
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConvergent access to bis-1,2,4-triazinyl-2,2′-bipyridines (BTBPs) and 2,2′-bipyridines via a Pd-catalyzed Ullman-type reaction†
Gabrielle D. Waters and
Jesse D. Carrick*
and
Jesse D. Carrick*
Department of Chemistry, Tennessee Technological University, Cookeville, TN 38505-0001, USA. E-mail: jcarrich@tntech.edc
First published on 16th March 2020
Abstract
Multidentate, soft-Lewis basic, complexant scaffolds have displayed significant potential in the discrete speciation of the minor actinides from the neutron-absorbing lanthanides resident in spent nuclear fuel. Efforts to devise convergent synthetic strategies to targets of interest to improve liquid–liquid separation outcomes continue, but significant challenges to improve solubility in process-relevant diluents to effectively define meaningful structure–activity relationships remain. In the current work, a synthetic method to achieve the challenging 2,2′-bipyridine bond of the bis-1,2,4-triazinyl-2,2′-bipyridine (BTBP) complexant class leveraging a Pd-catalyzed Ullman-type coupling is reported. This convergent strategy improves upon earlier work focused on linear synthetic access to the BTBP complexant moiety. Method optimization, relevant substrate scope and application, as well as a preliminary mechanistic interrogation are reported herein.
1 Introduction
Remediation of hazardous radionuclides produced as byproducts of the nuclear fuel cycle has been an area of intense research focus in recent decades as the need for improved efficiency of carbon-neutral alternative energy sources has increased. Minor actinides, such as 241Am have high levels of radioactivity and a half-life of over 400 years. Developing efficient techniques for the separation of the minor actinides from lanthanides is at the forefront of innovation due to the potential for the evolution of advanced recycling of spent nuclear fuel (SNF) via the partition and transmutation strategy. Issues that arise in separating minor actinides from lanthanides are the similarity in electronic properties which present challenges for liquid–liquid separations.1 Soft-Lewis basic, multi-dentate complexants, including tri- and tetra- on various scaffolds, which adhere to the CHON principle have demonstrated significant potential in this area2 in the last thirty years.3The 2,2′-bipyridine construct has been utilized extensively as a ligand for various metal-mediated processes,4 in addition to more contemporary areas of research such as photoredox catalysis.5 With an ongoing interest to develop ligands for catalytic transformations based on N-donor atoms, this moiety will continue to find relevance in synthetic method development,6 materials,7 and separations.8 Previous work in this laboratory focused on the goal of affording bis-1,2,4-triazyinyl-2,2′-bipyridines (BTBPs) directly from the premetalated zinc reagent via Negishi coupling9 without success (Fig. 1). This method was suitable for the production of hemi-1,2,4-triazinyl-2,2′-bipyridines (hemi-TBPs, 4), but was unable to be adapted to BTBPs even with extensive further optimization attempts. Traditional methods for accessing BTBPs often involve linear synthetic routes from the corresponding bis-carbonitrile 7 (ref. 10) and commonly employ toxic reagents. Condensation reactions with 1,2-dicarbonyls are frequently utilized, but symmetric complexants with limited functional group diversity are necessarily afforded. These constructs limit the opportunity to develop structure–activity relationships towards improved performance. These nonconvergent approaches also present limitations for complexant diversity.
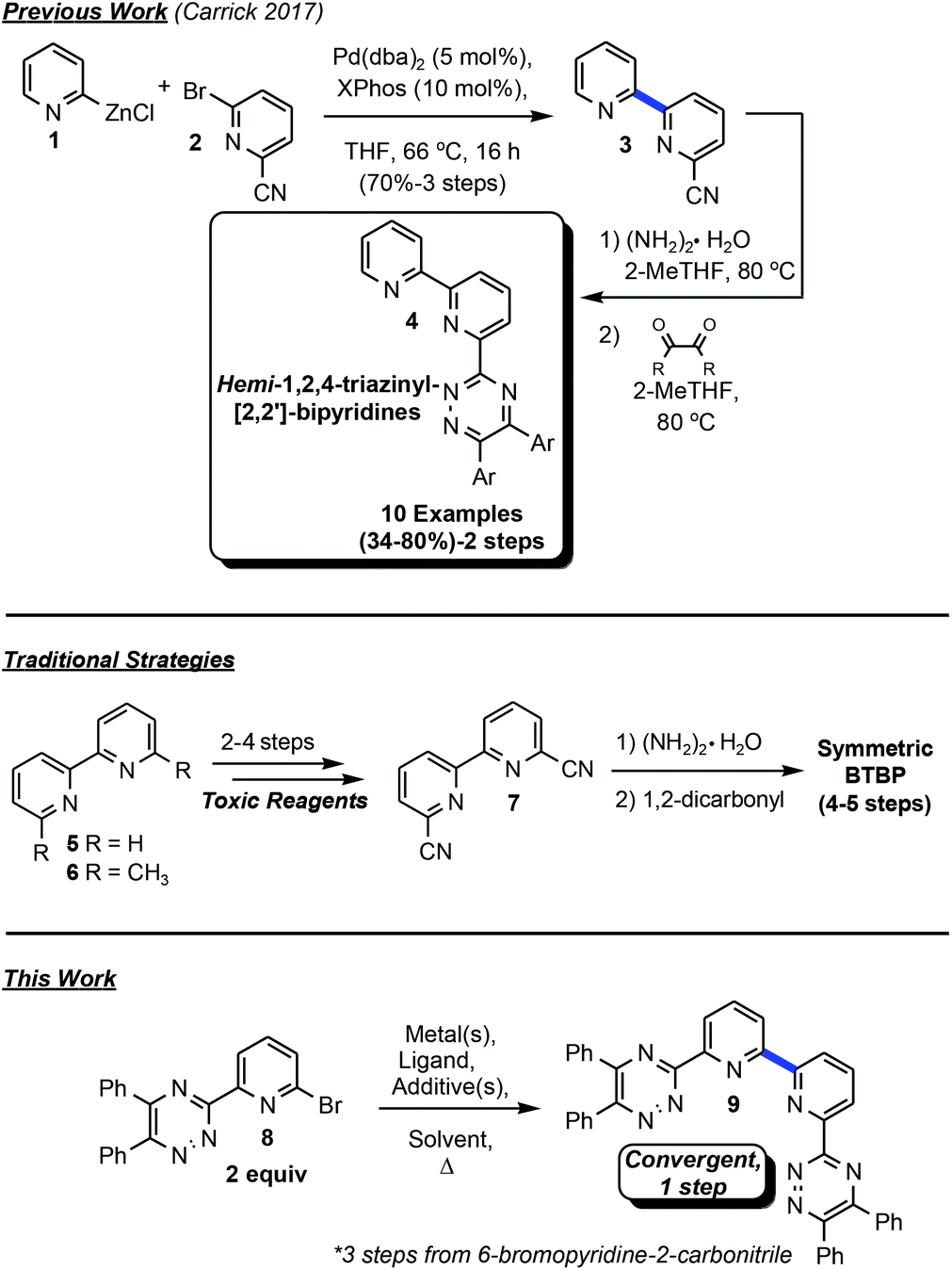 | ||
| Fig. 1 HTBP and BTBP access synthetic summary. | ||
Treatment of 2,2′-bipyridine under oxidative conditions followed by trimethylsilylcyanide11 or, alternatively, selenium-mediated oxidation of 6,6′-dimethyl-2,2′-bipyridine, subsequent condensation with hydroxylamine, tosylation, and finally elimination afford entry into 7.12 Commercial sources of 7 can frequently be cost prohibitive for development work.13 Undeterred by our previous lack of success in merging two discrete 6-bromo-2-pyridinyl-1,2,4-triazines and the construction of the 2,2′-bipyridine bond towards BTBPs, we set out upon a different course to employ metal-mediated strategies for direct bond formation via an Ullman-type14 homocoupling strategy. Method discovery, optimization, application to BTBPs and 2,2′-bipyridines, and preliminary efforts toward understanding the complex mechanistic underpinnings of this transformation are reported herein.
