δ 8.62 (d, J = 1.7 Hz, 1H), 8.57 (dt, J = 7.9, 1.5 Hz, 1H), 7.52 (t, J = 7.8 Hz, 1H), 7.34 (d, J = 9.0 Hz, 1H), 7.15–7.11 (m, 1H), 6.40 (t, J = 7.2 Hz, 1H), 4.77 (td, J = 10.0, 4.5 Hz, 1H), 3.58–3.55 (m, 1H), 3.11–3.04 (m, 1H), 2.08–2.05 (m, 1H), 1.94 (s, 3H), 1.77–1.73 (m, 1H), 1.70–1.66 (m, 1H), 1.63–1.57 (m, 1H), 1.33–1.20 (m, 3H), 1.14–1.04 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 170.8, 150.4 (d, 1JC–F = 265.9 Hz), 143.9 (d, 4JC–F = 3.8 Hz), 135.2, 134.8, 131.4, 130.8, 130.7, 129.1, 125.1 (d, 3JC–F = 6.3 Hz), 124.9 (d, 4JC–F = 2.5 Hz), 119.0, 113.9 (d, 3JC–F = 5.0 Hz), 112.5, 93.3 (d, 2JC–F = 17.6 Hz), 77.3, 61.1 (d, 5JC–F = 2.2 Hz), 31.2, 30.7, 24.0, 21.0; HRMS (ES+) calculated for C22H22FN4O2 [M + H]+: 393.1721, found: 393.1719.O str.), 1654 (CN), 1568 (NH bend), 1434 (CC); 1H NMR (500 MHz, CDCl3) δ 8.45 (t, J = 1.9 Hz, 1H), 8.24 (dt, J = 7.8, 1.3 Hz, 1H), 7.44–7.42 (m, 1H), 7.34 (d, J = 9.0 Hz, 1H), 7.31–7.27 (m, 1H), 7.11–7.07 (m, 1H), 6.36 (t, J = 7.2, Hz, 1H), 4.75 (td, J = 10.0, 4.5 Hz, 1H), 3.52 (s, 1H), 3.08–3.06 (m, 1H), 2.08–2.04 (m, 1H), 1.89 (s, 3H), 1.79–1.76 (m, 1H), 1.69–1.66 (m, 1H), 1.61–1.58 (m, 1H), 1.32–1.20 (m, 3H), 1.10–1.08 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 170.8, 150.5 (d, 1JC–F = 265.7 Hz), 143.9 (d, 4JC–F = 3.7 Hz), 136.0, 135.4, 130.4, 130.1, 129.8, 125.8, 125.7, 124.7 (d, 1JC–F = 6.3 Hz), 122.6, 113.8 (d, 4JC–F = 5.0 Hz), 93.1 (d, 2JC–F = 17.6 Hz), 77.6, 60.9 (d, 5JC–F = 2.1 Hz), 31.1, 30.7, 24.1, 24.0, 21.1; HRMS (ES+) calculated for C21H2279BrN3O2 [M + H]+: 446.0874, found: 446.0875.General procedure for the preparation of imidazo[1,2-a]pyridines 7b–c, 7e, 18 and ring-closed products 15, 17a–c
An appropriate aminocyclohexyl acetate derivative (14 or 16) and powdered KOH (4 eq.) were stirred in MeOH or tert-butanol (3–5 ml) at room temperature for 4 h. After removing solvent in vacuo, the crude residue was diluted with DCM (15–20 ml) and then washed successively with distilled water (2 × 10 ml) and saturated brine solution (10 ml). After drying over Na2SO4, the solvent was removed in vacuo and the crude mixture was purified by silica gel flash column chromatography, eluting the title compound with 25–50% EtOAc/hexane.C); Mp: 183–185 °C; 1H NMR (500 MHz, CDCl3) δ 7.60–7.56 (m, 1H), 7.53–7.48 (m, 2H), 7.38–7.34 (m, 2H), 7.05 (dd, J = 8.9, 7.2 Hz, 1H), 6.79 (dd, J = 7.2, 1.1 Hz, 1H), 3.85–3.78 (m, 1H), 3.27–3.21 (m, 1H), 2.70–2.63 (m, 1H), 2.43–2.38 (m, 1H), 1.92–1.86 (m, 1H), 1.61–1.54 (m, 2H), 1.50–1.44 (m, 1H), 1.16–1.07 (m, 2H), 1.11–0.89 (m, 1H), 0.78–0.71 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 