
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective convergent synthesis of 2-arylidene thiazolo[3,2-a]pyrimidines as potential anti-chikungunya agents†
Mohamed Fares *ab,
Patrick M. McCoskerac,
Muhammad A. Alsherbinyd,
Anthony C. Willise,
Timothy Clarkc,
Johan Neytsf,
Dirk Jochmansf and
Paul A. Keller*a
*ab,
Patrick M. McCoskerac,
Muhammad A. Alsherbinyd,
Anthony C. Willise,
Timothy Clarkc,
Johan Neytsf,
Dirk Jochmansf and
Paul A. Keller*a
aSchool of Chemistry & Molecular Bioscience, Molecular Horizons, University of Wollongong, Illawarra Health & Medical Research Institute, Wollongong, NSW 2522, Australia. E-mail: Ph.fares@yahoo.com; keller@uow.edu.au
bSchool of Chemistry, The University of Sydney, NSW 2006, Australia
cDepartment of Chemistry and Pharmacy, Computer-Chemistry-Center (CCC), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Nägelbachstrasse 25, 91052 Erlangen, Germany
dNICM Health Research Institute, Western Sydney University, Westmead, NSW 2145, Australia
eResearch School of Chemistry, The Australian National University, Canberra, ACT 2601, Australia
fKU Leuven (University of Leuven), Department of Microbiology and Immunology, Rega Institute for Medical Research, Laboratory of Virology and Chemotherapy, B-3000, Leuven, Belgium
First published on 31st January 2020
Abstract
Convergent and convenient regioselective synthesis of novel thiazolo[2,3-a]pyrimidine derivatives was accomplished using the one-pot reaction of 6-ethylthiouracil, bromoacetic acid, anhydrous sodium acetate, acetic anhydride, acetic acid and suitable aldehyde. X-ray crystallographic study reveals the presence of the Z configuration of only one regioisomer confirmed by computational studies as being the most likely isomer present.
Arboviruses mainly use arthropod vectors for transmission between hosts and they are considered a growing global health threat.1,2 This arises from the increasing number of travellers, from 450 million in 1990 to nearly 1.4 billion in 2018, and consequently travel-related diseases incidence has increased significantly.3 Chikungunya virus (CHIKV) (Alphavirus genus, family Togaviridae) is one of the prevalent tropical alphaviruses that is transmitted by Aedes mosquitoes.4,5 After an incubation period of 2–4 days, symptoms start with the onset of high fever and severe persistent joint pain that may last for weeks to years.6,7 Other symptoms of chikungunya fever (CHIKF) include headache, vomiting, rash, myalgia (muscle pain), photophobia and maculopapular rash.6,8 Unlike other arbovirus infections, around 15% of the patients may encounter “silent infections”.6
A variety of strategies have been applied to develop anti-CHIKV treatments, including vaccines, and for small molecule inhibitors, random screening, computer aided drug design, ligand-based drug design, high throughput screening and genome-wide loss of function screen.9–17 Non-steroidal anti-inflammatory drugs (NSAID) have been used to alleviate the symptoms of the CHIKF because of the lack of effective prevention or curing of the viral infection.18 However, so far there is no vaccine or approved medication to prevent or treat CHIKV infection.