2 Results and discussion
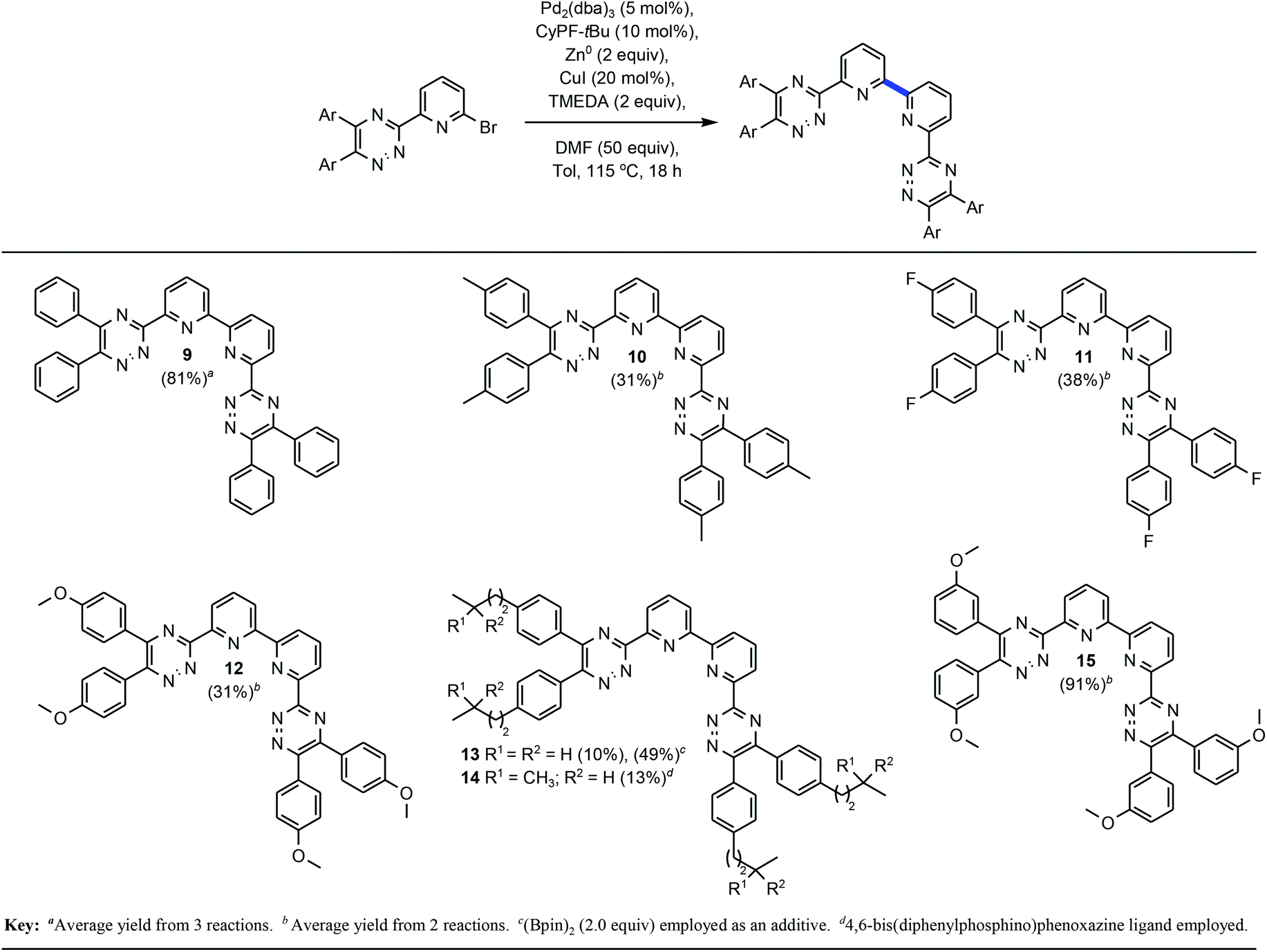
2.1 Method optimization
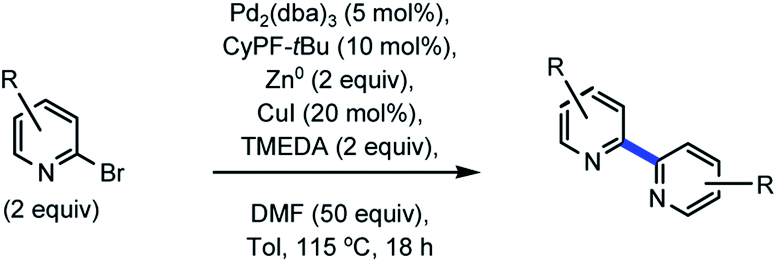
Table 1 delineates method optimization towards the desired BTBP end products. Initial reaction conditions began with screening a variety of nickel catalysts. Conditions pursuant to the Weix procedures15 with NiBr2·3H2O and manganese metal under reductive conditions afforded the proto-dehalogenated variant of 8 [MTPPhen] (entry 1).16 A similar observation was made with entries 3 and 5. This result is suggestive that the starting material had successfully accomplished an oxidative addition or, potentially a two-electron reduction with manganese or magnesium metal, but the subsequent transmetalation and reductive elimination steps did not transpire consequently affording the protodemetalated or dehalogenated 8. Evaluation of various other nickel catalysts with additives had similar results, or resulted in no conversion of 1 (entries 3–4).17 Kumada coupling18 was explored on an example that utilized isopropyl magnesium chloride to premetalate 8 via Mg–X exchange19 (entry 5) in concert with (2R)-1-[(1R)-1-[bis(1,1-dimethylethyl)phosphino]ethyl]-2-(dicyclohexylphosphino)ferrocene (CyPF-tBu) developed in the Hartwig laboratory20 and utilized in earlier Pd-catalyzed amination work from this group,21 but no conversion of 8 towards 9 was observed.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Expt. | Catalyst (mol%) | Ligand (mol%) | Metal (equiv.) | Additive (equiv.) | Solvent (M) | Temp. (°C) | Time (h) | Conv.b (yield)c |
| a Tetramethylethylenediamine (TMEDA), cyclopentyl methyl ether (CPME), dimethylformamide (DMF), dimethylacetamide (DMAc), toluene (Tol), or methyl tert-butyl ether (MTBE) as solvent. CyPF-tBu = 1-dicyclohexylphosphino-2-di-t-butylphosphinoethylferrocene.b Conversion determined from integration of select resonances in the 1H NMR spectrum without internal standard.c Isolated, purified yield where applicable.d Protodehalogenated MTPPhen afforded. | ||||||||
| 1 | NiBr2·3H2O (5%) | Phenanthroline (5%) | Mn0 (2.0) | — | DMF (0.05) | 40–140 | 48 | 0d |
| 2 | NiBr2·3H2O (5%) | — | Mg0 (1.0) | — | CPME (0.3) | 40 | 24 | 0 |
| 3 | NiCl2(dppp) (5%) | — | Zn0 (2.0) | TFA (0.1) | DMAc (0.3) | 150 | 24 | 0d |
| 4 | NiCl2·6H2O (5%) | — | Zn0 (1.2) | LiCl (1.0), AcOH (0.01) | DMF (0.5) | 60 | 12 | 0 |
| 5 | Pd2(dba)3 (5%) | CyPF-tBu (10%) | iPrMgCl (1.65) | CuI (2.0) | THF (0.3) | 66 | 24 | 0 |
| 6 | Pd2(dba)3 (5%) | XPhos (10%) | Zn0 (2.0) | TFA (0.1) | DMAc (0.3) | 66–110 | 48 | 0d |
| 7 | Pd(OAc)2 (5%) | RuPhos (10%) | Zn0 (2.0) | TFA (0.1) | Tol (0.3) | 110 | 24 | 0 |
| 8 | Pd(OAc)2 (5%) | XantPhos (10%) | Zn0 (2.0) | TFA (0.1), CuI (0.5) | DMF (0.3) | 115 | 48 | 0 |
| 9 | Pd2(dba)3 (5%) | CyPF-tBu (10%) | Zn0 (2.0) | TFA (0.1) | Tol (0.3) | 110 | 24 | 0 |
| 10 | Pd(dppf)2Cl2 (5%) | — | Zn0 (2.0) | TEA (3.0), CuI (0.2) | MTBE (0.25) | 55 | 36 | 0 |