143.9, 138.1, 133.6, 133.5, 132.6, 129.8, 129.6, 127.8, 126.8, 126.0, 123.7, 117.0, 114.2, 74.6, 65.3, 33.4, 30.3, 24.4, 24.0; HRMS (ES+) calculated for C19H20N3OCl2 [M + H]+: 376.0983, found: 376.0974.C); 1H NMR (500 MHz, CDCl3) δ 7.60–7.54 (m, 2H), 7.51–7.47 (m, 1H), 7.37–7.33 (m, 2H), 7.02–6.95 (m, 2H), 3.90 (s, 1H), 3.27–3.19 (m, 1H), 2.70–2.60 (m, 1H), 2.33 (s, 1H), 1.90–1.80 (m, 1H), 1.57–1.44 (m, 3H), 1.15–1.06 (m, 2H), 0.98–0.90 (m, 1H), 0.85–0.76 (m, 1H); 13C NMR (126 MHz, CDCl3): δ 144.0, 138.5, 133.7, 133.5, 132.5, 129.8, 129.6, 128.0, 126.8, 124.0, 118.8, 117.5, 112.2, 74.7, 64.7, 33.4, 30.1, 24.4, 24.1; HRMS (ES+) calculated for C19H20BrClN3O [M + H]+: 420.0473, found: 420.0465.C); 1H NMR (400 MHz, DMSO-d6) δ 8.46 (s, 1H), 7.97 (s, 1H), 7.62–7.52 (m, 3H), 7.44–7.39 (m, 2H), 7.23–7.18 (m, 1H), 7.11 (d, J = 6.8 Hz, 1H), 4.36 (d, J = 4.7 Hz, 1H), 4.24 (d, J = 3.2 Hz, 1H), 3.13–3.05 (m, 1H), 1.69–1.60 (m, 1H), 1.48–1.37 (m, 1H), 1.33–1.21 (m, 2H), 1.09–0.92 (m, 2H), 0.79–0.57 (m, 2H); 13C NMR (101 MHz, DMSO) δ 165.6, 140.7, 134.4, 133.2, 132.6, 131.8, 129.3, 129.2, 127.9, 126.6, 121.7, 118.8, 113.6, 71.9, 59.3, 32.6, 29.0, 23.0, 22.8.; HRMS (ES+) calculated for C20H22N4O2Cl [M + H]+: 385.1431, found: 385.1418.C–H), 2937 (C–H), 1638 (CN); 1H NMR (500 MHz, CDCl3) δ 7.67 (d, J = 7.6 Hz, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.35 (t, J = 7.5 Hz, 1H), 7.30–7.26 (m, 1H), 7.17 (d, J = 9.0 Hz, 1H), 6.92 (t, J = 8.1 Hz, 1H), 6.17 (d, J = 7.2 Hz, 1H), 4.13–4.07 (m, 1H), 3.84 (s, 1H), 3.42–3.35 (m, 1H), 2.29–2.23 (m, 1H), 2.06–2.00 (m, 1H), 1.87–1.81 (m, 1H), 1.76–1.62 (m, 2H), 1.50–1.40 (m, 1H), 1.39–1.23 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 147.6, 142.6, 133.4, 133.0, 132.4, 129.8, 128.8, 128.2, 127.1, 124.3, 123.6, 111.7, 96.6, 88.4, 60.4, 32.8, 31.6, 23.9, 23.6 ppm; HRMS (ES+) calculated for C19H19N3OCl [M + H]+: 340.1217, found: 340.1205.N), 1445 (CC); 1H NMR (500 MHz, CDCl3) δ 8.67–8.65 (m, 2H), 8.31–8.28 (m, 2H), 7.35 (dd, J = 8.9, 1.0 Hz, 1H), 7.25–7.22 (m, 1H), 6.40 (dd, J = 7.3, 1.0 Hz, 1H), 4.72 (dd, J = 9.4, 6.2 Hz, 1H), 2.96–2.90 (m, 1H), 2.69–2.61 (m, 1H), 2.46–2.41 (m, 1H), 2.06–1.94 (m, 3H), 1.85–1.76 (m, 1H), 1.73–1.63 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 163.5, 148.7, 147.0, 145.8, 140.4, 138.7, 129.1, 128.0, 127.1, 123.4, 112.3, 98.7, 83.8, 36.6, 31.6, 23.3, 20.9 ppm; HRMS (ES+) calculated for C19H17N4O3 [M + H]+: 349.1295, found: 349.1287.N), 1450 (CC); 1H NMR (500 MHz, CDCl3) δ 9.43 (t, J = 2.0 Hz, 1H), 8.75 (d, J = 7.8 Hz, 1H), 8.17 (dd, J = 8.2, 2.4 Hz, 1H), 7.59 (t, J = 8.9, 1H), 7.34 (d, J = 8.9 Hz, 