
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntrinsic defect engineered Janus MoSSe sheet as a promising photocatalyst for water splitting
Yimin Xua,
Yongsheng Yaoa,
Wenjin Yinbc,
Juexian Caoa,
Mingyang Chencd and
Xiaolin Wei *a
*a
aDepartment of Physics and Laboratory for Quantum Engineering and Micro-Nano Energy Technology, Xiangtan University, Xiangtan 411105, Hunan, China. E-mail: xlw@xtu.edu.cn
bSchool of Physics and Electronic Science, Hunan University of Science and Technology, Xiangtan 411201, China
cBeijing Computational Science Research Center, Beijing 100084, China
dSchool of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, China
First published on 16th March 2020
Abstract
The Janus MoSSe sheet has aroused significant attention due to its band edge position and intrinsic dipole moment, making it a strong candidate for water splitting photocatalysis. However, weak water adsorption seriously prevents its further application. Here, first-principles calculations are used to explore the effect of intrinsic defects on water adsorption and conversion at the Janus MoSSe sheet. First-principles calculation results clearly show that intrinsic defects (Svac, Moanti, and Moint) can effectively alter the interaction between water and the MoSSe sheet. Except for Svac defects, the adsorption energy of water at Moanti or Moint defects can be significantly increased by −1.0 to −1.5 eV with respect to the weak water adsorption on a pristine MoSSe sheet of about −0.24 eV. More importantly, the energy barrier for water conversion can be dramatically lowered by 48% to 0.7 eV at Moanti or Moint defects, together with a more stable final state. Such significant enhancement of the adsorption energy is attributed to the red shift of water energy levels, resulting from the strong interaction between O2p orbitals and Mo3d orbitals. It is shown that the intrinsic defects have the potential to change the photocatalytic reactivity of the surface, and thus this may serve as an important way to design photocatalysts for water splitting.
Introduction
Photocatalytic water splitting is gaining increased interest, since it is a promising “green chemistry” approach for the direct conversion of water into clean energy H2 driven by solar light.1–3 However, low-efficiency production of H2 seriously prevents its further applications. Thus, it is urgent to search for a photocatalyst with high stability and efficiency. As a potential photocatalyst, several key criteria should be satisfied as follows. Firstly, a suitable band gap larger than 1.23 eV is required for the photocatalyst to harvest the solar light.4 Then, the photocatalyst must have decent band edge positions for overall water splitting, with the conduction band minimum (CBM) higher than the reduction potential (EH+/EH2 = −4.44 eV) and valence band maximum (VBM) lower than the oxidation potential (EO2/EH2O = −5.67 eV). In addition, separation of the photogenerated electron–hole pair is also a crucial factor to reduce the possibility of carrier recombination.5Based on the above criteria, various semiconductors including TiO2, ZnO, CdS, Fe2O3, Bi2WO6, Cu2O, etc. have been proposed as photocatalysts to increase the efficiency for water splitting.1,6 However, some of these photocatalysts such as TiO2 are inactive for overall water splitting into O2 and H2 in the absence of sacrificial reagents because of a large band gap and low quantum efficiency.7 Recently, low dimensional materials have also aroused extensive attention due to the fundamental properties such as large surface area, tunable electronic structure, and abundant reactive sites.8–12 For example, researchers have found that two dimensional g-C3N4, and transition metal dichalcogenides (TMDs) have suitable band gaps and robust absorption in the solar spectrum, which is suitable for water splitting.13–15 Especially, it has reported that TMDs such as MoS2 exhibits a direct band gap of about 1.9 eV as it is tailored into single layer, and the band edge can across the reduction and oxidation potential of water splitting.16,17 However, the carrier mobility of electron and hole in the pristine MoS2 is relatively low, leading to the low efficiency in water splitting activity and hindering its further applications.18–20
Except for the traditional TMDs, forming a Janus structure is also an efficient strategy to enhance the physical and chemical properties of the materials.21,22 For example, Guo et al. found that the Janus group III chalcogenide structure could greatly enhance the piezoelectric effect associated with the intrinsic dipole perpendicular to the plane structure.23 Furthermore, the enhanced dipole of the Janus structure can generate an intrinsic electric field, which facilitates the separation of the photogenerated carriers in the photocatalytic reactions.24 On the other hand, some researches have proposed some new Janus structures with excellent photocatalytic property. For example, Yang et al. have proposed a novel family of Janus single-layer group-III monochalcogenides (J–MX, J = Ga, M = In, X = S and Se) using first-principles calculations, and found their band gaps are suitable for water photocatalytic splitting.25 And Gao et al. also proposed eight IV–V compound materials showing excellent photocatalysts for water splitting reactions with high efficiency of visible light.26
Recently, Lu et al. have grown a Janus monolayer of transition metal dichalcogenides named MoSSe through breaking the out-of-plane structural symmetry of the single layer MoS2.27 Janus MoSSe is structurally viewed as MoS2 with all of S atoms on one side replaced by Se atoms, and exhibits an electronic structure similar to that of MoS2.28 Unlike MoS2, an intrinsic dipole exists along the vertical direction of the MoSSe structure, and Yin et al. have found the carrier mobility of Janus MoSSe structure could be greatly affected by the dipole and layer thickness by DFT calculation.29 Further HSE06 result proposed by Ma et al. shows that the Janus MoSSe sheet can be a potential water splitting photocatalyst with wide solar-absorption range and retard carrier recombination.30 Based on the above results, other works on photocatalytic water splitting have been performed. For example, using HSE06+SOC method, Din et al. found that the appropriate band alignments for visible light water splitting can be achieved through forming MoSSe/GeC heterostructure.31 Further study showed that despite the band gaps of MoSSe/XN (X = Al, Ga) heterostructures being smaller than 1.23 eV, the band edge positions are always suitable for visible-infrared water splitting.32 On the other hand, Guan et al. found that few layer Janus MoSSe are suitable for visible light water splitting through thickness administrating.33 Therefore, it can be deduced that Janus MoSSe related materials can be excellent photocatalysis for water splitting. Unfortunately, the adsorption energy of molecular water at pristine MoSSe sheet is about 0.16 eV, suggesting an extremely weak van der Waal (vdW) interaction similar to CO2 adsorption at TiO2 surface,34,35 which will seriously hinder the process of water conversion.
So far, to enhance the adsorption of molecular species at the surface, there are several approaches such as surface modification, transition metal or nonmetal doping, and defect introduction.36–39 Among these methods, introducing intrinsic defect may be superior to other approaches.40 First, intrinsic defects are inevitable during the material growth and are also difficult to eliminate. Furthermore, defect introduction can not only vary the reactivity of the local surface structure, but also change the electronic structure of the material. For example, Zhang et al. reported that the band gap and work function of monolayer Janus MoSSe can be modified by simple hydrogenation method.41 On the other hand, during the photon related process, the presence of defects can change the quantum yield of photonic excitation by tuning the energy band structure, modifying electron–hole separation, and carrier transport property.42 For example, it has been reported that chalcogenide vacancy exists at TMDs.43 Chaurasiya et al. have found that intrinsic defects can effectively alter the adsorption behaviors of NO2, NO, and NH3 on Janus MoSSe sheet.44 However, the detailed role of the intrinsic defects on introducing the adsorption state and enhancing the photoreaction of water is still unknown.
