
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ab initio study on the excited states of pyrene and its derivatives using multi-reference perturbation theory methods†
Soichi Shirai * and
Shinji Inagaki
* and
Shinji Inagaki
Toyota Central R&D Laboratories, Inc., Nagakute, Aichi 480-1192, Japan. E-mail: shirai@mosk.tytlabs.co.jp
First published on 31st March 2020
Abstract
Low-lying singlet excited states of pyrene derivatives originated from the 1La and 1Lb states of pyrene have decisive influences on their absorption and fluorescence emission behaviors. Calculation of these excited states with quantitative accuracy is required for the theoretical design of pyrene derivatives tailored to target applications; this has been a long-standing challenge for ab initio quantum chemical calculations. In this study, we explore an adequate computational scheme through calculations of pyrene and its phenyl-substituted derivatives using multi-reference perturbation theory (MRPT) methods. All valence π orbitals on the pyrene moiety were assigned to the active orbitals. Computational load was reduced by restricting the electron excitations within the active orbitals in the preparation of reference configuration space. A generalized multi-configuration quasi-degenerate perturbation theory (GMCQDPT) was adopted to treat the reference space other than the complete active space. The calculated 1La and 1Lb excitation energies of pyrene are in good agreement with the experimental values. Calculations of 1,3,6,8-tetraphenyl pyrene suggest that the energetic ordering of 1La and 1Lb is inverted through tetraphenyl substitution and its lowest singlet excited state is the 1La parentage of pyrene, which is consistent with the experimentally deduced scheme. These results are not readily obtained by MRPT calculations with a limited number of active orbitals and single-reference theory calculations. Diphenyl pyrenes (DPPy) were also calculated at the same level of theory to investigate the dependence on the substitution positions of phenyl groups.
1. Introduction
Pyrene is one of the most well-studied polycyclic aromatic hydrocarbons (PAHs) because of its characteristic photophysical properties, such as prominent absorption bands and a fluorescence emission.1 These properties can be tailored to meet specific requirements through modifications of its chemical structure;2–11 therefore, pyrene derivatives are recognized as promising materials for organic light-emitting devices.12–25 Pyrene is also well known for the formation of an excimer.26–29 An aromatic excimer is a dimeric complex of the same aromatic molecules that is formed in the excited state. The fluorescence emission of an excimer was firstly observed for a pyrene solution by Förster and Kasper in 1954.27 The excimer fluorescence emission band is significantly red-shifted, broad, and structureless, so that it is clearly distinguishable from that of the monomer. Given the substantial distinction between monomer and excimer emissions, as well as their sensitivity to the surrounding environments, pyrene is used to study the molecular structural properties of polymers30–34 and macromolecules, such as proteins35,36 and DNA.37–42 It is also used as a probe in chemosensors that detect particular metal ions43–51 and molecules.52–55 The properties of an excimer are closely related to those of the monomer.56–58 Thus, the excited states of pyrene and its derivatives have been vigorously investigated and still continue to attract much interest from scientists with respect to both fundamental studies and practical applications.59–64Pyrene has two important excited states, 1La and 1Lb in Platt's notation.65 These excited states are closely relevant to the absorption and fluorescence emission behavior of pyrene. Excitation from the ground state to the 1La state gives a prominent absorption band around 340 nm in the UV/Vis spectrum because of its large oscillator strength. In contrast, the oscillator strength of the 1Lb state is negligibly small; the 1Lb absorption band is barely visible in the absorption spectrum.66 The 1Lb state is the lowest singlet excited state and the 1La state is the second lowest; therefore, the fluorescence emission of pyrene is of 1Lb parentage according to Kasha's rule.67 In substituted pyrene derivatives, the excitation energies and absorbance of these excited states are perturbed by the substituents. In some cases, even an energetic ordering of the 1La and 1Lb states is inverted as a result of the energy-level shifts of these excited states. 1,3,6,8-Tetraphyenyl pyrene (TPPy) is recognized as a typical example.68,69 TPPy exhibits a relatively short fluorescence lifetime (τ ≈ 3 ns)68,69 and a high fluorescence quantum yield (qF = 0.9)70 compared to pyrene (τ > 300 ns and qF = 0.32).68,69,71 These conspicuous changes in the photophysical properties can be attributed to an inversion of the energetic ordering of the 1La and 1Lb states through tetraphenyl substitution. Assuming the 1La–1Lb inversion, the fluorescence emission from TPPy originates from the 1La-derived excited state of pyrene with a large oscillator strength, which is consistent with the experimental results.68–70 Although this hypothetical mechanism based on the experimental findings was supported by theoretical studies that employed semi-empirical methods68,69,72 and has been widely accepted, the 1La–1Lb inversion in TPPy still remains to be corroborated by ab initio calculations. Given such a background, a dependable computational scheme that enables accurate calculations of these excited states is required for the theoretical design of pyrene derivatives customized to target applications; therefore, the prediction of their energetic ordering is of particular importance.
Calculation of the 1La and 1Lb excited states of pyrene with quantitative accuracy has been a long-term challenge for ab initio quantum chemistry.73 Time-dependent density functional theory (TDDFT)74–78 has been recognized as an efficient approach to the excited states of large molecules79,80 and is thus utilized in investigations of pyrene and its derivatives. Behind numerous successful results, comprehensive assessments indicate that TDDFT calculations yield inconsistencies in accuracy between the 1La and 1Lb excitation energies; in the worse cases, even the energetic ordering is incorrectly predicted.73,81,82 Similar problems are known for TDDFT calculations of some other PAHs.83–90 The second-order approximate coupled cluster singles and doubles (CC2) method is also widely used in the calculation of large molecules as an ab initio wave function approach.91 The energetic ordering of 1La and 1Lb of pyrene given by CC2 calculation is generally consistent with the experimental results. However, the incorporation of multi-configuration characters of excited states using single-reference methods such as CC2 is possibly insufficient, and could thus be responsible for the calculation errors. In this regard, multi-reference methods are expected to make up for this shortcoming.92 Bito et al. conducted multi-reference configuration interaction (MRCI) calculations as pioneering work,93 followed by theoretical studies that utilized multi-reference perturbation theory (MRPT) methods,73,81,94 such as multi-configuration quasi-degenerate perturbation theory (MCQDPT)95 and complete active space second-order perturbation theory (CASPT2).96 These methods adopt a complete active space self-consistent field (CASSCF) wavefunction97 as their reference. The CAS involves all electron configurations generated by distributing active electrons among active orbitals. Consequently, the dimension rapidly increases with the number of active orbitals and electrons, which makes the routine computation impossible. Therefore, in these studies, a limited number of π orbitals and π electrons were selected from a total of 16 valence π orbitals and 16 valence π electrons to construct the reference CAS. Although the energetic ordering of 1La and 1Lb is correctly predicted in most of these calculations, absolute errors in the excitation energies of 0.2–0.7 eV remain, and the calculation accuracy is inconsistent between the 1La and 1Lb states. The discrepancies at the CASSCF level are much larger because dynamical electron correlation is not sufficiently incorporated.