1H), 7.22 (dd, J = 8.9, 7.3 Hz, 1H), 6.39 (d, J = 7.3 Hz, 1H), 4.73–4.69 (m, 1H), 2.96–2.91 (m, 1H), 2.69–2.62 (m, 1H), 2.44–2.41 (m, 1H), 2.05–1.94 (m, 3H), 1.85–1.75 (m, 1H), 1.71–1.64 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 162.9, 148.6, 148.4, 145.6, 138.6, 135.6, 134.1, 129.0, 127.8, 126.4, 123.7, 122.4, 112.1, 98.7, 83.8, 36.6, 31.7, 23.4, 21.0 ppm; HRMS (ES+) calculated for C19H17N4O3 [M + H]+: 349.1295, found: 349.1290.N), 1445 (CC); 1H NMR (400 MHz, CDCl3) δ 8.81 (t, J = 1.7 Hz, 1H), 8.66 (dt, J = 8.0, 1.5 Hz, 1H), 7.60 (dt, J = 7.7, 1.5 Hz, 1H), 7.53 (t, J = 7.8 Hz, 1H), 7.32 (d, J = 8.9 Hz, 1H), 7.22 (dd, J = 8.9, 7.3 Hz, 1H), 6.38 (d, J = 7.3 Hz, 1H), 4.71–4.68 (m, 1H), 2.94–2.88 (m, 1H), 2.65–2.61 (m, 1H), 2.42–2.40 (m, 1H), 2.01–1.93 (m, 3H), 1.80–1.78 (m, 1H), 1.71–1.64 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 162.8, 148.6, 145.6, 138.8, 135.1, 132.6, 132.4, 131.0, 128.9, 127.8, 126.3, 119.2, 112.3, 112.1, 98.6, 83.8, 36.5, 31.6, 23.3, 20.9 ppm; HRMS (ES+) calculated for C20H17N4O [M + H]+: 329.1397, found: 329.1394.N), 1559 (NH bend), 1448 (CC); 1H NMR (400 MHz, CDCl3) δ 8.48 (d, J = 8.6 Hz, 2H), 8.24 (d, J = 8.6 Hz, 2H), 7.19–7.07 (m, 2H), 5.97 (d, J = 7.2 Hz, 1H), 4.49 (br s, 1H), 4.07 (s, 3H), 3.54–3.49 (m, 1H), 2.79–2.74 (m, 1H), 2.04–1.95 (m, 1H), 1.70–1.61 (m, 1H), 1.56–1.47 (m, 2H), 1.28–1.17 (m, 3H), 1.04–0.93 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 152.0, 146.3, 144.0, 141.5, 133.6, 128.2, 127.8, 125.9, 123.5, 110.7, 88.6, 75.2, 65.5, 56.4, 34.2, 30.1, 24.4, 24.3; HRMS (ES+) calculated for C20H23N4O4 [M + H]+: 383.1714, found: 383.1704.General procedure for the preparation of imidazo[1,2-a]pyridines 20a–g
To a chilled solution of 2-trans-hydroxycyclohexyl formamide rac-9 (287 mg, 2 mmol) (prepared as described previously) in freshly distilled DCM (10–15 ml) was added a stirrer bar, dried p-TsCl (382 mg, 2 mmol) and DABCO (225 mg, 2 mmol). The sealed reaction mixture was stirred under chilled conditions for 1 h and allowed to warm to room temperature with stirring for a further 3 h. Thereafter, an appropriate aromatic aldehyde 12 (2 mmol), 2-amino-2-fluoropyridine 13a (2 mmol) and anhydrous Na2SO4 (282 mg, 2 mmol) were added. The closed reaction mixture was heated at 50–60 °C in an oil bath for 10–12 h and later cooled to room temperature. Thereafter, the mixture was washed consecutively with distilled water (2 × 15 ml) and saturated brine solution (10 ml). After drying with anhydrous Na2SO4, the organic was evaporated in vacuo to leave a crude residue which was purified on flash silica gel by eluting with 