The recent advances and growing knowledge about the arboviruses expand our understanding of the CHIKV life cycle, pathogenesis and infection.1–6 Intensive research in the past 10 years has led to the identification of new chemical scaffolds that might be potential anti-CHIKV agents. Rhodanine I is a thiazolidin-4-one derivative, presenting a scaffold that exhibited broad antiviral activity against HCV, HIV-1 and Chikungunya (Fig. 1).19 A series of rhodanine derivatives was tested for their anti-CHIKV activity by a cytopathic effect (CPE) reduction assay (Fig. 1). The 2-methyl analogue II emerged as the best compound and displayed excellent activity with submicromolar EC50 (EC50 = 0.42 μM) against CHIKV. A docking study also showed good interaction between the ligand and CHIKV nsp2 protease.19
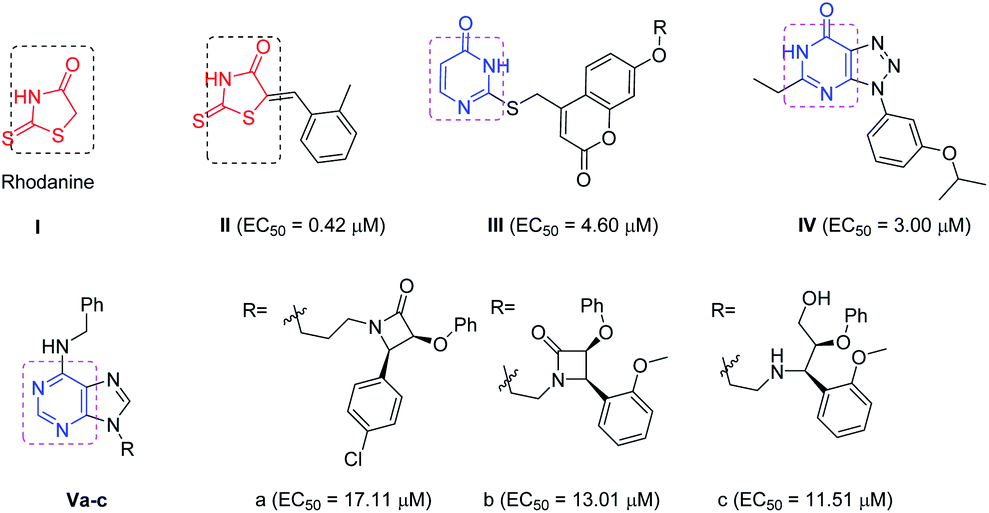 | ||
| Fig. 1 Examples of heterocyclic classes showing anti-CHIKV activity, including thiazolidin-4-one I and II, pyrimidine III and pyrimidine fused rings IV and IVa–c. | ||
Among the main heterocycles that form the basis of small molecule inhibitors of CHIKV are the pyrimidine and fused pyrimidine rings, e.g. III, IV and Va–c (Fig. 1).15,20,21 Recent reports indicated that hybridization between uracil–coumarin–arenes III resulted in the discovery of a novel anti-CHIKV scaffold that was found to impede CHIKV replication.20 In an effort to find new leads as CHIKV inhibitors, a random screening identified the [1,2,3]triazolo[4,5-d]pyrimidin-7(6H)ones as potential candidates.21 Iterative optimization cycles led to the most potent compound IV with EC50 = 3 μM and a selectivity index greater than 600 (Fig. 1).21 In 2012, purine-β-lactams Va and Vb and purine-amino propanol Vc (Fig. 1) conjugates were screened against nine different viruses including CHIKV. The amino propanol derivative Vc (EC50 = 11.51–17.11 μM) showed relatively higher activity other than the purine-β-lactam hybrids. Therefore, the β-lactam was not essential for activity.15
Recently, hybridization of two or more bioactive fragments has emerged as a concept for the exploration of novel multitarget acting inhibitors as well as novel anti CHIKV agents (Fig. 2).15,20,22,23 Therefore, in our ongoing efforts to discover new scaffolds for CHIKV infection, we investigated a molecular hybridization approach with the fusion of the uracil and the rhodanine pharmacophoric moieties (Fig. 2).22
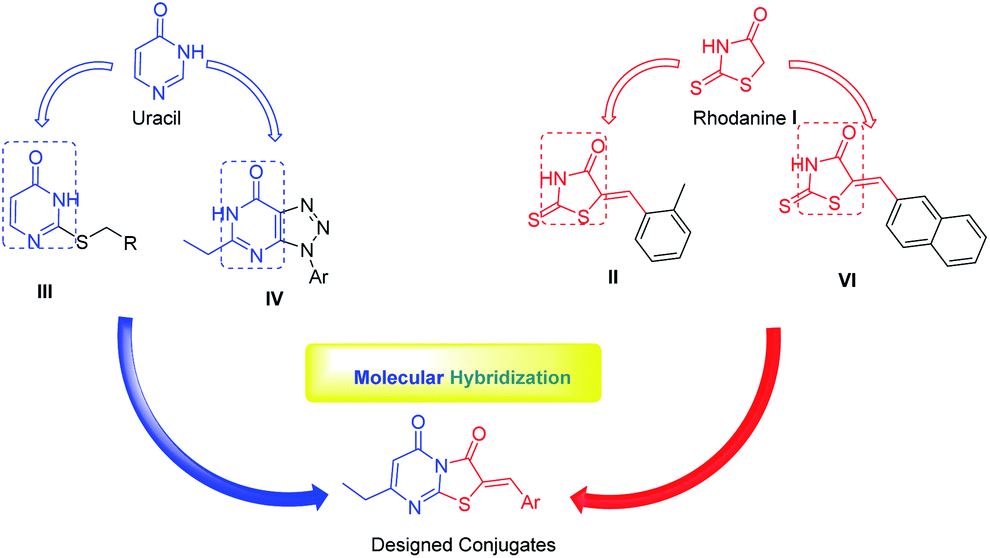 | ||
| Fig. 2 Design of the targeted uracil–rhodanine conjugates. | ||
The synthetic strategy started with the condensation of ethyl propionylacetate 1 and thiourea 2 to afford the known 6-ethyl thiouracil 3 with 69% yield.24,25 Although the synthesis of the thiazolopyrimidine derivatives from the 6-ethylthiouracil substrate has not previously been reported, this multicomponent reaction (MCR) was achieved using an analogous procedure26–29 by reacting 3 with the aldehyde, chloroacetic acid, sodium acetate anhydrous, acetic anhydride and acetic acid in one pot and at reflux for 4 h (Scheme 1).
