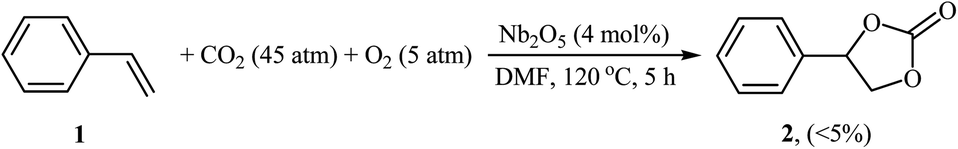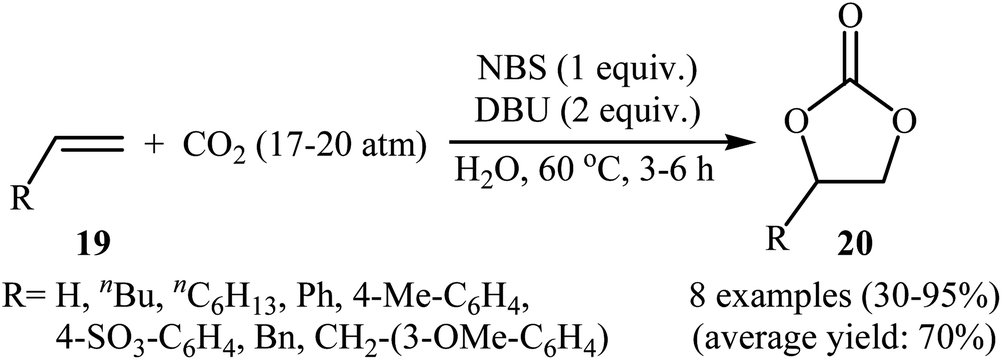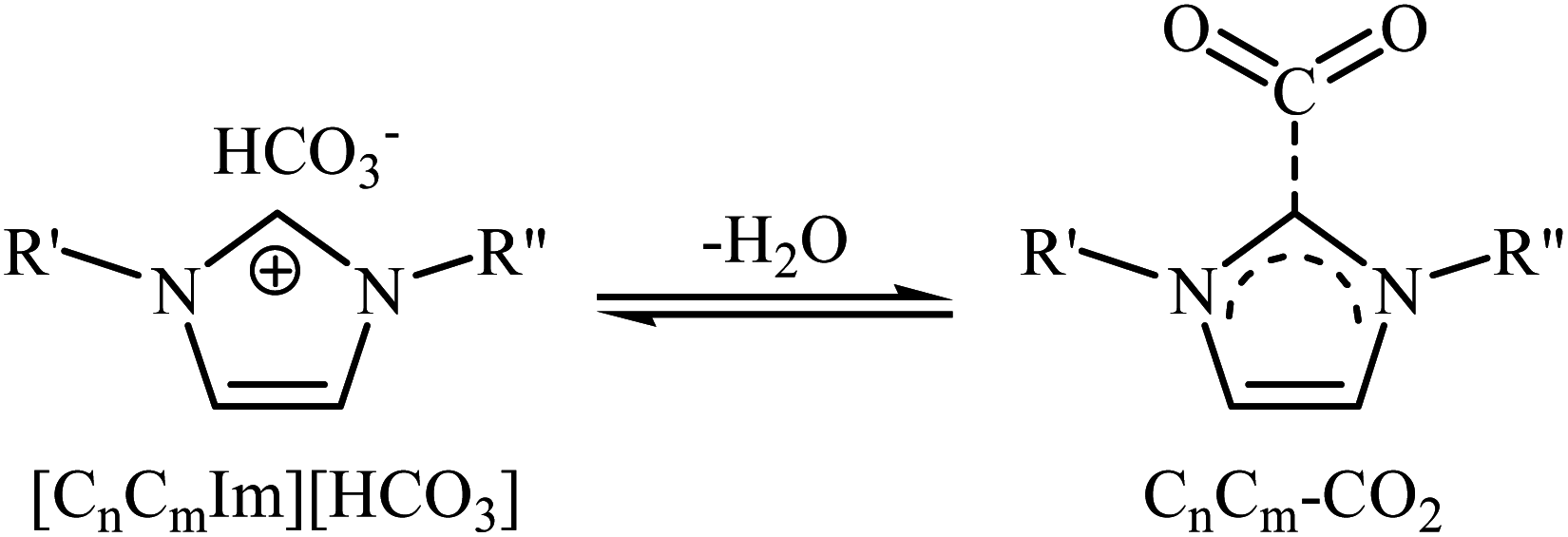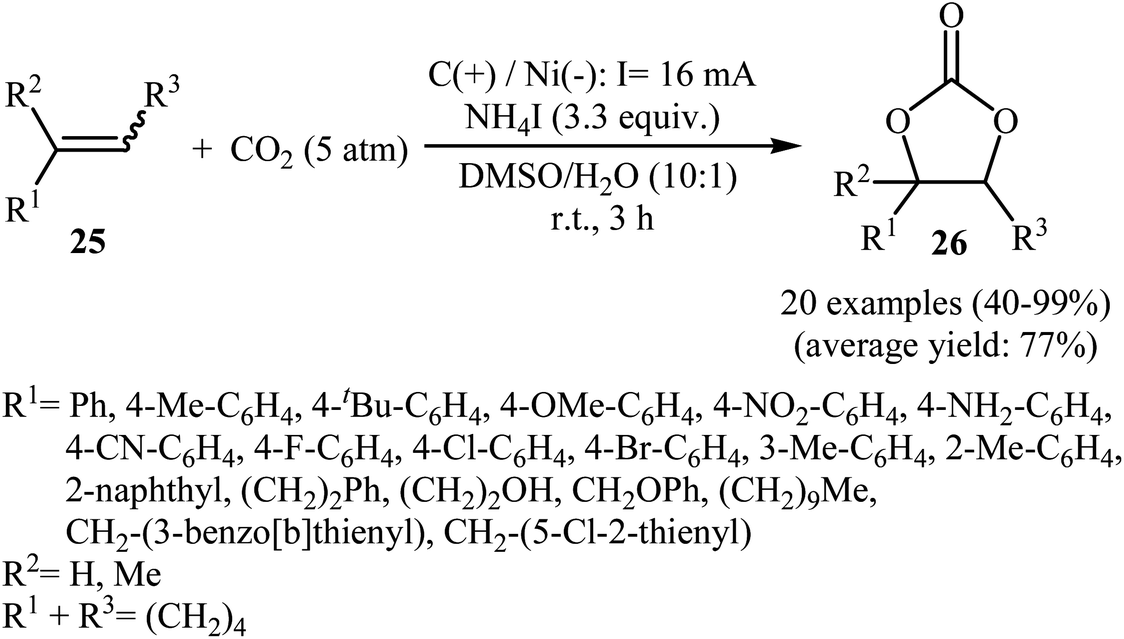DOI:
10.1039/C9RA10755J
(Review Article)
RSC Adv., 2020,
10, 9103-9115
Oxidative carboxylation of olefins with CO2: environmentally benign access to five-membered cyclic carbonates
Received
20th December 2019
, Accepted 18th February 2020
First published on 3rd March 2020
Abstract
The purpose of this focus review is to provide a comprehensive overview of the direct synthesis of five-membered cyclic carbonates via oxidative carboxylation of the corresponding olefins and carbon dioxide with particular attention on the mechanistic features of the reactions. The review is divided into two main sections. The first section is a discussion of the single-step reactions, while the second consists of an overview of one-pot, two-step sequential reactions.
1. Introduction
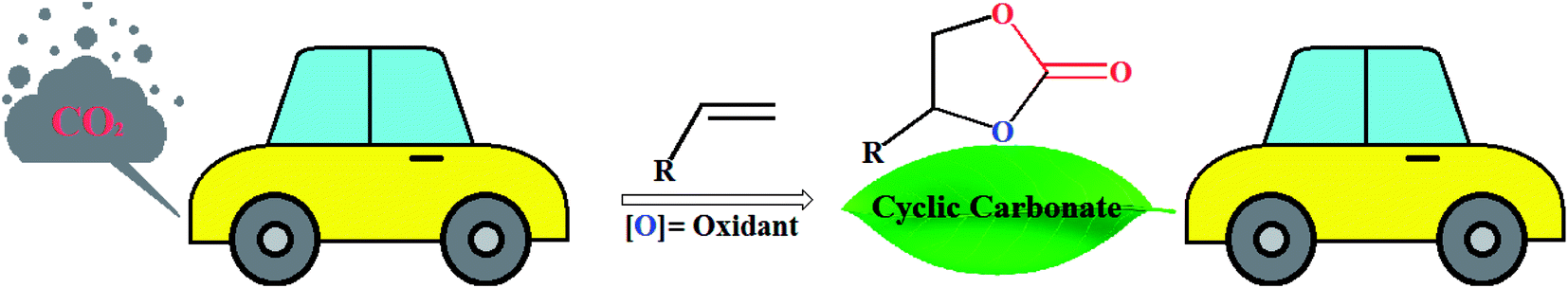
The use of carbon dioxide (CO2) as a building block for the synthesis of high-value fine chemicals is receiving growing interest in the context of sustainable and green chemistry because it is a cheap, plentiful, non-toxic, non-flammable and renewable compound.1–10 However, due to its high thermodynamic stability and chemical inertness, the efficient fixation of this greenhouse gas into chemical products is a challenging issue as decarboxylation reactions can readily occur.11 A strategy to overcome the low reactivity of CO2 is based on the reactions with high free energy substrates.12 The cycloaddition of CO2 and energy-rich epoxides stands as one of the most attractive and efficient strategies for CO2 conversion and utilization.13 This reaction is exothermic and generates five-membered cyclic carbonates, which are extremely useful solvents in the chemical industries, electrolytes in Li-ion batteries, and intermediates in various organic and polymer synthesis.14 Interestingly, this synthetic strategy is one of the few industrially relevant reactions utilizing CO2.15 Nevertheless, most epoxides are toxic and mutagenic and there is evidence that they cause cancer.16 Recently, the direct synthesis of cyclic carbonates from alkenes and CO2, so-called one-pot “oxidative carboxylation”, have attracted considerable attention owing to the lower cost and greater availability of alkenes as compared to the corresponding epoxides.17 The strategies for this appealing synthetic approach can be typically classified into two categories: (i) one-pot, two-step sequential reaction consisting of oxidation followed by carboxylation; and (ii) direct oxidation and carboxylation in just a single-step.18 In 2011, Sun and colleagues highlighted these reactions in their interesting review paper entitled “Direct synthetic processes for cyclic carbonates from olefins and CO2”.19 In connection with our recent review articles on CO2 fixation reactions,20 herein, we will try to provide a comprehensive and updated overview of recent developments in the synthesis of five-membered cyclic carbonates through the oxidative carboxylation of olefins with CO2 (Fig. 1). The review is divided into two major sections. The first section focuses exclusively on single-step conversion of alkenes to cyclic carbonates, while the second covers stepwise processes. The sections were classified based on the type of catalysts (e.g., metal-catalyzed reactions, metal-free reactions, electrocatalytic reactions) which may help a good understanding of the reaction mechanisms.
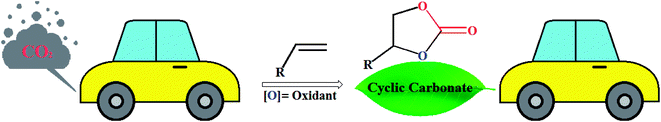 |
| | Fig. 1 Oxidative carboxylation of olefins with CO2. | |
2. Single-step approaches
2.1. Metal-catalyzed reactions
Synthesis of cyclic carbonates through the metal-catalyzed oxidative carboxylation of olefins has experienced considerable growth in recent years as evidenced by the number of published articles. One of the earliest reports describes the usefulness of metal catalysts for this conversion was published by Aresta and colleagues in 2000,21 who showed that the reaction of styrene 1 with CO2 (45 atm) in the presence of molecular oxygen as an oxidant and a catalytic amount of Nb2O5 in DMF at 120 °C afforded the corresponding styrene carbonate 2 in only 5% along with styrene oxide, benzaldehyde, and benzoic acid side products (Scheme 1). Although only one poor yield example was disclosed, this paper represents the first example of a rare-earth metal-catalyzed synthesis of carbonates from olefins and CO2. Noteworthy, other metal oxides such as ZnO, Fe2O3, MoO3, Ta2O5, La2O5, and V2O5 as well as silica, alumina and molecular sieves were also found to promote this CO2 fixation reaction; albeit, in lower yields. It should also be mentioned that previously the same authors demonstrated the usefulness of rhodium catalysts for this reaction.22 The authors suggested that this reaction may proceeds through the formation of styrene oxide intermediate via oxidation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond with O2 followed by cycloaddition with CO2. Subsequently, they found that the reaction temperature had a significant impact on the success of this transformation.23 Slightly increasing the temperature considerably increased the reaction rate. For example, performing the coupling of CO2 with styrene oxide at 135 °C, in the presence of above-mentioned catalytic system, gave the styrene carbonate in up to 80% yield, while for reaction at 110 °C, the desired product was obtained in only 11.2% yield. A similar example of the oxidative carboxylation of olefins by a metal catalyst was disclosed by Srivastava, Srinivas, and Ratnasamy in 2003, when they reported about the use of titanosilicate molecular sieve (TS-1; Si/Ti = 36) and DMAP combination to promote the reaction of olefins (i.e., allyl chloride, styrene) with CO2 (6.9 atm) in the presence of H2O2 as an epoxidizing agent in acetone.24 Although the reactions were carried out under a relatively mild condition, the product yields were still poor. In 2005, Arai's research team improved the efficiency of this reaction in the terms of yield (42%), temperature (80 °C) and time (4 h) by utilizing Au/SiO2–ZnBr2–TBAB (tetra-n-butylammonium bromide) combination as a catalytic system in the absence of any organic solvent.25 They suggested that Au/SiO2 was active for the epoxidation of styrene, and ZnBr2 and TBAB cooperatively catalyzed the subsequent cycloaddition of CO2 to the in situ generated epoxide.