25–80% EtOAc/Hex to afford title compounds 20a–g.N), 1555 (NH bend), 1434 (CC); 1H NMR (300 MHz, CDCl3) δ 8.49–8.41 (m, 2H), 7.71–7.66 (m, 2H), 7.39 (d, J = 9.0 Hz, 1H), 7.20–7.13 (m, 1H), 6.41 (t, J = 7.2 Hz, 1H), 3.97 (br. s, 1H), 3.57–3.53 (m, 1H), 2.84–2.81 (m, 1H), 2.07–1.93 (m, 2H), 1.69–1.65 (m, 1H), 1.59–1.50 (m, 2H), 1.31–1.20 (m, 2H), 1.07–0.97 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 150.6 (d, 1JC–F = 267.1 Hz), 144.1 (d, 4JCF = 2.5 Hz), 138.3, 135.9, 132.1, 128.5, 128.0, 125.6 (d, 3JCF = 12.0 Hz), 119.2, 113.9 (d, 3JC–F = 5.0 Hz), 110.8, 93.5 (d, 2JC–F = 17.6 Hz), 75.3, 65.0 (d, 5JC–F = 2.6 Hz), 34.4, 30.1, 24.4, 24.2 ppm; HRMS (ES+) calculated for C20H20FN4O [M + H]+: 351.1616, found: 351.1617.N), 1504 (NH bend), 1430 (CC); 1H NMR (300 MHz, CDCl3) δ 7.79 (td, J = 7.6, 1.8 Hz, 1H), 7.43–7.34 (m, 2H), 7.30–7.25 (m, 1H), 7.20–7.09 (m, 2H), 6.40 (t, J = 7.3 Hz, 1H), 3.58 (br. s, 1H), 3.37–3.31 (m, 1H), 3.18 (br s, 1H), 2.69–2.63 (m, 1H), 2.02–1.98 (m, 1H), 1.63–1.58 (m, 2H), 1.51–1.44 (m, 1H), 1.27–1.12 (m, 2H), 1.03–0.93 (m, 1H), 0.79–0.65 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 159.6 (d, 1JC–F = 245.7 Hz), 150.5 (d, 1JC–F = 268.4 Hz), 144.2 (d, 4JC–F = 3.8 Hz), 133.2, 131.9 (d, 4JC–F = 3.8 Hz), 129.9 (d, 3JC–F = 7.6 Hz), 126.5 (d, 4JC–F = 2.5 Hz), 124.73, 124.69 (d, 4JC–F = 3.2 Hz), 121.6 (d, 3JC–F = 15.1 Hz), 115.9 (d, 1JC–F = 22.7 Hz), 113.7 (d, 3JC–F = 5.0 Hz), 93.4 (d, 2JC–F = 17.6 Hz), 74.2, 65.5, 33.2, 30.9, 24.6, 24.0 ppm; HRMS (ES+) calculated for C19H20F2N3O [M + H]+: 344.1582, found: 344.1569.N), 1512 (NH bend), 1435 (CC); 1H NMR (500 MHz, CDCl3) δ 8.19 (dd, J = 8.3, 1.9 Hz, 1H), 8.04 (dd, J = 8.3, 2.0 Hz, 1H), 7.41 (t, J = 8.1 Hz, 1H), 7.35 (d, J = 8.9 Hz, 1H), 7.15–7.10 (m, 1H), 6.38 (t, J = 7.3 Hz, 1H), 3.91 (s, 1H), 3.58–3.53 (m, 1H), 2.87–2.81 (m, 1H), 2.07–1.99 (m, 2H), 1.69–1.65 (m, 1H), 1.59–1.52 (m, 2H), 1.29–1.24 (m, 2H), 1.06–1.02 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 157.5 (d, 1JC–F = 247 Hz), 150.5 (d, 1JC–F = 265.9 Hz), 144.1 (d, 4JC–F = 3.8 Hz), 136.4 (d, 3JC–F = 5.0 Hz), 134.6 (d, 3JC–F = 7.6 Hz), 130.3, 125.2 (d, 3JC–F = 10.1 Hz), 124.5 (d, 4JC–F = 1.3 Hz), 123.9 (d, 4JC–F = 3.8 Hz), 119.4 (d, 2JC–F = 17.6 Hz), 115.6 (d, 2JC–F = 22.7 Hz), 113.8 (d, 3JC–F = 5.0 Hz), 93.2 (d, 2JC–F = 17.6 Hz), 75.2, 64.9 (d, 5JC–F = 2.7 Hz), 34.4, 29.9, 24.8, 24.4, 24.2 ppm; HRMS (ES+) calculated for C19H19ClF2N3O [M + H]+: 378.1179, found: 378.1167.N), 1515 (NH bend), 1439 (CC); 1H NMR (500 MHz, CDCl3) δ 8.55–8.51 (m, 2H), 8.29–8.26 (m, 2H), 7.38 (d, J = 9.0 Hz, 1H), 7.18–7.13 (m, 1H), 6.41 (td, J = 7.3, 0.9 Hz, 