Recent advances in theory and computational techniques have yielded significant progress. Freidzon et al. demonstrated that the 1La and 1Lb excitation energies of pyrene can be accurately predicted98 using extended MCQDPT (XMCQDPT) method.99 Nenov et al. performed second-order perturbation theory restricted active space (RASPT2) calculations100 with the reference configuration space constructed using 16 π orbitals and 16 π electrons.101 The calculated excitation energies were in good agreement with the experimental values, even though the computational load was reduced by restricting the electron excitations within the active orbitals. Most recently, Noble et al. conducted multi-state CASPT2 calculations with the Cholesky decomposition technique102,103 in their study of electronic relaxations from the S3 state of pyrene.104 The reference configuration space was prepared using 16 π orbitals and 16 π electrons, and the results were quite accurate. Lischka et al. carried out the calculations of paradigmatic aromatic molecules including pyrene using both multi-reference and single-reference methods.105 In their results, the 1La and 1Lb excitation energies of pyrene calculated using the DFT/MRCI method106 were in good agreement with the experimental values. The same authors successfully applied their calculation scheme to the study of large-sized aromatic dimers.107 These studies suggest that the excited states of pyrene derivatives could also be calculated with quantitative accuracy by utilizing these advanced methods. Successful results with the full π valence reference space also imply that the incorporation of multi-configuration character is of key importance for accurate calculation.
In this study, we explore an adequate computational scheme that is suitable for the excited states of pyrene derivatives through calculations of pyrene and its phenyl substituted-derivatives shown in Fig. 1. Generalized multi-configuration quasi-degenerate perturbation theory (GMCQDPT)108 was adopted in addition to the original MCQDPT. In contrast to the conventional multi-reference theories where the reference space is limited to the CAS, GMCQDPT allows more general types of reference. Accordingly, it has the potential to reduce the computational load without a significant decrease of accuracy and could enable the handling of pyrene derivatives with large system sizes.
 | ||
| Fig. 1 Molecular structures of (a) pyrene, (b) 1,3,6,8-tetraphenylpyrene (TPPy), (c) 1,6-diphenylpyrene (1,6-DPPy), and (d) 2,7-diphenylpyrene (2,7-DPPy). | ||
2. Computational details
2.1. Reference wave functions for GMCQDPT
In the preparation of the reference wave function for GMCQDPT, the molecular orbitals are divided into three groups: inactive, active, and external orbitals. Whereas the inactive orbitals are always doubly occupied, external orbitals are always vacant. The electrons involved in the active orbitals are regarded as active electrons and the electron configurations are generated by distributing active electrons among active orbitals. The procedure so far is the same as that adopted in the construction of the CAS. Although any types of reference configuration space are available in GMCQDPT, the following three are proposed as typical examples: multi-reference determinant list plus excitations (MRX), restricted active space (RAS), and occupation restricted multiple active space (ORMAS).109–111 In this study, the MRX type of reference was employed because of its simplicity. In the MRX framework, parent configurations and the electron excitation level, n, are defined. The excited configurations having n or less excited electrons from the parent configurations are involved into the reference space, in addition to the parent configurations themselves. If n is equal to the number of active electrons, then the reference space is identical with the CAS. The dimension of the reference space can be reduced by limiting the number of parent configurations and specifying n less than the number of active electrons.In the GMCQDPT calculations of pyrene in this study, all 16 valence π orbitals were selected as active orbitals. Similarly, in the calculations of phenyl-substituted derivatives, the 16 π orbitals on their pyrene moieties were treated as active orbitals. The Hartree–Fock type ground state configuration with eight doubly-occupied π orbitals and eight unoccupied π* orbitals was defined as a parent configuration, and the n value was set at 2, 3 and 4. Henceforth, these reference spaces are denoted as MRX(n). GMCQDPT calculations were performed in the following two steps as well as the conventional CASSCF–MCQDPT procedure: multi-configuration self-consistent field (MCSCF) calculation with MRX(n) configuration space was firstly performed; the second-order perturbation calculation was subsequently conducted employing the obtained MCSCF function as its reference.
2.2. Calculations