| 11 | Pd2(dba)3 (5%) | CyPF-tBu (10%) | Zn0 (2.0) | TFA (0.1), CuI (0.2) | DMF (0.3) | 115 | 48 | 10 |
| 12 | Pd2(dba)3 (5%) | CyPF-tBu (10%) | Zn0 (2.0) | CuI (0.2) | DMF (0.3) | 115 | 36 | 30 |
| 13 | Pd2(dba)3 (5%) | CyPF-tBu (10%) | Zn0 (2.0) | CuI (0.2) | DMF (0.3) | 150 | 18 | 85(43) |
| 14 | Pd2(dba)3 (5%) | CyPF-tBu (10%) | Zn0 (2.0) | CuI (0.2), 1,4-dioxane (2.0) | DMF (0.3) | 115 | 18 | 22 |
| 15 | Pd2(dba)3 (5%) | CyPF-tBu (10%) | Zn0 (2.0) | CuI (0.2), TMEDA (2.0) | DMF (0.3) | 115 | 18 | 99(60) |
| 16 | Pd2(dba)3 (5%) | t-Bu-BrettPhos (10%) | Zn0 (2.0) | CuI (0.2), TMEDA (2.0) | DMF (0.3) | 115 | 18 | 63 |
| 17 | Pd2(dba)3 (5%) | dppf (10%) | Zn0 (2.0) | CuI (0.2), TMEDA (2.0) | DMF (0.3) | 115 | 18 | 95(51) |
| 18 | Pd2(dba)3 (5%) | CyPF-tBu (10%) | Zn0 (2.0) | CuI (0.2), TMEDA (2.0) | DMSO (0.3) | 115 | 18 | 0 |
| 19 | Pd2(dba)3 (5%) | CyPF-tBu (10%) | Zn0 (2.0) | CuI (0.2), TMEDA (2.0) | CPME (0.3) | 105 | 18 | 0 |
| 20 | Pd2(dba)3 (5%) | CyPF-tBu (10%) | Zn0 (2.0) | Cu0 (0.2), TMEDA (2.0) | DMF (0.3) | 115 | 18 | <10 |
| 21d | Pd2(dba)3 (2.5%) | CyPF-tBu (5%) | Zn0 (2.0) | CuI (0.2), TMEDA (2.0) | DMF (0.3) | 115 | 18 | <10 |
| 22 | Pd2(dba)3 (5%) | CyPF-tBu (5%) | Zn0 (2.0) | CuI (0.2), TMEDA (2.0) | DMF (0.3) | 115 | 18 | 15 |
| 23 | Pd2(dba)3 (5%) | CyPF-tBu (5%) | Zn0 (2.0) | CuI (0.2), TMEDA (2.0), DMF(50) | Tol (0.3) | 115 | 18 | 99(81) |
With preliminary experiments proving unproductive, palladium catalysts were evaluated with an incorporation of zinc towards facilitation of a one-pot Negishi coupling22 with various Buchwald23 ligands (entries 7–9). No desired product was afforded. The catalyst/ligand combinations explored in entries 7–9 were previously successful for the development of Suzuki–Miyaura cross-coupling24 as well as Buchwald–Hartwig amination on similar substrates.21 From past work involving Sonogashira coupling,25 copper(I) iodide was a competent additive for successful Pd-catalyzed alkynylation of 8. Entry 10 was a direct evaluation of successful Sonogashira conditions, which did not afford 9. For entries 9–11, copper(I) iodide was added to the reaction vessel after 24 hours. This was a turning point for reaction development as conversion of 8 towards 9 was observed spectroscopically by 1H NMR for entry 11 that incorporated knowledge from the current endeavor, in addition to previous work on Pd-catalyzed amination. Looking to improve the conversion, trifluoroacetic acid (TFA) was excluded from entry 12. The conversion increased three-fold, and suggested that protic acid was an inhibitor of the desired transformation and could have been accelerating protodemetalation. Increasing the temperature from 115 to 150 °C resulted in 85% conversion (entry 13). A series of experiments which evaluated ligands described to stabilize zinc complexes26 were subsequently incorporated to evaluate potential enhancement of the desired transformation.