1H), 4.01 (s, 1H), 3.60–3.57 (m, 1H), 2.88–2.82 (m, 1H), 2.05–1.99 (m, 1H), 1.80 (s, 1H), 1.70–1.49 (m, 3H), 1.32–1.22 (m, 2H), 1.09–0.98 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 150.6 (d, 1JC–F = 265.9 Hz), 146.8, 144.4 (d, 4JC–F = 3.8 Hz), 140.7, 136.0, 128.1, 126.0 (d, 4JC–F = 2.5 Hz), 125.5 (d, 3JC–F = 7.6 Hz), 123.6, 114.1 (d, 3JC–F = 5.0 Hz), 93.4 (d, 2JC–F = 17.6 Hz), 75.3, 65.0 (d, 5JC–F = 2.5 Hz), 34.5, 30.0, 24.4, 24.2 ppm; HRMS (ES+) calculated for C19H20FN4O3 [M + H]+: 371.1514, found: 371.1510.N), 1516 (NH bend), 1446 (CC1H NMR (500 MHz, CDCl3) δ 8.68–8.66 (m, 1H), 8.55 (dt, J = 8.1, 1.5 Hz, 1H), 7.55 (dt, J = 7.8, 1.5 Hz, 1H), 7.49 (t, J = 7.8, Hz, 1H), 7.39 (d, J = 9.0 Hz, 1H), 7.19–7.15 (m, 1H), 6.42 (t, J = 7.1 Hz, 1H), 4.04 (s, 1H), 3.58–3.52 (m, 1H), 2.85–2.81 (m, 1H), 2.05–1.90 (m, 2H), 1.69–1.63 (m, 1H), 1.57–1.47 (m, 2H), 1.31–1.21 (m, 2H), 1.04–0.97 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 150.6 (d, 1JC–F = 267.1 Hz), 143.9 (d, 4JC–F = 3.8 Hz), 135.4, 134.8, 131.8, 131.2, 130.9, 129.1, 126.0 (d, 3JC–F = 6.3 Hz), 125.1 (d, 3JC–F = 6.3 Hz), 119.0, 113.5 (d, 4JC–F = 5.0 Hz), 112.3, 93.7 (d, 2JC–F = 17.6 Hz), 75.0, 64.8 (d, 5JC–F = 2.6 Hz), 34.4, 30.0, 24.4, 24.2 ppm; HRMS (ES+) calculated for C20H20FN4O [M + H]+: 351.1616, found: 351.1615.N), 1446 (CC); 1H NMR (500 MHz, CDCl3) δ 7.87 (dd, J = 7.9, 1.2 Hz, 1H), 7.77 (dd, J = 7.8, 1.5 Hz, 1H), 7.67 (td, J = 7.7, 1.4 Hz, 1H), 7.47 (td, J = 7.7, 1.3 Hz, 1H), 7.39 (dd, J = 9.0, 0.9 Hz, 1H), 7.18–7.13 (m, 1H), 6.42 (t, J = 7.3 Hz, 1H), 3.83–3.77 (m, 1H), 3.34–3.30 (m, 1H), 2.78 (br s, 1H), 2.70–2.61 (m, 1H), 1.92–1.88 (m, 1H), 1.61–1.55 (m, 1H), 1.54–1.42 (m, 2H), 1.19–1.10 (m, 2H), 1.00–0.93 (m, 1H), 0.77–0.68 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 150.5 (d, 1JC–F = 267.1 Hz), 144.3 (d, 4JC–F = 3.8 Hz), 137.7, 136.7, 133.4, 132.6, 131.1, 128.2, 125.7 (d, 4JC–F = 2.5 Hz), 125.3 (d, 3JC–F = 6.3 Hz), 119.1, 114.1 (d, 3JC–F = 5.0 Hz), 112.4, 93.5 (d, 2JC–F = 17.6 Hz), 74.3, 65.0 (d, 5JC–F = 1.9 Hz), 33.5, 30.3, 24.2, 24.1 ppm; HRMS (ES+) calculated for C20H20FN4O [M + H]+: 351.1616, found: 351.1615.N), 1432 (CC); 1H NMR (500 MHz, CDCl3) δ 7.62–7.57 (m, 1H), 7.52–7.47 (m, 1H), 7.44–7.34 (m, 3H), 7.17–7.14 (m, 1H), 6.43 (t, J = 7.3, 1H), 3.53–3.36 (m, 1H), 3.30–3.25 (m, 1H), 3.08–2.71 (m, 1H), 2.66–2.59 (m, 1H), 2.00–1.94 (m, 1H), 1.66–1.58 (m, 2H), 1.53–1.44 (m, 1H), 1.20–1.10 (m, 2H), 1.02–0.93 (m, 1H), 0.73–0.64 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 150.5 (d, 1JC–F = 267.2 Hz), 143.6 (d, 4JC–F = 3.2 Hz), 135.7, 133.2, 132.7, 132.6, 129.8, 129.7, 127.0, 126.2 (d, 4JC–F = 2.9 Hz), 124.8 (d, 3JC–F = 6.8 Hz), 113.8 (d, 3JC–F = 5.0 Hz), 93.6 (d, 2JC–F = 17.7 Hz), 74.1, 65.4, 33.2, 31.0, 24.6, 24.0 ppm; HRMS (ES+) calculated for C19H20ClFN3O [M + H]+: 360.1273, found: 360.1272.Crystallography