The molecular geometrical structures were optimized using DFT with the B3LYP functional.112,113 The ground and excited states were subsequently calculated at the optimized geometries. The absence of an imaginary number frequency was confirmed for all optimized structures by vibrational analyses. In addition to the GMCQDPT calculations, conventional CASSCF–MCQDPT calculations were also performed for comparison; CAS(4πe, 4πo), CAS(8πe, 8πo), and CAS(12πe, 12πo) were used as reference spaces. CAS{(2m)πe, (2m)πo} (m = 2, 4, and 6) was constructed using the m highest occupied and the m lowest unoccupied π orbitals on the pyrene moiety. The ground state, and the 1La and 1Lb excited states were averaged with even weights in the MCSCF calculations, and these three states were simultaneously perturbed in the MRPT calculations. Equations of motion coupled cluster singles and doubles (EOM-CCSD),114,115 and TDDFT calculations with B3LYP, CAM-B3LYP,116 and ωB97XD117 functionals were also performed. The DPPy derivatives were calculated using GMCQDPT with MRX(4) and TDDFT. The cc-pVDZ basis set was used throughout the calculations.118 The molecular symmetries assumed were D2h, D2, C2, and C2h for pyrene,119 TPPy,120 1,6-DPPy, and 2,7-DPPy,121 respectively. MCSCF and MRPT calculations were performed using the GAMESS program.122,123 Other calculations were conducted using Gaussian09.1243. Results and discussion
3.1. Calculation results
The calculated excitation energies and oscillator strengths are summarized in Table 1 along with available experimental values.119–121,125–128 According to the previous studies, the 1La state of pyrene is characterized by two singly excited configurations: one is the highest occupied molecular orbital (HOMO) → the lowest unoccupied molecular orbital (LUMO) single excitation, and the other is the HOMO−1 → LUMO+1 single excitation. In contrast, HOMO−1 → LUMO and HOMO → LUMO+1 single excitations are dominant in the 1Lb state.119,129 The excited states were then identified on the basis of these configurations and related molecular orbitals. Fig. 2 shows the natural orbitals from HOMO−1 to LUMO+1 obtained using MCSCF with the MRX(4) configuration space. All natural orbitals from the MCSCF wave functions are presented in Fig. S1–S3.† Electronic state total energies and MCSCF excitation energies are listed in Table S1.†| Molecule | Method | Reference space | Number of electron configurations | 1La | 1Lb | ||
|---|---|---|---|---|---|---|---|
| Excitation energy (eV) | Oscillator strengtha | Excitation energy (eV) | Oscillator strengtha | ||||
| a Values are rounded off to four decimal places.b Ref. 125; adiabatic transition energies in vapor.c Ref. 126; gas-phase fluorescence excitation spectrum.d Ref. 127; λmax in acetonitrile.e Ref. 119; gas-phase fluorescence excitation spectrum.f Ref. 128; λmax in CH2Cl2.g Ref. 120; λmax in CH2Cl2.h Ref. 121; λmax in dilute CH2Cl2 (ca. 1 × 10−5 M). | |||||||
| Pyrene | GMCQDPT | MRX(2) | 4349 | 3.75 | 0.2030 | 3.17 | 0.0000 |
| MRX(3) | 84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 317 317 |
3.82 | 0.2511 | 3.31 | 0.0000 | ||
| MRX(4) | 853785 |
3.84 | 0.2308 | 3.40 | 0.0000 | ||
| MCQDPT | CAS(4πe, 4πo) | 28 | 3.55 | 0.2170 | 2.83 | 0.0007 | |
| CAS(8πe, 8πo) | 3684 | 3.75 | 0.3997 | 3.26 | 0.0000 | ||
| CAS(12πe, 12πo) | 640432 |
3.83 | 0.3668 | 4.01 | 0.0001 | ||
| EOM-CCSD | 4.45 | 0.3443 | 3.82 | 0.0001 | |||
| TD-B3LYP | 3.68 | 0.2534 | 3.74 | 0.0001 | |||
| TD-CAM-B3LYP | 3.98 | 0.3169 | 3.95 | 0.0001 | |||
| TD-ωB97XD | 3.99 | 0.3216 | 3.96 | 0.0001 | |||
| Exptl. | 3.85b, 3.87c, 3.71d | 3.41b, 3.37c,e | |||||
| TPPy | GMCQDPT | MRX(2) | 4349 | 3.11 | 0.2748 | 2.96 | 0.0000 |
| MRX(3) | 84317 |
3.27 | 0.3607 | 3.12 | 0.0001 | ||
| MRX(4) | 853785 |
3.07 | 0.2853 | 3.24 | 0.0000 | ||
| MCQDPT | CAS(4πe, 4πo) | 28 | 2.92 | 0.3832 | 2.60 | 0.0028 | |
| CAS(8πe, 8πo) | 3684 | 3.15 | 0.5360 | 3.04 | 0.0003 | ||
| CAS(12πe, 12πo) | 640432 |
3.69 | 0.5381 | 3.58 | 0.0000 | ||
| EOM-CCSD | 3.91 | 0.9563 | 3.65 | 0.0001 | |||
| TD-B3LYP | 3.13 | 0.7128 | 3.49 | 0.0003 | |||
| TD-CAM-B3LYP | 3.47 | 0.8555 | 3.76 | 0.0008 | |||
| TD-ωB97XD | 3.50 | 0.8649 | 3.77 | 0.0007 | |||
| Exptl. | 3.24f, 3.15g | ||||||
| 1,6-DPPy | GMCQDPT | MRX(4) | 1138281 |
3.39 | 0.2438 | 3.31 | 0.0169 |
| TD-B3LYP | 3.35 | 0.6050 | 3.61 | 0.0025 | |||
| TD-CAM-B3LYP | 3.69 | 0.7010 | 3.85 | 0.0085 | |||
| TD-ωB97XD | 3.71 | 0.7009 | 3.86 | 0.0093 | |||
| 2,7-DPPy | GMCQDPT | MRX(4) | 853785 |
3.64 | 0.1487 | 3.30 | 0.0001 |
| TD-B3LYP | 3.59 | 0.1276 | 3.38 | 0.0000 | |||
| TD-CAM-B3LYP | 3.92 | 0.2198 | 3.71 | 0.0000 | |||
| TD-ωB97XD | 3.94 | 0.2544 | 3.74 | 0.0000 | |||
| Exptl. | 3.63h | ||||||
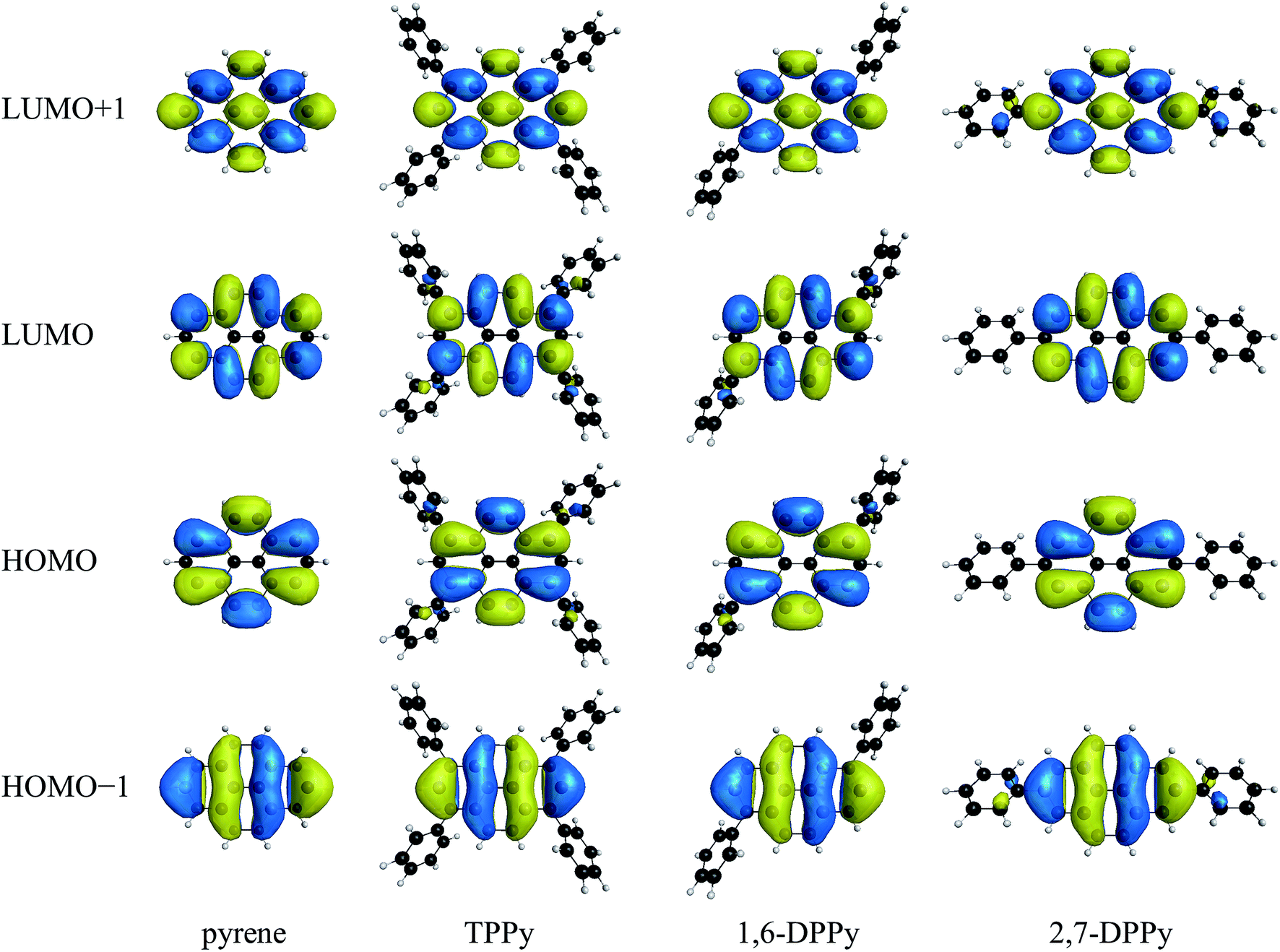 | ||
| Fig. 2 Natural orbitals of pyrene, TPPy, 1,6-DPPy, and 2,7-DPPy from the MCSCF wave functions with the MRX(4) configuration space. | ||
Overall, provided that the GMCQDPT method along with MRX(4) is adopted, the excitation energies of pyrene and TPPy can be accurately calculated, and the 1La–1Lb inversion through tetraphenyl substitution is predicted. The calculated oscillator strength of the 1La state for TPPy is consistently larger than that of pyrene, whereas that of the 1Lb state is still vanishingly small.