The purpose of this work is to unveil the effects of intrinsic defects on water splitting at Janus MoSSe sheet. Particular attention will be focused on the following questions: (i) What is the most stable configuration of the Janus MoSSe sheet with intrinsic defects like? (ii) What effects do the intrinsic defects have on water adsorption and conversion? (iii) What factors are crucial to govern the interaction between water and substrate? Our results clearly show that Moanti defect favors forming two Mo–S bonds and two Mo–Mo bonds, while Moint favors be located over lattice Mo site. The adsorption and conversion of water can be strongly enhanced by intrinsic defects. Thus, intrinsic defect introduction exhibits great potential for the design of photocatalysis for water splitting.
Computational method
The first-principles calculations were conducted by the spin-polarized density functional theory (DFT) within periodic boundary conditions, as implemented in the CP2K/Quickstep package.45 The CP2K implementation of DFT employs the hybrid Gaussian and plane wave (GPW) basis sets and the norm conserving Goedecker–Teter–Hutter pseudopotentials to describe the ion–electron interactions.46 The Gaussian functions consisting of a double-ζ plus polarization (DZVP) basis set was used for all calculations. Perdew–Burke–Ernzerhof (PBE) functional was employed for the exchange-correlation term based on the generalized gradient approximation (GGA).47 The energy cutoff for the real space grid was set at 500 Ry, which yields total energies converged to at least 0.001 eV per atom. Reciprocal space sampling was restricted to the Γ-point, due to the rather large size of the used simulation cells. To consider the weak interaction between water and Janus MoSSe sheet, vdW contribution was included with the Grimme's scheme (DFT-D3).48 A vacuum spacing of 15 Å was placed between each adjacent images. Transition states along the reaction pathways were searched by Climbing Image Nudged Elastic Band (CI-NEB) approach.49 The interaction between molecule and the substrate was described by the adsorption energy, which is expressed as,| Ead = Esub+mol − Esub − Emol | (1) |
Results and discussions
The adsorption of water molecule at defective Janus MoSSe sheet
The primitive cell of Janus MoSSe sheet is a hexagonal structure as denoted by the green line see Fig. 1(a), which consists of S–Mo–Se atomic tri-layers with the upper Se, middle Mo, and lower S atoms as shown in Fig. 1(b). The relaxed cell of Janus MoSSe is in P3m1 space group, and the Mo atom is in six-fold, and the S (Se) atom is in three-fold coordinated. Here, a (4 × 4) supercell is used to model the Janus MoSSe sheet for water adsorption as shown in Fig. 1. For comparison, the molecular water (M-H2O) adsorbed at pristine Janus MoSSe and MoS2 sheet are firstly considered. The S side of the MoSSe is firstly examined, and there are two possible adsorption configurations existing after relaxation, denoted as P-M1 and P-M2 respectively, as shown in Fig. 1. In P-M1, the H2O is adsorbed at hollow site surrounded by three Mo atoms with H atoms pointing to S atoms (Fig. 1(a)). In P-M2, the H2O is adsorbed vertically over a single lattice Mo (Fig. 1(c)). The calculated adsorption energy for P-M1 is −0.243 eV, which is 0.04 eV more negative than that of P-M2, suggesting P-M1 is slightly more stable. This result agrees well with the previous PBE prediction of 0.16 eV.30 Similar to S side, the water adsorption on Se side of MoSSe is about −0.240 eV, which is a little smaller than that of S side. As for MoS2 sheet, the adsorption of water is smaller than the above two with the value at about −0.219 eV. In all, we can find the adsorption energy of water on these three cases are quite close, where S side of MoSSe sheet has the largest water adsorption energy still a weak vdW interaction. The larger adsorption of water consistent with the shorter H–S bond length. Therefore, in the text, we choose S side of MoSSe as the typical one to investigate the water behavior.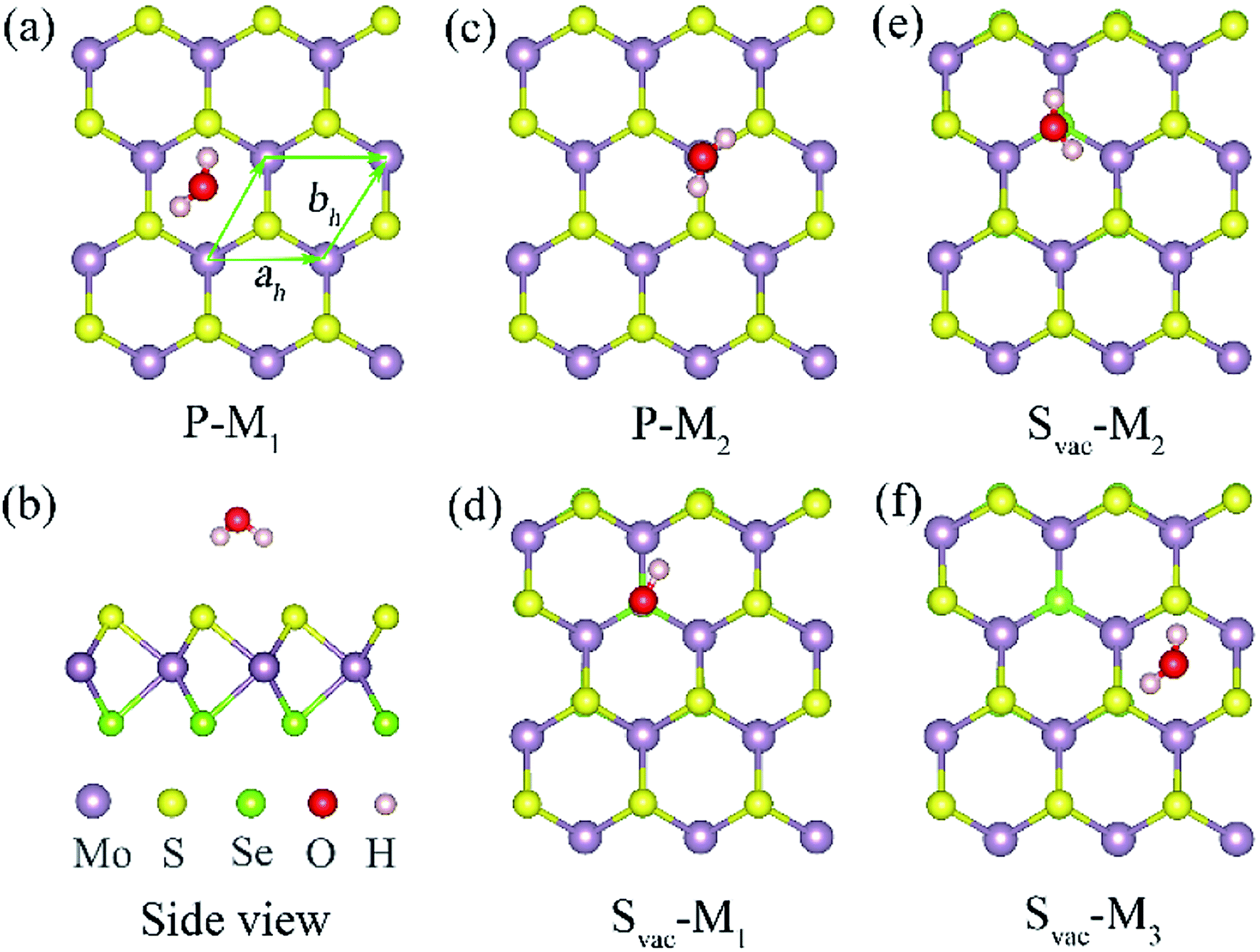 | ||
| Fig. 1 Optimized structures for single water molecule adsorbed at a (4 × 4) Janus MoSSe sheet. (a) and (c) top views of possible water adsorption configurations, and (b) the corresponding side view of (a) at pristine case. (d)–(f) the top views of possible water adsorption structures at Svac Janus MoSSe sheet. The primitive cell is denoted in green line. The Mo atom is light violet ball, S atom is yellow ball, Se is light green ball, O atom is red ball, and H atom is light pink ball for water adsorbed Janus MoSSe sheet, hereinafter. | ||
It has been reported that defect introduction is an effectively approach to alter the surface reactivity. Here, intrinsic defects including S vacancy (Svac), Mo antidefect (Moanti), and Mo interstitial (Moint) are considered for the Janus MoSSe sheet. It should be noted that only S side is considered, since the property of Se side is similar to that of S side. Firstly, one Svac defect is introduced in the (4 × 4) supercell. Three typical water adsorption configurations are obtained from the geometry optimization calculations, denoted as Svac-Mn (n = 1, 2, 3) as shown in Fig. 1. In Svac-M1, one H atom of M-H2O is located at the Svac, and the other H atom is pointing to the adjacent S atom in Fig. 1(d). In Svac-M2, the O atom of M-H2O resides at the Svac with the two H atoms pointing outwards. In Svac-M3, the M-H2O is absorbed at a hexagonal hollow site that is away from Svac. The corresponding calculated adsorption energies for Svac-Mn are provided in Table 1. It is shown that the M-H2O at Svac-M1 has the most negative adsorption energy of −0.321 eV, which is about 0.08 eV more negative than the M-H2O adsorption energy for the pristine sheet. This more negative adsorption energy of Svac-M1, as compared to P-M1, is consistent with substantially shorter Mo–H distance of 3.19 Å for Svac-M1 (vs. 4.29 Å for P-M1). To know the difference between MoSSe and MoS2 sheet, the water adsorbed at Svac defect MoS2 sheet is also examined. The result shows that the adsorption energy is about −0.297 eV at Svac MoS2, which is a little lower than that at Svac_MoSSe sheet. Thus, introducing Svac defect will slightly enhance the water adsorption on the Janus MoSSe sheet.
