
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of platinum nanoparticles using iron(II) as reductant and photosensitized H2 generation on an iron storage protein scaffold†
Brenda S. Benavides,
Silvano Valandro and
Donald M. Kurtz Jr*
and
Donald M. Kurtz Jr*
Department of Chemistry, University of Texas at San Antonio, San Antonio, Texas, USA. E-mail: Donald.kurtz@utsa.edu
First published on 4th February 2020
Abstract
The quest for efficient solar-to-fuel conversion has led to the development of numerous homogeneous and heterogeneous systems for photochemical stimulation of 2H+ + 2e− → H2. Many such systems consist of a photosensitizer, an H2-evolving catalyst (HEC), and sacrificial electron donor often with an electron relay between photosensitizer and HEC. Colloidal platinum remains a popular HEC. We report here a novel, simple, and high yield synthesis of Pt nanoparticles (Pt NPs) associated with human heavy chain ferritin (Hfn). The formation of the Pt NPs capitalizes on Hfn's native catalysis of autoxidation of Fe(II)(aq) (ferroxidase activity). Fe(II) reduces Pt(II) to Pt(0) and the rapid ferroxidase reaction produces FeO(OH), which associates with and stabilizes the incipient Pt NPs. This Pt/Fe-Hfn efficiently catalyzes photosensitized H2 production when combined with Eosin Y (EY) as photosensitizer and triethanolamine (TEOA) as sacrificial electron donor. With white light irradiation turnover numbers of 300H2 per Pt, 250H2 per EY were achieved. A quantum yield of 18% for H2 production was obtained with 550 nm irradiation. The fluorescence emission of EY is quenched by TEOA but not by Pt/Fe-Hfn. We propose that the photosensitized H2 production from aqueous TEOA, EY, Pt/Fe-Hfn solution occurs via a reductive quenching pathway in which both the singlet and triplet excited states of EY are reduced by TEOA to the anion radical, EY−˙, which in turn transfers electrons to the Pt/Fe-Hfn HEC. Hfn is known to be a remarkably versatile scaffold for incorporation and stabilization of noble metal and semiconductor nanoparticles. Since both EY and Hfn are amenable to scale-up, we envision further refinements to and applications of this photosensitized H2-generating system.
Introduction
The quest for efficient solar-to-fuel conversion has led to the development of numerous homogeneous and heterogeneous systems for photochemical stimulation of 2H+ + 2e− → H2.1–8 These systems are typically modular, consisting of a redox active photosensitizer (PS), sacrificial electron donor (SED), H2 evolving catalyst (HEC), and often an electron relay (typically methyl viologen (MV2+)) between PS and HEC. Although intensive efforts have been devoted to development of HECs using earth-abundant elements, colloidal platinum continues to be used due to its ease of synthesis, stability, catalytic efficiency, and versatility.5,9–12The three most commonly used visible light-absorbing PSs for H2 generation in aqueous solutions have been transition metal ion–polypyridyl complexes,13–15 porphyrins,16 and xanthene dyes.7,10,17–19 Among these PSs, xanthene dyes are the most amenable to scale-up and usually give higher H2 quantum yields than either metal–polypyridyl complexes or porphyrin-type. Photosensitized H2 generation from a combination of the inexpensive xanthene dye, Eosin Y (2,4,5,7-tetrabromofluorescein, EY), triethanolamine (TEOA) as SED, polymer-coated Pt NPs as HEC, and MV2+ as an electron relay was reported as long ago as 1983.20 Deposition of colloidal Pt on surfaces or within films that are presumed to facilitate photosensitized electron transfer reactions of EY function relatively efficiently for H2 generation without an added electron relay.11,19,21–23 Depending on the system, the photochemistry has been reported to occur by either oxidative or reductive quenching of 3EY*, via the cation radical, EY+˙, or the anion radical, EY−˙, respectively.24,25 Oxidative quenching is likely to predominate in the presence of a large excess of an electron relay molecule. More recently, TEOA has been reported to reductively quench the singlet excited state, 1EY*, in a concentration-dependent fashion.26
Various cage-like proteins have been shown to enclose catalytically active noble metal nanoparticles (NPs), including Pt in aqueous solution.27 The iron storage protein, ferritin, has proven to be remarkably versatile in this regard.28,29 The ∼8 nm hollow cavity of ferritins, represented by that of human ‘heavy chain’ ferritin (Hfn) in Fig. 1, can store up to an average of ∼3000 irons per 24-mer as either amorphous ferric oxyhydroxide (FeO(OH)) or the mineralized form, ferrihydrite.30,31 Ferritins incorporate iron by catalyzing the autoxidation of Fe(II)(aq), referred to as ferroxidase activity. In the absence of iron, Pt NPs have been shown to form within the ferritin cavity upon borohydride reduction of either tetrachloroplatinate(II) or hexachloroplatinate(IV).32–34 We have found only one report of the use of ferritin 24-mer-encapsulated Pt NPs for photosensitized H2 generation.35 That system used tris(2,2′-bipyridine)ruthenium(II)chloride as PS and MV2+ as electron relay. A bacterial homolog called bacterioferritin can bind a zinc(II)-porphyrin PS and enclose Pt NPs, but this construction was inactive for photosensitized H2 generation unless the 24-mer was dissociated into dimers.36
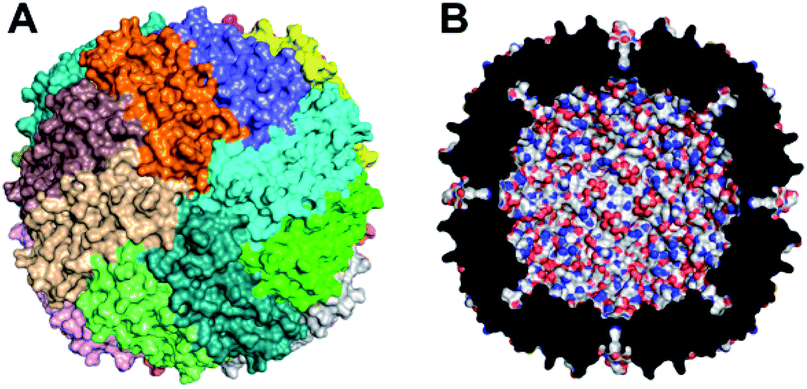 | ||
| Fig. 1 Structural features of Hfn. (A) Outer surface with the 24 identical subunits distinguished by color. (B) Cross section of the 24-mer showing the ∼8 nm internal cavity. Drawings used coordinates from Protein Data Bank entry 3ajo.37 | ||
We report here a novel, simple, and high yield synthesis of Pt NP-associated Hfn, which capitalizes on the ferroxidase activity of ferritins and incorporates Fe as well as Pt. We show that this Pt/Fe-Hfn 24-mer also efficiently catalyzes H2 production using EY as PS and TEOA as SED without an added electron relay.