The TDDFT calculations also predict a downward shift of the 1La energy level without significant change of the 1Lb energy level. However, in contrary to the GMCQDPT results, the TDDFT calculations suggest that the 1La state is the lowest singlet excited state. As mentioned above, the 1Lb excitation energy of pyrene is significantly overestimated by the TDDFT calculations. The 1La energy level calculated using TD-B3LYP is estimated to be lower than the 1Lb energy level even for non-substituted pyrene. The gap between 1La and 1Lb is extremely underestimated using TD-CAM-B3LYP and TD-ωB97XD; because of this imbalanced alignment of the 1La and 1Lb energy levels for non-substituted pyrene, the energetic ordering of the 1La and 1Lb states is easily inverted through the downward shift of the 1La energy level. Meanwhile, the TDDFT calculations suggest an increase of the 1La oscillator strength through the diphenyl substitution as well as the GMCQDPT calculations. The oscillator strengths of the 1Lb state are much smaller than those of the 1La state, as with the cases of pyrene and TPPy. Yet, 1,6-DPPy exhibits the largest 1Lb oscillator strength among the molecules calculated here.
The results of TDDFT calculations show a different behavior from the GMCQDPT results; the 1Lb energy level is more largely shifted downward without significant change of the 1La energy level. The behavior is also in contrast to the case of 1,6-DPPy which exhibits a largely downward shift of the 1La energy level without a major change in the 1Lb energy level. Meanwhile, a decrease of the 1La oscillator strength through diphenyl substitution is predicted using TDDFT, as with GMCQPDT.
3.2. Discussion
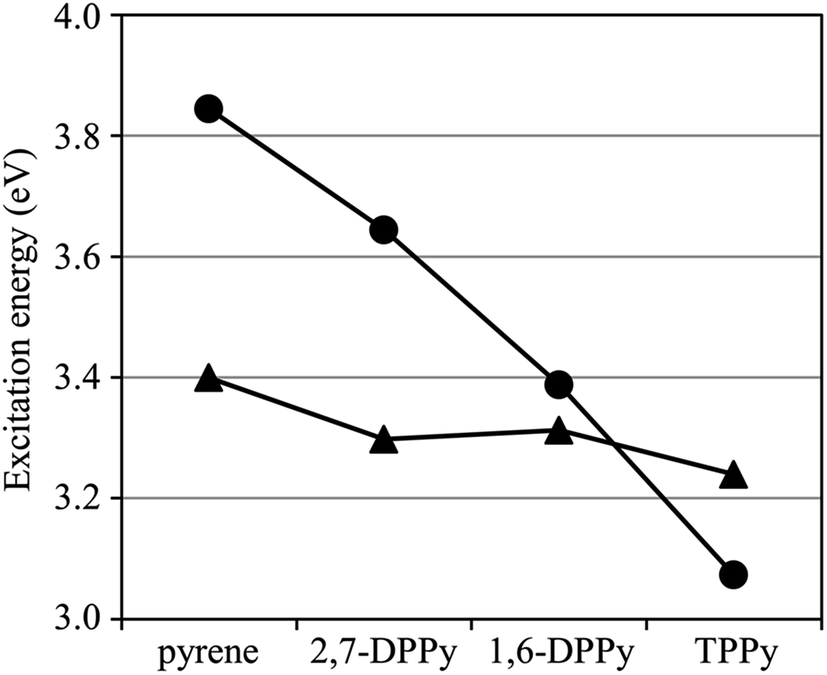 | ||
| Fig. 3 Excitation energies of the 1La (●) and 1Lb (▲) states calculated using GMCQDPT with MRX(4). | ||
The accuracy of the MRPT calculation reveals a strong dependence on the reference configuration space. Therefore, the requirements for the reference space to achieve reliable calculations can be determined from these results. The excitation energies with MCQDPT are highly dependent on the reference CAS, whereas those with GMCQDPT vary only in a narrow range. Thus, the calculation accuracy is sensitive to the number of active orbitals. There is no guarantee that MRPT with less active orbitals will be successful, even though its reference space is the CAS. In contrast, the incorporation of multiply-excited configurations into the reference space leads to a systematic improvement within the framework of a full π valence reference space. RASPT2 calculations by Nenov et al. implied that the reference space with incorporation of up to quadruple excitations is necessary to compute the excitation energies of pyrene with high accuracy.101 It should be noted that involving quadruply-excited configurations is also critically important for prediction of the 1La–1Lb inversion.
The improvement of the calculation accuracy through the expansion of the reference space implies a significant multi-configuration character of these excited states. Therefore, the main configurations of the excited states in the MCSCF wave functions and their weights were analyzed (Table 2). The total weights of the main configurations for 1La and 1Lb, denoted by σa and σb, are also presented in Table 2. A smaller σ value indicates a greater contribution of the electron configurations other than the main configurations. Therefore, the σ value is correlated with the degree of the multi-configuration character of the excited state. The weights of the main configurations are relatively reduced as the contributions of more electron configurations are incorporated; therefore, the σ values are decreased with expansion of the reference space. Let us review the results obtained for pyrene with MRX(n); the σb values are generally smaller than the σa values and more largely varied depending on the reference space. Such behavior can be attributed to the significant multi-configuration character of 1Lb compared to 1La, which is consistent with a high dependence of the calculated 1Lb excitation energy on the reference space (Table 1). The calculations with MRX(4) give the smallest σ values among the examined reference spaces for both La and Lb; this is expected to be more suitable to deal with the complexity of these excited states. Although the σ values with CAS have similar trends to that with MRX(n), their fluctuations with the reference space are more pronounced and the values are generally larger than those with MRX(4). These results suggest that multi-configuration character is only insufficiently incorporated with these CAS references, even at the maximum CAS(12πe, 12πo). The σ values and their trends for TPPy, 1,6-DPPy and 2,7-DPPy are quite similar to those of pyrene, which indicates a comparable degree of the multi-configuration character of their excited states.