C bond with O2 followed by cycloaddition with CO2. Subsequently, they found that the reaction temperature had a significant impact on the success of this transformation.23 Slightly increasing the temperature considerably increased the reaction rate. For example, performing the coupling of CO2 with styrene oxide at 135 °C, in the presence of above-mentioned catalytic system, gave the styrene carbonate in up to 80% yield, while for reaction at 110 °C, the desired product was obtained in only 11.2% yield. A similar example of the oxidative carboxylation of olefins by a metal catalyst was disclosed by Srivastava, Srinivas, and Ratnasamy in 2003, when they reported about the use of titanosilicate molecular sieve (TS-1; Si/Ti = 36) and DMAP combination to promote the reaction of olefins (i.e., allyl chloride, styrene) with CO2 (6.9 atm) in the presence of H2O2 as an epoxidizing agent in acetone.24 Although the reactions were carried out under a relatively mild condition, the product yields were still poor. In 2005, Arai's research team improved the efficiency of this reaction in the terms of yield (42%), temperature (80 °C) and time (4 h) by utilizing Au/SiO2–ZnBr2–TBAB (tetra-n-butylammonium bromide) combination as a catalytic system in the absence of any organic solvent.25 They suggested that Au/SiO2 was active for the epoxidation of styrene, and ZnBr2 and TBAB cooperatively catalyzed the subsequent cycloaddition of CO2 to the in situ generated epoxide.
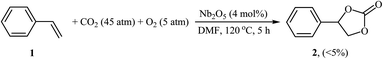 |
| | Scheme 1 Aresta's synthesis of styrene carbonate 2. | |
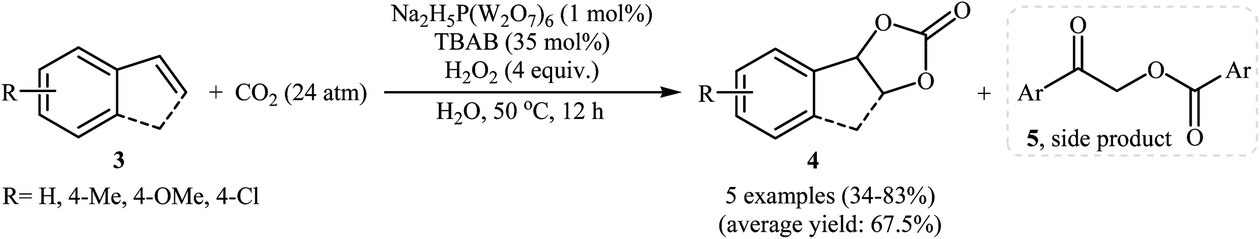
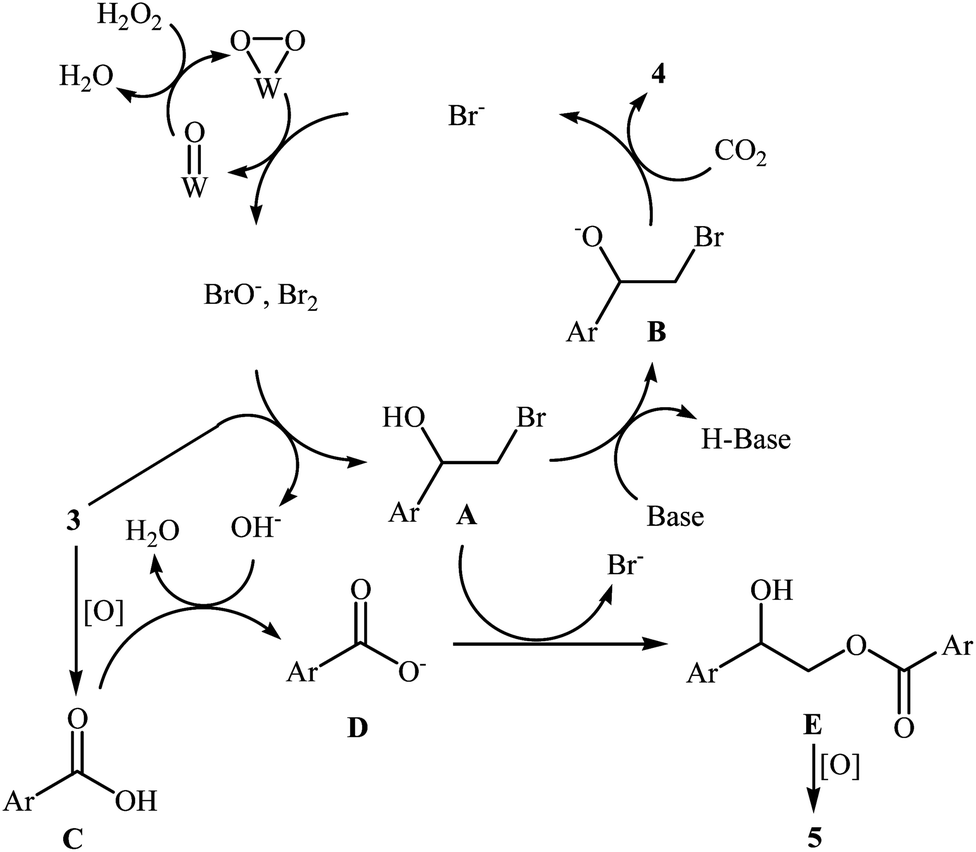
With the aim of designing a general and practical protocol to five-membered cyclic carbonates through metal-catalyzed oxidative carboxylation of olefins, the group of L.-N. He was able to reveal that a series of functionalized carbonates 4 could be obtained in moderate to high yields from the reaction of various olefins 3 with CO2 (24 atm) in water employing a binary catalyst system composed of sodium phosphotungstate [Na2H5P(W2O7)6] and TBAB with H2O2 as oxidant and NaHCO3 as base (Scheme 2).26 Interestingly, both terminal and internal olefins were compatible with the reaction conditions. However, the procedure was unsuccessful for cyclohexene. A possible reaction mechanism for this transformation involves the initial bromohydroxylation of olefin 3 by TBAB and H2O2 in the presence of Na2H5P(W2O7)6 to form bromohydrin intermediate A. Then, deprotonation of this intermediate by a base, followed by a cycloaddition reaction with CO2 affords the desired product 4 (Scheme 3). On the other hand, carboxylic acid C could be generated under the reaction conditions, which underwent deprotonation by either a base or hydroxyl anion generated in situ to form carboxylate anion D. This intermediate further reacted with intermediate A to form undesired side product 5 through the intermediate E. In a related investigation, Sun and co-workers also found that styrene was convert to the styrene carbonate in 38% yield, via oxidative carboxylation using nano-gold supported R201 resin as a catalyst and tert-n-butyl hydroperoxide (TBHP) as an oxidant at 80 °C.27 In their subsequent study, they reinvestigated the same reaction utilizing a three-component catalyst composed of Au/Fe(OH)3, ZnBr2, and Bu4NBr.28 The target styrene carbonate was obtained in 53% yield.
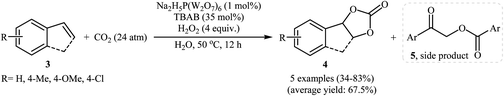 |
| | Scheme 2 Na2H5P(W2O7)6-catalyzed oxidative carboxylation of olefins 3 and CO2. | |
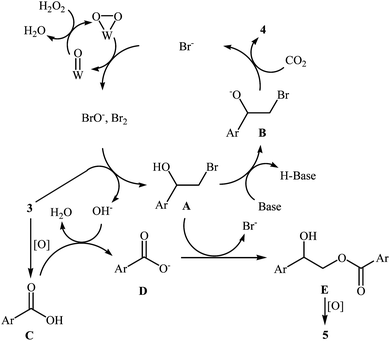 |
| | Scheme 3 Mechanism that accounts for the formation of cyclic carbonates 4. | |
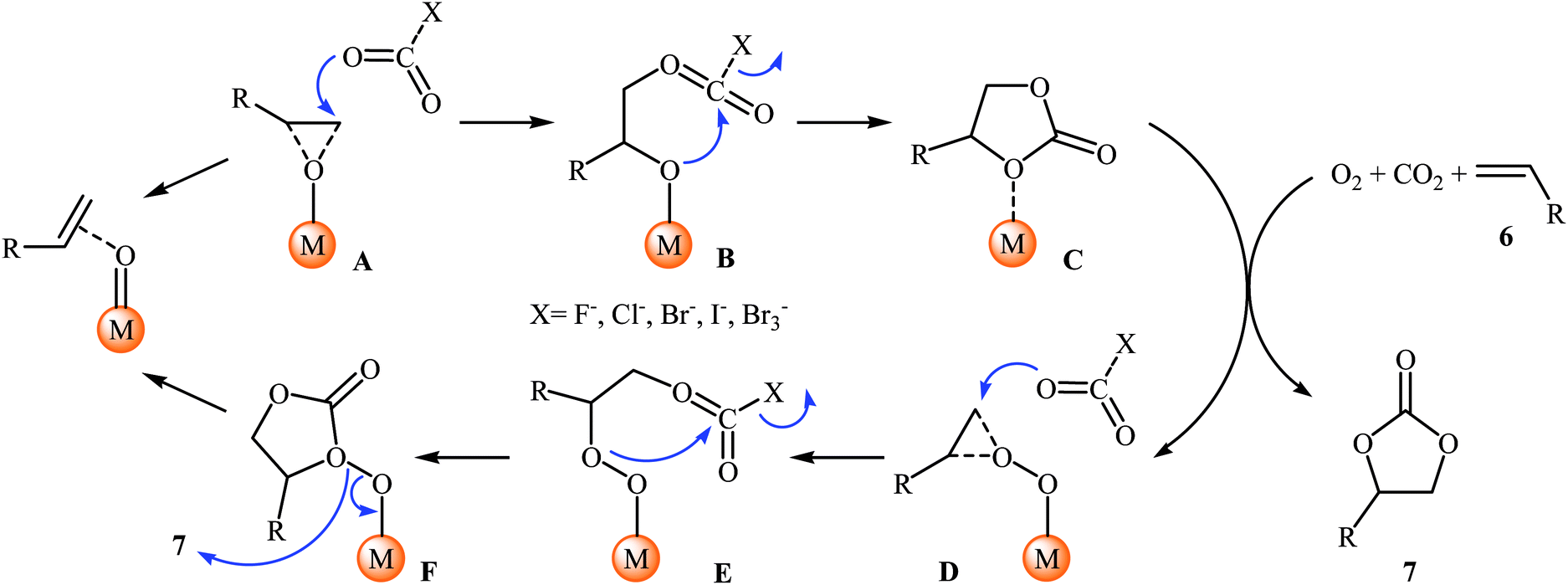
Shortly afterwards, Bai and Jing reported their results on the Ru-catalyzed three-component reaction of olefins 6, CO2, and O2.29 Optimal condition for this oxidative carboxylation was the combination of 4 mol% of dioxo(tetraphenylporphyrinato)ruthenium(VI) [Ru(TPP)(O)2], 8 mol% of phenyltrimethylammonium tribromide (PTAT) or TBAI, and DCM as solvent, at 30 °C. The cyclization reaction proceeded cleanly under milder conditions than those described in previous reports, and the target carbonates 7 were obtained in modest to good yields (Scheme 4). Noteworthy, under these reaction conditions, 100% selectivity was observed for styrene, although bromination addition species of other olefins were obtained as by-products. Interestingly, and in contrast with the results obtained by group of He, cyclohexene was found to be suitable substrate in this procedure. A plausible reaction mechanism proposed by the authors involves the initial formation of the epoxide intermediate A by the transfer of an oxygen atom from the catalyst to starting olefin 6. Next, ring opening of the epoxide through nucleophilic attack by CO2 which was activated by quaternary onium salt affords intermediate B that undergoes intramolecular cyclization to give the new intermediate C. This intermediate reacts with oxygen and olefin to form the target cyclic carbonate 7 and the intermediate D, which after nucleophilic attack by active CO2 affords a new intermediate E. Finally, this intermediate undergoes intramolecular cyclization to yield another molecule of target compound 7 and regenerate the oxometalloporphyrin catalyst through the intermediate F (Scheme 5).