2.2 BTBP scope
Addition of N,N,N′,N′-tetramethylethylenediamine (TMEDA), afforded nearly full conversion (entry 15) to the BTBP scaffold 9 and an initial 60% isolated yield from automated flash-column chromatography. When 1,4-dioxane was utilized as a stabilizing ligand 22% conversion was observed (entry 14).27 Screening of additional ligands for palladium (entries 16 and 17) validated CyPF-tBu as the most potent ligand in this transformation with the best impurity profile.Alternative solvents, including DMSO and CPME (entries 18 and 19) were evaluated and demonstrated DMF's profound influence as the superior solvent. Entry 20 evaluated the impact of a Cu0 intermediate versus CuX+ in the reaction sequence. Given the minimal yield obtained, it is hypothesized that CuX+ intermediates play a key transmetallating role in the desired outcome of the transformation. Decreasing the catalyst and ligand loading by half resulted in little to no conversion (entry 21). When entry 21 was performed with toluene as the solvent and DMF as an additive approximately 50% conversion was achieved with a worse impurity profile. Similarly, changing the zinc loading from 2 to 0.2 resulted in lower conversion (entry 22) further emphasizing the probability of facilitated two-electron reduction of the sp2 C–Br bond as a fundamental step in the transformation. Further optimization of entry 22 substantiated that utilizing DMF as an additive and toluene as the reaction solvent, or cosolvation, the isolated yield could be dramatically improved (entry 23). This set of conditions formed the baseline of which other examples would be explored. As part of due diligence, the appropriate control experiments which systematically and singularly removed, the catalyst, ligand, zinc, copper(I) iodide, and DMF with all other reagents remaining were performed. As expected, all control experiments resulted in no conversion of 8 and underscore the synergy of all reagents towards the formation of 9. With a seemingly viable method for the convergent construction of BTBP complexants realized, our focus progressed towards the evaluation of the substrate scope and transformation limits. Scheme 1 above highlights the BTBP scope evaluated for the developed method. Preparation of the required Br-MTP starting materials has been previously disclosed.21b,24a Commercially available benzils were utilized for the construction of the starting materials for 9–12, and 15, whereas synthesis of the benzil was required for 13–14. Purification of the desired end products via automated flash chromatography presented numerous challenges. Incorporation of DMF into the crystal lattices of solid end products contributed to difficulties in purification and characterization of pure materials.
 | ||
| Scheme 1 Evaluation of BTBP scope with diversified 6-bromo-[1,2,4]-triazinyl pyridines. | ||
The developed transformation was tolerant electron-withdrawing functionality (11) affording the desired convergent product in 38% yield. Alkoxy-substituted-1,2,4-triazines have found utility in separations processes of minor actinides28 and were of significant interest in the context of the current work. Pleasingly, the 4,4′-, as well as the 3,3′-dimethoxy-BTP proved competent examples for the transformation albeit in markedly different isolated yields which were most succinctly attributed to purification issues. Aliphatic, inductively electron-donating alkyl substituents on the Br-MTP starting materials proved challenging for this transformation. Although, the desired products were afforded in the case of 10 and 13–14 yields were modest in all four cases and additional attempts at optimization did not prove fruitful.29 An additional aliphatic example, 6,6′-bis-[5,6-bis(4-(3,3-dimethylbutyl)phenyl)-[1,2,4]triazin-3-yl]-2,2′-bipyridine, was successful by the described method, but purification challenges, poor solubility, and tendency towards degradation did not facilitate inclusion.
Given the high yield of 9, it was postulated that this specific product may be favorably interacting with the metal complex intermediates facilitating greater turnovers. As such, incorporation of 9 as additive did not accelerate the improvement of yield in the context of the aliphatic examples. It was, however, subsequently demonstrated that the addition (2.0 equiv.) of bispinacolatodiboron (B(pin)2) could facilitate an improved isolated yield of 13 as a direct correlation of earlier work,24a,30 but not in the case of aliphatic example 14. After thoroughly evaluating the BTBP scope of the 2,2′-bipyridine bond formation via this method, our focus shifted towards applications of 2,2′-bipyridines with excision of the 1,2,4-triazine moiety (Table 2).