| System | M-H2O | Ead | H–S (Å) | H–Mo (Å) | O–H (Å) | ∠HOH (degree) |
|---|---|---|---|---|---|---|
| Pristine | P-M1 | −0.243 | 2.661 | 4.290 | 0.972 | 103.414 |
| P-M2 | −0.239 | 2.645 | 0.972 | 103.35 | ||
| M-H2O | 0.97 | 104.131 | ||||
| Svac | Svac-M1 | −0.321 | 2.985 | 3.190 | 0.971 | 103.187 |
| Svac-M2 | −0.307 | 2.879 | 3.397 | 0.972 | 103.262 | |
| Svac-M3 | −0.230 | 2.658 | 4.290 | 0.972 | 103.492 |
Other than Svac defect, anti-defect is also considered by replacing one S atom by a Mo atom in the (4 × 4) supercell of the Janus MoSSe sheet. Three kinds of Moanti defects are allowed for by the (4 × 4) supercell, and the corresponding relaxed structures are denoted as Moanti-n (n = 1, 2, 3) as shown in Fig. 2. In Moanti-1, the Mo substitute resides at the exact position of the replaced S and forms three equal Mo–Mo bonds (Fig. 2(a)). In Moanti-2, the Mo substitute is slightly displaced towards the hollow center, forming two Mo–S and two Mo–Mo bonds (Fig. 2(b)). In Moanti-3 the Mo substitute is slightly displaced towards the adjacent lattice Mo, forming two Mo–S bonds and a Mo–Mo bond in Fig. 2(c). Among these three configurations, Moanti-2 defect is found to be more stable, as suggested by the predicted relative energies (Table 2).
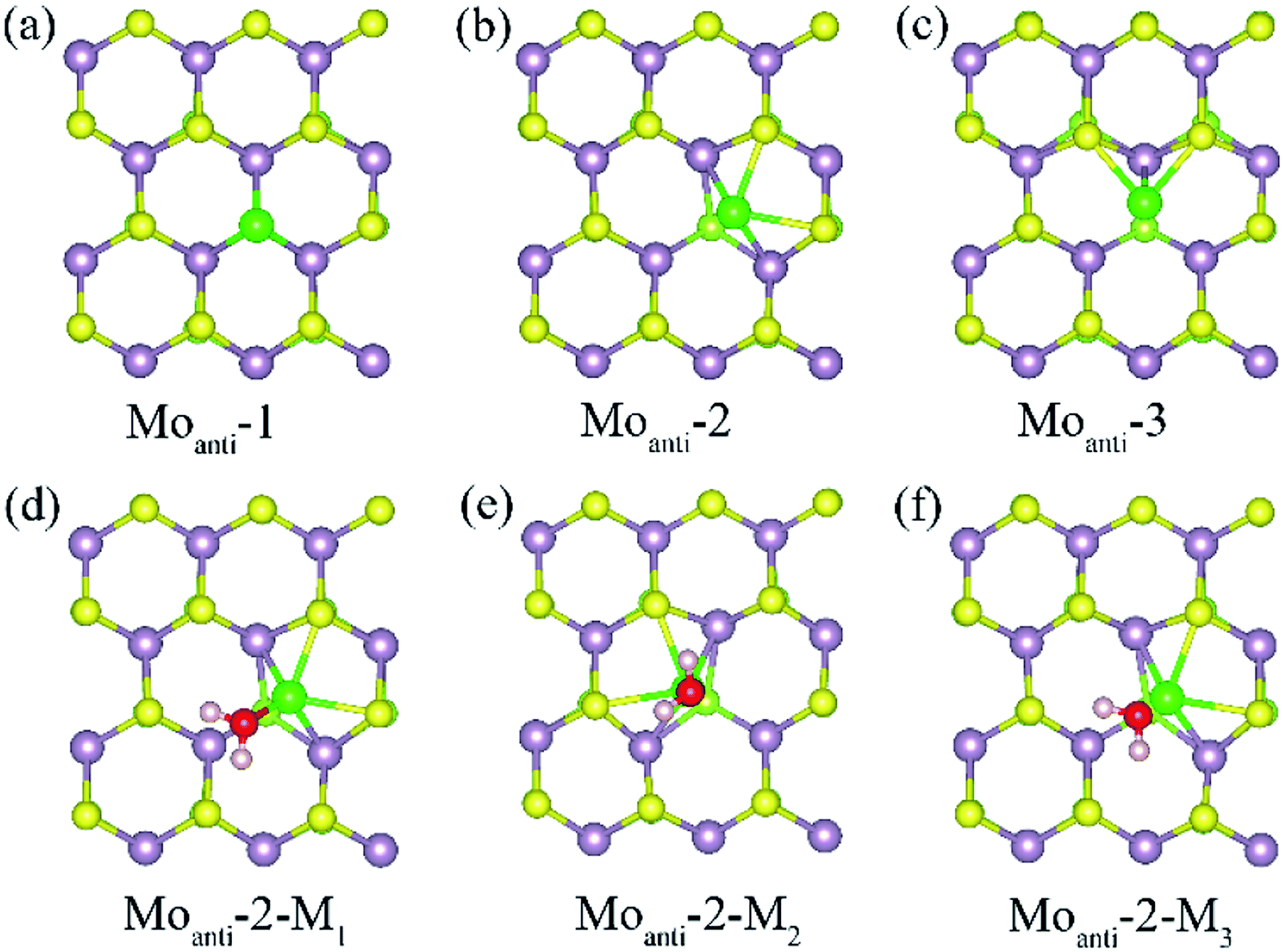 | ||
| Fig. 2 (a)–(c) The possible Moanti defect at Janus MoSSe sheet. (d)–(f) The water molecule adsorption at Moanti-2 structure. The Moanti atom is in green ball. | ||
| Defect type | Localized sites | Relative energy | Mo–Mo (Å) | Mo–S (Å) |
|---|---|---|---|---|
| Moanti | Moanti-1 | 0.450 | 2.522 | 3.115 |
| Moanti-2 | 0 | 2.430 | 2.443 | |
| Moanti-3 | 0.366 | 2.439 | 2.457 | |
| Moint | Moint-1 | 0.127 | 2.881 | 2.194 |
| Moint-2 | 0 | 2.443 | 2.430 |
The adsorption of molecular water at the Moanti-2 site is investigated. Three adsorption configurations are resulting from the geometry optimization calculations, denoted as Moanti-2-Mn (n = 1, 2, 3), as shown in Fig. 2. In Moanti-2-M1, the M-H2O is tiltedly adsorbed at Moanti atom site with H atoms pointing to the two nearest S atom. In Moanti-2-M2, the M-H2O resides right atop the Moanti atom with H atoms pointing to two nearest S atoms. Moanti-2-M3 is structurally close to Moanti-2-M1 except for the pointing directions of the H atoms. The corresponding adsorption energy and relaxed geometry parameters for the three configurations are shown in Table 3. It can be found that the adsorption energies of M-H2O at all three modes are more negative than −1.0 eV. Among the three configurations, Moanti-2-M1 has the most negative M-H2O adsorption energy (−1.42 eV), together with the shortest Mo–O distance of ∼2.2 Å. Relative to M-H2O at pristine sheet, it is unexpected to find