| Molecule | Method | Reference space | 1La | 1Lb | ||||
|---|---|---|---|---|---|---|---|---|
| H → L | H−1 → L+1 | σa | H → L+1 | H−1 → L | σb | |||
| Pyrene | GMCQDPT | MRX(2) | 0.7295 | 0.1563 | 0.8858 | 0.4089 | 0.3971 | 0.8060 |
| MRX(3) | 0.6808 | 0.1458 | 0.8265 | 0.3775 | 0.3594 | 0.7369 | ||
| MRX(4) | 0.6577 | 0.1185 | 0.7762 | 0.3318 | 0.3267 | 0.6586 | ||
| MCQDPT | CAS(4πe, 4πo) | 0.7906 | 0.2024 | 0.9930 | 0.5465 | 0.4451 | 0.9916 | |
| CAS(8πe, 8πo) | 0.7900 | 0.1163 | 0.9063 | 0.4259 | 0.3996 | 0.8255 | ||
| CAS(12πe, 12πo) | 0.7370 | 0.0818 | 0.8188 | 0.3550 | 0.3605 | 0.7154 | ||
| TPPy | GMCQDPT | MRX(2) | 0.7699 | 0.1189 | 0.8888 | 0.4247 | 0.3829 | 0.8076 |
| MRX(3) | 0.7250 | 0.1033 | 0.8283 | 0.3901 | 0.3487 | 0.7388 | ||
| MRX(4) | 0.6923 | 0.0866 | 0.7789 | 0.3405 | 0.3195 | 0.6600 | ||
| MCQDPT | CAS(4πe, 4πo) | 0.8638 | 0.1270 | 0.9907 | 0.5879 | 0.4040 | 0.9918 | |
| CAS(8πe, 8πo) | 0.8388 | 0.0680 | 0.9068 | 0.4377 | 0.3716 | 0.8093 | ||
| CAS(12πe, 12πo) | 0.7730 | 0.0478 | 0.8208 | 0.3563 | 0.3415 | 0.6978 | ||
| 1,6-DPPy | GMCQDPT | MRX(4) | 0.6330 | 0.0947 | 0.7277 | 0.3186 | 0.3040 | 0.6226 |
| 2,7-DPPy | GMCQDPT | MRX(4) | 0.6238 | 0.1465 | 0.7703 | 0.3301 | 0.3285 | 0.6587 |
The GMCSCF wave function with MRX(n) was constructed in terms of a linear combination of electron configurations and the GMCSCF calculations were carried out with a state-averaging scheme. Consequently, the GMCSCF orbital energies do not have clear physical meanings. The orbital energies obtained using the DFT calculations are dependent on the exchange-correlation functionals. Therefore, we analyzed the Hartree–Fock orbital energies collected in Table 3 where the energy gap between the p and q orbitals is denoted by Δp/q; the energy shifts from the corresponding value of pyrene are shown in parentheses. The molecular orbitals obtained from the Hartree–Fock calculations are shown in Fig. S4.† In TPPy and 1,6-DPPy, the HOMO and LUMO energy levels are largely shifted compared to the HOMO−1 and LUMO+1 levels; the HOMO energy level is shifted upward, whereas the LUMO energy level is shifted downward. In 2,7-DPPy, in contrast, the HOMO−1 and LUMO+1 energy levels of 2,7-DPPy are largely shifted without significant changes of HOMO and LUMO energies. These variations of the orbital energies are consistent with the orbital distributions shown in Fig. 2 and S4.† The HOMO and LUMO of pyrene have large coefficients at positions 1, 3, 6, and 8; therefore, these orbitals are sensitive to phenyl substitution at these positions. In contrast, the HOMO−1 and LUMO+1 have large coefficients at positions 2 and 7; the phenyl substitution at positions 2 and 7 has a large impact on these orbitals. The electronic interactions between pyrene moieties and phenyl substituents can be also visually confirmed in the molecular orbitals of pyrene derivatives; the HOMO and LUMO of TPPy and 1,6-DPPy are partially extended to the phenyl substituents, whereas HOMO−1 and LUMO+1 are extended in 2,7-DPPy. The orbital extensions are more noticeable in the Hartree–Fock orbitals (Fig. S4†). As a result of the energetic changes of the orbitals, the ΔHOMO/LUMO values are in the order of pyrene ≈ 2,7-DPPy > 1,6-DPPy > TPPy, which is in good agreement with that of the 1La excitation energies obtained using GMCQDPT with MRX(4) (Table 1). Although the ΔHOMO/LUMO value of 2,7-DPPy is almost the same as that with pyrene, ΔHOMO−1/LUMO+1 of 2,7-DPPy is much smaller than that of pyrene. The HOMO−1 → LUMO+1 single excitation is another main configuration of the 1La states; therefore, a reduced ΔHOMO−1/LUMO+1 could be responsible for the slightly decreased 1La excitation energy. A similar correlation between the Δ values and excitation energies can be also found for the TDDFT results (Table 1). Thus, as for 1La, the calculated excitation energies are well correlated with the Δ values related to the main configurations.
| Orbital | Py | TPPy | 1,6-DPPy | 2,7-DPPy |
|---|---|---|---|---|
| a Values in parentheses are variations from the corresponding orbital energy of pyrene. | ||||
| LUMO+1 | +2.58 | +2.45 | +2.50 | +1.93 |
| (−0.13) | (−0.08) | (−0.65) | ||
| LUMO | +1.62 | +1.32 | +1.46 | +1.55 |
| (−0.30) | (−0.16) | (−0.07) | ||
| HOMO | −7.03 | −6.64 | −6.82 | −7.12 |
| (+0.39) | (+0.21) | (−0.09) | ||
| HOMO−1 | −8.07 | −7.95 | −8.04 | −7.48 |
| (+0.12) | (+0.03) | (+0.59) | ||
| ΔHOMO/LUMO | +8.65 | +7.96 | +8.28 | +8.67 |
| (−0.69) | (−0.37) | (+0.02) | ||
| ΔHOMO−1/LUMO+1 | +10.65 | +10.40 | +10.54 | +9.41 |
| (−0.25) | (−0.11) | (−1.24) | ||
| ΔHOMO/LUMO+1 | +9.61 | +9.09 | +9.32 | +9.05 |
| (−0.52) | (−0.29) | (−0.56) | ||
| ΔHOMO−1/LUMO | +9.69 | +9.27 | +9.50 | +9.03 |
| (−0.42) | (−0.19) | (−0.66) | ||
The calculated ΔHOMO/LUMO+1 and ΔHOMO−1/LUMO values of phenyl substituted derivatives are lower than those of pyrene, which is consistent with their lower 1Lb excitation energies obtained using GMCQDPT with MRX(4). The fluctuations of the ΔHOMO/LUMO+1 and ΔHOMO−1/LUMO values are rather milder than those of the ΔHOMO/LUMO and ΔHOMO−1/LUMO+1 values. This behavior is also consistent with the insensitivity of the 1Lb excitation energies to phenyl substitution compared to the 1La excitation energies, which results in a reduction of the 1La–1Lb gap in 1,6-DPPy and the 1La–1Lb inversion in TPPy (Fig. 3). However, the ΔHOMO/LUMO+1 and ΔHOMO−1/LUMO values are in the order of pyrene > 1,6-DPPy > TPPy > 2,7-DPPy, which is not in agreement with the ordering of the 1Lb excitation energies with GMCQDPT: pyrene > 1,6-DPPy ≈ 2,7-DPPy > TPPy (Table 1). The 1Lb state has significant multi-configuration character, as suggested from the less σ values (Table 2), which could be responsible for the weak correlation between the Δ values and the 1Lb excitation energies, and also for smaller fluctuations of the 1Lb excitation energies. In contrast to the GMCQDPT results, the 1Lb excitation energies with TDDFT exhibit a stronger correlation with the ΔHOMO/LUMO+1 and ΔHOMO−1/LUMO values; these values are in the same order: pyrene > 1,6-DPPy > TPPy > 2,7-DPPy. Since TDDFT is a one-particle theory, the excitation energies obtained using TDDFT are susceptible to the energies of one-electron orbitals. Instead, the 1Lb excitation energy tends to be overestimated because of insufficient incorporation of multi-configuration character.
The results of the TDDFT calculations are similar to those obtained from the GMCQDPT calculations; the calculated 1La oscillator strengths are in the order of TPPy > 1,6-DPPy > pyrene; the calculated 1Lb oscillator strengths are nearly zero except for 1,6-DPPy. Overall, the effects of substituents on the 1La excitation energy, the 1La and 1Lb oscillator strengths can be reasonably evaluated using TDDFT; however, the 1Lb excitation energy cannot be accurately predicted; in the worse cases, even the energetic ordering is incorrectly predicted. Thus, a multi-reference treatment is essential for an accurate calculation of the 1Lb excitation energy, the ordering of the 1La and 1Lb states, and the energy gap between these states.