Results and discussion
Characterization of Pt-Hfn and Pt/Fe-Hfn
Hfn containing Pt NPs (Pt-Hfn) was prepared via sodium borohydride reduction of Pt(II) salt pre-incubated with Hfn following a published procedure.36 Dynamic light scattering (DLS) of Pt-Hfn (Fig. 2) showed a hydrodynamic diameter of 13.5 nm, which is close to that expected for the outer diameter of the Hfn 24-mer.37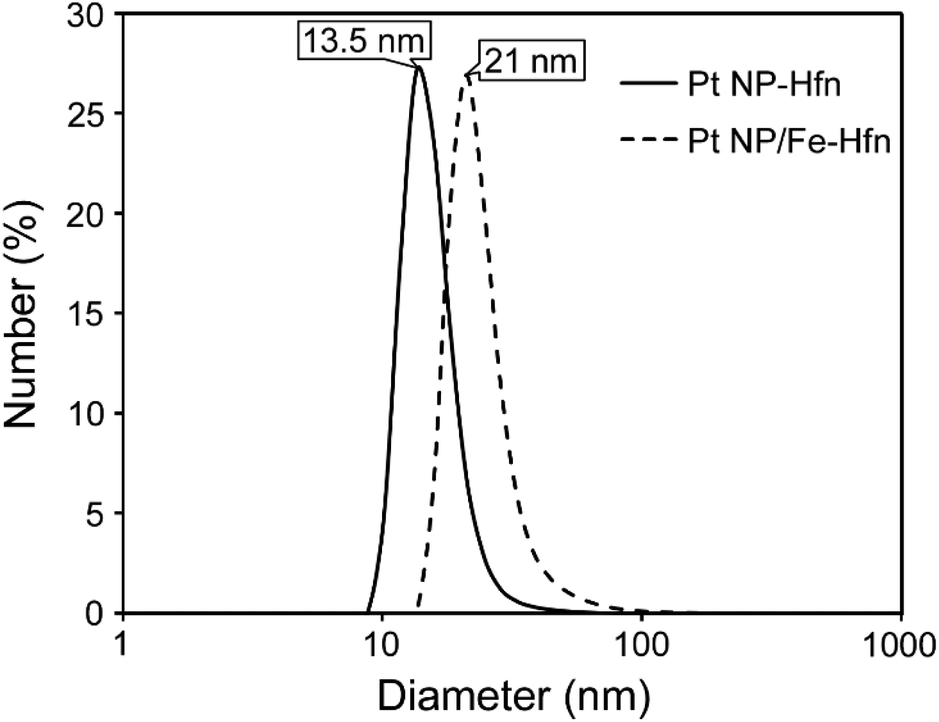 | ||
| Fig. 2 DLS of Pt-Hfn and Pt/Fe-Hfn in 0.3 M TEOA pH 9. | ||
The TEM of Pt-Hfn (Fig. 3) showed electron dense particles (average of 3 nm) largely filling the inner cavity of the protein shell. The most common diameter of the Pt-Hfn shell from TEM was 12 nm (range 11–13 nm). These results are consistent with previous reports of Pt NPs incorporated into ferritins via borohydride reduction of Pt(II) or Pt(IV) salts.32–34
 | ||
| Fig. 3 TEM of uranyl acetate-stained Pt-Hfn (top panel) and Pt/Fe-Hfn (bottom panel). Inset in the Pt/Fe-Hfn panel shows a separately obtained TEM of a portion of the same grid at higher magnification. | ||
Hfn containing Pt NPs and FeO(OH) (Pt/Fe-Hfn) was prepared by a process involving repeated additions of ferrous ammonium sulfate to an aerobic Pt(II) salt/Hfn mixture.38,39 Inductively coupled plasma-optical emission spectrometry (ICP-OES) gave 600 Pt and 1100 Fe per Pt/Fe-Hfn 24 mer. The hydrodynamic diameter of 21 nm for Pt/Fe-Hfn (Fig. 3) is larger than expected for the Hfn 24-mer. TEM (Fig. 3 Pt/Fe-Hfn panel) furnishes an explanation for the differing sizes. The Pt/Fe-Hfn showed individual 24-mer-sized protein shells containing multiple small (∼1 nm) electron dense Pt NPs lining the inner walls of the shell but also coating the outer surface (inset to Pt/Fe-Hfn panel of Fig. 3). Given our method of preparation and iron content of the Pt/Fe-Hfn, we presume voids between Pt NPs within the 24-mer contain much less electron dense colloidal or mineralized FeO(OH). The larger hydrodynamic diameter of Pt/Fe-Hfn could indicate formation of dimers of 24-mers, consistent with dimers observed by TEM (inset to the Fig. 3 Pt/Fe-Hfn panel).
Effects of Pt/Fe-Hfn on EY spectroscopic properties
The UV-vis absorption spectrum of 72 μM EY in 0.3 M TEOA pH 9 (Fig. 4) shows the features expected for the photochemically active dianionic form (λmax 304, 343, and 517 nm),40–42 and these features are unperturbed by the presence of Pt/Fe-Hfn. Since binding to proteins normally shifts the absorption maxima of EY,42 this lack of perturbation indicates no stable interaction between EY and the Pt/Fe-Hfn protein particles under our conditions.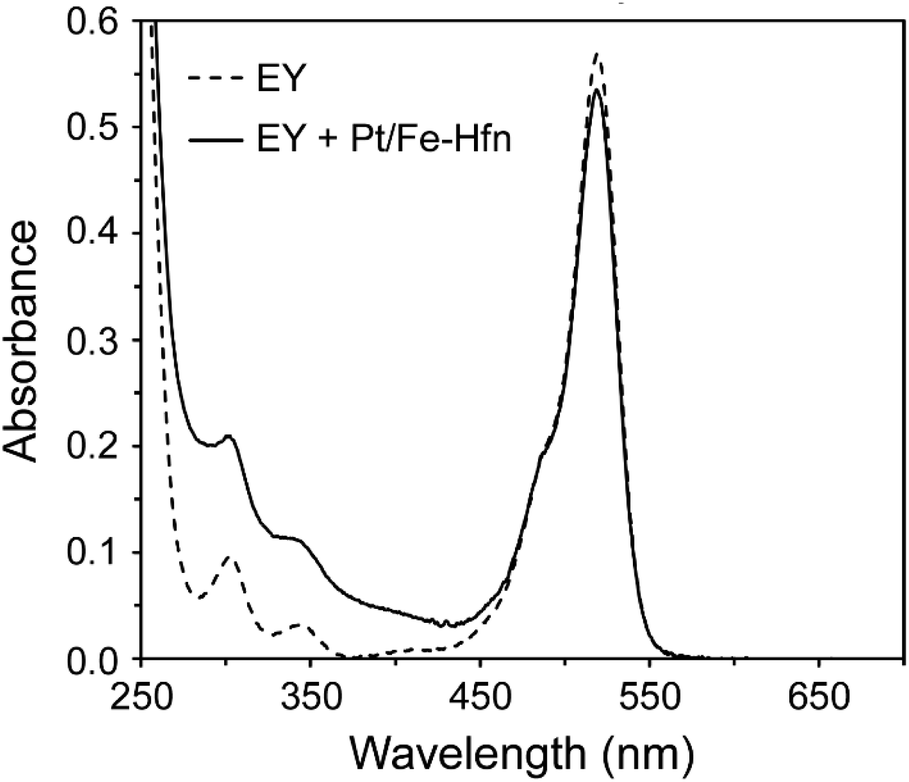 | ||
| Fig. 4 UV-vis absorption spectra in 0.3 M TEOA pH 9 of 72 μM EY and 72 μM EY + Pt/Fe-Hfn at 60 μM Pt. | ||
TEOA is known to dynamically quench the luminescence of photoexcited EY in anaerobic aqueous solutions.26,43 We investigated whether Pt/Fe-Hfn had any effect on this quenching. 1EY* shows a characteristic fluorescence emission spectrum with λmax at ∼540 nm in neutral to basic aqueous solution.44 Fig. 5 shows the expected quenching of EY fluorescence by TEOA but little or no quenching by Pt/Fe-Hfn. These observations indicate that reductive quenching of 1EY* can occur in our H2-generating system.