that the adsorption energy is dramatically increased by −1.18 eV. Furthermore, the difference between MoSSe and MoS2 sheet are checked. Compared with Moanti MoSSe, the water adsorption at Moanti MoS2 is about 0.036 eV larger than that at MoSSe sheet, suggesting Moanti defect at MoSSe has larger effect on water adsorption. Therefore, Moanti defect can strongly alter the adsorption behavior of water at Janus MoSSe sheet, which would be a potential way to promote the photocatalytic water splitting.
| System | M-H2O | Ead | H–S (Å) | O–Mo (Å) | O–H (Å) | ∠HOH (°) |
|---|---|---|---|---|---|---|
| Moanti-2 | Moanti-M1 | −1.415 | 2.324 | 2.199 | 0.937 | 102.650 |
| Moanti-M2 | −1.061 | 2.905 | 2.228 | 0.978 | 106.704 | |
| Moanti-M3 | −1.203 | 2.610 | 2.228 | 0.981 | 104.163 | |
| Moint-2 | Moint-M | −1.202 | 2.418 | 2.295 | 0.975 | 103.501 |
Interstitial defect is another common intrinsic defect type for transition metal dichalcogenides. The models for Janus MoSSe with Moint defect are constructed by adding one Mo atom to the (4 × 4) supercell. Two types of Moint defects are obtained from the geometry optimization calculations, denoted as Moint-1 and Moint-2, as shown in Fig. 3. For Moint-1, the Moint atom is located at hollow site forming three Mo–S bonds. In Moint-2, the Moint atom is located right atop the lattice Mo atom forming three Mo–S bonds. Although the Moint atom at Moint-2 seems repulsively with lattice Mo atom, the calculated relative energy shows Moint-2 is about 0.13 eV lower than that of Moint-1, suggesting Moint-2 is more stable.
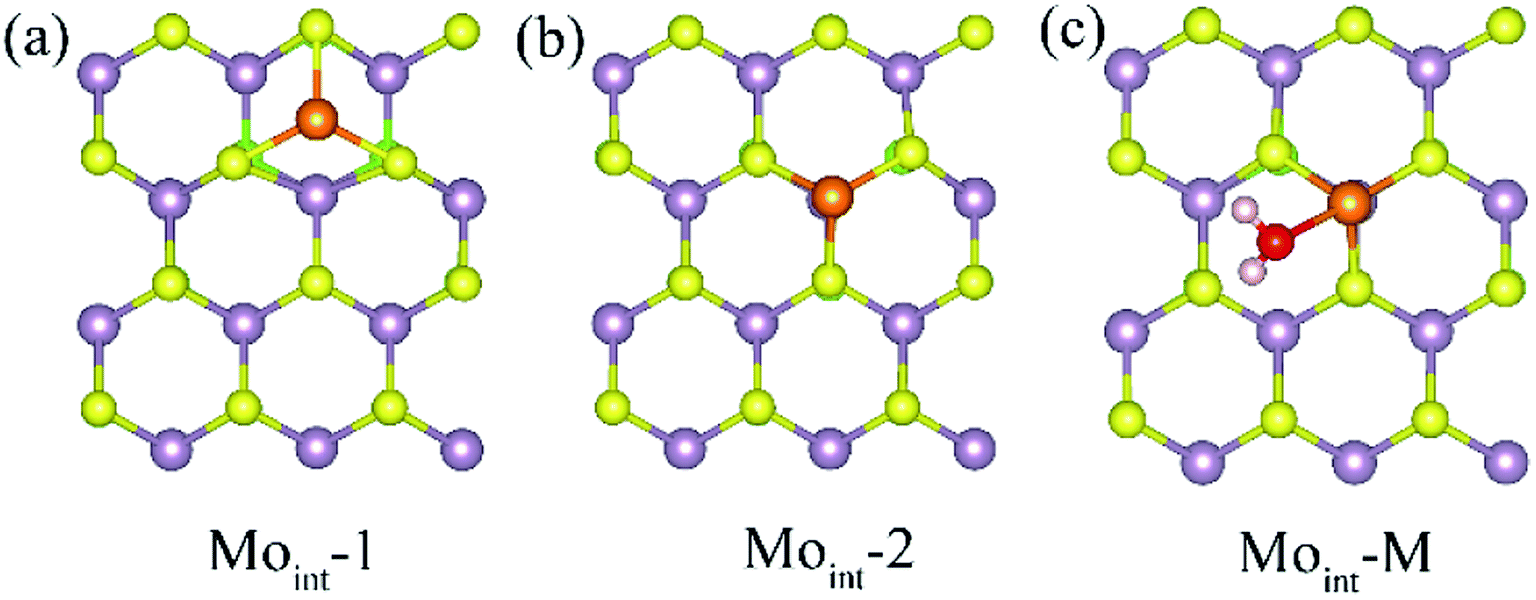 | ||
| Fig. 3 (a) and (b) The top view of possible Moint defect at Janus MoSSe sheet. (c) The adsorption of molecular water at Moint MoSSe sheet. The Moint atom is light orange ball. | ||
Based on the comparatively more stable Moint-2 structure, the behavior of M-H2O adsorption at Moint defective MoSSe sheet is investigated, and the corresponding calculated result is displayed in Table 3. Only one type of M-H2O adsorption configuration is found, denoted as Moint-M (Fig. 3). The corresponding M-H2O adsorption energy for Moint defective sheet is about −1.2 eV, comparable to that for the Moanti defective sheet. Furthermore, the difference between MoSSe and MoS2 sheet are checked. Compared with Moint MoSSe, the water adsorption at Moint MoS2 is close to that at MoSSe sheet, suggesting Moanti defect at both MoSSe and MoS2 sheet has identical effect on water adsorption. Thus, Moint defect can also effectively enhance water adsorption at defective MoSSe sheet. In all, the Svac defect can slightly increase the water adsorption, while Moanti and Moint can significantly enhance the interaction between water molecule and substrate.