|
|||
|---|---|---|---|
| Entry | Product | (Conv.a/yieldb) | |
| a Conversion determined from integration of selected resonances in the 1H NMR spectrum without internal standard.b Isolated, purified yield.c Attempted four times, starting material conversions was not achieved.d No conversion of starting material.e Reaction was continued for 60 h without product formation.f Less than 2 mol% conversion of starting material was observed. Isolation was not attempted. | |||
| 1 |  |
16 (86)a | |
| 2 | 17 R = 4,4′-CH3 | 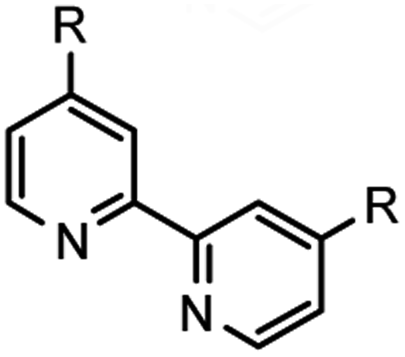 |
17 (95)a |
| 3 | 18 R = 4,4-CF3 | 18 (99)a | |
| 4 | 19 R = 6,6′-CN | 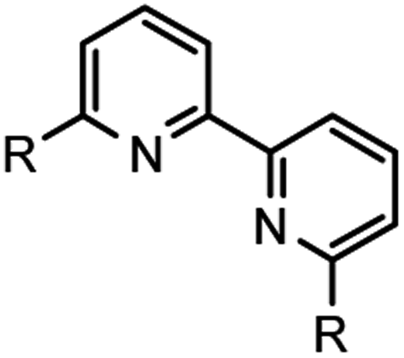 |
19 (70)a |
| 5 | 20 R = 6,6′-OCH3 | 20 (22)a,c | |
| 6 | 21 R = 6,6′-CO(OCH3) | 21 (10)a,d | |
| 7 | 22 R = 6,6′-CH3 | 22 (0)a,e | |
| 8 | 23 R = 6,6′-Ph | 23 (0)a,f | |
2.3 Bipyridine scope
Table 2 below summarizes our evaluation of the developed transformation in the context of less functionalized, and more ubiquitous 2,2′-bipyridines. Purification challenges existed within the confines of the successful examples. Formation of 16–18 (entries 1–3) was successful without substantive deviation from the developed procedure and each was observed in high conversion. Product 16 was isolated in 62% yield, whereas isolation of the product in high purity remained unrealized in the cases of 17 and 18 even though clean starting material conversion was observed spectroscopically in both cases. Access to 2,2′-bipyridines with electron-withdrawing functionality can present challenges for successful synthesis.31 In the case of 18 numerous work up conditions were attempted using existing protocols,17 acid, or base in addition to modified chromatographic procedures without success. When electron-withdrawing functionality was present at the 2-position on the 6-bromopyridine starting material mixed results were observed (entries 4–6). Good isolated yield of 19 was realized underscoring this approach as a complement to previously reported results in comparable yield.32 The resonance donating ability of the 6-methoxy substituent is perceived to negatively influence the coupling outcome towards 20 (entry 5) in a similar way to entry 7. Entry 6 afforded no conversion towards 21, but not as the result of addition to the ester carbonyl group of any of the reaction intermediates. While strongly, inductively electron-withdrawing substituents in the 4-position were compatible substrates (entry 3), similar results were not observed in the specific case of entry 6.A striking contradiction was observed in entry 7 when R = CH3. While the methyl substituent in the 4-position was a competent substrate in the transformation affording high conversion to the desired product 17, transposition of this substituent to the 6-position failed to afford 22. Interestingly, the 2-phenyl-6-bromopyridine example which failed to afford the homocoupled product 23, indirectly substantiates that the 1,2,4-triazine may play a critical role in stabilization of one, or more transition states toward the production of end products as excision of this moiety from the substrate in entry 8 did not afford any conversion of starting material, decomposition, or otherwise. Construction of biphenyls, specifically biphenyl, 4,4′-dimethoxybiphenyl, 4,4′-difluorobiphenyl, and 4,4′-dimethyldiphenyl to address inductively electron neutral, donating, and withdrawing functionality were attempted during the course of the investigation. All examples failed to afford conversion of starting material via the method described.
2.4 Utilization of BTBP in Ullman-type coupling
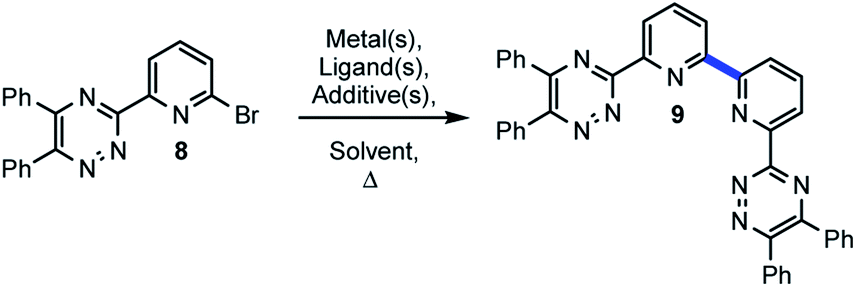
Given the high conversion and isolated yield of 9 (Scheme 1) we were curious if this product could be serving as a ligand for Pd and accelerating the homocoupling reaction in this case. An experiment was designed and executed whereby 9 would substitute for CyPF-tBu for a challenging example under the optimized conditions (Scheme 1).33 Pursuant to the aforementioned, 9 was utilized in concert with the remaining reaction parameters delineated in Table 1 to afford 11 in 60% conversion and 20% isolated yield. When CyPF-tBu was utilized as the ligand 11 was afforded in higher conversion and isolated yield (38%, Scheme 2).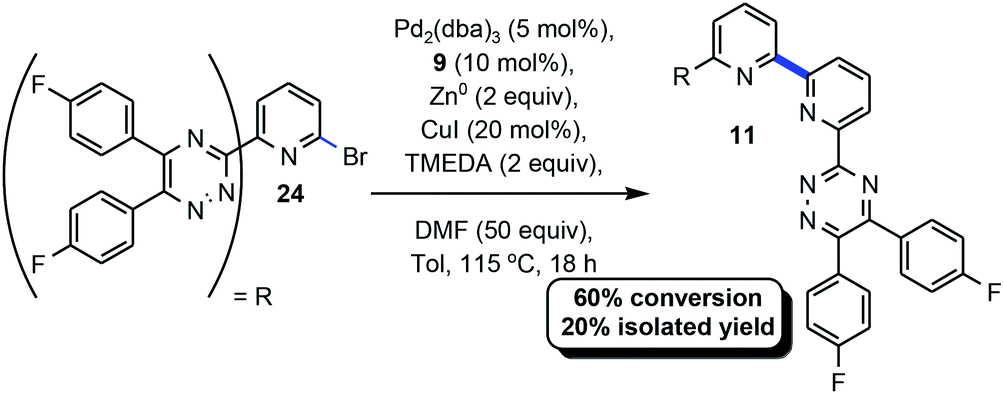 | ||
| Scheme 2 BTBP ligand in Ullmann-type coupling. | ||
2.5 Probing the mechanism for Ullman-type coupling
The standard transformation optimized in Table 1 above employed a standard reaction vial which was not purged with inert gas prior to performance of the experiment. Scheme 3 probed the potential role oxygen or other single-electron processes may play in this system. An experiment which involved complete removal of oxygen under an Ar atmosphere afforded 0% of 9 suggesting that oxygen may play a role in oxidizing one or more of the metalated intermediates. Interestingly, and seemingly contradictive, when the experiment was conducted in a pure oxygen atmosphere 9 was not afforded. Finally, incorporation of a super stoichiometric amount of the free radical scavenger, 2,2,6,6-tetramethyl-1-piperidinyloxy free radical (TEMPO), did not lead to the formation of 9.34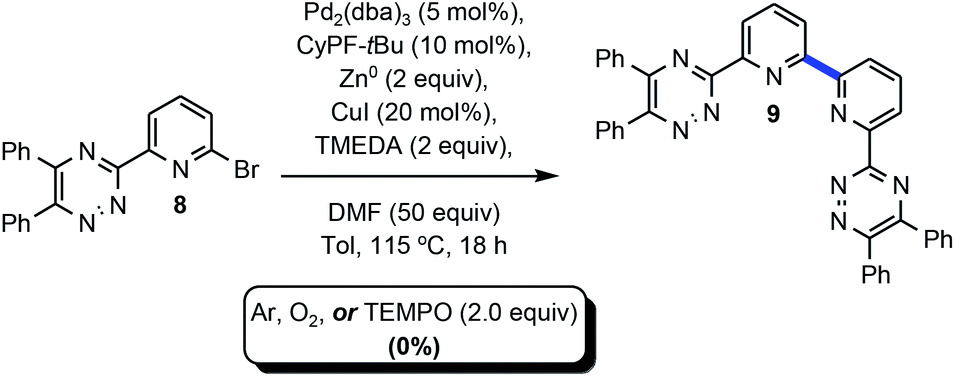 | ||