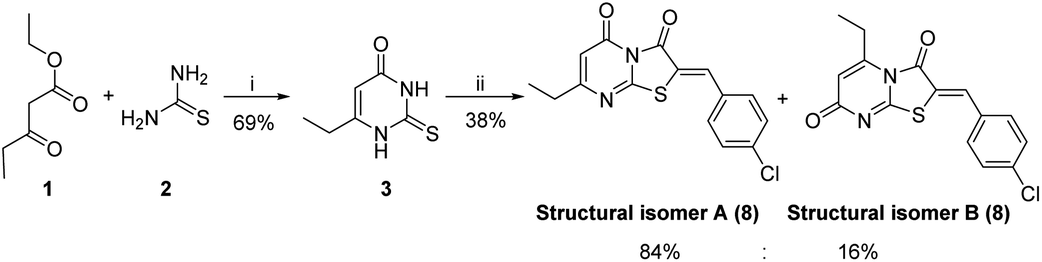 | ||
| Scheme 1 Reagents and conditions: (i) NaOEt, C2H5OH, 6 h, reflux; (ii) ClCH2COOH, anhydrous CH3COONa, (CH3CO)2, CH3COOH, 4-chlorobenzaldehyde, reflux, 4 h. | ||
These conditions consistently gave low yields and two regioisomers (e.g. chlorobenzaldehyde 8 A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B 16%:84%), the latter determined by analysis of the 1H NMR spectrum (Fig. 3) – attempts to separate the isomers by column chromatography failed.
:B 16%:84%), the latter determined by analysis of the 1H NMR spectrum (Fig. 3) – attempts to separate the isomers by column chromatography failed.
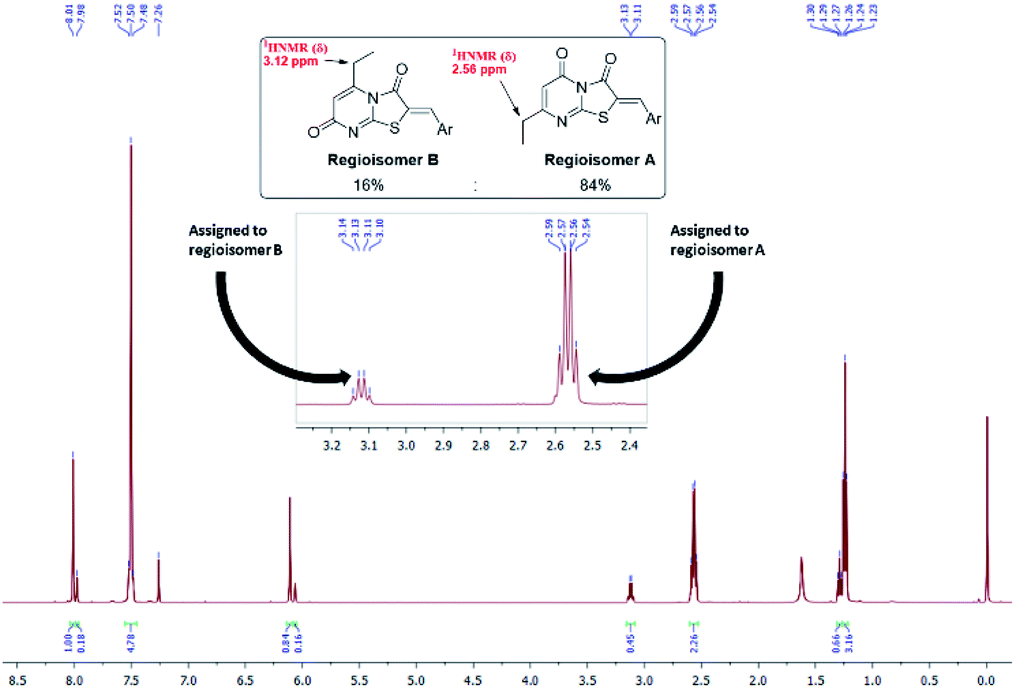 | ||
| Fig. 3 1H NMR spectrum of the MCR product of the two regioisomers of 8 in a ratio of 1:5. | ||
Optimization of the reaction involved varying the mode of addition, replacement of the chloroacetic acid with bromoacetic acid, and lowering the reaction temperature. This aimed to favour the formation of the kinetic regioisomer (Z)-A, assumed to be the desired product, over the thermodynamic regioisomer (Z)-B (Scheme 2), and resulted in the crystallization of only one regioisomer from the reaction mix (acetic acid) in each case. This may be due to the low reactivity and the less regioselectivity of the chloroacetic acid compared to bromoacetic acid.