The adsorption behavior of dissociated water at defective Janus MoSSe sheet
Above result shows the intrinsic defects have a prominent effect on the adsorption of water molecule at Janus MoSSe sheet. In order to know whether these defects could affect the water splitting, the adsorption behavior of dissociated water (D-H2O) is also explored. According to the possible atomic site, two types of D-H2O modes are existing for each defective MoSSe sheet as shown in Fig. 4. As for Svac defective sheet, the relaxed configuration of D-H2O can be denoted as Svac-Dn (n = 1, 2) for short. For example, the H atom of D-H2O in Svac-D1 is forming H–S next to Svac defect, while the OH is located at the site of Svac forming three Mo–O bonds. In Svac-D1, it forms two Mo–O bonds for Svac-D2. The case for Moanti defect is quite different because of the unsaturated bonds of lattice Mo atom near Moanti defect. Thus, it can form two different configurations of D-H2O with the H linked to lattice S atom in Moanti-D1 or to lattice Mo atom in Moanti-D2. As for D-H2O at Moint defect sheet, the H of D-H2O is linked to S atom next to Moint atom in Moint-D1, while both the OH and H atoms are directly absorbed to Moint atom in Moint-D2.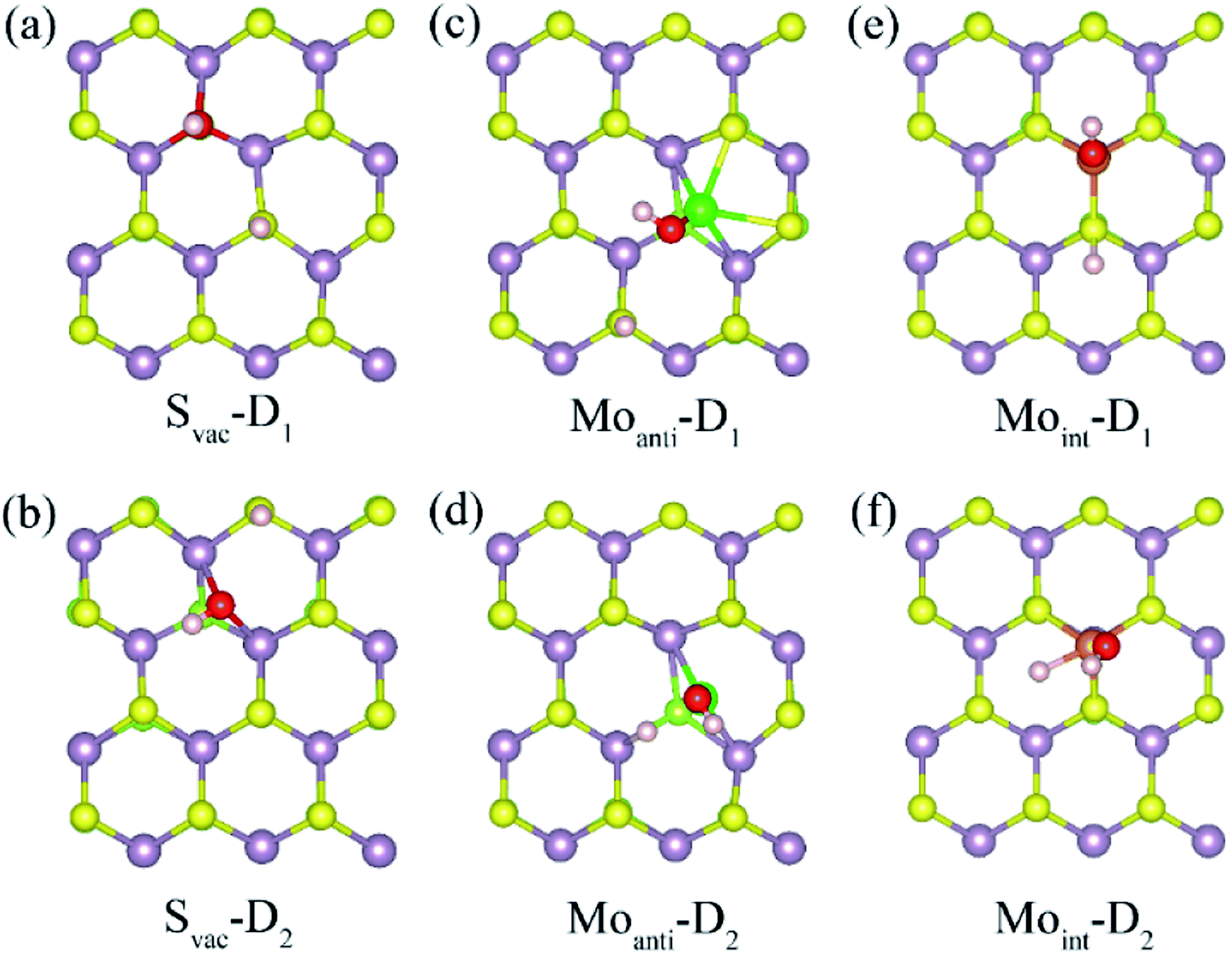 | ||
| Fig. 4 The relaxed structures of dissociated water at defective Janus MoSSe sheet. The top view of dissociated water at Svac (a) and (b), Moanti (c), (b), and Moint (e), (f) defective Janus MoSSe sheet. | ||
The calculated adsorption energy, energy difference, and the corresponding relaxed geometry parameters are displayed in Table 4. Surprisingly, it can be found that the adsorption energy of D-H2O at Svac Janus MoSSe sheet is positive, where Svac-D3 has a relative lower adsorption energy at about 0.658 eV. In order to check whether the M-H2O favors to form D-H2O, the energy difference between M-H2O and D-H2O is provided. The result shows that the energy difference is 0.985 eV for water at Svac defect sheet, showing M-H2O is more stable. Thus, the water molecule may be difficult to split at Svac Janus MoSSe sheet. As for water on Moanti defect sheet, the adsorption energies for D-H2O are negative, with Moanti-D2 lower adsorption energy at about −1.625 eV. Moreover, it is interesting to find that the energy difference is a negative value of −0.21 eV, indicating D-H2O is more stable. Thus, the molecular water may be favored to dissociate at Moanti defect sheet. Similar to the case at Moanti defect sheet, the adsorption energy for D-H2O at Moint defective Janus MoSSe sheet are all negative. And the energy difference is about −0.993 eV, which is larger than the case at Moanti sheet at about −0.21 eV. The lower adsorption energy of D-H2O is related with shorter Mo–O bond length. For example, the bond length of Mo–O at Svac-D1 sheet is changing from 2.117 Å to 1.91 Å for Moanti-D2, even shorten to 1.892 Å for Moint-D2. Therefore, the Moanti and Moint defects can greatly enhance the adsorption energy of D-H2O, which may promote the process of photocatalytic water splitting.