Crystal data for 17a: C19H16N4O3 (M =348.36 g mol−1): monoclinic, space group C2/c (no. 15), a = 20.8879(15) Å, b = 7.2791(5) Å, c = 21.5015(15) Å, β = 96.086(5)°, V = 3250.8(4) Å3, Z = 8, T = 173.15 K, μ(MoKα) = 0.100 mm−1, Dcalc = 1.424 g cm−3, 10492 reflections measured (3.81° ≤ 2θ ≤ 49.998°), 2857 unique (Rint = 0.1096, Rsigma = 0.1327) which were used in all calculations. The final R1 was 0.0444 (I > 2σ(I)) and wR2 was 0.0847 (all data). CCDC 1970362.
Crystal data for 17c: C20H16N4O (M =328.37 g mol−1): monoclinic, space group P21/n (no. 14), a = 12.0432(7) Å, b = 7.3016(4) Å, c = 18.3346(12) Å, β = 96.116(4)°, V = 1603.07(17) Å3, Z = 4, T = 173.15 K, μ(MoKα) = 0.088 mm−1, Dcalc = 1.361 g cm−3, 11067 reflections measured (3.866° ≤ 2θ ≤ 49.98°), 2831 unique (Rint = 0.1479, Rsigma = 0.2114) which were used in all calculations. The final R1 was 0.0410 (I > 2σ(I)) and wR2 was 0.0687 (all data). CCDC 1970363.
Conclusions
In summary, we have successfully developed a methodology for the synthesis of novel tetracyclic derivatives through intramolecular nucleophilic aromatic substitution of fluorine at position 5 of the imidazo[1,2-a]pyridine ring. In addition, we have demonstrated the improved utility, convenience and practicality of the pTsCl/DABCO-catalysed isocyanide in situ generation protocol over the conventional POCl3/Et3N protocol. Furthermore, we have managed to improve and simplify the existing pTsCl/DABCO N-formamide dehydration methodology to such an extent that it can be employed efficiently and cheaply in the absence of any preliminary purging of the reaction with nitrogen. Of particular importance is that it has also proved to be effective for preparing functionalised isocyanides in situ without the requirement for protection of reactive hydroxyl groups.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the National Research Foundation (NRF) and the Medical Research Council (MRC) of South Africa and the University of the Witwatersrand through the School of Chemistry and Research Office for their support and financial assistance. The assistance of Mr Thapelo Mbhele with the acquisition of HRMS data is gratefully acknowledged.Notes and references
- (a) A. Boltjes and A. Dömling, Eur. J. Org. Chem., 2019, 7007 CrossRef CAS; (b) J. Koubachi, S. El Kazzouli, M. Bousmina and G. Guillaumet, Eur. J. Org. Chem., 2014, 5119 CrossRef CAS.