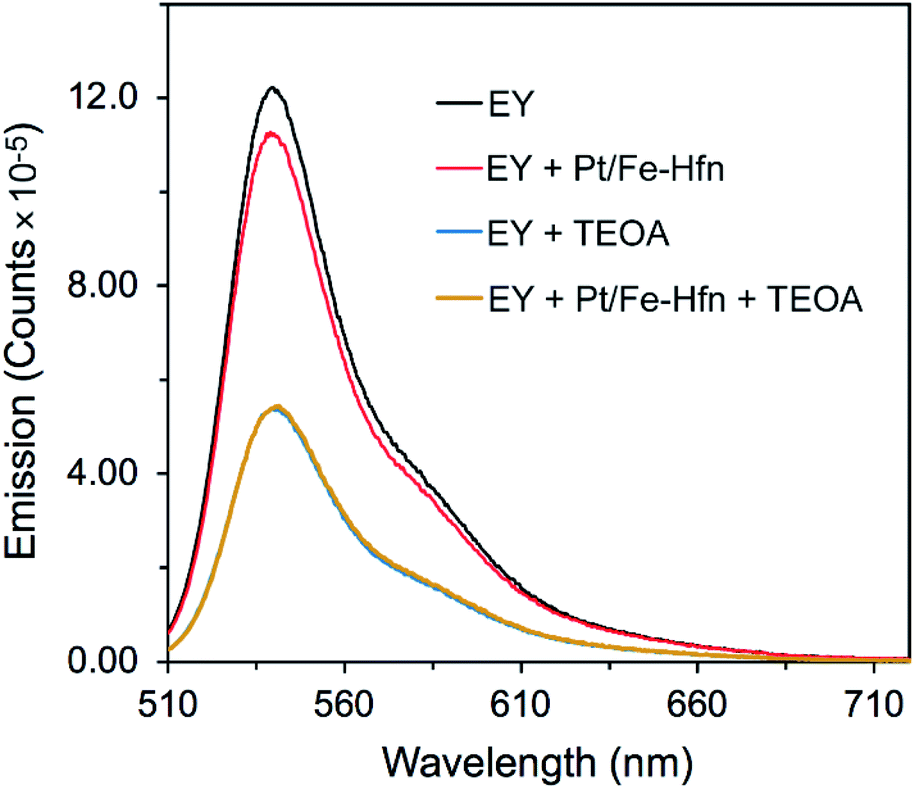 | ||
| Fig. 5 Luminescence emission spectra of EY in anaerobic aqueous solutions with the various additives listed in the figure labels. λex was at 490 nm. All solutions were at pH 9. EY was ∼2 μM. When present TEOA was 0.3 M and Pt/Fe-Hfn was at 60 μM Pt. | ||
Photosensitized generation of H2
We optimized the TEOA, Pt, and EY concentrations starting from those reported by Wang et al.43 but omitting MV2+. Time courses for photosensitized H2 production using white light irradiation are shown in Fig. 6. The six-hour turnover numbers (TONs) for Pt NP and Pt/Fe-Hfn (inset to Fig. 6) were similar to each other. As reported previously for polymer-coated Pt NPs,43 we found 72 μM EY to be optimal for photosensitized H2 production. The leveling off of H2 production by 6 h irradiation is most likely due to gradual debromination of EY, resulting ultimately in the completely debrominated form, fluorescein, which is typically a much less efficient PS.41,44 Fig. S1† shows that, in fact, substitution of fluorescein in place of EY leads to a relatively low level of photosensitized H2 generation. Omission of any single component (EY, TEOA, Pt-Hfn or Pt/Fe-Hfn) resulted in no photosensitized H2 generation. The time course for photosensitized H2 production of Pt/Fe-Hfn under the same conditions but irradiated using a 550 ± 25 nm filter is shown in Fig. S2,† and these data were used to determine the quantum yield. The 6 h H2 quantum yield for Pt/Fe-Hfn using chemical actinometry was 18%.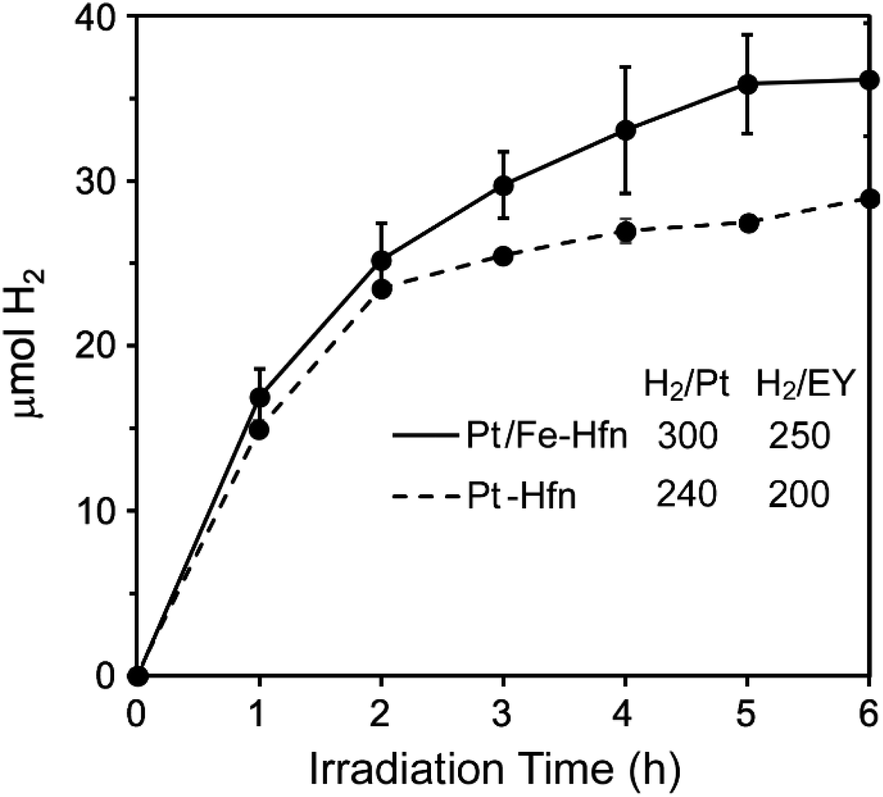 | ||
| Fig. 6 Time courses for photosensitized H2 production from a solution of either Pt-Hfn or Pt/Fe-Hfn at 60 μM Pt and 72 μM EY in 0.3 M TEOA pH 9 upon white light irradiation.‡ H2/Pt and H2/EY values represent 6 h TONs (mol H2/mol Pt or mol EY). | ||
When Fe-Hfn was substituted in place of Pt/Fe-Hfn at an iron concentration of ∼230 μM in 0.3 M TEOA pH 9 and 72 μM EY, less than 0.6 μmol H2 was generated over 6 h irradiation. Attempts to load the pre-formed Pt-Hfn with iron by the aerobic multiple-aliquot ferrous ammonium sulfate procedure or single addition of ∼1500 iron as ferrous ammonium sulfate per 24-mer to the anaerobic Pt-Hfn/EY/TEOA reaction solution immediately prior to irradiation gave less than half the 6 h yield of H2 and TONs per Pt or EY compared to those shown in Fig. 6.