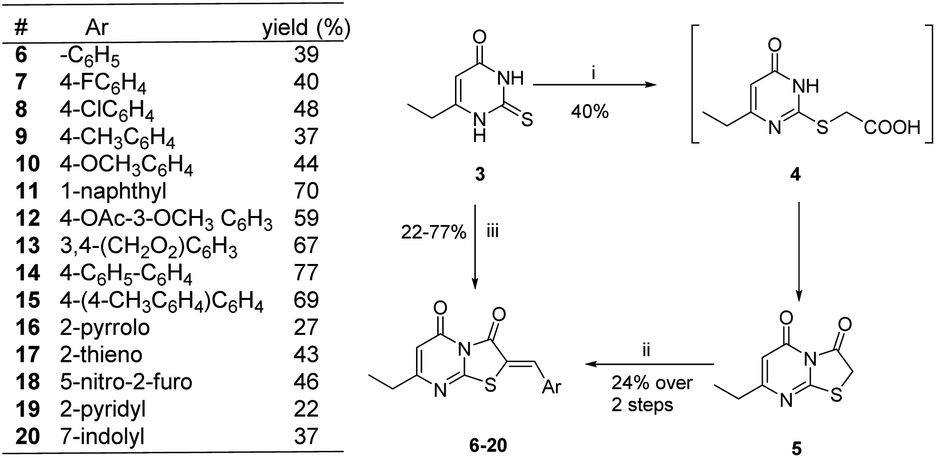 | ||
| Scheme 2 Reactions and conditions: (i) BrCH2COOH, anhydrous CH3COONa, (CH3CO)2, CH3COOH, 60 °C, 3 h; (ii) benzaldehyde, anhydrous CH3COONa, CH3COOH, 60 °C, 2 h. (iii) BrCH2COOH, anhydrous CH3COONa, (CH3CO)2, CH3COOH, aldehyde, 60 °C, 4 h. | ||
From the convergent reaction conditions, four possible isomers can be formed: the Z or E configurations of structural isomers A or B – (Z)-A, (E)-A, (Z)-B and (E)-B (Fig. 4). Analysis of the 1H NMR spectra showed a quartet at 2.48–2.57 ppm assigned to methylene hydrogens of the ethyl substituent – these were assigned to structural isomer A as the corresponding resonances of isomer B are expected to resonate more downfield (∼3 ppm) as a result of a deshielding anisotropic effect of the adjacent carbonyl group of the thiazole ring.
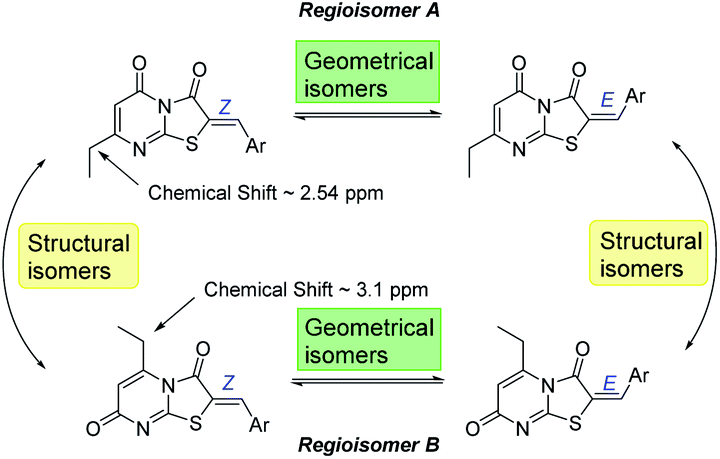 | ||
| Fig. 4 The four possible isomers that can be produced from the MCR reaction. | ||
The Z absolute configuration of this class of molecules was confirmed by X-ray crystallographic analysis of derivative 12 (Fig. 5). This illustrated the planarity of the thiazolopyrimidine rings with the aryl group, which may be attributed to two intramolecular hydrogen bonds. The Z-configuration is presumably stabilized by the two intramolecular hydrogen bonds between the arylidene H and the oxygen atom of the thiazole carbonyl group (HB1) and between the sulfur atom and the aryl hydrogen (HB2). The distance of both hydrogen bonds is 2.49 and 2.50 Å, for HB1 and HB2, respectively, and the angles are 102.81° and 132.94° for HB1 and HB2 respectively.