 |
| | Scheme 4 Jing's synthesis of cyclic carbonates 7. | |
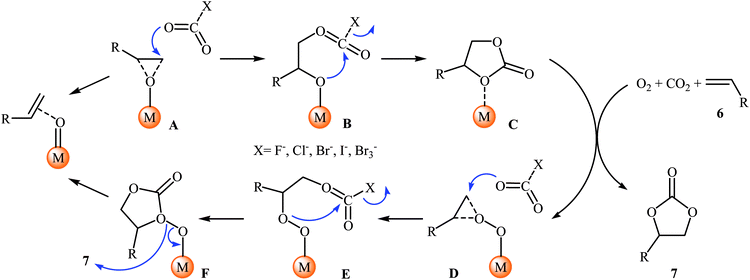 |
| | Scheme 5 Proposed mechanism for Rh-catalyzed three-component reaction of olefins 6, CO2, and O2. | |
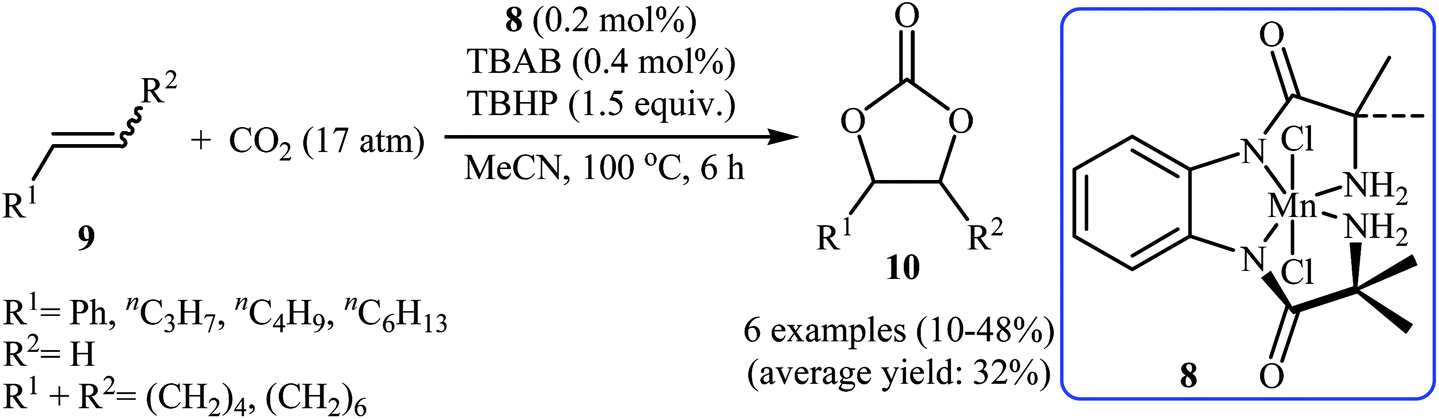
In 2015, Ghosh's research team developed a hexacoordinate manganese(III)-complex 8 catalyzed direct synthesis of cyclic carbonates 10 via oxidative carboxylation of the corresponding olefins 9 in the presence of TBAB as a co-catalyst.30 Screening of various oxidants such as H2O2, PhIO, TBHP, MeCO3H, Py-N-oxide, and m-CPBA; 1.5 equiv. TBHP was found to be ideal for each equivalent of olefin at 100 °C. The reactions were carried out in dry acetonitrile under 17 atm pressure of CO2 and provided the target carbonates 10 with yield ranging from 10% to 48% along with considerable amounts of undesired epoxides (Scheme 6). Of note, the above-mentioned temperature was crucial for this reaction, both increase and decrease of the temperature was decreased the yield of carbonate products in favor of the corresponding epoxides. Based on a series of control experiments, the authors speculated that the oxygen transfer takes place via an active metal-oxo species (Scheme 7).
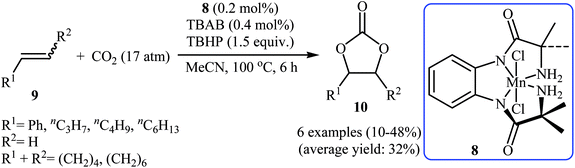 |
| | Scheme 6 Oxidative carboxylation of olefins 9 and CO2 catalyzed by a hexacoordinate manganese(III)-complex. | |
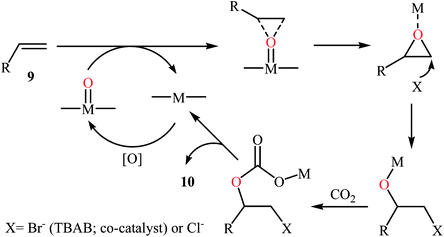 |
| | Scheme 7 Mechanistic proposal for the reaction in Scheme 6. | |
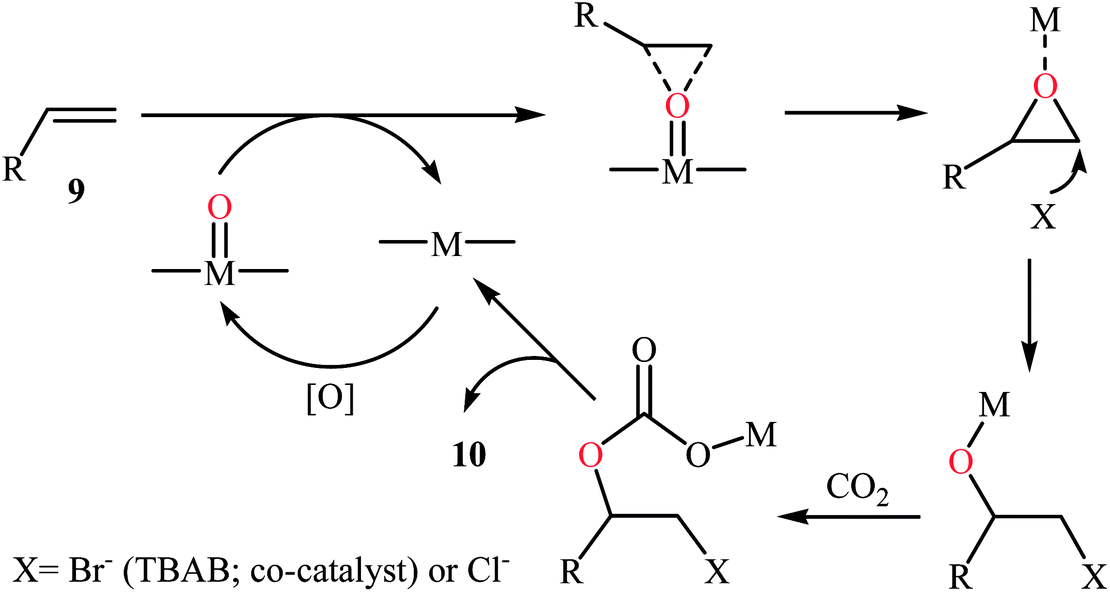
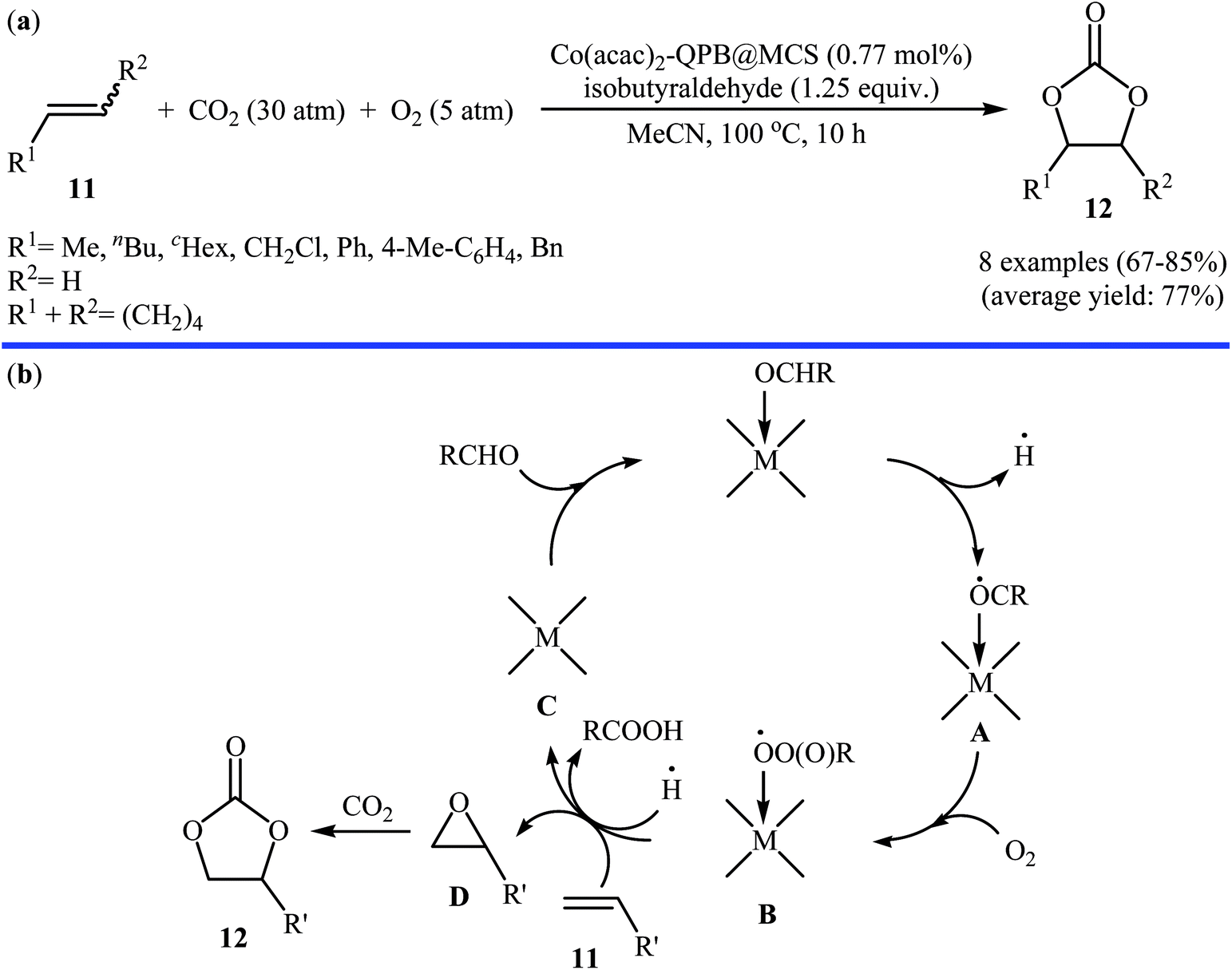
Concurrently, Jain and co-workers designed and synthesized a magnetic chitosan immobilized cobalt(II) acetylacetonate and triphenylphosphonium bromide nano-composite [Co(acac)2-QPB@MCS] via a three-step procedure through coating of in situ generated Fe3O4 nanoparticles with chitosan followed by immobilization of Co(acac)2 on the magnetic chitosan (MCS) surface and subsequent functionalization of generated Co(acac)2@MCS with 3-bromopropyltrimethoxysilane (BPTMS) and triphenylphosphine (Scheme 8).31 The prepared composite was well characterized by various techniques, including TEM, HRTEM, XPS, ICP-AES, FTIR and found that has spheroidal morphology with average size of 50–100 nm and 1.8 wt% loading of cobalt. The nanocomposite system was successfully utilized as an efficient catalyst for high yielding synthesis of various cyclic carbonates 12 through three-component reaction of olefins 11, CO2 (30 atm), and O2 (5 atm) in the presence of isobutyraldehyde as a sacrificial reductant in MeCN (Scheme 9a). Interestingly, the catalyst could be easily separated from the reaction mixture by means of an external magnet and reused several times without the loss of activity. Furthermore, leaching of cobalt species was not detected by ICP-AES analysis. The authors investigated the reaction mechanism by performing the reaction of styrene in the absence of CO2 and monitored the progress of this reaction by GC, which confirmed the formation of styrene epoxide as the major product. Thus, they suggested that this reaction starts with the generation of isopropylacyl radical from isobutyraldehyde via an electron transfer to the cobalt(II) complex which then undergoes addition with oxygen to give oxometal-complex radical and then peroxide-metal-complex radical. Next, peroxo–metal intermediate reacts with olefin to give styrene oxide and benzaldehyde as the by-product. Finally, the styrene oxide intermediate further reacts with CO2 to give corresponding cyclic carbonate (Scheme 9b).