EY limits H2 generation
We further tested that decomposition of EY and not the Fe/Pt-NP Hfn was the limiting factor in H2 generation by adding more EY after 6 h of irradiation, where H2 generation had levelled off in the initial reaction mixture (Fig. 6). As shown in Fig. 7, the additional EY prevented the 6 h levelling off of H2 generation, the rate of which decreased again between 9 and 12 h, but with an increased overall H2 yield.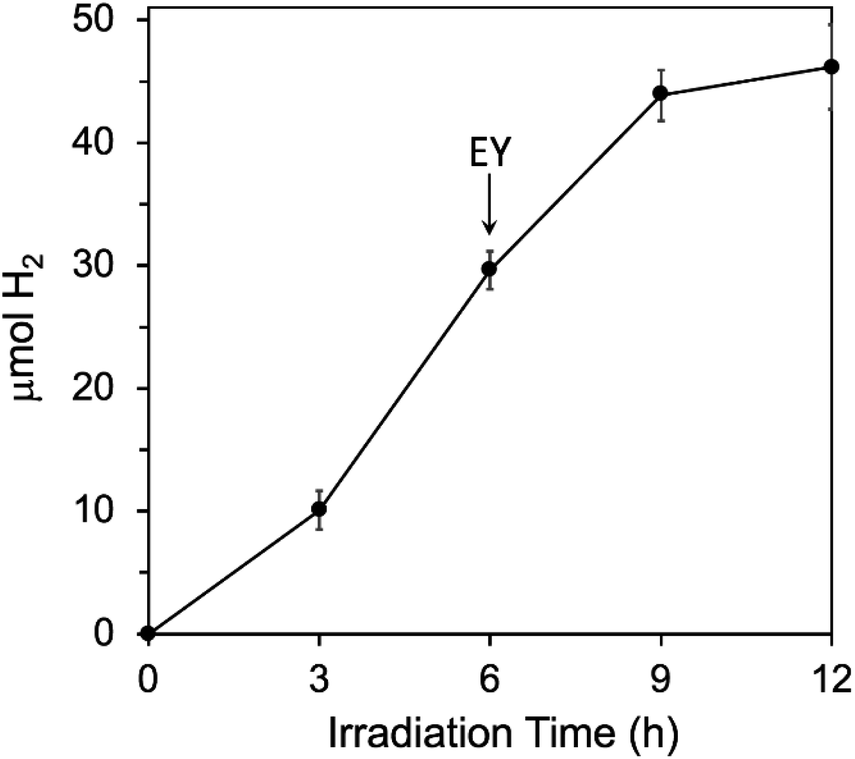 | ||
| Fig. 7 Effect of adding more EY during photosensitized H2 generation conditions are the same as listed in the Fig. 6 legend, except that an additional 72 μM more EY was added at 6 h, as indicated by the arrow. | ||
Air tolerance
3EY* generates singlet O2 from ground state triplet O2 with quantum yield ∼0.5 in aqueous solution.45 Nevertheless, in the absence of quenchers EY undergoes relatively slow photodegradation in aerobic aqueous solutions.41 A more recent report shows that EY−˙ produced from reductive quenching by TEOA can be quantitatively recycled back to EY via an oxidative photoreaction with O2, which generates superoxide.46 Despite the photoinduced generation of these reactive oxygen species, photosensitized H2 generation by the TEOA, EY, Pt/Fe-Hfn system, showed some tolerance to air exposure. The time course for H2 production upon white light irradiation of an air-saturated reaction mixture is shown in Fig. 8. H2 production leveled off after 2 h at approximately one-third that from an equivalent anaerobic reaction mixture (Fig. 6). Fig. S3 and S4† show GC traces corresponding to the time course in Fig. 8. These traces clearly show the presence of O2 in the headspace through at least 4 h of irradiation. A subsequent addition of another 72 μM of EY at 4 h irradiation time led to a restart of photosensitized H2 production, consistent with degradation of EY being the limiting factor.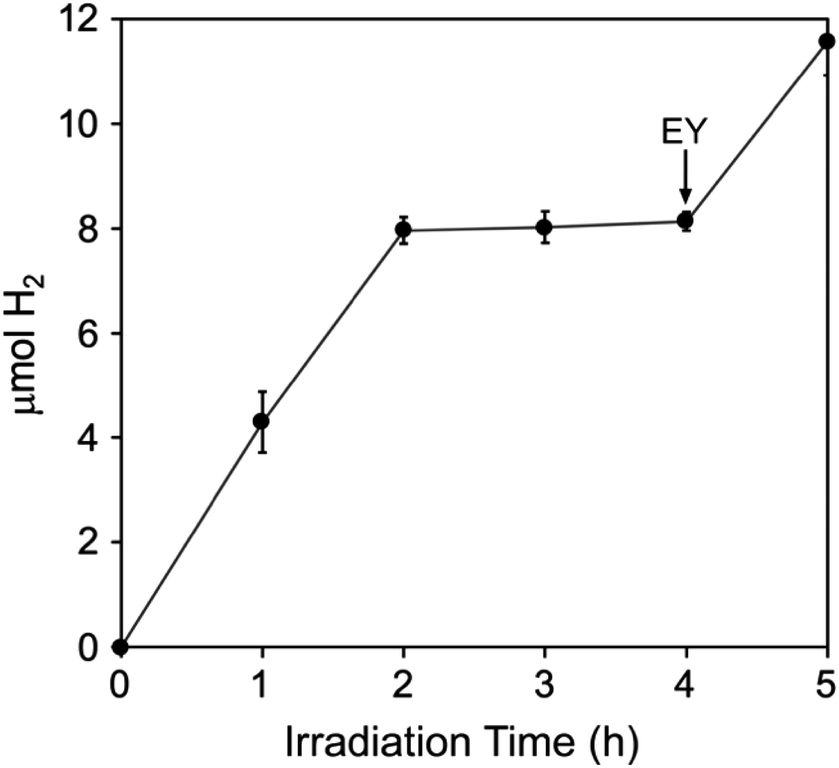 | ||
| Fig. 8 Photosensitized H2 production upon white light irradiation of an aerobic Pt/Fe-Hfn solution of composition identical to that listed in the Fig. 6 legend. The arrow indicates addition of 72 μM more EY.‡ | ||
Iron release
Fig. 9 shows that iron was gradually released upon white light irradiation of a Pt/Fe-Hfn solution under the same conditions as for photosensitized H2 generation. Approximately 30% of initial iron content remained in the Pt/Fe-Hfn after 6 h of irradiation. This iron release presumably occurred upon photosensitized reduction of Fe(O)OH to Fe(II), as demonstrated in our previous work using Zn protoporphyrin IX as photosensitizer.38 Assuming the iron is released as Fe(II), the ∼300 mol H2/mol Pt TON shown in Fig. 6 translates to ∼2% of photogenerated reducing equivalents being devoted to iron reduction relative to H2 production during 6 h irradiation.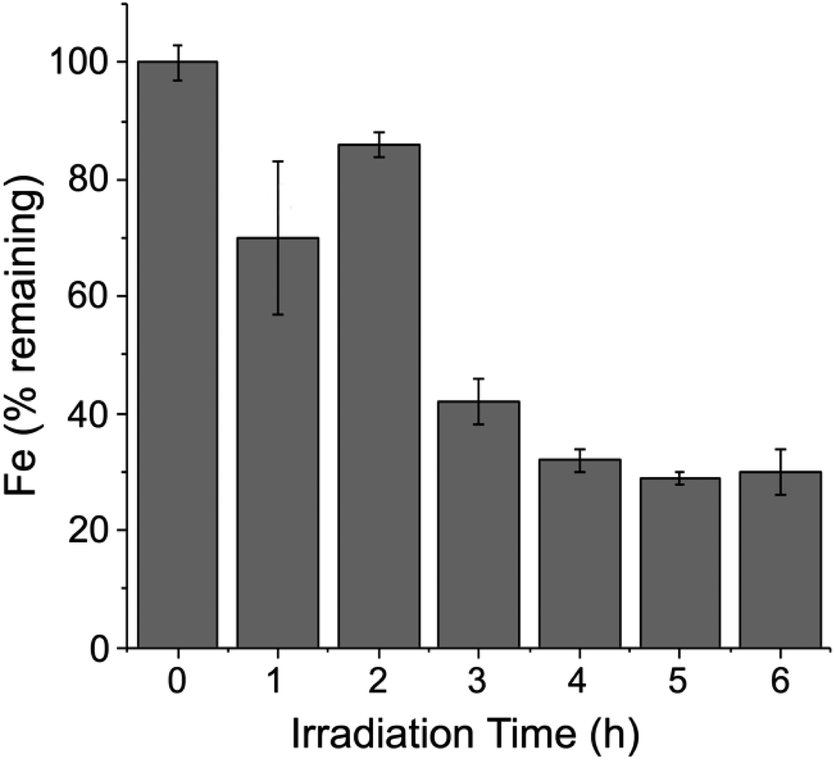 | ||
| Fig. 9 Time course for iron remaining with the Pt/Fe-Hfn during white light irradiation. Conditions were identical to those described in the legend to Fig. 6.‡ | ||