- L. Dyminska, Bioorg. Med. Chem., 2015, 23, 6087 CrossRef CAS PubMed.
- T. O'Malley, T. Alling, J. V. Early, H. A. Wescott, A. Kumar, G. C. Moraski, M. J. Miller, T. Masquelin, P. A. Hipskind and T. Parisha, Antimicrob. Agents Chemother., 2018, 62, e02439-17 CrossRef PubMed.
- A. M. Palmer, S. Chrismann, G. Münch, C. Brehm, P. J. Zimmermann, W. Buhr, J. Senn-Bilfinger, M. P. Feth and W. A. Simon, Bioorg. Med. Chem., 2009, 17, 368 CrossRef CAS PubMed.
- F. Göktaş, N. Cesur, D. Şatana and M. Uzun, Turk. J. Chem., 2014, 38, 581 CrossRef.
- N. M. Shukla, D. B. Salunke, E. Yoo, C. A. Mutz, R. Balakrishna and S. A. David, Bioorg. Med. Chem., 2012, 20, 5850 CrossRef CAS PubMed.
- K. B. Puttaraju and K. Shivashankar, RSC Adv., 2013, 3, 20883 RSC.
- A. K. Bagdi, S. Santra, K. Monir and A. Hajra, Chem. Commun., 2015, 51, 1555 RSC.
- (a) K. Groebke, L. Weber and F. Mehlin, Synlett, 1998, 661 CrossRef CAS; (b) C. Blackburn, B. Guan, P. Fleming, K. Shiosaki and S. Tsai, Tetrahedron Lett., 1998, 39, 3635 CrossRef CAS; (c) H. Bienaymé and K. Bouzid, Angew. Chem., Int. Ed., 1998, 37, 2234 CrossRef.
- (a) M. L. Bode, D. Gravestock and A. L. Rousseau, Org. Prep. Proced. Int., 2016, 48, 89 CrossRef CAS; (b) A. L. Rousseau, P. Matlaba and C. J. Parkinson, Tetrahedron Lett., 2007, 48, 4079 CrossRef CAS.
- (a) A. Gautier, Liebigs Ann. Chem., 1867, 142, 289 CrossRef; (b) A. W. Hofmann, Liebigs Ann. Chem., 1867, 144, 114 CrossRef.
- P. Doz, I. Ugi, U. Fetzer, U. Eholzer, H. Knupfer and K. Offermann, Angew. Chem., Int. Ed., 1965, 4, 472 CrossRef.
- G. Skorna and I. Ugi, Angew. Chem., Int. Ed., 1977, 16, 259 CrossRef.
- I. Ugi and R. Meyr, Angew. Chem., 1958, 70, 702 CrossRef CAS.
- X. Wang, Q.-G. Wang and Q.-L. Luo, Synthesis, 2015, 47, 49 CAS.
- S. K. Guchhait, G. Priyadarshani, V. Chaudhaury, D. S. Seladiya, T. M. Shah and N. P. Bhogayta, RSC Adv., 2013, 3, 10867 RSC.
- M. L. Bode, D. Gravestock, S. S. Moleele, C. W. van der Westhuyzen, S. C. Pelly, P. A. Steenkamp, H. C. Hoppe, T. Khan and L. A. Nkabinde, Bioorg. Med. Chem., 2011, 19, 4227 CrossRef CAS PubMed.
- N. Grenouillat, B. Vauzeilles and J.-M. Beau, Heterocycles, 2007, 73, 891 CrossRef CAS.
- D. T. Whitaker, K. S. Whitaker and C. R. Johnson, p-Toluenesulfonyl Chloride, in Encyclopedia of Reagents for Organic Synthesis, John Wiley and sons, 2001 Search PubMed.
- (a) P. G. Gassman and T. L. Guggenheim, J. Am. Chem. Soc., 1982, 104, 5849 CrossRef CAS; (b) P. G. Gassman and T. L. Guggenheim, Org. Synth., 1990, 7, 294 Search PubMed.
- S. M. Creedon, H. K. Crowley and D. G. McCarthy, J. Chem. Soc., Perkin Trans. 1, 1998, 1015 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of all NMR spectra and crystallographic data. CCDC 1970362 and 1970363. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra10447j |
| This journal is © The Royal Society of Chemistry 2020 |