| Scheme 3 Efforts toward understanding the operative reaction mechanism. | ||
3 Conclusions
In summary, the authors have disclosed a Pd-catalyzed Ullman-type coupling for the convergent synthesis of functionalized BTBPs which advances access to these important materials for separations beyond traditionally linear approaches. The examples describe highlight strategic opportunities to acquire diversified complexants for the definition structure–activity relationships in separations systems. Preliminary mechanistic evaluation of the transformation suggests a synergistic transformation dependent on cooperativity of Pd2+, Zn0, and Cu1+. Evaluation of prepared complexants in separations experiments of simulated spent nuclear fuel are ongoing and will be reported in due course.4 Experimental
4.1 General considerations
All reagents were purchased from U.S. chemical suppliers, stored according to published protocols, and used as received unless indicated otherwise. All experiments were performed in oven- or flame-dried glassware. Reaction progress was monitored using thin-layer chromatography on glass-backed silica gel plates and/or 1H NMR analysis of crude reaction mixtures. Rf values for compounds that resulted in a concentrically observed spot on normal phase silica gel are reported using the conditions listed. Melting point data listed is for a single, uncorrected experiment unless noted otherwise. All reported yields listed are for pure compounds and corrected for residual solvent, if applicable, from 1H NMR spectroscopy, unless otherwise indicated. In cases where multiple examples were attempted, the yield data for the highest discrete yield is included and average yield data is reported above in Table 2 or Scheme 3. Infrared spectral data was acquired from the (form) listed. All 1H and 13C NMR data was acquired from a 500 MHz or 900 MHz multinuclear spectrometer with broad-band N2 cryo- (500 MHz) or liquid He cryoprobe (900 MHz). Chemical shifts are reported using the δ scale and are referenced to the residual solvent signal: CDCl3 (δ 7.26) and (CD3)2S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (2.50) for 1H NMR and CDCl3 (δ 77.15), (CD3)2SO (δ 39.52), and (CD3)2CO (29.84) for 13C{1H} NMR.35 Splittings are reported as follows: (br) = broad, (s) = singlet, (d) = doublet, (t) = triplet, and (m) = multiplet. 13C NMR spectra obtained on a 500 or 900 MHz spectrometer with broadband cryoprobe were corrected for ringdown using linear back prediction. High resolution mass spectrometry (HRMS) data was obtained utilizing electron impact ionization (EI) with a magnetic sector (EBE trisector), double focusing-geometry mass analyzer.
O (2.50) for 1H NMR and CDCl3 (δ 77.15), (CD3)2SO (δ 39.52), and (CD3)2CO (29.84) for 13C{1H} NMR.35 Splittings are reported as follows: (br) = broad, (s) = singlet, (d) = doublet, (t) = triplet, and (m) = multiplet. 13C NMR spectra obtained on a 500 or 900 MHz spectrometer with broadband cryoprobe were corrected for ringdown using linear back prediction. High resolution mass spectrometry (HRMS) data was obtained utilizing electron impact ionization (EI) with a magnetic sector (EBE trisector), double focusing-geometry mass analyzer.
Method A. This method was utilized with all entries in Scheme 1 except 10 and 14, as well as all entries in Table 2. To an 8 mL reaction vial equipped with a magnetic stir bar at ambient temperature was charged Zn0 (2.0 equiv.), TMEDA (2.0 equiv.), and 3-(6-bromo-pyridin-2-yl)-5,6-diphenyl-[1,2,4]triazine (0.025 g, 0.064 mmol, 0.5 equiv.). Toluene and DMF were added successively to a separate 20 mL vial and 1/3 of the total mixture (0.3 M) was combined with the first vial and agitated for 0.5 hours. In a separate 8 mL reaction vial at ambient temperature was charged Pd2(dba)3 (5 mol%), CyPF-tBu (10 mol%), CuI (20 mol%), 3-(6-bromo-pyridin-2-yl)-5,6-diphenyl-[1,2,4]triazine (0.025 g, 0.064 mmol, 0.5 equiv.) in 1/3 of the DMF and Tol mixture. Once vial 1 had been agitated for 0.5 hours the vials were combined and the vial charged with Zn0 was rinsed with the last 1/3 of the mixture. The resulting slurry was heated at 115 °C for the time indicated upon which time the crude reaction was concentrated to afford the crude mixture which was absorbed on Celite and purified using automated flash column chromatography under the discrete conditions for each described compound to afford the pure compound in the listed yield.