Ferroxidase activity and Pt NP formation
Previous preparations of Pt NPs within ferritins have typically used commercial horse spleen apoferritin (HSAF) and borohydride as reducing agent.32–34 HSAF consists of a combination of ‘heavy’ and ‘light’ protein subunits in the 24-mer. The Hfn used in this work contains only the ‘heavy’ chain subunits. The heavy chains contain ferroxidase centers (FCs), which are located in the protein shell. The FC in each of the 24 subunits binds two Fe(II) and catalyzes their autoxidation. The resulting Fe(III) in the FC migrates into the inner cavity of the protein shell, where it forms polynuclear FeO(OH) nucleation sites.31,47 The FC then binds two more Fe(II) and repeats the cycle. Additional Fe(II) can migrate through pores on the protein shell and are oxidatively incorporated onto the growing FeO(OH) surface. This process is diagrammed in Scheme 1.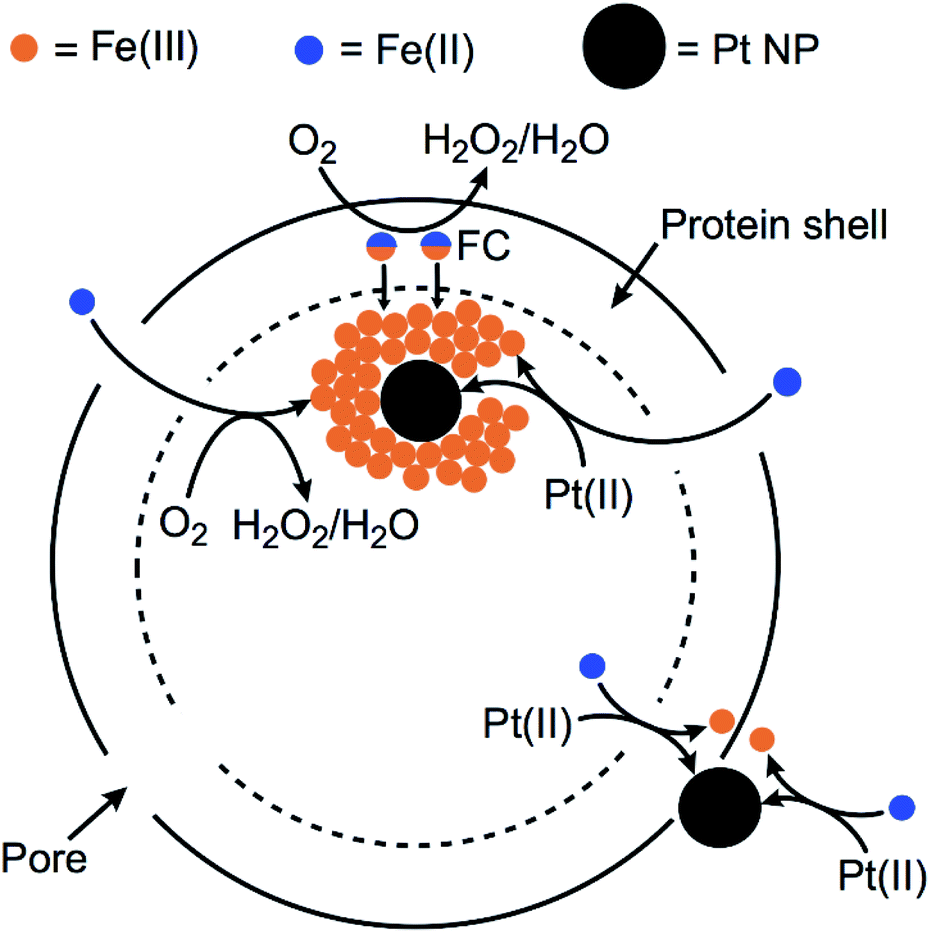 | ||
| Scheme 1 Proposed formation of Pt NPs and FeO(OH) with Hfn. | ||
Scheme 1 also diagrams likely pathways for formation and stabilization of the Pt NP in Pt/Fe-Hfn. Our results show that Pt NPs can be incorporated into Hfn in air-saturated solutions upon adding aliquots of Fe(II) salt to Hfn that had been pre-incubated with the Pt(II) salt. When we titrated the Pt(II) salt/Hfn solution with Fe(II) salt under anaerobic conditions, the resulting Pt(0) formed a bulk precipitate with little or no formation of Pt NPs associated with Hfn. This observation is consistent with a requirement for the ferroxidase reaction for formation of the Pt NPs. We suggest that the added iron serves as both reductant and Pt NP coating. Fe(II) reduces Pt(II) to Pt(0) and the rapid ferroxidase reaction produces FeO(OH), which associates with the incipient Pt NPs. This association would inhibit aggregation to form the larger Pt NPs obtained when using borohydride as reductant (Fig. 3, Pt-Hfn panel). Some Pt NPs also associate with the outer protein surface of the Pt/Fe-Hfn (inset to Fig. 3 Pt/Fe-Hfn panel). How these outer surface Pt NPs form and whether they are associated with FeO(OH) is not known. HSAF and light chain human ferritin were reported to show increased ferroxidase activity after incorporation of Pt NP via the Pt(II) salt/borohydride reduction procedure.34,48 Using our protocol, the Pt NPs form during addition of Fe(II) and are therefore less likely to significantly contribute to the ferroxidase activity.
FePt alloy NPs were reported to form at elevated temperature in a more heat stable ferritin using a mixture of ferrous ammonium sulfate, potassium tetrachloroplatinate and sodium borohydride as reducing agent.49 However, this report explicitly stated that this procedure was unsuccessful using Hfn. In any case our more oxidizing conditions at room temperature seem unlikely to result in formation of FePt mineral phases via reduction of the M(II) salts.