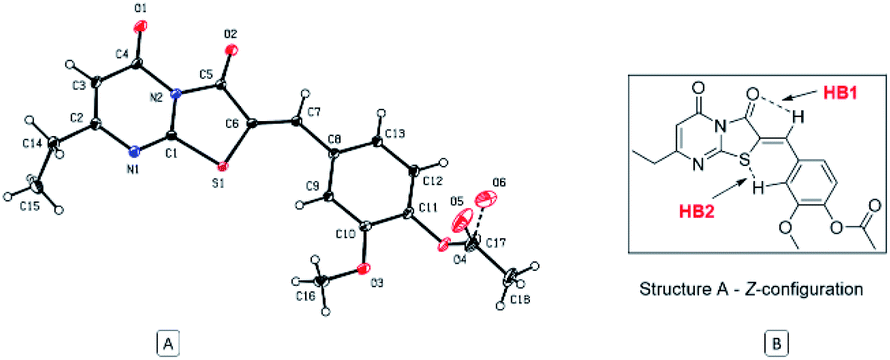 | ||
| Fig. 5 (A) ORTEP diagram of compound 12, (B) representation of stabilizing hydrogen bonds of the regioisomer (Z)-A. | ||
To investigate the observed preference for the Z isomer, density functional theory (DFT) calculations were performed using the widely applied M06-2X functional with a Dunnings aug-cc-pVDZ basis set. Compound 6 (Ar = C6H6) was selected for the theoretical study and all structures were optimised with modelled acetic acid solvation (SMD). Analysis of the proposed mechanism indicated a possible alcohol intermediate and dehydration step preceded the final Z/E isomer formation, and to keep the model simple, the calculations have focussed on these molecules only, maintaining chirality where applicable (Fig. S1†). Conformer analysis of the alcohol intermediate indicated a Gibbs free energy of reaction (ΔrG0{298 K}) range of +0.35 to −2.30 kcal mol−1 with the Z arranged C1 being the lowest and the E arranged C2 the highest energy conformers respectively (Fig. S2†). Analysis of the optimised geometries gave no indication of hydrogen bond stabilisation for the alcohol, despite attempts at manually inputting the conformation. This indicated steric and geometric arrangement of the alcohol intermediate is more significant for the energy term.
A stacked ring conformer (C3) was also observed with a favourable ΔrG0{298 K} of −1.30 kcal mol−1. To better account for this interaction, Grimme's dispersion (D3) and geometric counterpoise (gCP) corrections30 were applied to the energy term, and the stacked C3 was observed to increase by +1.29 kcal mol−1, whereas C1/C2 increased by +0.87 and +0.85 kcal mol−1 respectively. The similar increase to the energy term for C1/C2 suggested there are no differences in dispersion effects to account for in these conformers. However, the greater energy change observed with C3 was indicative the ring stacking is not a favourable intramolecular force. Based on the subtle differences in energies of the conformers, C1 is the apparent favoured alcohol intermediate conformer, which is arranged to dehydrate to the Z isomer.
The transition states of the proposed dehydration reactions were calculated and the saddle point ΔrG0{298 K} were found to be +32.58 and +30.05 kcal mol−1 for the E T1 and Z T2 arranged transition states respectively (Fig. S3a†). With the D3-gCP corrections ΔrG0{298 K} were +33.51 and +30.90 kcal mol−1 for T1 and T2 respectively. The similar increases in energy (+0.93 and +0.85 kcal mol−1) indicated negligible differences in dispersion force correction required when comparing T1 and T2. What was particularly interesting to note, in contrast to the alcohol intermediate, is the preference of the water leaving hydrogen bound to the carbonyl. Despite attempts in manually positioning the water to leave adjacent to the sulfur, and in turn have H–S interaction, the transition state only converged to allow hydrogen bonding of the water with the thiazole carbonyl. Subsequently, it was proposed the principal factor dictating the favoured transition state is the arrangement of the aryl group, as the water has apparent preference for leaving trans to the sulfur, in favour of Z isomer formation.