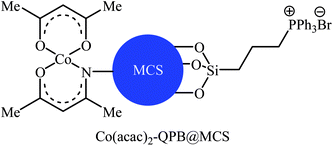 |
| | Scheme 8 Chemical structure of Co(acac)2-QPB@MCS. | |
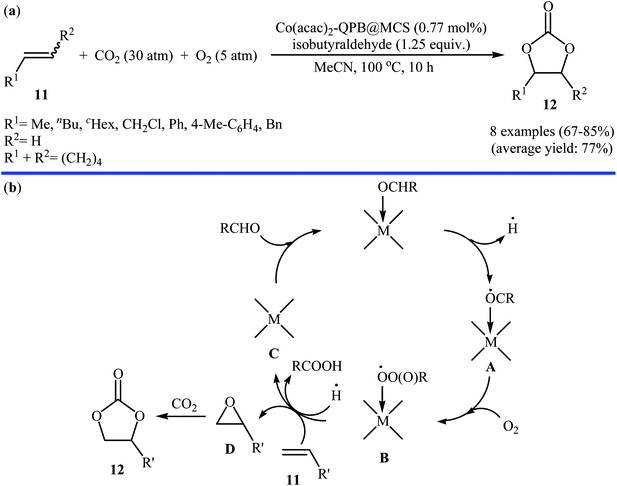 |
| | Scheme 9 (a) Synthesis of cyclic carbonates 12 from olefins 11 and CO2 catalyzed by Co(acac)2-QPB@MCS; (b) proposed mechanism for the formation of cyclic carbonates 12. | |
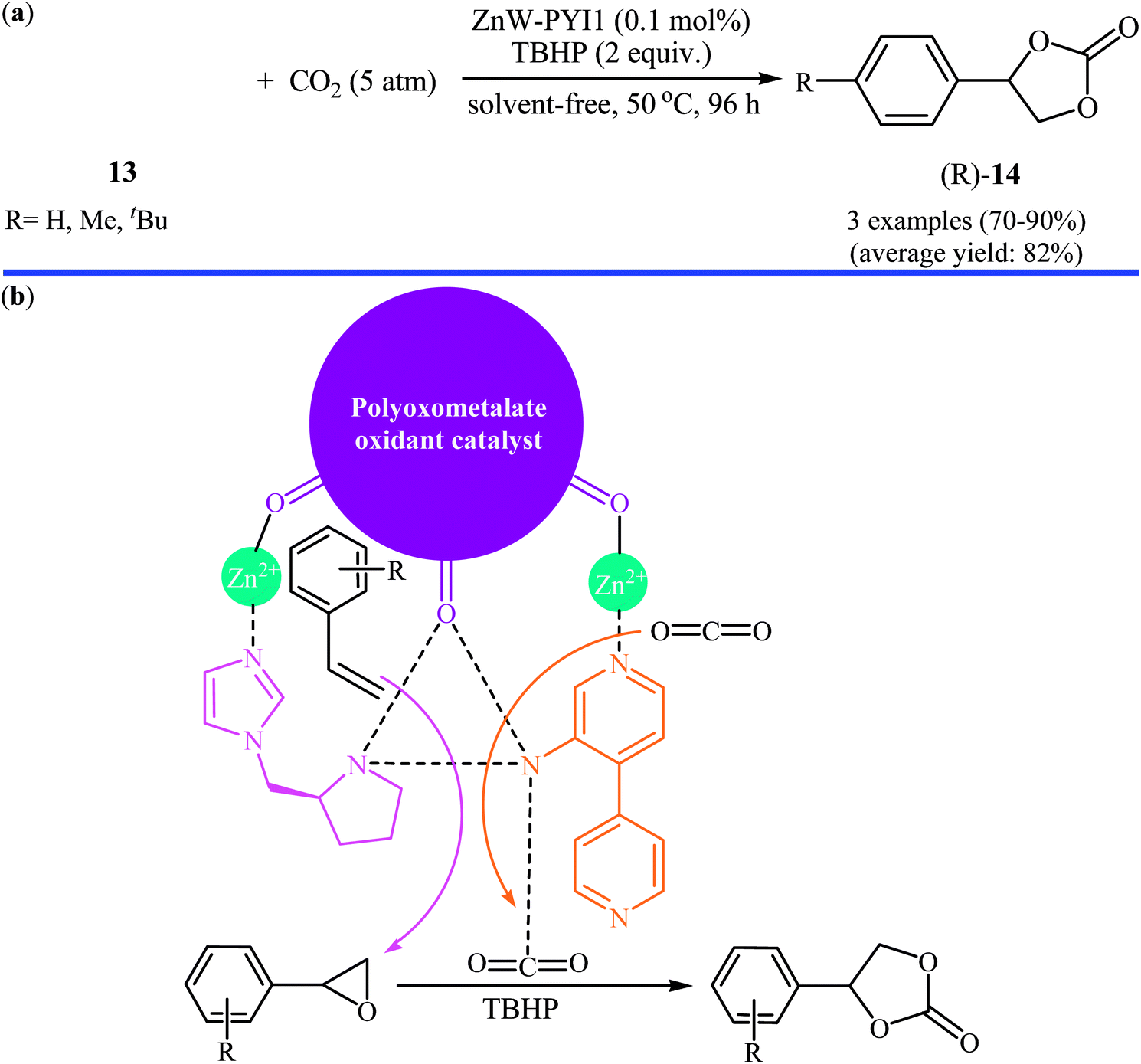
In the same year, Han and Qi along with their co-workers used Keggin-type [ZnW12O40]6− anion and the chiral L- and D-pyrrolidin-2-ylimidazole (PYI) ligands for the synthesis of polyoxometalate-based metal–organic frameworks denoted ZnW-PYI1 and ZnW-PYI2, respectively.32 The oxidative carboxylation of an small series of styrene derivatives 13 in the presence of ZnW-PYI1/TBHP combination proceeded smoothly at 50 °C and afforded the corresponding (R)-aryl(ethylene carbonates) 14 in good to excellent yields with moderate to high enantioselectivity (Scheme 10a). Cinnamaldehyde did not work well in the reaction and therefore no other internal olefins were examined in the protocol. It was found that the catalyst could be recovered from the reaction suspension by a simple filtration and reused for at least three successive times with moderate loss of activity and without tangible decrease in selectivity. It is worth mentioning that the ZnW-PYI2 solids exhibited similar catalytic activities but gave products with opposite chiralities. The authors suggested that this tandem reaction proceeds through the asymmetric epoxidation of the styrene derivative by the synergistic action of the ZnW anion and PYI ligand in the presence of TBHP. The epoxide intermediate can then react with activated CO2 to ultimately yield the cyclic carbonate (Scheme 10b).
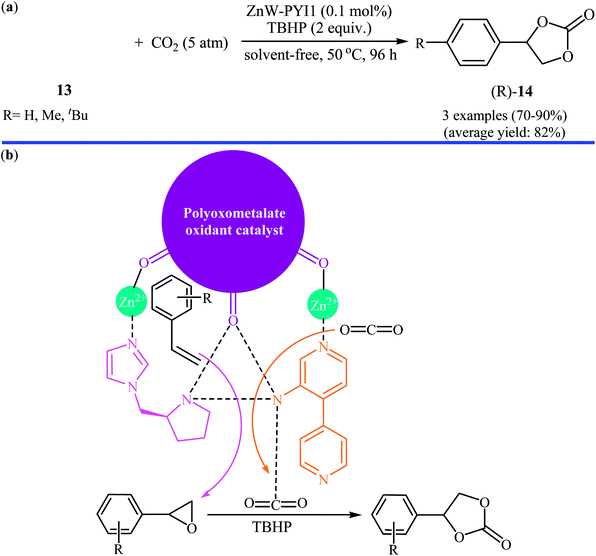 |
| | Scheme 10 (a) ZnW-PYI1-catalyzed oxidative carboxylation of styrene derivatives 13; (b) proposed mechanism for the formation of aryl(ethylene carbonates) 14. | |
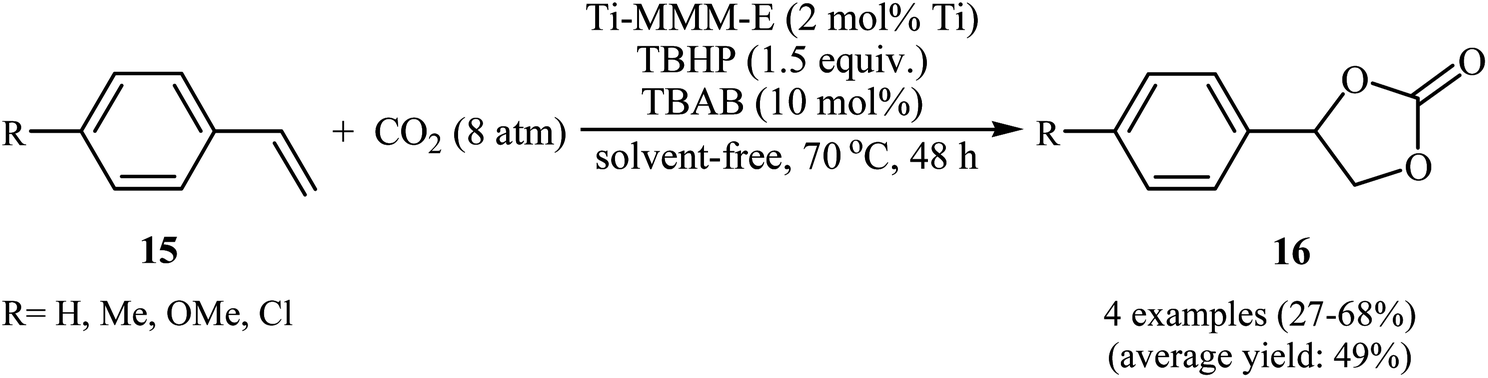
Not long after, the group of Kholdeeva prepared a mesoporous titanium–silicate (Ti-MMM-E) by evaporation-induced self-assembly (EISA) methodology in EtOH/H2O/HCl solution using tetraethoxy titanium (TEOT) modified with acetylacetone as titanium precursor, tetraethyl orthosilicate (TEOT) as silica precursor and cetyltrimethylammonium bromide (CTAB) as template.33 The catalytic activity of this mesoporous material was evaluated in the oxidative carboxylation of aromatic terminal olefins 15 and CO2 (8 atm) in the presence of TBHP and TBAB at 70 °C under solvent-free conditions (Scheme 11). The obtained results from this study revealed that the expected styrene carbonates 16 were formed in moderate to good yields ranging from 27% to 64%. However, both cyclic and acyclic aliphatic olefins were incompatible in this reaction.
 |
| | Scheme 11 Solvent-free synthesis of cyclic carbonates 16 by a Ti-containing catalyst. | |
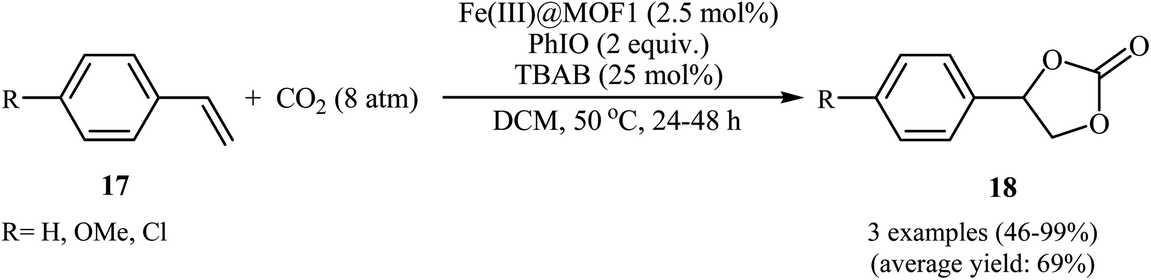
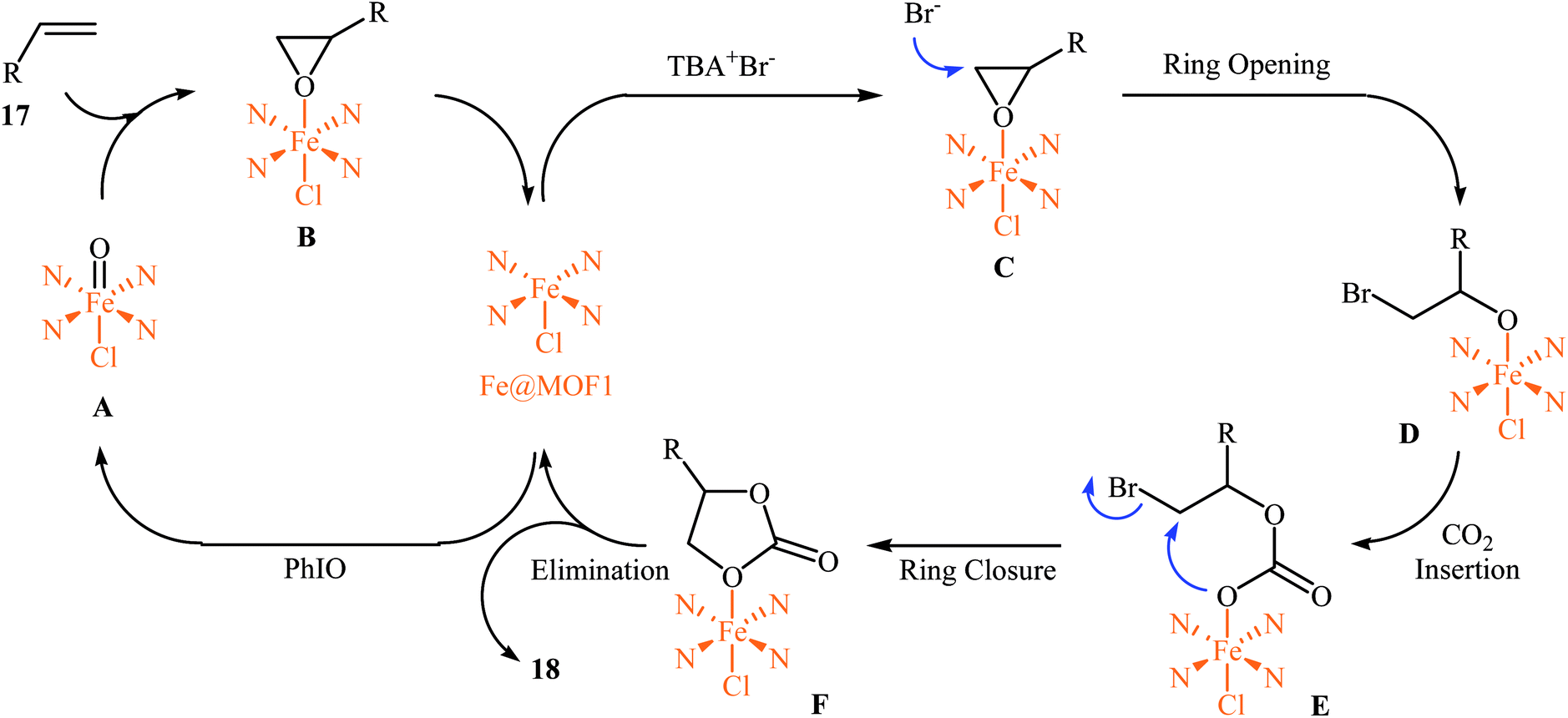
In 2018, Gándara and Nguyen designed and synthesized three new 3D lanthanide-based metal–organic frameworks, [Nd2(BIPA-TC)1.5]·8H2O, [Eu-(H2BIPA-TC)(BIPA-TC)0.5·8H2O][NH2(CH3)2], and [Tb-(H2BIPA-TC)(BIPA-TC)0.5·6H2O][NH2(CH3)2] (namely MOF-590, MOF-591, and MOF-592, respectively) from the corresponding metal(III) nitrate hydrates and benzoimidephenanthroline tetracarboxylic acid (H4BIPA-TC), a tetratopic linker, under solvothermal conditions.34 Compared to other MOFs, MOF-590 was found to be more effective catalyst for the oxidative carboxylation of olefins and CO2. Thus, in the presence of only 0.18 mol% of this MOF, 8 mol% of TBAB, and 1.9 equiv. of TBHP under atmospheric pressure of CO2 and solvent-free condition, styrene was converted to the styrene carbonate in 87% yield within 10 h. The recycling test established that this MOF catalyst could be recovered and reused over five reaction cycles with no remarkable loss in activity. In addition, MOF-592 was demonstrated more effective adsorbent for the separation of CO2 from a gas mixture of CO2 and N2. The same authors also demonstrated the construction of styrene carbonate in high yield (80%) utilizing zirconium-based metal–organic framework (Zr6O4(OH)4(H2CPB)1.5(CH3CO)6; denoted MOF-892) as a catalyst under a similar strategy.35 Inspired by these works, Nagaraja and co-workers synthesized a novel Mn(II)-porphyrin metal–organic framework, [{Mn2(TCPP)·2H2O}·DMF]n (MOF1) through the reaction of Mn(NO3)2·6H2O and H4TCPP ligand under the solvothermal conditions.36 Single crystal X-ray structural determination revealed that MOF1 crystallizes in the monoclinic crystal system with a centrosymmetric P21/n space group and features a 3D architecture constituted by binuclear Mn(II) nodes and non-metallated porphyrin ligand. Interestingly, the non-metallated nature of porphyrin ring was exploited for the implantation of Lewis acidic Fe(III) ions to generate Fe(III)@MOF1 material. The catalytic activity of this hybrid system was tested for oxidative carboxylation of styrene derivatives 17 and CO2 (8 atm) in the presence of TBAB as a co-catalyst and PhIO as an oxidant in DCM at 50 °C. With a low catalyst loading (2.5 mol%), both electron-rich and electron-poor styrenes converted into the corresponding carbonates 18 in moderate to high yields (Scheme 12). It has been shown that the catalyst can be recycled and reused up to three cycles with almost no loss of activity and retaining of the structural rigidity. The authors proposed mechanism for this transformation is depicted in Scheme 13.