Photosensitization mechanism
While both oxidative and reductive quenching of 3EY* to EY+˙ and EY−˙, respectively, are thermodynamically feasible,24,25 our results are more consistent with the reductive quenching pathway shown in Scheme 2. TEOA is an efficient reductive quencher of not only 3EY, but also the shorter-lived 1EY*.26,43,44 Under conditions close to those used for the luminescence quenching results shown in Fig. 5, it has been estimated that TEOA reductively quenches about half of the photogenerated 1EY*.26 This reductive quenching competes with intersystem crossing of 1EY* to 3EY*. Our system contains a large excess of TEOA but no added electron relay between EY and HEC. Continuous irradiation, thus, likely generates a low steady state level of EY−˙.44 EY is too large to penetrate the pores of the Hfn protein shell. However, reducing agents such as reduced flavins, which do not penetrate the Hfn protein shell, are known to reduce FeO(OH) to Fe2+ inside the shell.50 We, therefore, propose that the highly reducing EY−˙ transfers electrons to Pt NPs on the outer surface and inside the protein shell, both of which function as HEC. The pathway shown in Scheme 2 would also apply to the borohydride-prepared Pt-Hfn, in which the Pt NPs are located only inside the protein shell.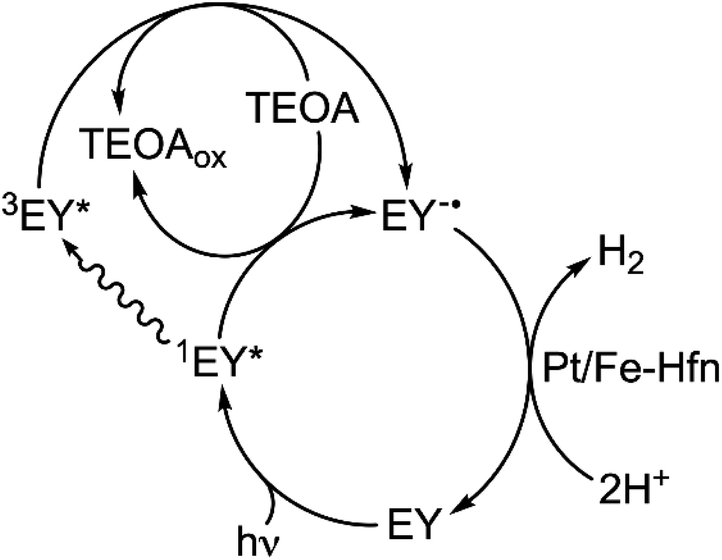 | ||
| Scheme 2 Proposed mechanism for photosensitized H2 production using TEOA/EY/Pt/Fe-Hfn. | ||
Comparisons with other TEOA/EY/Pt NP systems
Due to wide variations in solution volumes, heterogeneity, irradiation pathlengths and photon flux, comparing H2 generation rates among various systems using EY as photosensitizer is not straightforward. However, the 18% quantum yield and TON (250 mol H2/mol EY) for photosensitized H2 production by the TEOA, EY, Pt/Fe-Hfn system are comparable to those reported for other H2 generating systems using EY as PS, Pt as HEC and TEOA as SED.11,21–23,43Potential advantages of the TEOA, EY, Pt/Fe-Hfn system
We have not found previous reports of Fe(II)(aq) used for preparation of Pt NPs. In our Hfn system ferrous ammonium sulfate functions as the reducing agent for tetrachloroplatinate and occurs in aerobic aqueous solutions near neutral pH. Hfn is quite durable, and its production is amenable to scale-up.51 Our photosensitized H2-generating system also showed activity in air-saturated solution. This activity could be advantageous in Pt-containing ‘water splitting’ systems, which produce both H2 and O2.52 Pt is known to catalyze the back reaction of H2 with O2. The remarkably versatile ferritin scaffold can incorporate a variety of metal oxide and semiconductor NPs, can be adsorbed to electrochemically active surfaces, and is tolerant to a variety of modifications to the polypeptide chains.28,29,49,53–55 We can, thus, envision further refinements to and applications of our photosensitized TEOA, EY, Pt/Fe-Hfn H2-generating system.Conclusions
We have described a novel method for producing and stabilizing small Pt NPs via reduction of a Pt(II) salt by Fe(II)(aq) in an iron storage protein scaffold. The formation of the Pt NPs required the presence of both Fe(II)(aq) and O2. We conclude that this requirement stems from the native ferroxidase activity of the Hfn. The FeO(OH) produced from this activity associates with and stabilizes the Pt NPs. Photosensitized H2 production from aqueous TEOA, EY, Pt/Fe-Hfn solutions occurred with TON and quantum efficiency comparable to other systems using xanthene dye PSs and does not require an added electron relay between PS and Pt NPs. We propose a reductive quenching pathway in which TEOA reduces 1EY* and 3EY* to EY−˙, which then transfers electrons to the Pt/Fe-Hfn. A small portion of the photogenerated reducing equivalents is diverted towards reduction of FeO(OH), whereas the vast majority are funneled into catalytic H2 production on the Pt NPs.Experimental
Reagents and general methods
All chemicals were purchased from either Sigma-Aldrich or Fisher Scientific at the highest grades available. Stock solutions of EY or fluorescein were prepared in DMSO. All aqueous solutions were prepared in water that had been passed through a Milli-Q ultrapurification system (Merck Millipore, Inc.) to achieve a resistivity of 18 MΩ. Solutions containing EY were stored in aluminum foil-covered containers and manipulated in low room light.Protein overexpression and purification
The gene encoding Hfn was synthesized and inserted into the 5′ NcoI and 3′ BamHI restriction sites of expression plasmid pAG8H56 by GenScript, Inc. (Piscataway, NJ). E. coli BL21(DE3) competent cells (Invitrogen) were transformed with this Hfn-encoding plasmid. The proteins were expressed from 1 L cultures of the transformed strain in Luria-Bertani broth containing 100 mg L−1 ampicillin (LB/amp) at 37 °C. When the OD600 of the 1 L cultures reached 0.6–0.8, 1 mL of 100 mg mL−1 of isopropyl-beta-D-thiogalactoside was added to induce protein expression. These 1 L cultures were incubated for an additional four hours with shaking at 37 °C. Cells were then harvested by centrifugation at 4 °C and frozen at −80 °C.For isolation of Hfn the thawed cell pellet was resuspended in 25 mL of 50 mM 3-(N-morpholino)propanesulfonic acid (MOPS) pH 7.3 containing 250 mM NaCl, and 2 mM β-mercaptoethanol. The resuspended cells were lysed on ice by sonication and cellular debris was removed by centrifugation. The supernatant was heated at 60 °C for 15 min, followed by 30 min centrifugation at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g to remove precipitate. The supernatant was loaded on S200 Sephacryl® gel filtration XK16/100 column (GE HealthCare Life Sciences) that had been pre-equilibrated with 50 mM MOPS pH 7.3, 250 mM NaCl, 2 mM β-mercaptoethanol. Protein content of eluted fractions was assessed by glycine sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The Hfn-containing fractions were pooled and concentrated by buffer exchange into 50 mM MOPS pH 7.4 and stored at −80 °C.
000×g to remove precipitate. The supernatant was loaded on S200 Sephacryl® gel filtration XK16/100 column (GE HealthCare Life Sciences) that had been pre-equilibrated with 50 mM MOPS pH 7.3, 250 mM NaCl, 2 mM β-mercaptoethanol. Protein content of eluted fractions was assessed by glycine sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The Hfn-containing fractions were pooled and concentrated by buffer exchange into 50 mM MOPS pH 7.4 and stored at −80 °C.
Loading of Hfn with Fe only (Fe-Hfn), Pt only (Pt-Hfn), or Pt and Fe (Pt/Fe-Hfn)
All metal loading steps were carried out at room temperature under an aerobic atmosphere using Hfn solutions in 50 mM MOPS pH 7.4. Iron-only loaded Hfn (Fe-Hfn) was prepared using a published procedure involving repeated additions of ferrous ammonium sulfate to achieve a ratio of 2500 iron/24-mer.38,39 This procedure reproducibly resulted in Hfn containing ∼2300 Fe/24-mer, as determined using the protein and iron analyses methods described below.For preparations of Pt-containing Hfns a 100 mM aqueous stock solution of K2PtCl4 was prepared 12 hours prior to use. K2PtCl4 was added to 3.5 mL of 1 μM Hfn to achieve a Pt concentration of 1.6 mM. The Hfn/K2PtCl4 solution was incubated for 2 h. For preparation of Pt only loaded Hfn (Pt-Hfn) sodium borohydride was added from a freshly prepared aqueous solution to achieve 0.5 mol borohydride per mol K2PtCl4. The mixture was stirred for ∼60 min, then centrifuged at 10000×g for 5 min to remove an insoluble black precipitate. For preparation of Pt and iron-loaded Hfn (Pt/Fe-Hfn) an aliquot of an anaerobic aqueous stock solution of 0.02 M ferrous ammonium sulfate was added via syringe to 3.5 mL of an air-saturated 1.6 mM K2PtCl4/1 μM Hfn solution in a septum capped-vial to achieve 100 mol equivalents of added Fe(II) per Hfn 24-mer. The mixture was incubated at room temperature for 20 minutes and then centrifuged at 5500×g for 10 minutes to remove any precipitate. This process was repeated to achieve a mol ratio of ∼1500 added iron per Hfn 24-mer (extensive precipitation and loss of protein occurred beyond this ratio). If used within a few days of preparation, after the final centrifugation the Pt-Hfn or Pt/Fe-Hfn solutions were exchanged into 0.3 M TEOA pH 9 using desalting columns (Econo-Pac 10DG column, Bio-Rad), then incubated under an N2 atmosphere in a glovebox (Vacuum Atmospheres Co) for 8–12 h, and stored at 4 °C. For longer term storage Pt-Hfn or Pt/Fe-Hfn solutions were kept in 50 mM MOPS pH 7.4 and frozen at −80 °C. Just before use these frozen solutions were thawed, exchanged into 0.3 M TEOA by multiple concentration/redilution cycles using 100 K molecular weight cut-off centrifugal filter units (MillisporeSigma™Amicon™ Ultra) and transferred to the glove box, as described above.