Method B. This method was used for 10 in Scheme 1. Compound 14 in Scheme 1 followed this procedure as well with the exception of utilization of 4,6-bis(diphenylphosphino)phenoxazine as the ligand instead of CyPF-tBu. To an 8 mL reaction vial equipped with a magnetic stir bar at ambient temperature was charged Zn0 (2 equiv.), TMEDA (2 equiv.), and 3-(6-bromo-pyridin-2-yl)-5,6-diphenyl-[1,2,4]triazine (0.0167 g, 0.027 mmol, 0.33 equiv.). Combine the toluene and DMF in a separate 20 mL vial and take 1/3 of total mixture (0.3 M) to add to the first vial and let agitate for 0.5 hours. In a separate 8 mL reaction vial at ambient temperature was charged Pd2(dba)3 (5 mol%), CyPF-tBu (10 mol%), CuI (20 mol%), 3-(6-bromo-pyridin-2-yl)-5,6-diphenyl-[1,2,4]triazine (0.0167 g, 0.0269 mmol, 0.33 equiv.) in 1/3 of the total mixture. Once the first vial has agitated for 0.5 hours the vials were combined, the vial charged with zinc was rinsed with the last 1/3 of the mixture and added to the third vial containing the remainder of the 3-(6-bromo-pyridin-2-yl)-5,6-diphenyl-[1,2,4]triazine. The resulting slurry was heated at 115 °C. After two hours at the intended temperature, a third of the portion of the mixture in the last vial is added to the reaction mixture. This is repeated two more times in two hour intervals until all of reagent is added to the reaction vial. Upon the reaction finishing at which time the crude reaction was concentrated to afford the crude mixture which was absorbed on Celite and purified using automated flash column chromatography under the discrete conditions for each described compound to afford the pure compound in the listed yield.
4.2 Compound data
![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) max = 3080, 2941, 2920, 1607, 1578, 1563, 1496, 1479, 1353, 1181, 1075, 814, 791 cm−1; HRMS (EI): m/z: [M]+ calcd for C44H34N8 674.2906; found: 674.2907.max = 1603, 1578, 1564, 1519, 1511, 1408, 1396, 1386, 1356, 1159, 1148, 840, 805 cm−1; HRMS (EI): m/z: [M]+ calcd for C40H22F4N8 690.1904; found: 690.1881.max = 3079, 2954, 2923, 2851, 1605, 1576, 1565, 1516, 1494, 1359, 1250, 1173, 1031, 1016, 833, 801 cm−1; HRMS (EI): m/z: [M]+ calcd for C44H34N8O4 738.2703; found: 738.2720.max = 3059, 3032, 2956, 2928, 2857, 1609, 1580, 1567, 1492, 1466, 1379, 837, 802 cm−1; HRMS (EI): m/z: [M]+ calcd for C56H58N8 842.4784; found: 842.4758.max = 3060, 3033, 2952, 2926, 2868, 1608, 1580, 1568, 1488, 1468, 1376, 1351, 1185, 820, 797, 722 cm−1; HRMS (EI): m/z: [M–C3H7]+ calcd for C57H59N8 855.4857; found: 855.4835.max = 3064, 3003, 2956, 2932, 2835, 1599, 1580, 1503, 1487, 1452, 1428, 1376, 1362, 1289, 1266, 1241, 1038, 1019, 921, 862, 809, 787, 730, 701 cm−1; HRMS (EI): m/z: [M]+ calcd for C44H34N8O4 738.2703; found: 738.2668.
max = 3080, 2941, 2920, 1607, 1578, 1563, 1496, 1479, 1353, 1181, 1075, 814, 791 cm−1; HRMS (EI): m/z: [M]+ calcd for C44H34N8 674.2906; found: 674.2907.max = 1603, 1578, 1564, 1519, 1511, 1408, 1396, 1386, 1356, 1159, 1148, 840, 805 cm−1; HRMS (EI): m/z: [M]+ calcd for C40H22F4N8 690.1904; found: 690.1881.max = 3079, 2954, 2923, 2851, 1605, 1576, 1565, 1516, 1494, 1359, 1250, 1173, 1031, 1016, 833, 801 cm−1; HRMS (EI): m/z: [M]+ calcd for C44H34N8O4 738.2703; found: 738.2720.max = 3059, 3032, 2956, 2928, 2857, 1609, 1580, 1567, 1492, 1466, 1379, 837, 802 cm−1; HRMS (EI): m/z: [M]+ calcd for C56H58N8 842.4784; found: 842.4758.max = 3060, 3033, 2952, 2926, 2868, 1608, 1580, 1568, 1488, 1468, 1376, 1351, 1185, 820, 797, 722 cm−1; HRMS (EI): m/z: [M–C3H7]+ calcd for C57H59N8 855.4857; found: 855.4835.max = 3064, 3003, 2956, 2932, 2835, 1599, 1580, 1503, 1487, 1452, 1428, 1376, 1362, 1289, 1266, 1241, 1038, 1019, 921, 862, 809, 787, 730, 701 cm−1; HRMS (EI): m/z: [M]+ calcd for C44H34N8O4 738.2703; found: 738.2668.4.3 Special instructions
It should be noted that 11 demonstrated significant sensitivity to acetonitrile, acetone, and methanol. Pursuant to the aforementioned, these solvents should be avoided where possible for the purposes of chromatographic purification, transfer, and/or NMR analysis. Deuterated chloroform and dimethyl sulfoxide were suitable alternative solvents for spectroscopic or other characterization.Additionally, prepared materials readily incorporate residual N,N-dimethylformamide and ethyl acetate into the crystal lattice which were challenging to remove via standard reduced pressure techniques. Chromatography with acetonitrile effectively removed the N,N-dimethylformamide whereas azeotropic removal of ethyl acetate with acetonitrile in all cases except 11 proved successful.