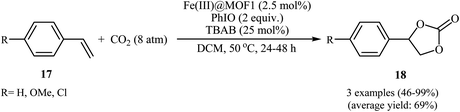 |
| | Scheme 12 Nagaraja's synthesis of styrene carbonates 18. | |
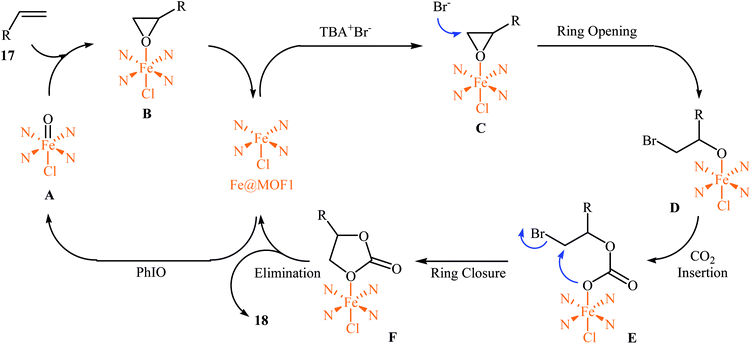 |
| | Scheme 13 Mechanism that accounts for the formation of carbonates 18. | |
2.2. Metal-free reactions
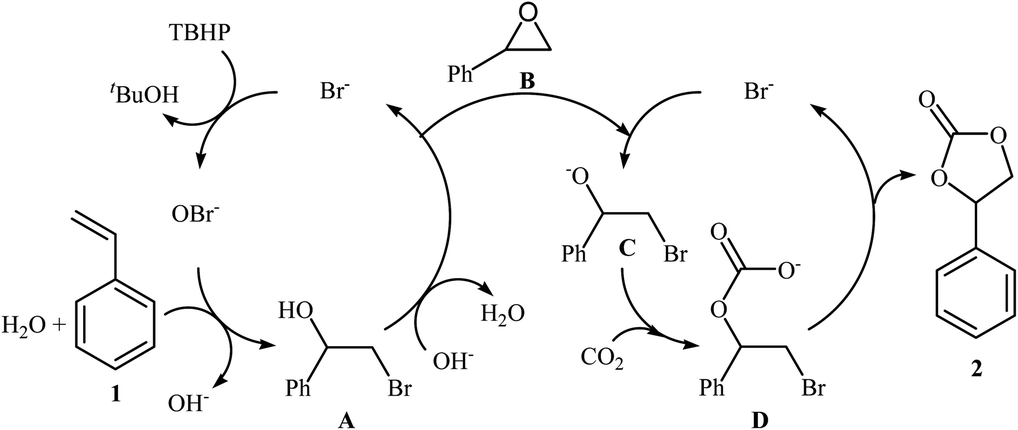
The direct metal-free oxidative carboxylation of olefins and CO2 provides a very valuable approach to a wide range of five-membered cyclic carbonates. This process generally rely on the use of halogen-containing catalysts/promoters to stabilize the ring-opening intermediates. One of the earliest mention of the synthesis of carbonates from olefins and CO2 under metal-free conditions can be found in a 2004 paper by Arai et al.37,38 although only one low-yielding example was described. Considering the catalyst, oxidant, temperature and CO2 pressure, the optimized conditions of this CO2 fixation reaction involved using TBAB as a catalyst and aq TBHP as an oxidant in supercritical CO2 at 80 °C. Under the optimized conditions, styrene carbonate was produced in 33% yield from the corresponding olefin within 6 h. A possible process for the formation of compound 2 should involve the formation of hypobromite OBr− via the oxidation of Br− with TBHP, followed by bromination of the styrene 1 in the presence of H2O to give the bromohydrin intermediate A, which after hydrobromination in the presence of in situ generated OH− converts to the epoxide B. Next, ring opening of the epoxide through nucleophilic attack by Br− affords an oxy anion species C. Finally, nucleophilic attack of this intermediate to the carbon atom in CO2 leads to the carbonate salt intermediate D that cyclize to the cyclic carbonate 2 (Scheme 14). In 2013, Balzhinimaev and co-workers reinvestigated this reaction by performing the process at room temperature under 8 atm pressure of CO2 in DCM.39 However, only trace amounts of the desired carbonate were obtained.
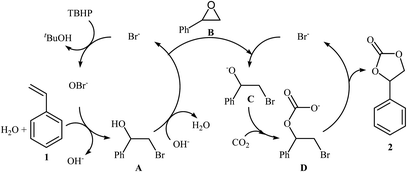 |
| | Scheme 14 Synthesis of styrene carbonate 2 from styrene 1 and CO2 with TBHP in the presence of TBAB. | |
Two years later, Eghbali and Li developed an elegant and efficient method for the synthesis of carbonates by the reaction of corresponding olefins with CO2 in the most environmentally friendly solvent, water.40 Thus, by using N-bromosuccinimide (NBS) together with 1,8-diazabicyclo[5.4.0]undecenc-7-ene (DBU) in neat water, the oxidative carboxylation of various terminal olefins 19 with CO2 (17–20 atm) afforded the corresponding five-membered cyclic carbonates 20 in moderate to excellent yields (Scheme 15). Of note, ethylene was also successfully served as a convenient substrate in this synthetic procedure. The results showed that the presence of DBU was crucial for the success of this reaction. Replacing DBU with some other bases (e.g., DMAP, DABCO, Hunig's base, Et3N, pyridine, 1-methylimidazole, N,N-dimethylaniline, N-methyl diphenylamine, N,N,N′,N′,N′′-pentamethyl-diethylene triamine, N,N,N′,N′-tetramethyldiamine methane) led to much lower yields or even no desired carbonate at all. Other sources of bromide ion such as NaBr and TBAB were also found to promote the reaction, albeit with somewhat lower efficiencies. Unfortunately, no comment was made by the authors regarding the possible mechanistic pathway of this reaction.
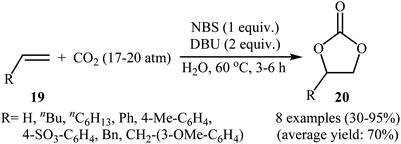 |
| | Scheme 15 NBS-mediated oxidative carboxylation of olefins 19 and CO2 in water. | |
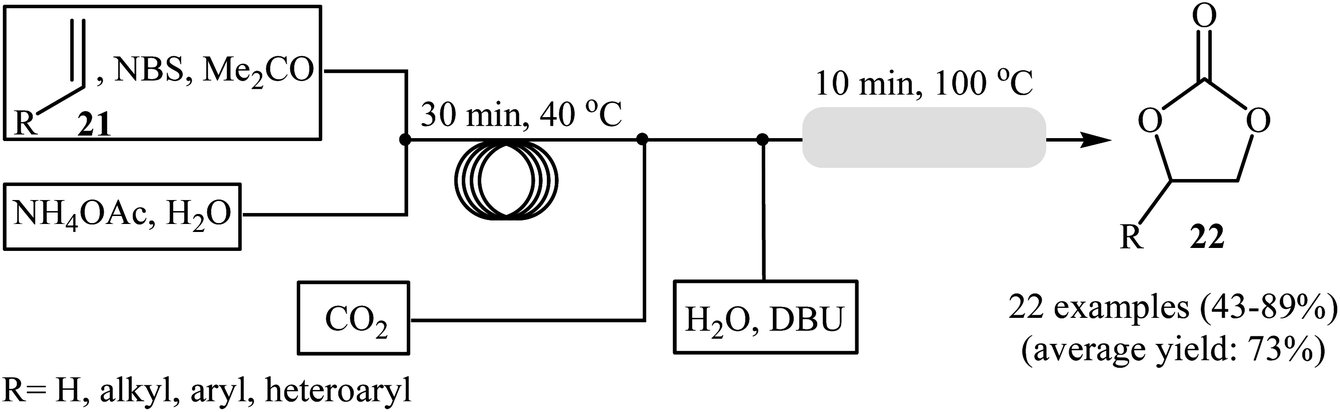
Drawing inspiration from these works, an interesting gas–liquid flow system for the synthesis of cyclic carbonates from olefins and CO2 in the presence of a combination of NBS and DBU was designed by Hatton and Jamison.41 As shown in Scheme 16, two separated streams: (i) olefin and NBS in MeCN and (ii) aqueous NH4OAc solution were introduced into the flow system and met at a T-mixer and the resulting stream was introduced into a PFA (perfluoroalkoxy) tubing reactor with a 30 min residence time at 40 °C. The resulting stream was then met with a flow of CO2 followed by a separate stream of aqueous DBU solution and the reaction mixture was heated at 100 °C while being passed through a packed-bed reactor (filled with stainless-steel powder) which served to facilitate mixing. Various aliphatic, aromatic, and heteroaromatic olefins 21 were tolerated under the conditions and provided their corresponding cyclic carbonates 22 in moderate to excellent yields. In the cases of aromatic olefins, the reactions were conducted at a slightly lower temperature to avoid diol formation. It is interesting to note that in all reported examples, no epoxide or dibromide by-product was observed, which were the main products under the batch condition. It is worthwhile to mention that besides CO2, Br3CCO2H42 and NaHCO3 (ref. 43) were also successfully utilized as C1 sources in this interesting synthetic procedure.
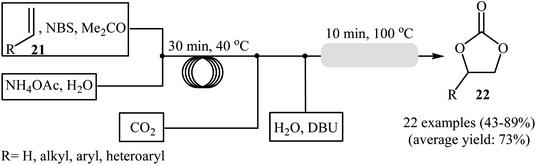 |
| | Scheme 16 Continuous flow synthesis of cyclic carbonates 22 developed by Hatton and Jamison. | |
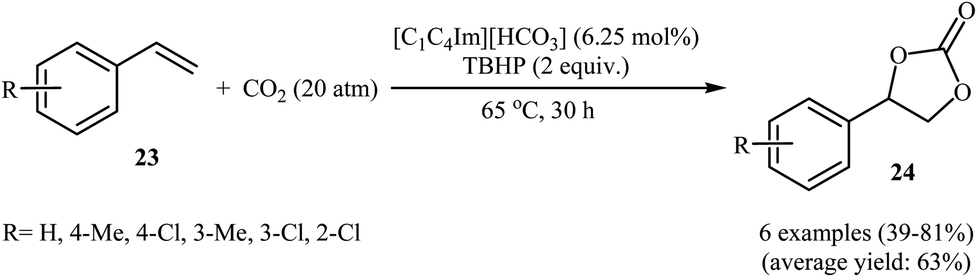
Very recently, Hu and Zhang along with their co-workers reported that imidazolium hydrogen carbonate ionic liquids ([CnCmIm][HCO3]) could effectively catalyze the oxidative carboxylation of olefins and CO2 under metal- and solvent-free conditions.44 When compared with other ionic liquids (i.e. [C1C2Im][HCO3], [C1C6Im][HCO3], [C1C12Im][HCO3]), [C1C4Im][HCO3] was found to be the best catalyst for this transformation. Under the optimized conditions (6.25 mol% catalyst, 2 equiv. TBHP, 20 atm, 65 °C) various styrene derivatives 23 react to give good yields of the corresponding carbonates 24 (Scheme 17). In contrast with the results obtained by Hatton–Jamison's work,41 electron-rich styrenes appeared to afford higher yield than electron-poor ones under this condition. To study the recyclability and reusability of this bifunctional catalyst, the oxidative carboxylation of simple styrene was considered as a model reaction. The results demonstrated that the catalyst could be easily separated from the reaction mixture by extraction and reused five consecutive cycles without significantly losing its catalytic performance. The authors found that [C1C4Im][HCO3] can spontaneously convert into a carbene–CO2 adduct; in fact, there is about 35% carbene–CO2 in this ionic liquid. Thus, they suggested that the active center of [C1C4Im][HCO3] for the epoxidation is the HCO3− anion and for cycloaddition is the carbene–CO2 adduct (Scheme 18).