UV-vis absorption spectra, luminescence emission spectra
UV-vis absorption spectra were obtained in 0.1 cm pathlength cells using an Ocean Optics Flame-S-UV-vis-ES spectrometer. Steady-state photoluminescence spectra were acquired on an Edinburgh FLS1000 Photoluminescence Spectrometer.Elemental analyses, protein quantification, and TEM
Protein was quantified by 660 nm Protein Assay (Thermoscientific Pierce). Samples for metal quantification were prepared by mixing 100 μL of Pt-Hfn or Pt/Fe-Hfn solutions with 500 μL freshly prepared aqua regia, and then further diluting to 7 mL total volume with water. Pt and Fe concentrations were quantified by inductively coupled plasma optical emission spectrometry (ICP-OES) at the University of Texas at San Antonio Department of Civil and Environmental Engineering. Transmission electron microscopy (TEM) used a JEOL 2010F field emission transmission electron microscope at 200 kV. 10 μL of Pt-Hfn or Pt/Fe-Hfn solutions containing ∼40 μg mLPt−1 in 50 mM MOPS pH 7.3 were placed onto ultrathin holey carbon-coated copper grids (Ted Pella) and allowed to dry at room temperature. Sizes of protein particles in solution were determined using dynamic light scattering (DLS). 1 mL samples of 1 μM Pt-Hfn or Pt/Fe-Hfn 24-mer in 0.3 M TEOA were placed in polystyrene cuvettes (Fisher Scientific), and DLS data were obtained on a Malvern Instruments Zetasizer Nano ZS. Both TEM and DLS were done at the University of Texas at San Antonio Department of Physics and Astronomy.Photosensitized generation of H2
All experiments were conducted at room temperature. Pt-Hfn or Pt/Fe-Hfn solutions in 0.3 M TEOA pH 9, aqueous 0.3 M TEOA solution pH 9, solid EY, and DMSO were brought into the glovebox. A 10 mM stock solution of EY in DMSO was prepared in the glove box. All stock solutions were left open to the N2 atmosphere for 8–12 h before use. Solutions for irradiation were prepared to contain Pt-Hfn or Pt/Fe-Hfn at a concentration of 60 μM Pt (∼0.1 μM Hfn 24-mer) in 0.3 M TEOA and 72 μM EY. 2 mL aliquots of each solution were transferred to three 6.5 mL glass vials (Chemglass). Each vial was capped with a rubber septum and Mininert® screw thread valve The capped vials were then removed from the glovebox and placed 10 cm from a 300 W halogen lamp focused through a slide projector lens with a HOYA 62 mm UV-IR multi-coated filter with wavelength cut-offs below 390 nm and above 700 nm. H2 content of the headspace was measured by gas chromatography, as described previously.36,57 For quantum yields solutions were irradiated identically to that described above but using a 550 ± 25 nm filter (Optical Filter Shop) covering the projector lens. The photon flux for H2 quantum yield was determined by chemical actinometry using solutions of Reineke's salt.58,59 The quantum yield percent was calculated as: [(2 × mol H2 produced) ÷ mol incident photons] × 100.Light-triggered iron release from Pt/Fe-Hfn
A 25 mL solution of Pt/Fe-Hfn (60 μM Pt) and 72 μM EY in 0.3 M TEOA pH 9 was prepared in the glovebox under a N2 atmosphere. The solution was split equally into seven 6.5 mL vials, then capped and removed from the glovebox. The vials were placed 10 cm from white light irradiations as described above. At selected irradiation times, individual vials were removed from the irradiation beam, and solutions were passed through desalting columns (Econo-Pac 10DG column, Bio-Rad); the first 3.0 mL of effluent, which contained the protein, were collected and prepared as described above to determine iron remaining with the protein by ICP-OES.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by funds from a University of Texas at San Antonio Lutcher Brown Endowed Chair (to D. M. K.), and National Institutes of Health grant MBRS/RISE GM060655 (to B. S. B). H. Shipley in the UTSA Department of Civil and Environmental Engineering, and K. Nash in the UTSA Department of Physics and Astronomy provided access to ICP-OES and DLS instrumentation, respectively.Notes and references
- Z. Han and R. Eisenberg, Acc. Chem. Res., 2014, 47, 2537–2544 CrossRef CAS.
- D. Z. Zee, T. Chantarojsiri, J. R. Long and C. J. Chang, Acc. Chem. Res., 2015, 48, 2027–2036 CrossRef CAS PubMed.
- B. Kandemir, S. Chakraborty, Y. Guo and K. L. Bren, Inorg. Chem., 2016, 55, 467–477 CrossRef CAS PubMed.
- S. Cao, L. Piao and X. Chen, Trends in Chemistry, 2019, 2, 57–70 CrossRef.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- G. F. Liao, J. S. Fang, Q. Li, S. H. Li, Z. S. Xu and B. Z. Fang, Nanoscale, 2019, 11, 7062–7096 RSC.
- W. Zhang, J. Hong, J. Zheng, Z. Huang, J. S. Zhou and R. Xu, J. Am. Chem. Soc., 2011, 133, 20680–20683 CrossRef CAS PubMed.
- C.-J. Wang, S. Cao, B. Qin, C. Zhang, T.-T. Li and W.-F. Fu, ChemSusChem, 2014, 7, 1924–1933 CrossRef CAS PubMed.
- D. Pedone, M. Moglianetti, E. De Luca, G. Bardi and P. P. Pompa, Chem. Soc. Rev., 2017, 46, 4951–4975 RSC.
- P. Chowdhury, G. Malekshoar and A. K. Ray, Inorganics, 2017, 5, 34 CrossRef.
- S. X. Min and G. X. Lu, J. Phys. Chem. C, 2012, 116, 19644–19652 CrossRef CAS.
- J. Kotani, R. Hanazaki, K. Ohkubo, Y. Yamada and S. Fukuzumi, Chem.–Eur. J., 2011, 17, 2777–2785 CrossRef PubMed.
- D. N. Xu, Q. Q. Chu, Z. Z. Wu, Q. Y. Chen, S. Q. Fan, G. J. Yang and B. Z. Fang, J. Catal., 2015, 325, 118–127 CrossRef CAS.
- Y. J. Wang, G. Chang, Q. Chen, G. J. Yang, S. Q. Fan and B. Fang, Chem. Commun., 2015, 51, 685–688 RSC.
- S. C. Yang, G. Chang, G. J. Yang, Y. J. Wang and B. Z. Fang, Catal. Sci. Technol., 2015, 5, 228–233 RSC.
- K. Ladomenou, M. Natali, E. Iengo, G. Charalampidis, F. Scandola and A. G. Coutsolelos, Coord. Chem. Rev., 2015, 304–305, 38–54 CrossRef CAS.