4. Conclusion
The absorption and fluorescence emission behavior of pyrene derivatives is characterized by the low-lying excited states derived from 1La and 1Lb of pyrene. In this study, pyrene, TPPy, 1,6-DPPy, and 2,7-DPPy as typical examples were calculated using MRPT methods to explore a dependable computational scheme for these excited states. When all valence π and π* orbitals on the pyrene moiety were incorporated into the reference space, the calculated 1La and 1Lb excitation energies of pyrene were in good agreement with the experimental values. The fluorescence emission of TPPy was predicted to have pyrene 1La parentage, which theoretically corroborates the experimental observations. A reference space involving singly, doubly, triply, and quadruply excited configurations was required to predict the 1La–1Lb inversion through tetraphenyl substitution. MRPT calculations with smaller reference spaces and single-reference theory calculations exhibit inconsistencies with these results, which suggests that adequate treatment of the multi-configuration character is essential. The effect of phenyl substitution was dependent not only on the excited state but also on the substitution position. The detailed mechanism was clarified by examination of the MCSCF wave functions. These calculations were successfully conducted at a tractable computational cost by the adoption of GMCQDPT to enable the handling of general types of reference configuration space.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was partially supported by the Advanced Catalytic Transformation Program for Carbon Utilization (ACT-C) of the Japan Science and Technology Agency (JST) (Grant number: JPMJCR12Y1). The authors are grateful to Dr Norihiro Mizoshita and Mr Yoshifumi Maegawa at Toyota Central R&D Labs.; this study was motivated by their studies on mesoporous light-emitting materials. The authors are also grateful to Dr Yuri Yamada at Toyota Central R&D Labs. for technical help.References
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef PubMed.
- B.-C. Wang, J.-C. Chang, H.-C. Tso, H.-F. Hsu and C.-Y. Cheng, J. Mol. Struct.: THEOCHEM, 2003, 629, 11–20 CrossRef.
- T. M. Figueira-Duarte, S. C. Simon, M. Wagner, S. I. Druzhinin, K. A. Zachariasse and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 10175–10178 CrossRef PubMed.
- K. Fujimoto, H. Shimizu, M. Furusyo, S. Akiyama, M. Ishida, U. Furukawa, T. Yokoo and M. Inouye, Tetrahedron, 2009, 65, 9357–9361 CrossRef.
- M. Sase, S. Yamaguchi, Y. Sagara, I. Yoshikawa, T. Mutai and K. Araki, J. Mater. Chem., 2011, 21, 8347–8354 RSC.
- A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Pålsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef PubMed.
- S.-S. Li, K.-J. Jiang, C.-C. Yu, J.-H. Huang, L.-M. Yang and Y.-L. Song, New J. Chem., 2014, 38, 4404–4408 RSC.
- Y. Niko, S. Sasaki, K. Narushima, D. K. Sharma, M. Vacha and G.-i. Konishi, J. Org. Chem., 2015, 80, 10794–10805 CrossRef.
- H. Örücü and N. Acar, Comput. Theor. Chem., 2015, 1056, 11–18 CrossRef.
- R. Liu, H. Ran, Z. Zhao, X. Yang, J. Zhang, L. Chen, H. Sun and J.-Y. Hu, ACS Omega, 2018, 3, 5866–5875 CrossRef.
- V. S. Vyas, S. V. Lindeman and R. Rathore, J. Photochem. Photobiol., A, 2019, 375, 209–218 CrossRef.
- T. Oyamada, S. Akiyama, M. Yahiro, M. Saigou, M. Shiro, H. Sasabe and C. Adachi, Chem. Phys. Lett., 2006, 421, 295–299 CrossRef.
- J. N. Moorthy, P. Natarajan, P. Venkatakrishnan, D.-F. Huang and T. J. Chow, Org. Lett., 2007, 9, 5215–5218 CrossRef.
- N. Mizoshita, Y. Goto, Y. Maegawa, T. Tani and S. Inagaki, Chem. Mater., 2010, 22, 2548–2554 CrossRef.
- S. Yamaguchi, I. Yoshikawa, T. Mutai and K. Araki, J. Mater. Chem., 2012, 22, 20065–20070 RSC.
- Y. Maegawa, N. Mizoshita, T. Tani and S. Inagaki, Chem. Lett., 2012, 41, 316–318 CrossRef.
- K. Sumi, Y. Niko, K. Tokumaru and G.-i. Konishi, Chem. Commun., 2013, 49, 3893–3895 RSC.
- D. Chercka, S.-J. Yoo, M. Baumgarten, J.-J. Kim and K. Müllen, J. Mater. Chem. C, 2014, 2, 9083–9086 RSC.
- T. H. El-Assaad, M. Auer, R. Castañeda, K. M. Hallal, F. M. Jradi, L. Mosca, R. S. Khnayzer, D. Patra, T. V. Timofeeva, J.-L. Brédas, E. J. W. List-Kratochvil, B. Wex and B. R. Kaafarani, J. Mater. Chem. C, 2016, 4, 3041–3058 RSC.
- H. Ju, K. Wang, J. Zhang, H. Geng, Z. Liu, G. Zhang, Y. Zhao and D. Zhang, Chem. Mater., 2017, 29, 3580–3588 CrossRef.
- D. H. Ahn, J. H. Jeong, J. Song, J. Y. Lee and J. H. Kwon, ACS Appl. Mater. Interfaces, 2018, 10, 10246–10253 CrossRef.
- H. Jung, S. Kang, H. Lee, Y.-J. Yu, J. H. Jeong, J. Song, Y. Jeon and J. Park, ACS Appl. Mater. Interfaces, 2018, 10, 30022–30028 CrossRef.
- H. Maeda, T. Suzuki and M. Segi, Photochem. Photobiol. Sci., 2018, 17, 781–792 RSC.
- N. Mizoshita and S. Inagaki, Macromol. Chem. Phys., 2018, 219, 1700596 CrossRef.
- J.-H. Lee, C.-H. Chen, P.-H. Lee, H.-Y. Lin, M.-k. Leung, T.-L. Chiu and C.-F. Lin, J. Mater. Chem. C, 2019, 7, 5874–5888 RSC.
- F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef.
- T. Förster and K. Kasper, Z. Phys. Chem., 1954, 1, 275–277 CrossRef.
- J. B. Birks, Rep. Prog. Phys., 1975, 38, 903–974 CrossRef.
- T. Azumi and S. P. McGlynn, J. Chem. Phys., 1964, 41, 3131–3138 CrossRef CAS.
- J. Duhamel, Acc. Chem. Res., 2006, 39, 953–960 CrossRef.
- M. Ingratta, J. Hollinger and J. Duhamel, J. Am. Chem. Soc., 2008, 130, 9420–9428 CrossRef.
- S. J. Teertstra, W. Y. Lin, M. Gauthier, M. Ingratta and J. Duhamel, Polymer, 2009, 50, 5456–5466 CrossRef.
- Z. Xu, N. J. Singh, J. Lim, J. Pan, H. N. Kim, S. Park, K. S. Kim and J. Yoon, J. Am. Chem. Soc., 2009, 131, 15528–15533 CrossRef.
- M. Danko, P. Hrdlovič, M. Brzezinski, A. Duda and T. Biela, Polymer, 2018, 156, 76–84 CrossRef.
- G. Bains, A. B. Patel and V. Narayanaswami, Molecules, 2011, 16, 7909–7935 CrossRef CAS PubMed.