| System | D-H2O | Ead | Edif | H–S | O–Mo | O–H |
|---|---|---|---|---|---|---|
| Svac | Svac-D1 | 0.664 | 0.985 | 1.359 | 2.177 | 0.983 |
| Svac-D2 | 0.896 | 1.217 | 1.36 | 2.201 | 1.003 | |
| Moanti-2 | Moanti-D1 | −0.800 | 0.615 | 1.362 | 1.914 | 0.978 |
| Moanti-D2 | −1.625 | −0.209 | 1.778 (H–Mo) | 1.916 | 0.977 | |
| Moint-2 | Moint-D1 | −1.414 | −0.211 | 1.389 | 1.901 | 0.971 |
| Moint-D2 | −2.195 | −0.993 | 1.679 (H–Mo) | 1.892 | 0.972 |
Discussion
The above result shows that the intrinsic defects can dramatically enhance the interaction between water and Janus MoSSe sheet. To check the role of these defects, the structural and electronic property of intrinsic defects are discussed. First of all, the structural parameter of the defective structure is examined, since introducing defect will induce local structure distortion. It can be found that the bond length of Mo–S around the defect will be strongly shrunk, where it is shorten by 0.02 Å for Svac and 0.17 Å for Moint defect sheet. Furthermore, these defects will introduce unsaturated Mo atom at the Janus MoSSe sheet, which will enhance the reactivity of sheet.Except for the local structure distortion, the electronic structures of the defective structure are also examined since defects provide unique electronic property, which are crucial to the reactivity of surface. Here, the projected partial electronic density of state (PDOS) for defective Janus MoSSe sheet adsorbed with molecular water are calculated together with pristine case for comparison, and the corresponding calculated result is shown in Fig. 5. It should be noted that only the electronic density of states for Mo, S, O, and H atoms are provided, since the water is adsorbed at S side. For pristine sheet, we can find that the PDOS of adsorbed water remains in three intact peaks similar to single water molecule. This strongly suggests that the adsorbed water is hardly interact with pristine Janus MoSSe sheet, corresponding to weak adsorption energy of −0.16 eV.30 As a Svac defect exists in the structure, the three peaks are almost preserved, but the location of the peaks have a little red shift toward lower energy level, suggesting water adsorption is more favored corresponding larger adsorption energy of −0.23 eV. The case for Moanti and Moint defects are much different. First of all, it is unexpected to find that the three peaks disappear, and the PDOS of adsorbed water disperses into a large area, suggesting a strong interaction between O2p orbital of water with Mo3d orbitals of Moanti or Moint defect. Furthermore, the PODS of the adsorbed water is dispersed in the arrange from −6.6 eV to −5.0 eV for Moanti defect, and −6.0 eV to −5.2 eV for Moint defect sheet. Thus, a dramatic red shift of the PDOS for the adsorbed water is relative to the water adsorbed at pristine Janus MoSSe sheet. This results in larger adsorption energy of water at Moanti or Moint defect Janus MoSSe sheet with the value of −1.4 eV or 1.2 eV. Therefore, the origin of enhanced adsorption energy of water at defective S side of sheet should be the orbitals interaction between O2p of water and Mo3d of defects, together with the red shift of water energy level.
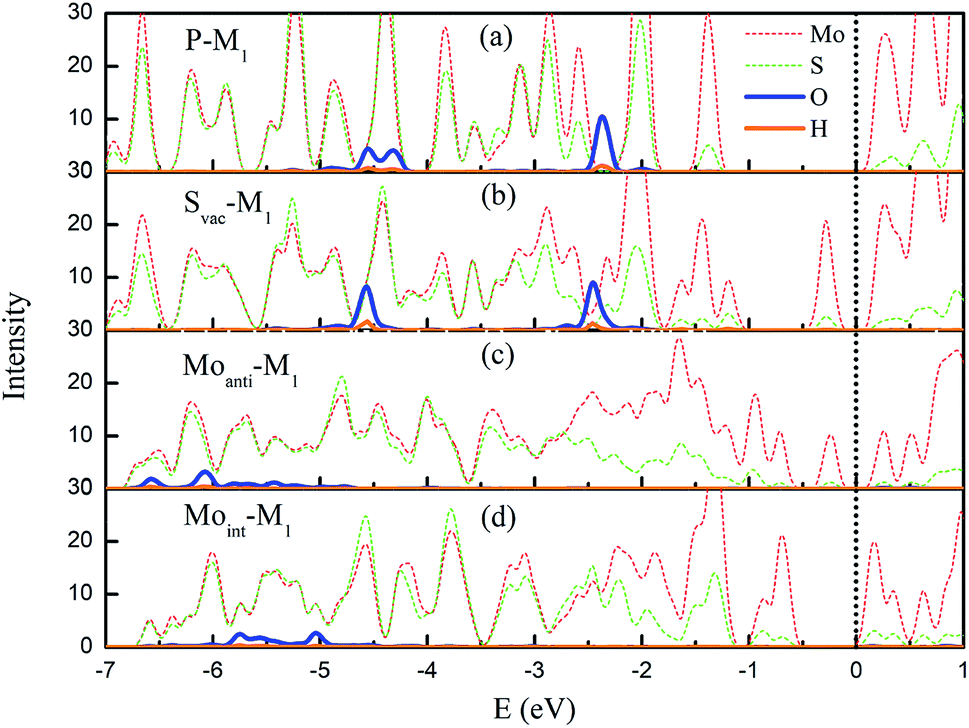 | ||
| Fig. 5 The projected partial electronic density of states (PDOS) of the M-H2O adsorbed at defective MoSSe sheet. The PDOS of H2O adsorbed at pristine Janus MoSSe sheet is also provided in (a) for comparison. (b)–(d) represent the PDOS of M-H2 adsorbed at Svac, Moanti, and Moint defective MoSSe sheet, respectively. The PDOS of Mo, S, O, and H atoms are in red (short dash), green (short dash), blue, and orange lines. The black dot line denotes the VBM, which is setting at zero eV. | ||
Conversion of water at defective Janus MoSSe sheet
As mentioned in introduction, Janus MoSSe sheet is a promising photocatalyst for water splitting, however it is limited by the weak water adsorption. Fortunately, the above result shows that Svac, Moanti and Moint intrinsic defects can greatly enhance the adsorption of water at Janus MoSSe sheet. Thus, it is of interest to know the effect of intrinsic defect on water conversion. Generally, it involves both the most stable M-H2O state and D-H2O state during the water splitting process, which can be expressed as:| H2O → H+ + OH− | (2) |
This reaction process involves two steps with the water molecule adsorbed at the Janus MoSSe sheet; consecutively, one H of M-H2O transfers to lattice S or Mo atom to form H–S or Mo–H bond, leaving OH bond at the sheet.
The calculated energy barrier of water conversion to H and OH at defective Janus MoSSe sheet are shown in Fig. 6. It should be mentioned that D-H2O is unable to form at pristine Janus MoSSe sheet. Thus, we only consider the water conversion at the defective sheet. The result shows that it requires 1.31 eV energy to overcome the transition state for water splitting at Svac defect sheet. During this process, the H atom of water towards to the adjacent lattice S atom through a transition state to form an H–S bond and an O–H bond as shown in Fig. 7(a). Additionally, the final state of D-H2O is a little lower than that of transition state, but 1.21 eV larger than initial state of M-H2O. Thus, water may be difficult to split at Svac defect Janus MoSSe sheet.