Conflicts of interest
The authors declare no competing interest.Acknowledgements
Financial support for this work was provided by an award from the U.S. Department of Energy, Basic Energy Sciences, Separations Program Award: DE-SC0018033. An award from the National Science Foundation (NSF) Major Research Instrumentation Program (1531870) is gratefully acknowledged for the acquisition of the University's 500 MHz multinuclear NMR spectrometer with broad-band N2 cryoprobe. The authors would like to thank Dr Qiaoli Liang, The University of Alabama, for acquisition of HRMS data, and Dr Markus W. Voehler for acquisition of the 13C NMR of 11, supported in part by grants for NMR instrumentation from the NSF (0922862), National Institutes of Health (S10 RR025677) and Vanderbilt University matching funds.References
-
(a) H. H. Dam, D. N. Reinhoudt and W. Verboom, Chem. Soc. Rev., 2007, 36, 367 RSC
; (b) K. L. Nash and M. Nilsson, in Reprocessing and Recycling of Spent Nuclear Fuel, ed. R. Taylor, Woodhead, Cambridge, UK, 2015, pp 3–23 Search PubMed
- Z. Kolarik, Chem. Rev., 2008, 108, 4208 CrossRef CAS PubMed
-
(a) Z. Kolarik, U. Müllich and F. Gassner, Solvent Extr. Ion Exch., 1999, 17, 1155 CrossRef CAS
- For a recent review on metal-mediated processes with bipys and related ligands see:
(a) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681 CrossRef CAS PubMed
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed
- Bipy method development review(s);
(a) C. Kaes, A. Katz and M. W. Hosseini, Chem. Rev., 2000, 100, 3553 CrossRef CAS PubMed
-
(a) H. Jia, B. Billmann, H. Onishi, J. Smiatek, S. Roeser, S. Wiemers-Meyer, R. Wagner, M. Winter and I. Cekic-Laskovic, Chem. Mater., 2019, 31, 4025 CrossRef CAS
-
(a) J. M. Muller, S. S. Galley, T. E. Albrecht-Schmitt and K. L. Nash, Inorg. Chem., 2016, 55, 11454 CrossRef CAS PubMed
- R. P. Downs, A.-L. Chin, K. M. Dean and J. D. Carrick, J. Heterocycl. Chem., 2017, 54, 3008 CrossRef CAS
-
(a) F. W. Lewis, L. M. Harwood, M. J. Hudson, A. Geist, V. N. Kozhevnikov, P. Distler and J. John, Chem. Sci., 2015, 6, 4812 RSC
-
(a) C. N. Noufele, C. T. Pham, A. Hagenbach and U. Abram, Inorg. Chem., 2018, 57, 12255 CrossRef CAS PubMed
- F. W. Lewis, L. M. Harwood, M. J. Hudson, M. G. B. Drew, A. Wilden, M. Sypula, G. Modolo, T.-H. Vu, J.-P. Simonin, G. Vidick, N. Bouslimani and J. F. Desreux, Procedia Chem., 2012, 7, 231 CrossRef CAS
- $97.00 for 250 mg or $214 per 1 g from prominent chemical vendors' websites, accessed 10/11/19 Search PubMed.
- For relevant reviews see:
(a) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed
-
(a) J. A. Buonomo, D. A. Everson and D. J. Weix, Synthesis, 2013, 45, 3099 CrossRef CAS PubMed
- S. Tai, S. V. Marchi and J. D. Carrick, J. Heterocycl. Chem., 2016, 53, 1138 CrossRef CAS
- L.-Y. Liao, X.-R. Kong and X.-F. Duan, J. Org. Chem., 2013, 79, 777 CrossRef PubMed
- For a review see:
(a) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed
- P. Knochel, C. Sämann, V. Werner and N. M. Barl, Heterocycles, 2014, 90, 827 CrossRef
-
(a) Q. Shen, S. Shekhar, J. P. Stambuli and J. F. Hartwig, Angew. Chem., Int. Ed., 2005, 44, 1371 CrossRef CAS PubMed
-
(a) S. Tai, S. V. Marchi, E. J. Dover and J. D. Carrick, J. Org. Chem., 2015, 80, 6275 CrossRef CAS PubMed
- For a recent review on the Negishi cross-coupling see:
(a) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9507 CrossRef PubMed
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed
-
(a) A.-L. Chin and J. D. Carrick, J. Org. Chem., 2016, 81, 1106 CrossRef CAS PubMed
- S. Chaudhuri and J. D. Carrick, J. Org. Chem., 2018, 83, 10261 CrossRef CAS PubMed
- S. L. Buchwald, P. Knochel, C. Stathakis, S. Bernhardt and J. R. Colombe, Org. Lett., 2013, 15, 5754 CrossRef PubMed
- Handbook of Functionalized Organometallics, ed. P. Knochel, Wiley-VCH, 2005, vol. 1, pp 251–333 Search PubMed
- T. G. Hill, A.-L. Chin, S. Tai, J. D. Carrick, D. D. Ensor and L. H. Delmau, Sep. Sci. Technol., 2018, 53, 1848 CrossRef CAS
- Purification issues with 10 inhibited high isolated yield. Compound 11 afforded high conversion of starting material, but also presented purification challenges. Compound 12 afforded 50% conversion of starting material with substantive purification issues. Product 13 afforded minimal conversion after 50 hours of reaction time in the absence of (Bpin)2. When the reaction sequence towards 14 was performed with Xantphos under the optimized conditions described in Table 1 no product was afforded necessitating the utilization of the alternative ligand.
- A. E. Lambert and J. D. Carrick, J. Heterocycl. Chem., 2018, 55, 1232 CrossRef CAS
- L. Hao, J. Oppenheimer, M. R. Smith and R. E. Maleczka, Tetrahedron Lett., 2016, 57, 2231 CrossRef
- J. Peng, X. Liu and Y. Kishi, Tetrahedron Lett., 2011, 52, 2172 CrossRef CAS
- A synthetic procedure to 25 can be found in ref. 24a.
-
(a) A. C. Albéniz, P. Espinet, R. López-Fernández and S. Ayusman, J. Am. Chem. Soc., 2002, 124, 11278 CrossRef PubMed
-
(a) G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00673d |
| This journal is © The Royal Society of Chemistry 2020 |