- A. Fujii, Y. Sekiguchi, H. Matsumura, T. Inoue, W.-S. Chung, S. Hirota and T. Matsuo, Bioconjugate Chem., 2015, 26, 537–548 CrossRef CAS PubMed.
- A. A. Martí, S. Jockusch, N. Stevens, J. Ju and N. J. Turro, Acc. Chem. Res., 2007, 40, 402–409 CrossRef PubMed.
- P. Conlon, C. J. Yang, Y. Wu, Y. Chen, K. Martinez, Y. Kim, N. Stevens, A. A. Marti, S. Jockusch, N. J. Turro and W. Tan, J. Am. Chem. Soc., 2008, 130, 336–342 CrossRef CAS.
- X. Su, X. Xiao, C. Zhang and M. Zhao, Appl. Spectrosc., 2012, 66, 1249–1261 CrossRef CAS.
- F. Wojciechowski, J. Lietard and C. J. Leumann, Org. Lett., 2012, 14, 5176–5179 CrossRef CAS.
- C. Wu, C. Wang, L. Yan and C. J. Yang, J. Biomed. Nanotechnol., 2009, 5, 495–504 CrossRef CAS.
- A. O. Krasheninina, S. D. Novopashina, K. E. Apartsin and G. A. Venyaminova, Molecules, 2017, 22, 2108 CrossRef.
- S. K. Kim, S. H. Lee, J. Y. Lee, J. Y. Lee, R. A. Bartsch and J. S. Kim, J. Am. Chem. Soc., 2004, 126, 16499–16506 CrossRef CAS PubMed.
- X.-L. Ni, S. Wang, X. Zeng, Z. Tao and T. Yamato, Org. Lett., 2011, 13, 552–555 CrossRef CAS PubMed.
- S. Das, A. Sahana, A. Banerjee, S. Lohar, D. A. Safin, M. G. Babashkina, M. Bolte, Y. Garcia, I. Hauli, S. K. Mukhopadhyay and D. Das, Dalton Trans., 2013, 42, 4757–4763 RSC.
- S. Sarkar, S. Roy, A. Sikdar, R. N. Saha and S. S. Panja, Analyst, 2013, 138, 7119–7126 RSC.
- W. Cho, H. J. Lee, G. Choi, S. Choi and M. Oh, J. Am. Chem. Soc., 2014, 136, 12201–12204 CrossRef CAS.
- S. S. Razi, R. Ali, P. Srivastava and A. Misra, RSC Adv., 2015, 5, 79538–79547 RSC.
- M. Shellaiah, Y. C. Rajan, P. Balu and A. Murugan, New J. Chem., 2015, 39, 2523–2531 RSC.
- M. Zhao, X. Zhou, J. Tang, Z. Deng, X. Xu, Z. Chen, X. Li, L. Yang and L.-J. Ma, Spectrochim. Acta, Part A, 2017, 173, 235–240 CrossRef CAS.
- Y. Upadhyay, T. Anand, L. T. Babu, P. Paira, G. Crisponi, A. K. Sk, R. Kumar and S. K. Sahoo, Dalton Trans., 2018, 47, 742–749 RSC.
- S. Zhang, L. Ding, F. Lü, T. Liu and Y. Fang, Spectrochim. Acta, Part A, 2012, 97, 31–37 CrossRef CAS.
- A. Ghosh, A. Sengupta, A. Chattopadhyay and D. Das, Chem. Commun., 2015, 51, 11455–11458 RSC.
- S. Lohar, D. A. Safin, A. Sengupta, A. Chattopadhyay, J. S. Matalobos, M. G. Babashkina, K. Robeyns, M. P. Mitoraj, P. Kubisiak, Y. Garcia and D. Das, Chem. Commun., 2015, 51, 8536–8539 RSC.
- K. Khownium, J. Romsaiyud, S. Borwornpinyo, P. Wongkrasant, P. Pongkorpsakol, C. Muanprasat, B. Boekfa, T. Vilaivan, S. Ruchirawat and J. Limtrakul, New J. Chem., 2019, 43, 7051–7056 RSC.
- S. Shirai, S. Iwata, T. Tani and S. Inagaki, J. Phys. Chem. A, 2011, 115, 7687–7699 CrossRef CAS PubMed.
- S. Shirai, S. Iwata, Y. Maegawa, T. Tani and S. Inagaki, J. Phys. Chem. A, 2012, 116, 10194–10202 CrossRef CAS PubMed.
- S. Shirai, Y. Kurashige and T. Yanai, J. Chem. Theory Comput., 2016, 12, 2366–2372 CrossRef CAS PubMed.
- Y. Numata, Y. Suzuki and I. Suzuka, J. Photochem. Photobiol., A, 2012, 237, 49–52 CrossRef CAS.
- K. Yamazaki, N. Niitsu, K. Nakamura, M. Kanno and H. Kono, J. Phys. Chem. A, 2012, 116, 11441–11450 CrossRef CAS PubMed.
- B. Saed and R. Omidyan, J. Phys. Chem. A, 2013, 117, 2499–2507 CrossRef CAS PubMed.
- M. J. Bertocchi, A. Bajpai, J. N. Moorthy and R. G. Weiss, J. Phys. Chem. A, 2017, 121, 458–470 CrossRef CAS PubMed.
- J. Merz, J. Fink, A. Friedrich, I. Krummenacher, H. H. Al Mamari, S. Lorenzen, M. Haehnel, A. Eichhorn, M. Moos, M. Holzapfel, H. Braunschweig, C. Lambert, A. Steffen, L. Ji and T. B. Marder, Chem.–Eur. J., 2017, 23, 13164–13180 CrossRef CAS.
- F. Lu, N. Kitamura, T. Takaya, K. Iwata, T. Nakanishi and Y. Kurashige, Phys. Chem. Chem. Phys., 2018, 20, 3258–3264 RSC.
- J. R. Platt, J. Chem. Phys., 1949, 17, 484–495 CrossRef CAS.
- J. Tanaka, Bull. Chem. Soc. Jpn., 1965, 38, 86–102 CrossRef CAS.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- C. Rullière, E. C. Colson and P. C. Roberge, Can. J. Chem., 1975, 53, 3269–3275 CrossRef.
- J. C. Rayez and C. Rulliere, Chem. Phys. Lett., 1979, 63, 289–292 CrossRef.
- I. B. Berlman, J. Phys. Chem., 1970, 74, 3085–3093 CrossRef CAS.
- I. B. Berlman, in Handbook of Fluorescence Spectra of Aromatic Molecules, ed. I. B. Berlman, Academic Press, 2nd edn, 1971, pp. 107–415, DOI:10.1016/B978-0-12-092656-5.50011-3.
- W. Sotoyama, H. Sato, M. Kinoshita, T. Takahashi, A. Matsuura, J. Kodama, N. Sawatari and H. Inoue, SID Symposium Digest of Technical Papers, 2003, 34, 1294–1297 CrossRef CAS.
- I. S. K. Kerkines, I. D. Petsalakis, G. Theodorakopoulos and W. Klopper, J. Chem. Phys., 2009, 131, 224315 CrossRef PubMed.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- E. K. U. Gross and W. Kohn, Adv. Quantum Chem., 1990, 21, 255–291 CrossRef.
- C. Jamorski, M. E. Casida and D. R. Salahub, J. Chem. Phys., 1996, 104, 5134–5147 CrossRef.
- M. Petersilka, U. J. Gossmann and E. K. U. Gross, Phys. Rev. Lett., 1996, 76, 1212–1215 CrossRef.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef.