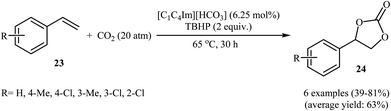 |
| | Scheme 17 Ionic liquid-catalyzed synthesis of cyclic carbonates 24 from olefins 23 and CO2. | |
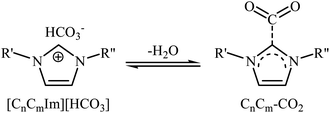 |
| | Scheme 18 The equilibrium between [CnCmIm][HCO3] and carbene–CO2. | |
2.3. Electrocatalytic reactions
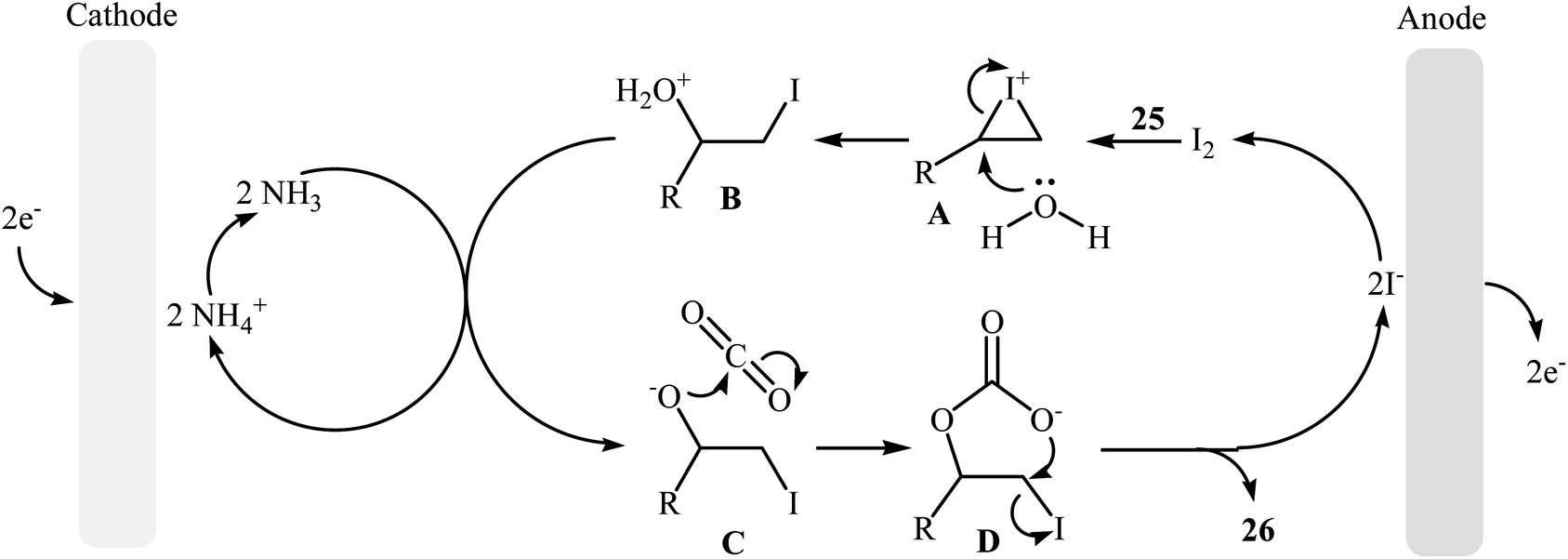
Electrocatalytic synthesis of cyclic carbonates directly from the corresponding olefins and CO2 has been rarely studied; indeed, only one example of such a reaction was reported in the literature by Yuan–Jiang and co-workers.45 By employing styrene as the model reactant, the reaction variables such as electrodes, electrolyte, and solvent were carefully studied. The results indicated that an undivided graphite/Ni-cell was more effective than other cells (e.g., graphite/Al, graphite/Zn, graphite/Cu, Al/Ni, Zn/Ni) and compared to other electrolytes NH4I was the best choice for the conversion. DMSO was also found to be the best solvent over DMF, THF, DCM, MeCN, dioxane, and acetone. Noteworthy, the presence of a small amount of H2O was crucial for the success of this reaction. Under the optimized conditions, a large variety of terminal and internal olefins 25 were tolerated well and provided the target carbonates 26 in moderate to quantitative yields (Scheme 19). The procedures could also be adapted to large scale synthesis of various important carbonates. On the basis of mechanism studies, the authors proposed that this oxidative carboxylation reaction starts with the generation of molecular iodine and ammonia at the anode (2I− → I2 + 2e−) and cathode (2NH4+ + 2e− → 2NH3 + H2), respectively. Next, I2 reacts with olefin 25 to form iodonium intermediate A, which after reaction with water gives iodohydrin intermediate B. Subsequently, this latter undergoes deprotonation with NH3 to yield the anionic intermediate C, followed by reaction with CO2 to generate intermediate D. Finally, the intramolecular cyclization of D affords the expected carbonate 26 and regenerates I− (Scheme 20).
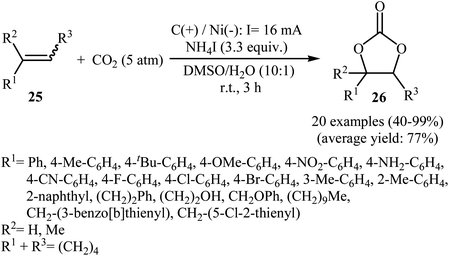 |
| | Scheme 19 Electrochemical conversion of CO2 with olefins 25 and water into cyclic carbonates 26. | |
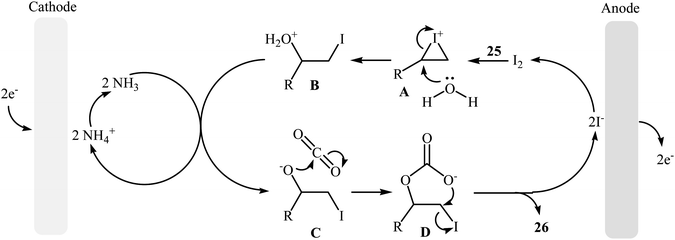 |
| | Scheme 20 Mechanistic proposal for the reaction in Scheme 19. | |
3. One-pot, two-step approaches
3.1. Metal-catalyzed reactions
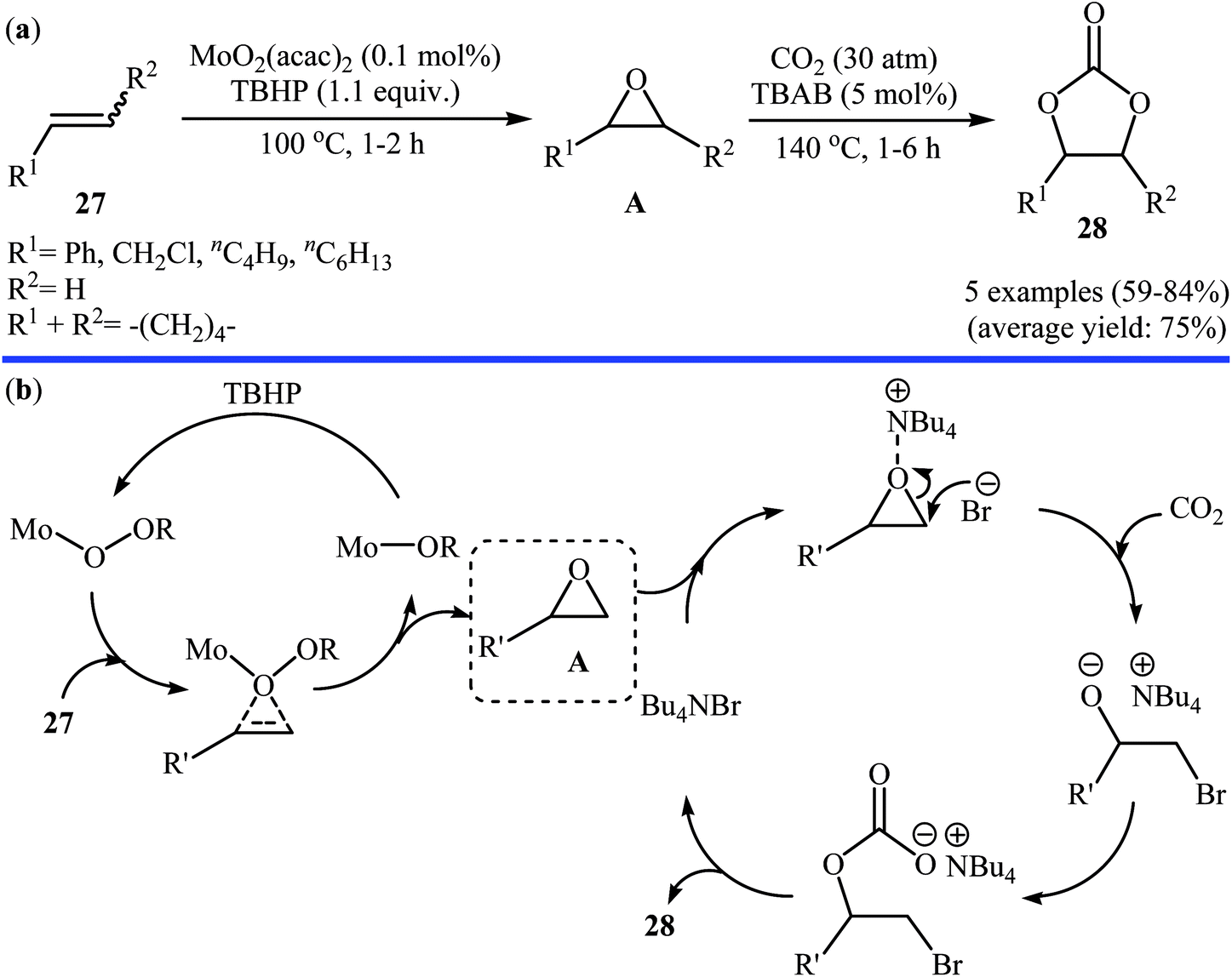
In 2007, Qiao and Yokoyama along with their colleagues developed a convenient one-pot two step methodology for the synthesis of styrene carbonate 2 form styrene 1 and CO2 employing methyltrioxorhenium(MTO)/urea hydrogen peroxide (UHP)/Br4[1-ethyl-3-methylimidazolium EMIm]Br4/Zn[EMIm]2Br4 combination as a catalytic system.46 Initially styrene, MTO, UHP, and [EMIm]Br4 were added to the reactor and after the epoxidation reaction (2 h), Zn[EMIm]2Br4 and CO2 (30 atm) were added to the same reactor to afford the expected carbonate in 83% within 2 h. Four years later, the group of Li–Hu synthesized a range of cyclic carbonates 28 in good to high yields from the corresponding olefins 27 through a similar one-pot two-step process in which after the epoxidation of the olefins by TBHP in the presence of MoO2 catalyst (100 °C, 1–2 h), CO2 and TBAB were added into the same reactor and heated to 140 °C for 1–6 h (Scheme 21a).47 Based on the literature, the authors proposed a mechanistic pathway to this reaction as shown in Scheme 21b. Later, Siewniak and co-workers revisited this reaction by performing the cycloaddition step in the presence of ZnBr2/PS-TBMAC (PS = polystyrene, TBMAC = tributylmethylammonium chloride) combination as a catalytic system.48
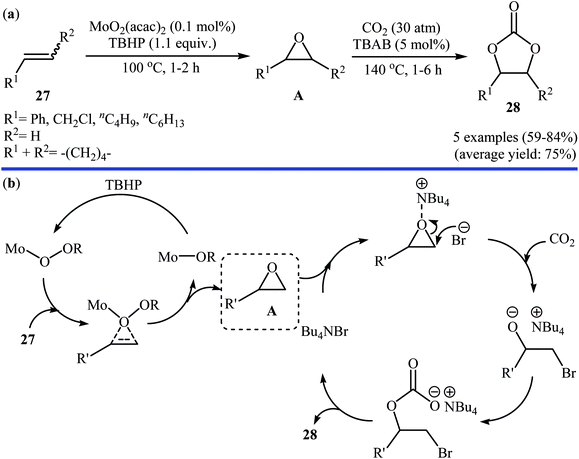 |
| | Scheme 21 (a) Synthesis of cyclic carbonates 28 from olefins 27 and CO2 catalyzed by MoO2(acac)2-TBAB; (b) proposed mechanism for the formation of carbonates 28. | |
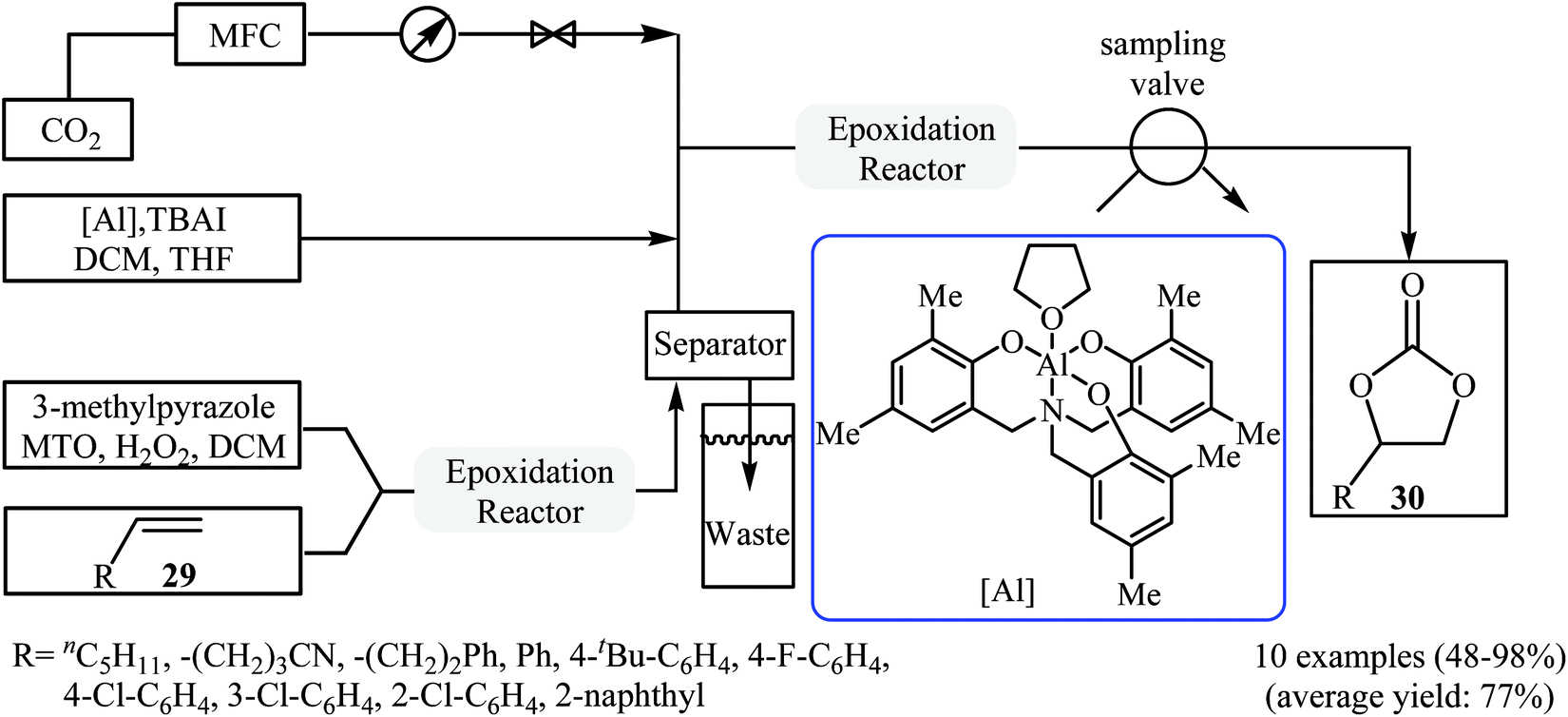
In 2017, Rioux and colleagues disclosed an interesting continuous flow synthesis of carbonates directly from the corresponding olefins.49 They demonstrated oxidative carboxylation of olefins using packed bed flow reactors in series to couple Re-catalyzed olefin epoxidation and Al-catalyzed epoxide carboxylation in a single sequence. Under optimal reaction conditions [first step: methyltrioxorhenium (1 mol%), 3-methylpyrazole (24 mol%), H2O2 (5.0 equiv.), 40 °C, 1 h; second step: CO2 (7.5 atm), [Al] catalyst (2 mol%), TBAI (10 mol%), 100 °C, 40 min] various aromatic and aliphatic terminal olefins 29 efficiently transformed to the corresponding cyclic carbonates 30 in moderate to almost quantitative yields (Scheme 22). However, internal olefins failed to participate in this reaction.