Subsequent calculation of the product (E)-6 and (Z)-6 isomers indicated ΔrH0{298 K} of −2.40 and −6.78 kcal mol−1 respectively, which indicts the Z isomer to be thermodynamically favoured, and D3-gCP corrections were in good agreement (Fig. 6 and S3b†). Comparison of the calculated (Z)-6 structure with the X-ray crystal structure of compound 12 indicated similar bond distances of HB1 = 2.43 Å and HB2 = 2.50 Å for the proposed intramolecular hydrogen bonds (Fig. 5).
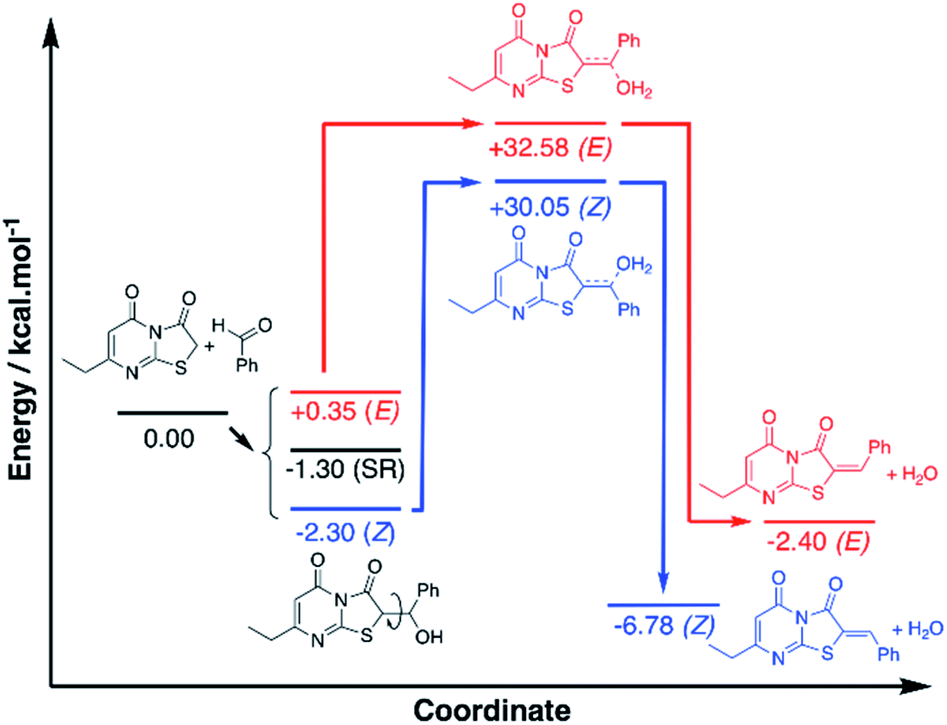 | ||
| Fig. 6 Energy profile for the formation of compound 6(Z) and 6(E), calculated with DFT M06-2X/aug-cc-pVDZ. Divergent reaction pathways are color coded red for E formation and blue for Z formation. SR = stacked rings conformer. | ||
Likewise, the bond angles were in good agreement for HB1 and HB2 calculated at 101.34° and 127.70°, compared with 102.81° and 132.94° from the crystal structure, respectively. In conclusion, although the energy differences are small between the investigated reaction pathways, there is a consistent preference for the Z forming pathway at each step investigated and this result was reciprocated by D3-gCP energy corrections (Fig. 6).
The compounds were screened for their antiviral activities in a viral-cell based assay against Chikungunya virus (Indian Ocean strain 899) (Table S1†).
Based on the biological results, compounds 6–20 showed different percentages of viral replication inhibition at 20 μg mL−1 with (Z)-7-ethyl-2-((4′-methyl-[1,1′-biphenyl]-4-yl)methylene)-5H-thiazolo[3,2-a]pyrimidine-3,5(2H)-dione showing the best activity with 58% inhibition of CHIKV replication. Derivatives 6, 11 and 20 showed 19, 28 and 34% inhibition of viral replication, while rest of the compounds showed fair or no activity. However, thiazolodione 15 can be considered as a potential lead compound in our future iterative cycles of optimization. The compound emerged as the most promising antiviral among the tested series with EC50 = 42 μM, with IC50 > 250 μM against the breast cancer cell line MCF-7 and the endothelial human sapiens cell line EA.hy926. Compound 15 is endowed with p-methylbiphenyl tail functionality which might interact favourably with the target. Our outlook will include optimization of compound 15 with the prime aim to find safe and effective anti-CHIKV agents.