- L. González, D. Escudero and L. Serrano-Andrés, ChemPhysChem, 2012, 13, 28–51 CrossRef CAS PubMed.
- A. D. Laurent, C. Adamo and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 14334–14356 RSC.
- Y. H. Park and B.-S. Cheong, Curr. Appl. Phys., 2006, 6, 700–705 CrossRef.
- E. L. Graef and J. B. L. Martins, J. Mol. Model., 2019, 25, 183 CrossRef.
- M. Parac and S. Grimme, Chem. Phys., 2003, 292, 11–21 CrossRef CAS.
- S. Grimme and M. Parac, ChemPhysChem, 2003, 4, 292–295 CrossRef CAS PubMed.
- Y.-L. Wang and G.-S. Wu, Int. J. Quantum Chem., 2008, 108, 430–439 CrossRef CAS.
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 3272–3277 CrossRef.
- N. Kuritz, T. Stein, R. Baer and L. Kronik, J. Chem. Theory Comput., 2011, 7, 2408–2415 CrossRef PubMed.
- R. M. Richard and J. M. Herbert, J. Chem. Theory Comput., 2011, 7, 1296–1306 CrossRef PubMed.
- M. Krykunov, S. Grimme and T. Ziegler, J. Chem. Theory Comput., 2012, 8, 4434–4440 CrossRef PubMed.
- B. Moore, H. Sun, N. Govind, K. Kowalski and J. Autschbach, J. Chem. Theory Comput., 2015, 11, 3305–3320 CrossRef PubMed.
- O. Christiansen, H. Koch and P. Jørgensen, Chem. Phys. Lett., 1995, 243, 409–418 CrossRef.
- H. Lischka, D. Nachtigallová, A. J. A. Aquino, P. G. Szalay, F. Plasser, F. B. C. Machado and M. Barbatti, Chem. Rev., 2018, 118, 7293–7361 CrossRef CAS PubMed.
- Y. Bito, N. Shida and T. Toru, Chem. Phys. Lett., 2000, 328, 310–315 CrossRef CAS.
- C.-H. Chin and S. H. Lin, Phys. Chem. Chem. Phys., 2016, 18, 14569–14579 RSC.
- H. Nakano, J. Chem. Phys., 1993, 99, 7983–7992 CrossRef CAS.
- K. Andersson, P. Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS.
- B. O. Roos, Adv. Chem. Phys., 1987, 69, 399–445 Search PubMed.
- A. Y. Freidzon, R. R. Valiev and A. A. Berezhnoy, RSC Adv., 2014, 4, 42054–42065 RSC.
- A. A. Granovsky, J. Chem. Phys., 2011, 134, 214113 CrossRef.
- P.-Å. Malmqvist, A. Rendell and B. O. Roos, J. Phys. Chem., 1990, 94, 5477–5482 CrossRef.
- A. Nenov, A. Giussani, B. P. Fingerhut, I. Rivalta, E. Dumont, S. Mukamel and M. Garavelli, Phys. Chem. Chem. Phys., 2015, 17, 30925–30936 RSC.
- F. Aquilante, T. B. Pedersen and R. Lindh, J. Chem. Phys., 2007, 126, 194106 CrossRef.
- F. Aquilante, P.-Å. Malmqvist, T. B. Pedersen, A. Ghosh and B. O. Roos, J. Chem. Theory Comput., 2008, 4, 694–702 CrossRef CAS.
- J. A. Noble, C. Aupetit, D. Descamps, S. Petit, A. Simon, J. Mascetti, N. Ben Amor and V. Blanchet, Phys. Chem. Chem. Phys., 2019, 21, 14111–14125 RSC.
- B. Shi, D. Nachtigallová, A. J. A. Aquino, F. B. C. Machado and H. Lischka, J. Chem. Phys., 2019, 150, 124302 CrossRef.
- S. Grimme and M. Waletzke, J. Chem. Phys., 1999, 111, 5645–5655 CrossRef CAS.
- B. Shi, D. Nachtigallová, A. J. A. Aquino, F. B. C. Machado and H. Lischka, Phys. Chem. Chem. Phys., 2019, 21, 9077–9088 RSC.
- H. Nakano, R. Uchiyama and K. Hirao, J. Comput. Chem., 2002, 23, 1166–1175 CrossRef CAS.
- J. Ivanic, J. Chem. Phys., 2003, 119, 9364–9376 CrossRef CAS.
- J. Ivanic, J. Chem. Phys., 2003, 119, 9377–9385 CrossRef CAS.
- L. Roskop and M. S. Gordon, J. Chem. Phys., 2011, 135, 044101 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- H. Koch and P. Jørgensen, J. Chem. Phys., 1990, 93, 3333–3344 CrossRef CAS.
- J. F. Stanton and R. J. Bartlett, J. Chem. Phys., 1993, 98, 7029–7039 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- M. Baba, M. Saitoh, Y. Kowaka, K. Taguma, K. Yoshida, Y. Semba, S. Kasahara, T. Yamanaka, Y. Ohshima, Y.-C. Hsu and S. H. Lin, J. Chem. Phys., 2009, 131, 224318 CrossRef PubMed.
- X. Feng, H. Tomiyasu, J.-Y. Hu, X. Wei, C. Redshaw, M. R. J. Elsegood, L. Horsburgh, S. J. Teat and T. Yamato, J. Org. Chem., 2015, 80, 10973–10978 CrossRef CAS.
- Y. L. Qiao, J. Zhang, W. Xu and D. B. Zhu, Tetrahedron, 2011, 67, 3395–3405 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- M. S. Gordon and M. W. Schmidt, in Theory and applications of computational chemistry, ed. C. E. Dykstra, G. Frenking, K. S. Kim and G. E. Scuseria, Elsevier, Amsterdam, 2005, pp. 1167–1189, DOI:10.1016/B978-044451719-7/50084-6.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. S. Becker, I. S. Singh and E. A. Jackson, J. Chem. Phys., 1963, 38, 2144–2171 CrossRef CAS.
- E. A. Mangle and M. R. Topp, J. Phys. Chem., 1986, 90, 802–807 CrossRef CAS.
- C. M. Jones and S. A. Asher, J. Chem. Phys., 1988, 89, 2649–2661 CrossRef CAS.
- V. de Halleux, J. P. Calbert, P. Brocorens, J. Cornil, J. P. Declercq, J. L. Bredas and Y. Geerts, Adv. Funct. Mater., 2004, 14, 649–659 CrossRef CAS.
- N. S. Ham and K. Ruedenberg, J. Chem. Phys., 1956, 25, 13–26 CrossRef CAS.
- M. K. Roos, S. Reiter and R. de Vivie-Riedle, Chem. Phys., 2018, 515, 586–595 CrossRef CAS.
- Y. Niko, S. Kawauchi, S. Otsu, K. Tokumaru and G.-i. Konishi, J. Org. Chem., 2013, 78, 3196–3207 CrossRef CAS.
- E. B. Guidez and C. M. Aikens, J. Phys. Chem. C, 2013, 117, 21466–21475 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Natural orbitals from MCSCF wave functions (Fig. S1–S3); total energies and MCSCF excitation energies (Table S1); Hartree–Fock orbitals (Fig. S4). See DOI: 10.1039/c9ra10483f |
| This journal is © The Royal Society of Chemistry 2020 |