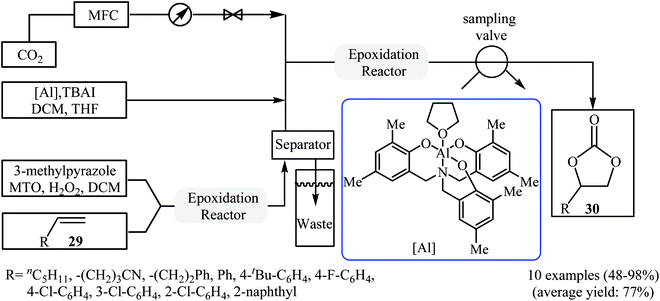 |
| | Scheme 22 The flow synthesis of cyclic carbonates 30 from olefins 29 and CO2 developed by Rioux. | |
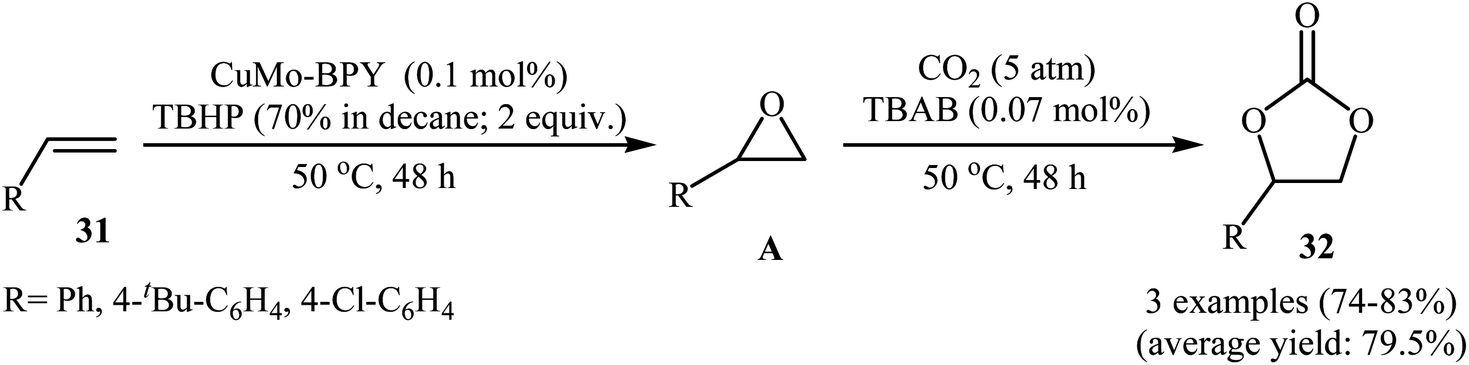
In 2018, Hutchings and co-workers described a one-pot, two-step sequential reaction for the synthesis of 1,2-decylene carbonate.50 The strategy consisted of the epoxidation of 1-decene in the presence of SiO2-supported gold catalyst under an air atmosphere and cycloaddition of CO2 (20 atm) with the formed epoxide using TBAB and ZnBr2. The single-step version of this reaction was also investigated. However, due to deactivation of the catalyst by TBAB, very low 1,2-decylene carbonate selectivity was observed. In this context, Han and Li have reported the high yielding synthesis of styrene carbonates 32 by epoxidation of the corresponding styrenes 31 employing TBHP as an oxidant in the presence of only 0.1 mol% of Cu3(μ3-OH)2(4,4′-BPY) (MoO4)2 (CuMo-BPY, BPY = 4,4′-bipyridine) as a MOF-based catalyst followed by subsequent CO2 (5 atm) insertion in the presence of TBAB (Scheme 23).51 However, the authors found some limitations in the protocol when they attempted to epoxidize the bulky olefins such as 3,5-di-tert-butyl-4′-vinyl biphenyl. Unfortunately, in these cases, unsatisfactory yields were observed. Thus it was suggested that epoxidation occurred in the channels of the MOF, not on the external surface. Furthermore, aliphatic olefins failed to participate in this convention.
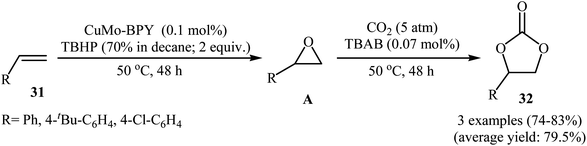 |
| | Scheme 23 Li's synthesis of cyclic carbonates 32. | |
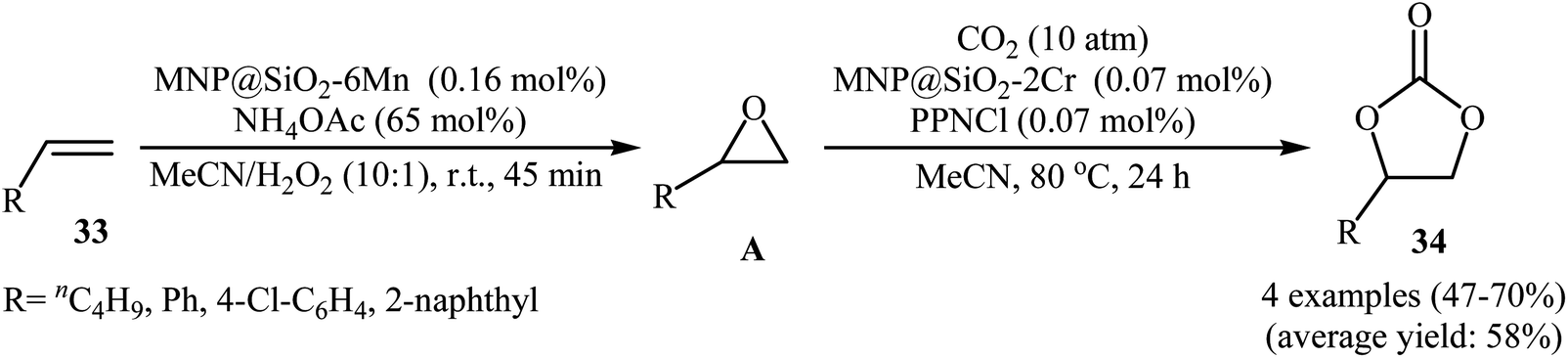
Very recently, Carrilho and Pereira along their co-workers reported an interesting heterobimetallic dual catalytic system based on the combination of two hybrid materials denoted MNP@SiO2-6Mn (6Mn = MnOAc-TDCPP; TDCPP = 5,10,15,20-tetra(2,6-dichlorophenyl)porphyrin) and MNP@SiO2-2Cr (2Cr = CrCl-pCF3TPP; pCF3TPP = 5,10,15,20-tetra(4-trifluoromethylphenyl) porphyrin) for direct transformation of olefins 33 into the corresponding cyclic carbonates 34 using CO2 and H2O2 (Scheme 24).52 The epoxidation step was carried out with the MNP@SiO2-6Mn catalyst in the presence of NH4OAc as axial ligand in MeCN at room temperature. After completion of the reaction (45 min), the solvent was evaporated and the resulting mixture was transferred to another recipient containing MNP@SiO2-2Cr and PPNCl co-catalyst under CO2 (10 atm) atmosphere at 80 °C to continue the cycloaddition step. Both aliphatic and aromatic olefins were compatible with the reaction condition and the respective cyclic carbonates were obtained in moderate to good yields. However, 4-methylstyrene did not take part in this transformation. Significantly, the solids could be reused in three consecutive sequential epoxidation/CO2 addition cycles, without significant loss of catalytic activity and just a slight decrease of selectivity.
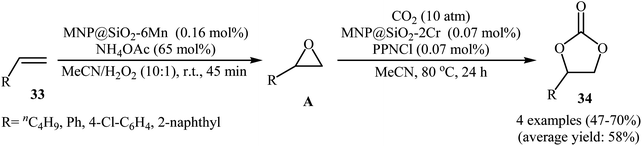 |
| | Scheme 24 Sequential transformation of olefins 33 and CO2 into cyclic carbonates 34 catalyzed by hybrid-metalloporphyrin magnetic nanoparticles. | |
3.2. Metal-free reactions
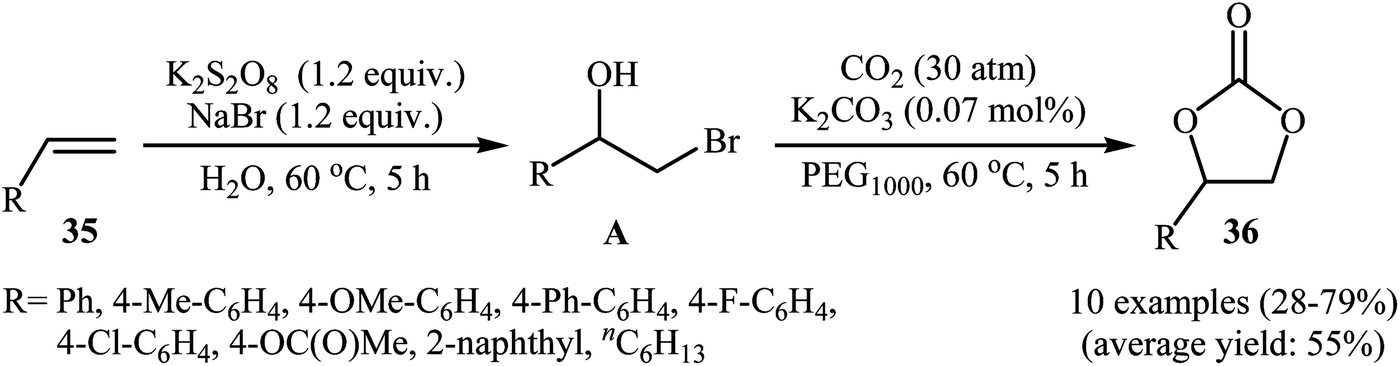
In 2016, L.-N. He and colleagues developed an efficient protocol for the metal-free synthesis of five-membered cyclic carbonates from the corresponding olefins via hydroxybromination–carboxylation reaction sequences.53 Thus, in the presence of 1 equiv. of K2CO3 as an inexpensive base in PEG1000, bromohydrins A, which were prepared through the hydroxybromination of olefins 35 by the composition of K2S2O8–NaBr, were treated with CO2 (30 atm) to give cyclic carbonates 36 in almost fair to good yields (Scheme 25). The results demonstrated that aromatic olefins provided better yields than aliphatic ones and electron-rich aromatic olefins gave relatively higher yields compared to the electron-deficient ones. To the best of our awareness, this is the first and only example of the metal-free synthesis of cyclic carbonates from olefins and CO2 through a one-pot two-step reaction.
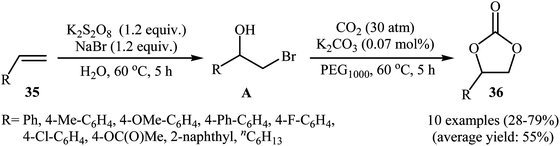 |
| | Scheme 25 Synthesis of cyclic carbonates 36 from olefins 35 and CO2 in the presence of K2S2O8. | |
4. Conclusion
Since the industrial revolution, which began in 1850, human sources of carbon dioxide emissions into the atmosphere have been growing dramatically (almost 3% each year). This has caused serious environmental issues such as global warming, climate change, and ocean acidification. Therefore, the capture, utilization and storage (CCS) of this greenhouse gas have been attracting extensive attention worldwide. Chemical fixation of CO2 is considered as a promising route for the utilization of CO2 because it can be used as a renewable and environmentally benign C1 feedstock for the synthesis of many value-added chemicals. However, high thermal and kinetic stability of CO2 limit its application and a large energy input is usually required for its activation. A strategy to overcome the low reactivity of CO2 is based on the reactions with high free energy substrates. The cycloaddition of CO2 and energy-rich epoxides stands as one of the most attractive and efficient strategies for CO2 conversion and utilization. However, most epoxides are toxic and mutagenic. Moreover, their preparation requires an additional step form the corresponding olefins. An alternative or complementary route involves the direct oxidative carboxylation of olefins. As illustrated, both terminal and internal olefins were compatible with this page of cyclic carbonates synthesis. In addition, aliphatic, aromatic, as well as heteroaromatic olefins were applicable to this reaction and at least three different kinds of catalysts (metal-, organo-, and electro-catalysts) have already been found as active catalysts for this CO2-fixation reaction. However, most of the reactions covered in this review have been performed under harsh conditions (high reaction temperature and/or high CO2 pressure). Therefore, it is still necessary to develop novel and highly efficient catalytic systems that can allow this conversion under milder conditions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was carried out with funding from the National Key R&D Program of China (Grant No. 2018YFC0808103) and Key Laboratory of Western Mine Exploitation and Hazard Prevention, Ministry of Education (Grant No. SKLCRKF1916) and National Natural Science Foundation of China (Grant No. 51874232).
References
- N. Kindermann, T. Jose and A. W. Kleij, Top. Curr. Chem., 2017, 375, 61–88 CrossRef PubMed.
- J. Song, Q. Liu, H. Liu and X. Jiang, Eur. J. Org. Chem., 2018, 696–713 CrossRef CAS.
- B. Zou and C. Hu, Curr. Opin. Green Sustain. Chem., 2017, 3, 11–16 CrossRef.
- Z. Zhang, T. Ju, J.-H. Ye and D.-G. Yu, Synlett, 2017, 28, 741–750 CrossRef CAS.
- T. Niemi and T. Repo, Eur. J. Org. Chem., 2019, 1180–1188 CrossRef CAS.
- Y. Zhang, X. Dai, H. Wang and F. Shi, Acta Phys.-Chim. Sin., 2018, 34, 845–857 CAS.
- N. A. Tappe, R. M. Reich, V. D'Elia and F. E. Kühn, Dalton Trans., 2018, 47, 13281–13313 RSC.
- A. Rafiee, K. R. Khalilpour, D. Milani and M. Panahi, J. Environ. Chem. Eng., 2018, 6, 5771–5794 CrossRef CAS.
- R. Dalpozzo, N. Della Ca, B. Gabriele and R. Mancuso, Catalysts, 2019, 9, 511 CrossRef.
-
(a) B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC;
(b) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Sci. Rev. Lett., 2018, 1, 23–30 Search PubMed.
-
(a) Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC;
(b) E. A. Mahmood, B. Azizi and S. Majedi, Chem. Sci. Rev. Lett., 2020, 3, 2–8 Search PubMed;
(c) S. Majedi, S. Majedi and F. Behmagham, Chem. Sci. Rev. Lett., 2019, 2, 187–192 Search PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
-
(a) C. Martin, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef CAS;
(b) A. Monfared, R. Mohammadi, A. Hosseinian, S. Sarhandi and P. D. K. Nezhad, RSC Adv., 2019, 9, 3884–3899 RSC.
-
(a) A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed;
(b) J. Sun, S.-i. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
-
(a) D. Wade, S. C. Airy and J. E. Sinsheimer, Mutat. Res. Genet. Toxicol., 1978, 58, 217–223 CrossRef CAS;
(b) C. Voogd, J. Van der Stel and J. Jacobs, Mutat. Res. Genet. Toxicol. Environ. Mutagen, 1981, 89, 269–282 CrossRef CAS.
- X. D. Lang and L. N. He, Chem. Rec., 2016, 16, 1337–1352 CrossRef CAS PubMed.
-
(a) J. A. Verdol, Cyclic carbonate from olefin, US Pat., 3025305, 1962;
(b) C. Fumagalli, G. Caprara and P. Roffia, Process for the preparation of alkylene carbonates, US Pat., 4009183, 1977;
(c) G. A. Wheaton, J.-L. Kao and M. N. Sheng, Preparation of alkylene carbonates from olefins, US Pat., 4224223, 1980;
(d) J.-L. Kao, G. A. Wheaton, H. Shalit and M. N. Sheng, Preparation of alkylene carbonates from olefins, US Pat., 4247465, 1981.
- J. Sun, L. Liang, J. Sun, Y. Jiang, K. Lin, X. Xu and R. Wang, Catal. Surv. Asia, 2011, 15, 49–54 CrossRef CAS.
-
(a) S. Arshadi, E. Vessally, M. Sobati, A. Hosseinian and A. Bekhradnia, J. CO2 Util., 2017, 19, 120–129 CrossRef CAS;
(b) S. Arshadi, E. Vessally, A. Hosseinian, S. Soleimani-amiri and L. Edjlali, J. CO2 Util., 2017, 21, 108–118 CrossRef CAS;
(c) E. Vessally, S. Soleimani-Amiri, A. Hosseinian, L. Edjlali and M. Babazadeh, J. CO2 Util., 2017, 21, 342–352 CrossRef CAS;
(d) E. Vessally, M. Babazadeh, A. Hosseinian, S. Arshadi and L. Edjlali, J. CO2 Util., 2017, 21, 491–502 CrossRef CAS;
(e) K. Didehban, E. Vessally, M. Salary, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 23, 42–50 CrossRef CAS;
(f) A. Hosseinian, S. Farshbaf, R. Mohammadi, A. Monfared and E. Vessally, RSC Adv., 2018, 8, 17976–17988 RSC;
(g) S. Farshbaf, L. Z. Fekri, M. Nikpassand, R. Mohammadi and E. Vessally, J. CO2 Util., 2018, 25, 194–204 CrossRef CAS;
(h) A. Hosseinian, S. Ahmadi, R. Mohammadi, A. Monfared and Z. Rahmani, J. CO2 Util., 2018, 27, 381–389 CrossRef CAS;
(i) E. Vessally, A. Hosseinian, M. Babazadeh, L. Edjlali and R. Hosseinzadeh-Khanmiri, Curr. Org. Chem., 2018, 22, 315–322 CrossRef CAS;
(j) E. Vessally, A. Hosseinian, L. Edjlali, M. Babazadeh and K. Didehban, Mini-Rev. Org. Chem., 2018, 15, 315–323 CrossRef CAS;
(k) S. Arshadi, A. Banaei, S. Ebrahimiasl, A. Monfared and E. Vessally, RSC Adv., 2019, 9, 19465–19482 RSC;
(l) X. Zhao, S. Yang, S. Ebrahimiasl, S. Arshadi and A. Hosseinian, J. CO2 Util., 2019, 33, 37–45 CrossRef CAS;
(m) S. Mohammadi, M. Musavi, F. Abdollahzadeh, S. Babadoust and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 49–55 Search PubMed;
(n) S. Shahidi, P. Farajzadeh, P. Ojaghloo, A. Karbakhshzadeh and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 37–44 Search PubMed;
(o) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed;
(p) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed.
- M. Aresta, A. Dibenedetto and I. Tommasi, Appl. Organomet. Chem., 2000, 14, 799–802 CrossRef CAS.
- M. Aresta, E. Quaranta and A. Ciccarese, J. Mol. Catal., 1987, 41, 355–359 CrossRef CAS.
- M. Aresta and A. Dibenedetto, J. Mol. Catal. A: Chem., 2002, 182, 399–409 CrossRef.
- R. Srivastava, D. Srinivas and P. Ratnasamy, Catal. Lett., 2003, 91, 133–139 CrossRef CAS.
- J. Sun, S.-i. Fujita, F. Zhao, M. Hasegawa and M. Arai, J. Catal., 2005, 230, 398–405 CrossRef CAS.
- J.-L. Wang, J.-Q. Wang, L.-N. He, X.-Y. Dou and F. Wu, Green Chem., 2008, 10, 1218–1223 RSC.
- D. Xiang, X. Liu, J. Sun, F.-S. Xiao and J. Sun, Catal. Today, 2009, 148, 383–388 CrossRef CAS.
- Y. Wang, J. Sun, D. Xiang, L. Wang, J. Sun and F.-S. Xiao, Catal. Lett., 2009, 129, 437–443 CrossRef CAS.
- D. Bai and H. Jing, Green Chem., 2010, 12, 39–41 RSC.
- P. Ramidi, C. M. Felton, B. P. Subedi, H. Zhou, Z. R. Tian, Y. Gartia, B. S. Pierce and A. Ghosh, J. CO2 Util., 2015, 9, 48–57 CrossRef CAS.
- S. Kumar, N. Singhal, R. K. Singh, P. Gupta, R. Singh and S. L. Jain, Dalton Trans., 2015, 44, 11860–11866 RSC.
- Q. Han, B. Qi, W. Ren, C. He, J. Niu and C. Duan, Nat. Commun., 2015, 6, 10007 CrossRef PubMed.
- N. V. Maksimchuk, I. D. Ivanchikova, A. B. Ayupov and O. A. Kholdeeva, Appl. Catal., B, 2016, 181, 363–370 CrossRef CAS.
- H. T. Nguyen, Y. Tran, H. N. Nguyen, T. C. Nguyen, F. Gándara and P. T. Nguyen, Inorg. Chem., 2018, 57, 13772–13782 CrossRef CAS PubMed.
- P. T. Nguyen, H. T. Nguyen, H. N. Nguyen, C. A. Trickett, Q. T. Ton, E. Gutiérrez-Puebla, M. A. Monge, K. E. Cordova and F. Gándara, ACS Appl. Mater. Interfaces, 2017, 10, 733–744 CrossRef PubMed.
- N. Sharma, S. S. Dhankhar, S. Kumar, T. D. Kumar and C. M. Nagaraja, Chem. - Eur. J., 2018, 24, 16662–16669 CrossRef CAS PubMed.
- J. Sun, S.-i. Fujita, B. M. Bhanage and M. Arai, Catal. Commun., 2004, 5, 83–87 CrossRef CAS.
- J. Sun, S.-i. Fujita, B. M. Bhanage and M. Arai, Catal. Today, 2004, 93, 383–388 CrossRef.
- O. V. Zalomaeva, N. V. Maksimchuk, A. M. Chibiryaev, K. A. Kovalenko, V. P. Fedin and B. S. Balzhinimaev, J. Energy Chem., 2013, 22, 130–135 CrossRef CAS.
- N. Eghbali and C.-J. Li, Green Chem., 2007, 9, 213–215 RSC.
- J. Wu, J. A. Kozak, F. Simeon, T. A. Hatton and T. F. Jamison, Chem. Sci., 2014, 5, 1227–1231 RSC.
- S. G. Davies, A. M. Fletcher, W. Kurosawa, J. A. Lee, G. Poce, P. M. Roberts, J. E. Thomson and D. M. Williamson, J. Org. Chem., 2010, 75, 7745–7756 CrossRef CAS PubMed.
- X. Yang, J. Wu, X. Mao, T. F. Jamison and T. A. Hatton, Chem. Commun., 2014, 50, 3245–3248 RSC.
- J. Liu, G. Yang, Y. Liu, D. Wu, X. Hu and Z. Zhang, Green Chem., 2019, 21, 3834–3838 RSC.
- X. Gao, G. Yuan, H. Chen, H. Jiang, Y. Li and C. Qi, Electrochem. Commun., 2013, 34, 242–245 CrossRef CAS.
- F. Ono, K. Qiao, D. Tomida and C. Yokoyama, Appl. Catal., A, 2007, 333, 107–113 CrossRef CAS.
- F. Chen, T. Dong, T. Xu, X. Li and C. Hu, Green Chem., 2011, 13, 2518–2524 RSC.
- A. Siewniak, K. Jasiak-Jaroń, Ł. Kotyrba and S. Baj, Catal. Lett., 2017, 147, 1567–1573 CrossRef.
- A. A. Sathe, A. M. Nambiar and R. M. Rioux, Catal. Sci. Technol., 2017, 7, 84–89 RSC.
- R. V. Engel, R. Alsaiari, E. Nowicka, S. Pattisson, P. J. Miedziak, S. A. Kondrat, D. J. Morgan and G. J. Hutchings, Top. Catal., 2018, 61, 509–518 CrossRef CAS PubMed.
- Z. Shi, G. Niu, Q. Han, X. Shi and M. Li, Mol. Catal., 2018, 461, 10–18 CrossRef CAS.
- L. D. Dias, R. M. Carrilho, C. A. Henriques, M. J. Calvete, A. M. Masdeu-Bultó, C. Claver, L. M. Rossi and M. M. Pereira, ChemCatChem, 2018, 10, 2792–2803 CrossRef CAS.
- J.-N. Xie, Z.-F. Diao, C. Qiao, R. Ma and L.-N. He, J. CO2 Util., 2016, 16, 313–317 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d
*d