In this paper, we shed the light to the convergent synthesis of a new series of 2-arylidene thiazolo[3,2-a]pyrimidines. Using simple synthons, namely 6-ethylthiouracil, bromoacetic acid and different aldehydes in a mixture of acetic acid/acetic anhydride and catalytic amount of anhydrous sodium acetate, a novel series of (Z)-7-ethyl-2-arylidine-5H-thiazolo[3,2-a]pyrimidine-3,5(2H)-diones was achieved. Optimization of the multi component reaction conditions by replacing chloroacetic acid with the more regioselective bromoacetic acid, lowering reaction temperature and changing the mode of addition and we were able to prepare one isomer at each case. X-ray crystal structure of compound 12 shows that only one regioselective isomer formed with the Z configuration that is potentially stabilized by two intramolecular hydrogen bonds. Antiviral activity evaluation demonstrated the tailed thiazolopyrimidine 15 as a candidate for future development.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MF thanks UOW for the University Postgraduate Award and International Postgraduate Tuition Award scholarships.References
- A. Cifuentes Kottkamp, E. De Jesus, R. Grande, J. A. Brown, A. R. Jacobs, J. K. Lim and K. A. Stapleford, J. Virol., 2019, 93, e00389-19 CrossRef PubMed.
- Y. S. Huang, S. Higgs and D. L. Vanlandingham, Front. Microbiol., 2019, 10, 22 CrossRef PubMed.
- C. B. F. Vogels, C. Ruckert, S. M. Cavany, T. A. Perkins, G. D. Ebel and N. D. Grubaugh, PLoS Biol., 2019, 17, e3000130 CrossRef PubMed.
- A. A. Rashad, S. Mahalingam and P. A. Keller, J. Med. Chem., 2014, 57, 1147–1166 CrossRef CAS PubMed.
- B. J. Matthews, O. Dudchenko, S. B. Kingan, S. Koren, I. Antoshechkin, J. E. Crawford, W. J. Glassford, M. Herre, S. N. Redmond, N. H. Rose, G. D. Weedall, Y. Wu, S. S. Batra, C. A. Brito-Sierra, S. D. Buckingham, C. L. Campbell, S. Chan, E. Cox, B. R. Evans, T. Fansiri, I. Filipovic, A. Fontaine, A. Gloria-Soria, R. Hall, V. S. Joardar, A. K. Jones, R. G. G. Kay, V. K. Kodali, J. Lee, G. J. Lycett, S. N. Mitchell, J. Muehling, M. R. Murphy, A. D. Omer, F. A. Partridge, P. Peluso, A. P. Aiden, V. Ramasamy, G. Rasic, S. Roy, K. Saavedra-Rodriguez, S. Sharan, A. Sharma, M. L. Smith, J. Turner, A. M. Weakley, Z. Zhao, O. S. Akbari, W. C. t. Black, H. Cao, A. C. Darby, C. A. Hill, J. S. Johnston, T. D. Murphy, A. S. Raikhel, D. B. Sattelle, I. V. Sharakhov, B. J. White, L. Zhao, E. L. Aiden, R. S. Mann, L. Lambrechts, J. R. Powell, M. V. Sharakhova, Z. Tu, H. M. Robertson, C. S. McBride, A. R. Hastie, J. Korlach, D. E. Neafsey, A. M. Phillippy and L. B. Vosshall, Nature, 2018, 563, 501–507 CrossRef CAS PubMed.
- O. Schwartz and M. L. Albert, Nat. Rev. Microbiol., 2010, 8, 491–500 CrossRef CAS PubMed.
- Y. Villero-Wolf, S. Mattar, A. Puerta-Gonzalez, G. Arrieta, C. Muskus, R. Hoyos, H. Pinzon and D. Pelaez-Carvajal, Sci. Rep., 2019, 9, 9970 CrossRef PubMed.
- P. Mahendradas, K. Avadhani and R. Shetty, J. Ophthalmic Inflammation Infect., 2013, 3, 35 CrossRef PubMed.
- L. J. Chang, K. A. Dowd, F. H. Mendoza, J. G. Saunders, S. Sitar, S. H. Plummer, G. Yamshchikov, U. N. Sarwar, Z. Hu, M. E. Enama, R. T. Bailer, R. A. Koup, R. M. Schwartz, W. Akahata, G. J. Nabel, J. R. Mascola, T. C. Pierson, B. S. Graham, J. E. Ledgerwood and V. R. C. S. Team, Lancet, 2014, 384, 2046–2052 CrossRef CAS.
- T. Agarwal, S. Asthana and A. Bissoyi, Indian J. Pharm. Sci., 2015, 77, 453–460 CrossRef CAS PubMed.
- P. T. Nguyen, H. Yu and P. A. Keller, J. Mol. Model., 2014, 20, 2216 CrossRef PubMed.
- R. K. Maheshwari, V. Srikantan and D. Bhartiya, J. Virol., 1991, 65, 992–995 CrossRef CAS.
- P. T. Nguyen, H. Yu and P. A. Keller, J. Mol. Graphics Modell., 2015, 57, 1–8 CrossRef CAS PubMed.
- A. Karlas, S. Berre, T. Couderc, M. Varjak, P. Braun, M. Meyer, N. Gangneux, L. Karo-Astover, F. Weege, M. Raftery, G. Schonrich, U. Klemm, A. Wurzlbauer, F. Bracher, A. Merits, T. F. Meyer and M. Lecuit, Nat. Commun., 2016, 7, 11320 CrossRef CAS PubMed.
- M. D'Hooghe, K. Mollet, R. De Vreese, T. H. Jonckers, G. Dams and N. De Kimpe, J. Med. Chem., 2012, 55, 5637–5641 CrossRef PubMed.
- K. M. Feibelman, B. P. Fuller, L. Li, D. V. LaBarbera and B. J. Geiss, Antiviral Res., 2018, 154, 124–131 CrossRef CAS PubMed.
- A. Gomez-SanJuan, A. M. Gamo, L. Delang, A. Perez-Sanchez, S. N. Amrun, R. Abdelnabi, S. Jacobs, E. M. Priego, M. J. Camarasa, D. Jochmans, P. Leyssen, L. F. P. Ng, G. Querat, J. Neyts and M. J. Perez-Perez, ACS Infect. Dis., 2018, 4, 605–619 CrossRef CAS PubMed.
- H. A. Rothan, H. Bahrani, A. Y. Abdulrahman, Z. Mohamed, T. C. Teoh, S. Othman, N. N. Rashid, N. A. Rahman and R. Yusof, Antiviral Res., 2016, 127, 50–56 CrossRef CAS PubMed.
- S. S. Jadav, B. N. Sinha, R. Hilgenfeld, B. Pastorino, X. de Lamballerie and V. Jayaprakash, Eur. J. Med. Chem., 2015, 89, 172–178 CrossRef CAS PubMed.
- J. R. Hwu, M. Kapoor, S. C. Tsay, C. C. Lin, K. C. Hwang, J. C. Horng, I. C. Chen, F. K. Shieh, P. Leyssen and J. Neyts, Antiviral Res., 2015, 118, 103–109 CrossRef CAS PubMed.
- A. Gigante, M. D. Canela, L. Delang, E. M. Priego, M. J. Camarasa, G. Querat, J. Neyts, P. Leyssen and M. J. Perez-Perez, J. Med. Chem., 2014, 57, 4000–4008 CrossRef CAS PubMed.
- M. Fares, W. M. Eldehna, S. M. Abou-Seri, H. A. Abdel-Aziz, M. H. Aly and M. F. Tolba, Arch. Pharm., 2015, 348, 144–154 CrossRef CAS PubMed.
- J. R. Hwu, W. C. Huang, S. Y. Lin, K. T. Tan, Y. C. Hu, F. K. Shieh, S. O. Bachurin, A. Ustyugov and S. C. Tsay, Eur. J. Med. Chem., 2019, 166, 136–143 CrossRef CAS PubMed.
- J. P. Spengler and W. Schunack, Arch. Pharm., 1984, 317, 425–430 CrossRef CAS PubMed.
- M. Fares, S. M. Abou-Seri, H. A. Abdel-Aziz, S. E. Abbas, M. M. Youssef and R. A. Eladwy, Eur. J. Med. Chem., 2014, 83, 155–166 CrossRef CAS PubMed.
- M. M. Mohamed, A. K. Khalil, E. M. Abbass and A. M. El-Naggar, Synth. Commun., 2017, 47, 1441–1457 CrossRef CAS.
- A.-S. M. Abdel-fattah, A. M. Negm and A. E. Mustafa Gaafar, Phosphorus, Sulfur, and Silicon and the Related Elements, 1992, 72, 145–156 CrossRef CAS.
- A. Hamouda, Der Pharma Chem., 2014, 6, 346–357 Search PubMed.
- E. Abdelghani, S. A. Said, M. Assy and A. M. A. Hamid, J. Iran. Chem. Soc., 2015, 12, 1809–1817 CrossRef CAS.
- H. Kruse and S. Grimme, J. Chem. Phys., 2012, 136, 154101 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1968317. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra00257g |
| This journal is © The Royal Society of Chemistry 2020 |