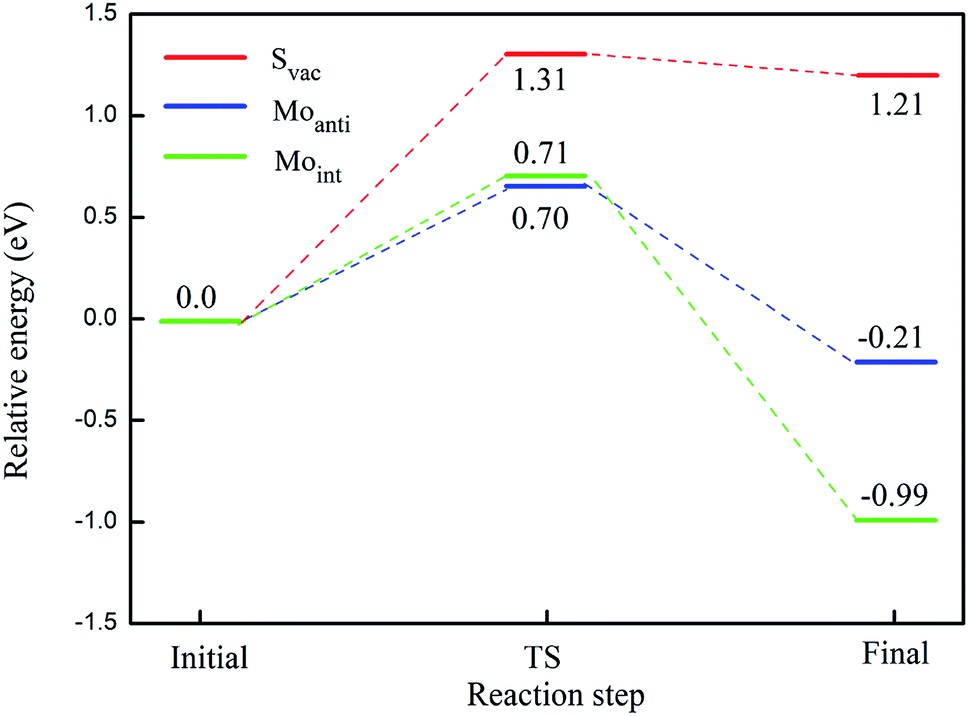 | ||
| Fig. 6 Illustration of reaction pathway via M-H2O to form H and OH bond. The sum of energies of the water and substrate is the zero reference for energy. The transition state is denoted as TS for short. The relative energy for water splitting at Svac, Moanti, and Moint defect sheet are in red, blue, and green lines, respectively. | ||
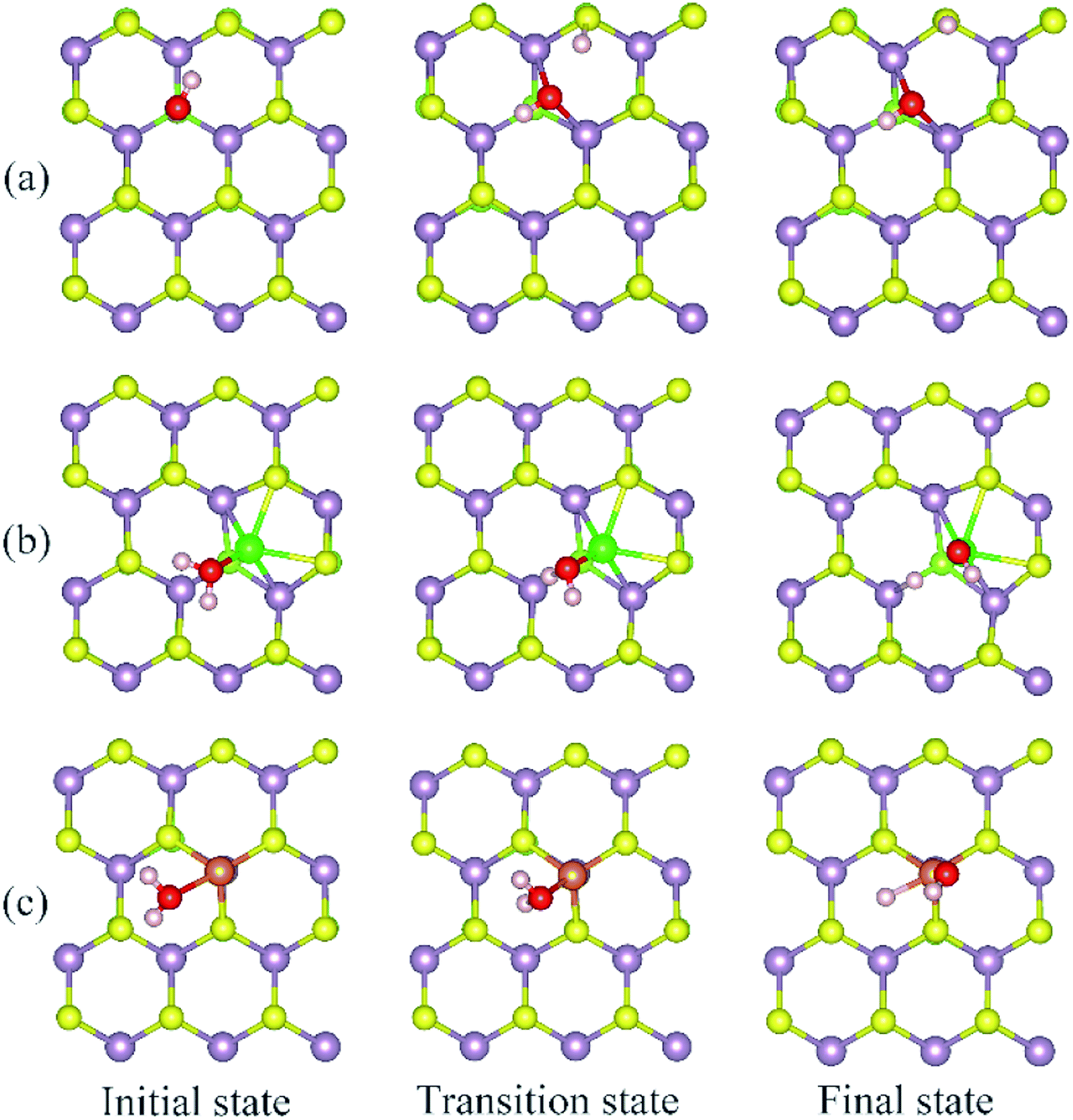 | ||
| Fig. 7 Selected configurations along the conversion pathway of water at defective Janus MoSSe sheet. The detailed defects are displayed in (a) Svac, (b) Moanti, and (c) Moint defective Janus MoSSe sheet. | ||
Compared to Svac defect, the behavior of other defects such as Moanti and Moint defects appears completely different. For example, for Moanti defect, one H atom of water links to unsaturated lattice Mo atom through a transition state as shown in Fig. 7(b). The corresponding energy barrier for the water splitting is about 0.71 eV, which is dramatically lower than that at Svac sheet. Furthermore, it is interesting to find that the relative energy of the final state is about 0.21 eV lower than the initial state. As for Moint defect, it has larger impact than Svac and Moanti defects on water splitting. The H atom of water towards to Moint atom forming Mo–H bond as shown in Fig. 7(c). The calculated result shows that it needs 0.7 eV to overcome the transition state, close to the case at Moanti sheet. Interestingly, the final state of D-H2O is about −0.99 eV, extremely lower than the initial state. Therefore, the Moanti and Moint defects have greatly impact on decreasing the energy barrier of water splitting, indicating introducing defect should be a promising approach to modify the reactivity of low dimensional materials.