- Q. F. Liu, B. R. Liu, Q. Zhang, J. P. Gao and J. J. Ma, Mater. Lett., 2018, 221, 46–50 CrossRef CAS.
- X. J. Zhang, Z. L. Jin, Y. X. Li, S. B. Li and G. X. Lu, J. Phys. Chem. C, 2009, 113, 2630–2635 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- H. Misawa, H. Sakuragi, Y. Usui and K. Tokumaku, Chem. Lett., 1983, 12, 1021–1024 CrossRef.
- X. Zhang, Z. Jin, Y. Li, S. Li and G. Lu, J. Power Sources, 2007, 166, 74–79 CrossRef CAS.
- Y. X. Li, M. M. Guo, S. Q. Peng, G. X. Lu and S. B. Li, Int. J. Hydrogen Energy, 2009, 34, 5629–5636 CrossRef CAS.
- Y. X. Pan, H. P. Cong, Y. L. Men, S. Xin, Z. Q. Sun, C. J. Liu and S. H. Yu, ACS Nano, 2015, 9, 11258–11265 CrossRef CAS PubMed.
- D. P. Hari and B. Konig, Chem. Commun., 2014, 50, 6688–6699 RSC.
- S. D.-M. Islam, T. Konishi, M. Fujitsuka, O. Ito, Y. Nakamura and Y. Usui, Photochem. Photobiol., 2000, 71, 675–680 CrossRef CAS PubMed.
- S. Li, H. Zhang, R. Lu and A. Yu, Spectrochim. Acta, Part A, 2017, 184, 204–210 CrossRef CAS PubMed.
- M. Uchida, S. Kang, C. Reichhardt, K. Harlen and T. Douglas, Biochim. Biophys. Acta, 2010, 1800, 834–845 CrossRef CAS PubMed.
- G. Jutz, P. van Rijn, B. Santos Miranda and A. Boker, Chem. Rev., 2015, 115, 1653–1701 CrossRef CAS PubMed.
- S. Abe, B. Maity and T. Ueno, Chem. Commun., 2016, 52, 6496–6512 RSC.
- N. D. Chasteen and P. M. Harrison, J. Struct. Biol., 1999, 126, 182–194 CrossRef CAS PubMed.
- K. H. Ebrahimi, P. L. Hagedoorn and W. R. Hagen, Chem. Rev., 2015, 115, 295–326 CrossRef PubMed.
- L. B. Zhang, L. Laug, W. Munchgesang, E. Pippel, U. Gosele, M. Brandsch and M. Knez, Nano Lett., 2010, 10, 219–223 CrossRef PubMed.
- Q. Y. Deng, B. Yang, J. F. Wang, C. G. Whiteley and X. N. Wang, Biotechnol. Lett., 2009, 31, 1505–1509 CrossRef CAS PubMed.
- A. Sennuga, J. van Marwijk and C. G. Whiteley, Nanotechnology, 2012, 23, 035102 CrossRef PubMed.
- Z. Varpness, C. Shoopman, J. W. Peters, M. Young and T. Douglas, in Biomolecular Catalysis: Nanoscale Science and Technology, ed. J. Kim, S. H. Kim and P. Wang, American Chemical Society, Washington, DC, 2008, vol. 982, ch. 17, pp. 263–272 Search PubMed.
- E. R. Clark and D. M. Kurtz Jr, Dalton Trans., 2016, 45, 630–638 RSC.
- T. Masuda, F. Goto, T. Yoshihara and B. Mikami, Biochem. Biophys. Res. Commun., 2010, 400, 94–99 CrossRef CAS PubMed.
- D. Cioloboc, C. Kennedy, E. N. Boice, E. R. Clark and D. M. Kurtz Jr, Biomacromolecules, 2018, 19, 178–187 CrossRef CAS PubMed.
- V. J. Wade, S. Levi, P. Arosio, A. Treffry, P. M. Harrison and S. Mann, J. Mol. Biol., 1991, 221, 1443–1452 CrossRef CAS PubMed.
- M. Majek, F. Filace and A. J. von Wangelin, Beilstein J. Org. Chem., 2014, 10, 981–989 CrossRef CAS PubMed.
- A. Alvarez-Martin, S. Trashin, M. Cuykx, A. Covaci, K. De Wael and K. Janssens, Dyes Pigm., 2017, 145, 376–384 CrossRef CAS.
- D. J. Gao, Y. Tian, F. H. Liang, D. H. Jin, Y. H. Chen, H. Q. Zhang and A. M. Yu, J. Lumin., 2007, 127, 515–522 CrossRef CAS.
- L. Wang, H. Zhao, Y. Chen, R. Sun and B. Han, Opt. Commun., 2016, 370, 122–126 CrossRef CAS.
- E. M. Arbeloa, C. M. Previtali and S. G. Bertolotti, J. Lumin., 2016, 180, 369–375 CrossRef CAS.
- L. V. Lutkus, S. S. Rickenbach and T. M. McCormick, J. Photochem. Photobiol., A, 2019, 378, 131–135 CrossRef CAS.
- A. Aguirre-Soto, K. Kaastrup, S. Kim, K. Ugo-Beke and H. D. Sikes, ACS Catal., 2018, 8, 6394–6400 CrossRef CAS.
- C. Pozzi, F. Di Pisa, C. Bernacchioni, S. Ciambellotti, P. Turano and S. Mangani, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 1909–1920 CrossRef CAS PubMed.
- L. Li, L. Zhang, U. Carmona and M. Knez, Chem. Commun., 2014, 50, 8021–8023 RSC.
- Y. J. Kang, M. Uchida, H. H. Shin, T. Douglas and S. Kang, Soft Matter, 2011, 7, 11078–11081 RSC.
- F. Bou-Abdallah, J. J. Paliakkara, G. Melman and A. Melman, Pharmaceuticals, 2018, 11, 120 CrossRef CAS PubMed.
- M. Liang, K. Fan, M. Zhou, D. Duan, J. Zheng, D. Yang, J. Feng and X. Yan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14005–14905 CrossRef.
- S. Bai, W. Yin, L. Wang, Z. Li and Y. Xiong, RSC Adv., 2016, 6, 57446–57463 RSC.
- J. M. Slocik, C. A. Crouse, J. E. Spowart and R. R. Naik, Nano Lett., 2013, 13, 2535–2540 CrossRef CAS PubMed.
- J. A. Han, Y. J. Kang, C. Shin, J. S. Ra, H. H. Shin, S. Y. Hong, Y. Do and S. Kang, Nanomedicine, 2014, 10, 561–569 CrossRef CAS PubMed.
- S. Kim, G. S. Kim, J. Seo, G. Gowri Rangaswamy, I. S. So, R. W. Park, B. H. Lee and I. S. Kim, Biomacromolecules, 2016, 17, 12–19 CrossRef CAS.
- R. A. Schaller, S. K. Ali, K. E. Klose and D. M. Kurtz Jr, Biochemistry, 2012, 51, 8563–8570 CrossRef CAS PubMed.
- E. R. Clark and D. M. Kurtz Jr, Inorg. Chem., 2017, 56, 4585–4594 CrossRef PubMed.
- E. E. Wegner and A. W. Adamson, J. Am. Chem. Soc., 1966, 88, 394–403 CrossRef CAS.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Boca Raton, 3rd edn, 2006 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Time course of H2 production with 550 nm irradiation; GC traces of H2 production in air-saturated solution; time course for H2 production using fluorescein as PS. See DOI: 10.1039/d0ra00341g |
| ‡ All Error bars represent standard deviations from the average for three simultaneously irradiated reaction vials. |
| This journal is © The Royal Society of Chemistry 2020 |