Conclusion
In summary, the first-principles calculations are carried out to explore the effect of intrinsic defects on water adsorption and conversion at Janus MoSSe sheet. The result shows that relative to pristine case, the adsorption energy of M-H2O increases to −0.32 eV under Svac defect, while D-H2O is difficult to form. Unlike Svac defect, the effect of Moanti and Moint defect is much stronger than Svac defect on water behavior at Janus MoSSe sheet. The water adsorption energy at both Moanti and Moint sheet can be increased by −1.0 eV. More importantly, the energy barrier for water splitting can be lowered by 40% to 0.7 eV at Moanti or Moint defects sheet, and the final state of D-H2O is stable than the initial state. This significant enhancement of the adsorption energy originates from the strong interaction between O2p orbital and Mo3d orbitals. These results presented here showed that the intrinsic defect has the potential to change the property of materials, thus it is an important way to design photocatalyst for water splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science Challenge Project (TZ2018004) and the National Natural Science Foundation of China (No. 51572016, U1930402, 11804090, 11847213, 11774298, and 21503012). It is supported by the Scientific Research Fund of Hunan provincial Education Department, China (Grant No. 17C0626, and 2019JJ50148), and Scientific Research Fund of University (No. E517558). This research is supported by a Tianhe-2JK computing time award at the Beijing Computational Science Research Center (CSRC) and the Special Program for Applied Research on Super Computation of the NSFC Guangdong Joint Fund (the second phase) under Grant No. U1501501.References
- Yin, B. Wen, C. Zhou, A. Selloni and L.-M. Liu, Surf. Sci. Rep., 2018, 73, 58 CrossRef CAS.
- K. Wenderich and G. Mul, Chem. Rev., 2016, 116, 14587 CrossRef CAS PubMed.
- J. Su, Y. Wei and L. Vayssieres, J. Phys. Chem. Lett., 2017, 8, 5228 CrossRef CAS PubMed.
- M. Kapilashrami, Y. Zhang, Y. S. Liu, A. Hagfeldt and J. Guo, Chem. Rev., 2014, 114, 9662 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919 CrossRef CAS PubMed.
- Y. Yang, et al., Adv. Mater., 2018, 30, 1704479 CrossRef PubMed.
- R. Li, Y. Weng, X. Zhou, X. Wang, Y. Mi, R. Chong, H. Han and C. Li, Energy Environ. Sci., 2015, 8, 2377 RSC.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159 CrossRef CAS PubMed.
- P. Miro, M. Ghorbani-Asl and T. Heine, Angew. Chem., Int. Ed. Engl., 2014, 53, 3015 CrossRef CAS PubMed.
- Z. Tan, et al., J. Am. Chem. Soc., 2016, 138, 16612 CrossRef CAS PubMed.
- R. Li, L. Zhang, L. Shi and P. Wang, ACS Nano, 2017, 11, 3752 CrossRef CAS PubMed.
- F. Shayeganfar, K. S. Vasu, R. R. Nair, F. M. Peeters and M. Neek-Amal, Phys. Rev. B, 2017, 95, 144109 CrossRef.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159 CrossRef CAS PubMed.
- R. K. Joshi, S. Shukla, S. Saxena, G. H. Lee, V. Sahajwalla and S. Alwarappan, AIP Adv., 2016, 6, 015315 CrossRef.
- W. Li, Z. Lin and G. Yang, Nanoscale, 2017, 9, 18290 RSC.
- Y.-N. Z. Hui Zhang, H. Liu and Li-M. Liu, J. Mater. Chem. A, 2014, 2, 15389 RSC.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575 CrossRef CAS PubMed.
- W. S. Yun, S. W. Han, S. C. Hong, I. G. Kim and J. D. Lee, Phys. Rev. B, 2012, 85, 033305 CrossRef.
- S. Fathipour, et al., Appl. Phys. Lett., 2014, 105, 192101 CrossRef.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147 CrossRef CAS PubMed.
- K.-A. N. Duerloo, M. T. Ong and E. J. Reed, J. Phys. Chem. Lett., 2012, 3, 2871 CrossRef CAS.
- L. Dong, J. Lou and V. B. Shenoy, ACS Nano, 2017, 11, 8242 CrossRef CAS PubMed.
- Y. Guo, S. Zhou, Y. Bai and J. Zhao, Appl. Phys. Lett., 2017, 110, 163102 CrossRef.
- X. Li, Z. Li and J. Yang, Phys. Rev. Lett., 2014, 112, 018301 CrossRef PubMed.
- H. Yang, P. Zhao, Y. Ma, X. Lv, B. Huang and Y. Dai, J. Appl. Phys., 2019, 52, 455303 Search PubMed.
- X. Gao, Y. Shen, Y. Ma, S. Wu and Z. Zhou, Inorg. Chem., 2019, 58, 12053 CrossRef CAS PubMed.
- A.-Y. Lu, et al., Nat. Nanotechnol., 2017, 12, 744 CrossRef CAS PubMed.
- C. Shang, B. Xu, X. Lei, S. Yu, D. Chen, M. Wu, B. Sun, G. Liu and C. Ouyang, Phys. Chem. Chem. Phys., 2018, 20, 20919 RSC.
- W.-J. Yin, B. Wen, Q.-X. Ge, G.-Z. Nie, X.-L. Wei and L.-M. Liu, J. Mater. Chem. C, 2018, 6, 1693 RSC.
- X. Ma, X. Wu, H. Wang and Y. Wang, J. Mater. Chem. A, 2018, 6, 2295 RSC.
- H. U. Din, M. Idrees, A. Albar, M. Shafiq, I. Ahmad, C. V. Nguyen and B. Amin, Phys. Rev. B, 2019, 100, 165425 CrossRef CAS.
- W. Yin, B. Wen, Q. Ge, D. Zou, Y. Xu, M. Liu, X. Wei, M. Chen and X. Fan, Prog. Nat. Sci.: Mater. Int., 2019, 29, 335 CrossRef CAS.
- Z. Guan, S. Ni and S. Hu, J. Phys. Chem. C, 2018, 122, 6209 CrossRef CAS.
- W.-J. Yin, M. Krack, B. Wen, S.-Y. Ma and L.-M. Liu, J. Phys. Chem. Lett., 2015, 6, 2538 CrossRef CAS PubMed.
- W.-J. Yin, B. Wen, Q.-X. Ge, X.-L. Wei, M. Chen and L.-M. Liu, Comput. Mater. Sci., 2018, 155, 424 CrossRef CAS.
- J. Sá, C. Garlisi, G. Palmisano, J. Czapla-Masztafiak, Y. Kayser and J. Szlachetko, Mater. Chem. Phys., 2018, 208, 281 CrossRef.
- F. Gunkel, et al., Phys. Rev. X, 2016, 6, 031035 Search PubMed.
- Y. Wang, J. Zhao, T. Wang, Y. Li, X. Li, J. Yin and C. Wang, J. Catal., 2016, 337, 293 CrossRef CAS.
- X. Ma, X. Yong, C.-c. Jian and J. Zhang, J. Phys. Chem. C, 2019, 123, 18347 CrossRef CAS.
- D. Merki, H. Vrubel, L. Rovelli, S. Fierro and X. Hu, Chem. Sci., 2012, 3, 2515 RSC.
- X. Zhang, et al., Mater. Res. Express, 2019, 6, 105055 CrossRef CAS.
- K. Zhang, et al., Nano Lett., 2017, 17, 6676 CrossRef CAS PubMed.
- Z. Wang, L. Chu, L. Li, M. Yang, J. Wang, G. Eda and K. P. Loh, Nano Lett., 2019, 19, 2840 CrossRef CAS PubMed.
- R. Chaurasiya and A. Dixit, Appl. Surf. Sci., 2019, 490, 204 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103 CrossRef CAS.
- M. Krack, Theor. Chem. Acc., 2005, 114, 145 Search PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- J. A. Stefan Grimme, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132(1), 154104 CrossRef PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |