
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight and magnetism dual-gated photoinduced electron transfer-reversible addition–fragmentation chain transfer (PET-RAFT) polymerization†
Lingjuan Hu‡
a,
Qi Wang‡a,
Xiaomeng Zhanga,
Haitao Zhaoa,
Zhe Cuia,
Peng Fua,
Minying Liua,
Nan Liu b,
Shengbao Heb,
Xinchang Pang*a and
Xiaoguang Qiao*a
b,
Shengbao Heb,
Xinchang Pang*a and
Xiaoguang Qiao*a
aHenan Joint International Research Laboratory of Living Polymerizations and Functional Nanomaterials, Henan Key Laboratory of Advanced Nylon Materials and Application, School of Materials Science and Engineering, Zhengzhou University, Zhengzhou, 450001, China. E-mail: Pangxinchang1980@163.com; joexiaoguang@hotmail.com
bChina National Tobacco Quality Supervision and Test Centre, Zhengzhou 450001, China
First published on 13th February 2020
Abstract
A novel raspberry-like γ-Fe2O3@carbon dot (CD) nanocatalyst was prepared and applied for photoinduced electron transfer-reversible addition–fragmentation chain transfer (PET-RAFT) polymerization. The nanocatalyst was found to be an efficient photocatalyst in visible light-regulated PET-RAFT polymerization owing to the oxidative quenching mechanism between the photoexcited γ-Fe2O3@CDs and the RAFT agent in the PET process. Notably, polymerization can be reversibly ceased in the absence of light or under an external magnetic field. The superparamagnetic nature and high saturation magnetization value (∼30.4 emu g−1) of the nanocatalyst contribute to convenient recycling of the nanocatalyst after polymerization. The PET-RAFT polymerization with the nanocatalyst before and after recycling was investigated, which displayed all the characteristics of controlled/living polymerization systems.
Introduction
In recent years, with the development of “living”/controlled radical polymerization,1–7 the temporal and spatial sequence regulation of polymers is mediated by external stimuli, such as microwaves,8 ultrasound,9 light10–13 or electric pulses.14 Among these stimuli, light shows the greatest potential for an ideal system because it is cheap, renewable, facile to manipulate, etc. Moreover, the introduction of light as an external stimulus in polymerization not only optimizes the reaction conditions, but also achieves the design, synthesis and precise control of complex systems.15–19 Photo-induced living radical polymerization have been reported in numerous studies,20–22 among which the photo-induced electron transfer-reversible addition–fragmentation chain transfer (PET-RAFT) polymerization is recognized to be a versatile technique for the synthesis of tailored polymers and fabrication of advanced materials. This so-called PET-RAFT is mediated by photocatalysts that can transfer electrons from excited states to chain transfer agents (CTAs) to generate radicals with the irradiation of light and initiate polymerization.23 Visible light,24 near-infrared light and far-red light-induced25 RAFT polymerizations at various wavelength bands have gained wide attention.The previously developed polymerized photocatalysts always require metal complexes, such as fac-[Ir(ppy)3],26 Ru(bpy)3Cl2,27 zinc porphyrins,28,29 and semiconducting nanoparticles.30 Despite the excellent catalytic properties, trace amounts of these photocatalysts remaining in the polymer matrix may cause decomposition of the final polymer products or lethal contamination.31,32 Therefore, Boyer et al. developed some metal-free catalysts such as Eosin Y and halogenated xanthene dyes to avoid metal contamination.33 Very recently, carbon dots (CDs) have been reported to be used as efficient metal-free photocatalysts in visible-light-regulated PET-RAFT polymerization.34 As new nanocarbon materials, CDs are well-known for their low toxicity, low cost, good solvent adaptability and excellent photostability. Although such metal-free PET-RAFT systems may be environment-friendly on the whole, the catalysts remaining in the polymer products still create some problems in the performance of the final products. Therefore, it is quite necessary to develop a simple method to separate and recycle the catalysts in order to avoid the negative effect on the final polymer products and realize the reusability of the catalysts simultaneously.
Boyer et al. conjugated Eosin Y to the surface of silica nanoparticles and applied it as a recyclable organic nanocatalyst for photo-induced RAFT polymerization.35 However, the described recycling process relies on centrifugation, which is strongly dependent on the size of silica particles. Magnetic separation is another common separation technique that has been widely used in a number of fields.36 Contrary to centrifugation, magnetic separation is simpler and more effective. Magnetic semiconductor nanoparticles (Zn0.64Fe2.36O4) have been reported to be recyclable oxytolerant catalysts for sunlight-photolyzed RAFT photopolymerization.37 The most direct and convenient way is the combination of a photocatalyst with superparamagnetic nanomaterials, such as γ-Fe2O3 and Fe3O4.38,39 For example, it has been reported that magnetic Fe3O4 combined with the ZnPTPP nanocomposites can be separated from the polymerization system and recycled by simple magnetic decantation.40 However, the designed catalyst has low saturation magnetization (∼12.9 emu g−1) because of the decrease in dipolar interactions caused by silica. Therefore, the process of recovering the catalyst must rely on extra centrifugation in addition to magnetic separation. To the best of our knowledge, the combination of superparamagnetic materials and CDs as catalysts in PET-RAFT polymerization has never been reported before.
In this paper, novel raspberry-like γ-Fe2O3@CD nanocomposites were fabricated and applied as recoverable efficient photocatalysts for PET-RAFT polymerization. The CDs have less effect on the saturation magnetization value of the γ-Fe2O3@CD nanocomposites (∼30.4 emu g−1), indicating the high magnetic responsiveness of the catalysts. The excellent efficiency of the hybrid catalysts in the PET-RAFT process was demonstrated by using different monomers. Notably, the polymerization process can be reversibly ceased in the absence of light or under an external magnetic field. After polymerization, the magnetic separation performance and catalysis recoverability of the raspberry-like γ-Fe2O3@CD nanoparticles were also evaluated.
Experimental sections
Materials
Methyl methacrylate (MMA, 99%), methyl acrylate (MA, 99%), n-butyl methacrylate (BA, 99%), triethylamine (≥99%), styrene (St, 99%), and anhydrous dimethyl sulfoxide (DMSO) were all purchased from Aladdin. The MMA monomers were purified by passing through a basic alumina column (100–200 mesh, purchased from Aladdin) to remove the inhibitors prior to use. Sodium thiosulfate pentahydrate (99%) and sodium citrate tribasic dehydrate (99%) were purchased from J&K Scientific. Blue LED light strips (λmax = 420 nm, 1 m, 6 W, 2 mW cm−2) were purchased from Rishang Optoelectronics Co., Ltd. for visible light-regulated RAFT polymerizations. 4-Cyanopentanoic acid dithiobenzoate (CPADB) was also purchased from Aladdin. For the preparation of γ-Fe2O3 nanoparticles, FeCl2 and FeCl3 precursors, ammonium hydroxide solution (NH4OH, 28–30 wt%) in water, nitric acid (HNO3, 1 M) and hydrochloric acid (HCl, 36 wt%) were purchased from Sigma-Aldrich. NH3·H2O, HNO3, acetone, ethanol and ether were purchased from Sinopharm Chemical Reagent Co. Ltd. The chemicals were used without any further purification. Deionized (DI) water was used from the laboratory.Characterizations
Transmission electron microscopy (TEM) analysis was performed with a Philips CM200 microscope at an accelerating voltage of 200 kV. X-ray diffraction (XRD) analysis was performed with an X'Pert PRO polycrystalline powder X-ray diffractometer from Panalytical. The monochromatic Cu Kα ray diffraction was used with a scanning speed of 0.5° min−1 and a diffraction angle range of 20–70°. The magnetization curves of the nanoparticles were characterized by a Quantum Design MPMS-5S superconducting quantum interference device (SQUID) magnetometer. 1H-NMR spectra were recorded using a Bruker spectrometer (300 MHz), and all the samples were analyzed using CDCl3 as the solvent. The molecular weights (Mn) of the polymers were measured by using a Waters gel permeation chromatograph (1515 HPLC pump) with THF as the solvent. THF was used as the eluent at a flow rate of 1.0 ml min−1 at 35 °C. The UV-vis absorption spectra of the samples were obtained using a Thermo Scientific EV300 UV-vis spectrophotometer. The Fourier transform infrared (FTIR) spectra of the samples were obtained by using a Bruker Spectrum FTIR system. The photoluminescence spectra of the samples were recorded on a Thermo Scientific Lumina spectrofluorometer.Experimental procedures
Results and discussion
Synthesis and characterization of γ-Fe2O3 and γ-Fe2O3@CD raspberry-like nanoparticles
As illustrated in Scheme 1a, the synthetic route for the preparation of the γ-Fe2O3@CD hybrid nanoparticles (NPs) was designed to be an in situ growth process. γ-Fe2O3 nanoparticles with a high positive charge (+41.9 mV) were first prepared through co-precipitation and strong oxidation by following a previously reported method.41 Five distinct diffraction peaks can be seen clearly and no other miscellaneous peaks appear in the XRD pattern of the sample, illustrating the successful synthesis of γ-Fe2O3 (Fig. S1†). Considering the strong negative charges, the precursors of CDs (−102.3 mV) were supposed to be adsorbed on the surface of γ-Fe2O3 and in situ grow to form raspberry-like nanocatalysts. | ||
| Scheme 1 (a) The design and synthesis of γ-Fe2O3@CD nanoparticles via the hydrothermal method. (b) Proposed mechanism of γ-Fe2O3@CD-catalyzed PET-RAFT polymerization under blue LED light irradiation. The chemical structures of RAFT agents in this study. | ||
The TEM images show that the obtained magnetic γ-Fe2O3 NPs exhibit approximately spherical morphological characteristics with a uniform size of ∼30 nm (Fig. 1a). γ-Fe2O3 NPs were then mixed with sodium citrate and sodium thiosulfate solutions in aqueous media. During the hydrothermal reaction process, the precursors (oligomers with rich SO3−, COO−, and OH− groups) of CDs appeared gradually and adsorbed on the surface of the γ-Fe2O3 nanoparticles owing to the positive and negative electrostatic effects (Fig. S2†). The precursors grew continually to form tiny nodules (CDs) on the surface of γ-Fe2O3, and raspberry-like hybrid nanoparticles were finally obtained (Fig. 1c and d). Compared with pure CDs (Fig. 1b, ∼5.3 nm), those on the γ-Fe2O3 surface had poor uniformity and regularity, which may be due to the confinement of growth and orientation. Even so, it could be seen that both γ-Fe2O3@CD and CD nanoparticles had the same UV absorption (Fig. 2a) and strong wide PL emission wavelengths ranging from 400 nm to 520 nm upon excitation using the wavelength of 360 nm, which was attributed to the homogeneous surface structure and size of S-doped CDs (Fig. 2b).42 These results illustrate the successful coating of CDs on the surface of the γ-Fe2O3 nanoparticles.
 | ||
| Fig. 1 TEM images of (a) superparamagnetic γ-Fe2O3, (b) sulfur-doped carbon dots, (c and d) superparamagnetic γ-Fe2O3@CD raspberry-like nanoparticles as the catalysts at different magnifications. | ||
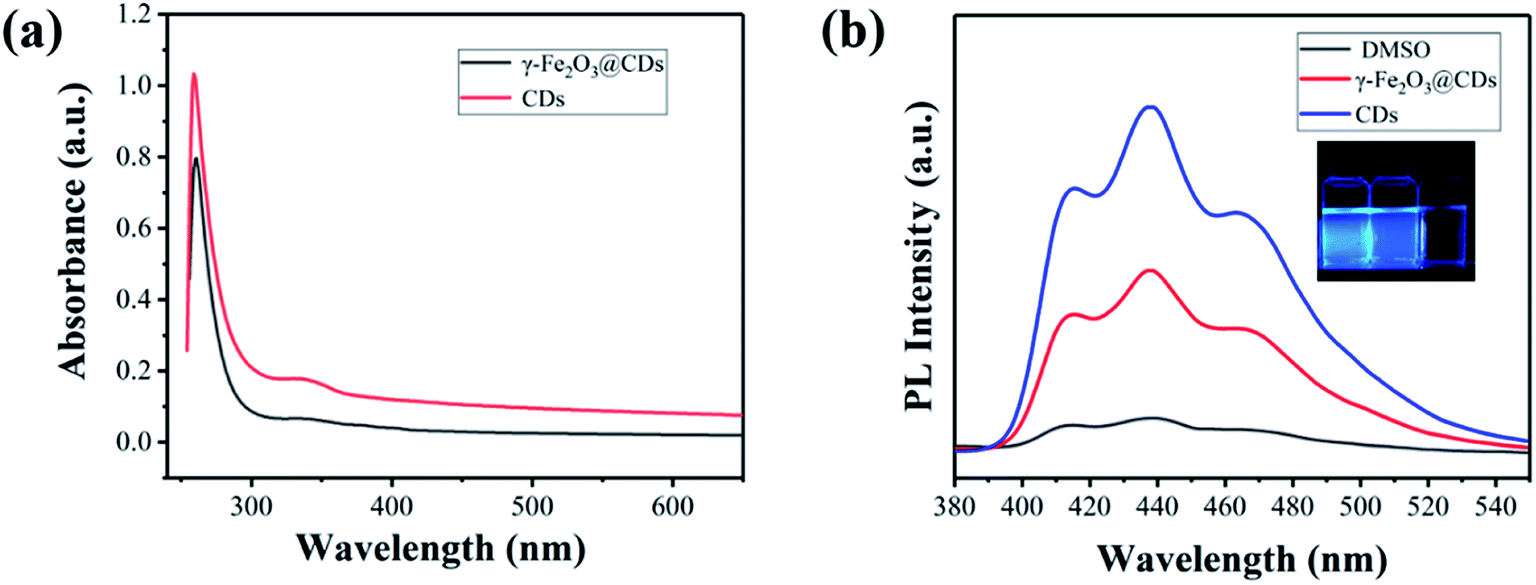 | ||
| Fig. 2 UV-vis absorption spectrum (a) and photoluminescence spectrum (b) of CDs and superparamagnetic γ-Fe2O3@CD nanoparticles dispersed in DMSO (the inset photographs represent CDs, γ-Fe2O3@CDs dispersed in DMSO and pure DMSO, respectively). | ||
PET-RAFT polymerization of MMA
As shown in Fig. 3, the PET-RAFT polymerization of MMA using γ-Fe2O3@CDs as a photoredox catalyst exhibits all the features of a controlled/living system. Fig. 3a shows the linear evolution of ln([M]0/[M]t) with the exposure time, and such first-order kinetics suggest a constant and steady concentration of growing radicals during polymerization. The GPC traces of PMMA obtained in the kinetic study showed the monomodal distribution of the molecular weight shifting with the exposure time (Fig. 3b). The evolution of Mn showed a linear increase with the increase in molar conversion and gave narrow molar mass distributions (Mw/Mn < 1.2) throughout the polymer, indicating a good control of the polymerization process (Fig. 3d). Notably, the polymerization process can be easily manipulated by switching the light or adopting a magnetic field. The entire polymerization process is mediated by the rapidly reversible activation and deactivation of the nanocatalyst in “ON/OFF” experiments. Fig. 3c shows good temporal control over the polymerization process. In the absence of light, no polymerization was observed, and the polymerization was reinitiated steadily once the light was switched “ON”. The experimental molecular weight linearly increased with monomer conversion; it was substantially consistent with the theoretical values and showed a narrow dispersity distribution (Mw/Mn ≈ 1.19, Fig. 3d). Similarly, the polymerization reaction stopped immediately once the magnetic nanocatalyst was separated from the system due to the highly limited access of CTA to CDs on the surface, which may influence the electron transfer between CDs on the surface of the γ-Fe2O3@CD nanocatalyst and the RAFT agent. The polymerization preferentially restarted once the magnetic field was removed (Fig. 3e) as the nanocatalyst could be well re-dispersed in the media according to its superparamagnetic properties. In addition, the living characters of the polymer were confirmed through a chain-extension experiment, in which the macro-RAFT agent (PMMA with Mn = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 g mol−1) extended to the polymer chain with Mn = 18700 g mol−1 and a narrow molar mass distribution (Mw/Mn = 1.15).
700 g mol−1) extended to the polymer chain with Mn = 18700 g mol−1 and a narrow molar mass distribution (Mw/Mn = 1.15).
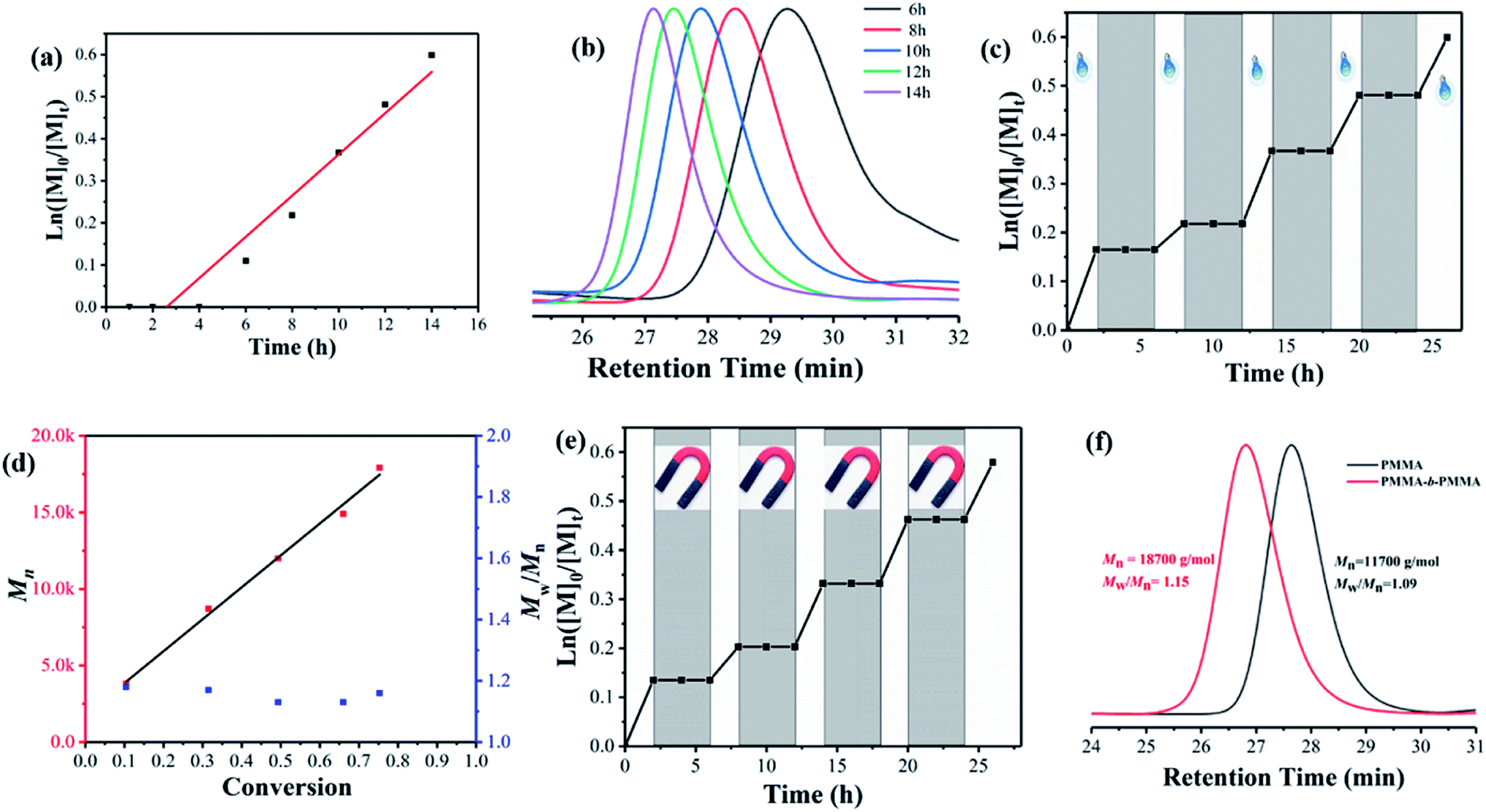 | ||
| Fig. 3 Kinetic study of PET-RAFT polymerization of MMA catalyzed by γ-Fe2O3@CD photocatalyst. (a) Plot of ln[M]0/[M]t versus exposure time t. (b) GPC traces in the kinetic study of PMMA synthesized via PET-RAFT polymerization. (c) Temporal control over polymerization upon switching the light. (d) Number-average molecular weight (Mn) and polydispersity index (PDI) of PMMA synthesized in the kinetic study. (e) Temporal control over polymerization upon adopting a magnetic field; (f) GPC traces of chain extension products for PMMA (black) and PMMA-b-PMMA (red). | ||
According to the polymerization results, a photoredox quenching mechanism was proposed to explain the effective catalysis of γ-Fe2O3@CDs in PET-RAFT polymerization (Scheme 1b). CDs on the surface of γ-Fe2O3@CDs were supposed to act as electron acceptors or electron donors via electron transfer.43–45 Photoexcited γ-Fe2O3@CDs could undergo charge transfer with the RAFT agent upon light irradiation. The reduced RAFT agent may generate free radicals and oxidize the excited γ-Fe2O3@CDs as a degenerative chain transfer agent. The generated free radicals can initiate polymerization and chain growth in a controlled manner, and the radicals may eventually interact with the oxidized γ-Fe2O3@CDs to “reset” the system and complete the catalytic cycle.46
In order to demonstrate the electron transfer between γ-Fe2O3@CDs and RAFT agents, a fluorescence quenching study was performed (Fig. S5†), showing that the fluorescence emission intensity decreased continuously as the concentration of the RAFT agent increased. In addition, the ratio of I0/I showed a nonlinear relationship with the concentration of the RAFT agent, demonstrating the electron transfer between the chain transfer agent and γ-Fe2O3@CDs. The addition of TEA in the polymerization process may remove the holes as sacrificial electron donors to accelerate the PET-RAFT polymerization rate. Moreover, such PET-RAFT polymerization systems with other types of monomers were investigated, as listed in Table 1. According to the controlled molecular weight and narrow molecular weight distributions (Mw/Mn < 1.2) for MA, BA and St, the versatility and universality of the nanocatalysts can be confirmed.
| Entrya | [M]:[CTA] |
Monomer | RAFT agent | Time (h) | Conv.b (%) | Mn,thb (g mol−1) | Mn,GPCc (g mol−1) | Mw/Mn |
|---|---|---|---|---|---|---|---|---|
| a The reactions were performed at room temperature with 50% v/v monomer concentration under blue LED light irradiation (6 W, λmax = 460 nm, 2 mW cm−2) in the absence of oxygen in DMSO ([M]0/[CTA]:[TEA] = 200:1:6).b The molecular weight was calculated using the following equation: Mn,NMR = [M]0/[CTA] × MMw × conv. + MCTAw, where [M]0, [CTA], MMw, conv. and MCTAw correspond to initial monomer concentration, initial CTA concentration, molar mass of monomer, monomer conversion determined from 1H NMR spectroscopy, and molar mass of CTA.c Molecular weight and polydispersity index were determined by THF GPC analysis calibrated using polystyrene standards.d The reaction was carried out in the dark.e The reaction was performed without γ-Fe2O3@CD nanoparticles.f The reaction was performed in the absence of TEA. |
||||||||
| 1 | 200:1 |
MMA | — | 24 | 0 | 0 | 0 | — |
| 2d | 200:1 |
MMA | CPADB | 48 | 0 | 0 | 0 | — |
| 3e | 200:1 |
MMA | CPADB | 24 | 17.65 | 3800 | 4500 | 1.19 |
| 4f | 200:1 |
MMA | CPADB | 24 | 30.2 | 6300 | 7200 | 1.17 |
| 5 | 200:1 |
MMA | CPADB | 24 | 76.42 | 15600 |
14200 |
1.18 |
| 6 | 200:1 |
MA | CPDTC | 24 | 84.43 | 14500 |
20600 |
1.12 |
| 7 | 200:1 |
BA | CPDTC | 24 | 48.32 | 12700 |
13300 |
1.07 |
| 8 | 200:1 |
St | CDTPA | 48 | 41.29 | 9000 | 5100 | 1.13 |
| 9 | 100:1 |
MMA | CPADB | 24 | 79.33 | 8200 | 7700 | 1.14 |
| 10 | 400:1 |
MMA | CPADB | 24 | 75 | 30300 |
28900 |
1.13 |
Recovery and reuse of the γ-Fe2O3@CD nanocatalyst
Fig. 4a shows the magnetization curves functioned with the magnetic field for pure γ-Fe2O3 and γ-Fe2O3@CD nanoparticles before and after recovery. The magnetization curves indicate excellent superparamagnetic properties, which are crucial for magnetism-gated PET-RAFT polymerization. As expected, the saturation magnetization value for γ-Fe2O3@CDs (∼30.4 emu g−1) was smaller than that for pure γ-Fe2O3 (∼33.1 emu g−1), and this may be due to the shielding effect of the exterior CD shell and the increased interparticle distances between the magnetic γ-Fe2O3 nanoparticles. On the other hand, such a small reduction in the saturation magnetization value (2.7 emu g−1) suggested that the CD shell was so thin that the shielding effect was very weak. Moreover, the recycled γ-Fe2O3@CD nanoparticles showed almost the same magnetization curves as the one before use, indicating that the CD layer on the surface of γ-Fe2O3 (or the raspberry-like hybrid structure) did not change or shield during polymerization and recycling. The γ-Fe2O3@CD nanocatalysts could be directly recycled by magnetic separation without centrifugation due to the high magnetic responsiveness (the inset picture in Fig. 4a). Fig. 4b shows that the recycled γ-Fe2O3@CD nanoparticles yield bright blue emission when illuminated under UV light (360 nm), indicating that the γ-Fe2O3@CD nanoparticles still have excellent fluorescence properties after recovery.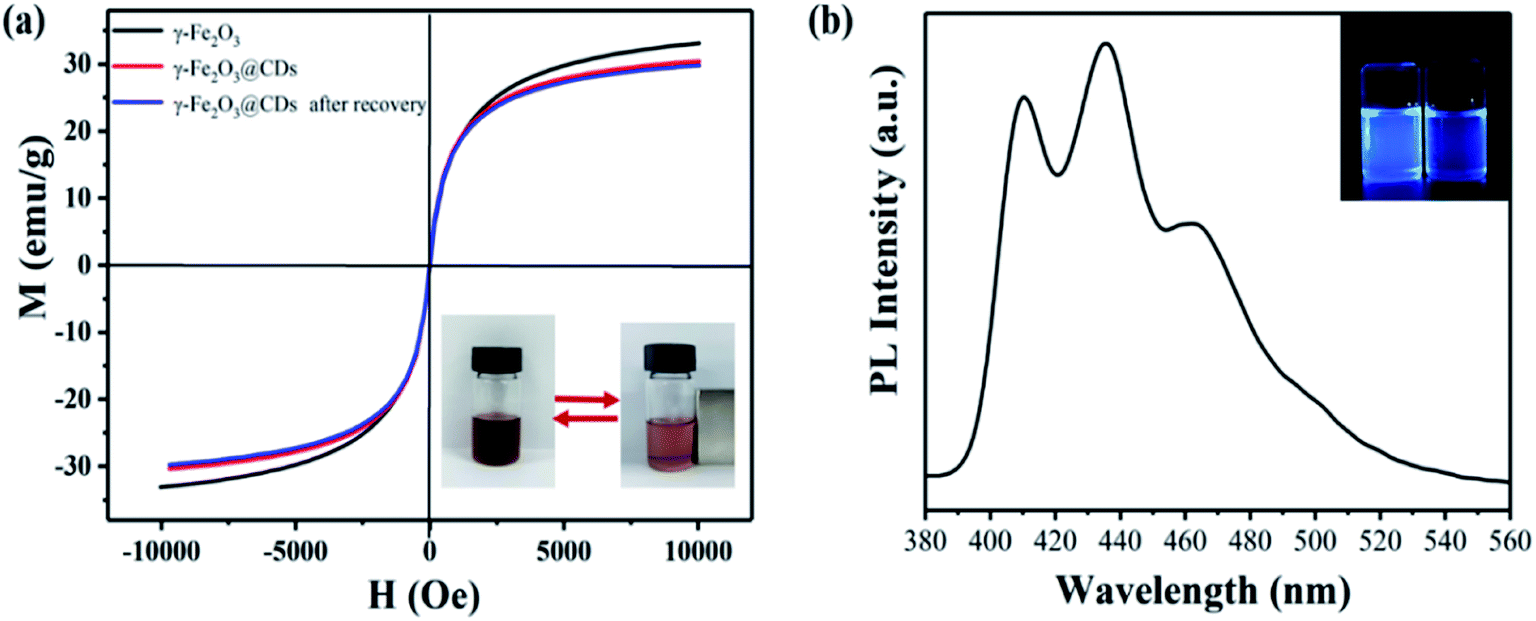 | ||
| Fig. 4 (a) Field-dependent magnetization of the synthesized γ-Fe2O3, γ-Fe2O3@CD and γ-Fe2O3@CD nanoparticles after recovery. (b) The photoluminescence spectrum of superparamagnetic γ-Fe2O3@CD nanoparticles after recovery (the inset picture is a fluorescence photograph of γ-Fe2O3@CDs in DMSO and pure DMSO under 360 nm irradiation). | ||
The recycled γ-Fe2O3@CD nanoparticles were then applied for PET-RAFT polymerization with MMA. As shown in Fig. 5, very similar polymerization kinetic results are obtained. Although the polymerization rate was slightly slower than that of the polymerization at the first time, the linear evolution of the kinetic plots was observed again. The linear increase in Mn on increasing monomer conversion and the narrow molar mass distribution (Mw/Mn < 1.2) confirmed the excellent control of polymerization. The GPC peaks of the polymer shifted to a higher molar mass with the increase in monomer conversion, which confirmed the living character of the polymerization. These results indicated the excellent catalytic properties and recoverability of the γ-Fe2O3@CD nanoparticles in PET-RAFT polymerization.
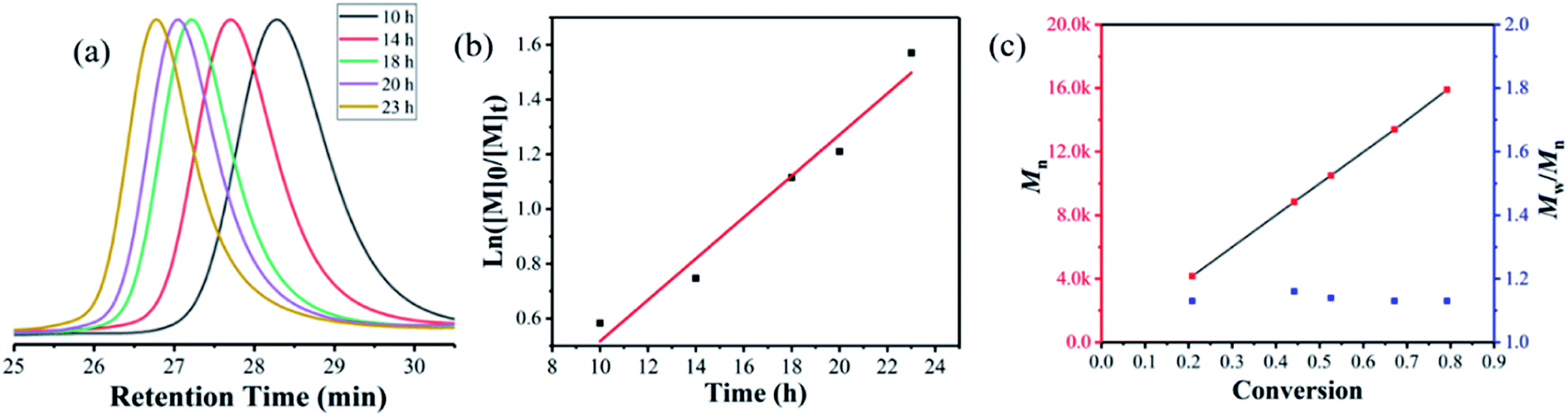 | ||
| Fig. 5 PET-RAFT polymerization of γ-Fe2O3@CD nanoparticles recovered for the second time by magnetic separation at room temperature under blue light irradiation (6 W, λmax = 460 nm, 2 mW cm−2) in the absence of oxygen at 25 °C in DMSO ([M]0/[CTA] = 200:1). (a) GPC traces in the kinetic study of PMMA synthesized via PET-RAFT polymerization for the second time. (b) The relationship between ln([M]0/[M]t) and reaction time; (c) Mn vs. monomer conversion and Mw/Mn versus conversion. | ||
In order to evaluate the stability of magnetically responsive γ-Fe2O3@CD nanoparticles after polymerization and recovery, a series of cycle experiments were performed for at least 5 times, as listed in Table 2. All the monomer conversions exceeded 50 mol% during cyclic polymerization, indicating the high recycling stability of the nanocatalysts (Fig. S9†). However, the conversion of second polymerization was slightly lower than that for the first one, which may be due to the partial aggregation of the γ-Fe2O3@CD nanoparticles during the recovery process. The high surface energy of the nanoparticles was suggested to be the reason for aggregation, which resulted in lower catalytic efficiency for polymerization. Relatively narrow molecular weight distributions (Mw/Mn ≤ 1.33) were observed for all polymerizations, demonstrating good control of the PET-RAFT polymerization with the nanocatalyst.
| No. of cycles | Time (h) | Conv.b (%) | Mn,thc (g mol−1) | Mn,GPCd (g mol−1) | Mw/Mn |
|---|---|---|---|---|---|
| a Reactions were performed in the absence of oxygen at room temperature with 50% v/v monomer concentrations with [MMA]:[CPADB] of 200:1.b Monomer conversion was determined by using 1H NMR spectroscopy.c Theoretical molecular weight was calculated using the following equation: Mn,NMR = [M]0/[CTA] × MMw × conv. + MCTAw, where [M]0, [CTA], MMw, conv. and MCTAw correspond to initial monomer concentration, initial CTA concentration, molar mass of monomer, monomer conversion determined from 1H NMR spectroscopy, and molar mass of CTA.d Molecular weight and polydispersity index (Mw/Mn) were determined by GPC analysis calibrated to polystyrene standards. |
|||||
| 1 | 24 | 79.17 | 16100 |
19000 |
1.13 |
| 2 | 24 | 65.32 | 13400 |
16800 |
1.23 |
| 3 | 24 | 62.40 | 12800 |
15500 |
1.22 |
| 4 | 24 | 60.81 | 12200 |
14800 |
1.33 |
| 5 | 24 | 63.52 | 13000 |
14200 |
1.17 |
Conclusions
In summary, recyclable novel dual stimuli-responsive raspberry-like γ-Fe2O3@CD photocatalysts have been designed and synthesized through an in situ growth process. The precursors of CDs (−102.3 mV) were adsorbed on the surface of γ-Fe2O3 (+41.9 mV) based on the positive and negative electrostatic interactions and in situ grew to form raspberry-like nanocatalysts. The PET-RAFT polymerization in the presence of the γ-Fe2O3@CD nanocatalyst exhibited all the characteristics of controlled radical polymerization with different monomers and displayed magnetic/visible light double regulated behavior. Due to the high magnetization values before (∼30.4 emu g−1) and after recycling (∼30.0 emu g−1), the nanocatalyst could be easily removed by simple magnetic separation from the polymer products and reused repeatedly, making such PET-RAFT processes more sustainable and less-contaminated. Moreover, the raspberry-like nanocatalyst is expected to be applied in various living radical polymerization systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the National Science Foundation of China (Grant No. U1804128, 51973201, to Xinchang Pang), Key R&D and Promotion Special Program of Henan Province (Grant No. 2018-966, to Xinchang Pang), the 111 project (D18023, to Xinchang Pang), the National Science Foundation for Young Scientists of China (Grant No. 51703206, to Zhe Cui), the National Key R&D Program of China (2017YFB0307600, to Minying Liu), the Postdoctoral Research grand in Henan Province (No. 1901007).Notes and references
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef PubMed.
- S. Shanmugam, J. Xu and C. Boyer, Macromol. Rapid Commun., 2017, 38, 1700143 CrossRef PubMed.
- B. Li, Y. Shi and Z. Fu, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1653–1663 CrossRef.
- H. Sun, L. Yang, M. P. Thompson, S. Schara, W. Cao, W. Choi, Z. Hu, N. Zang, W. Tan and N. C. Gianneschi, Bioconjugate Chem., 2019, 30, 1889–1904 CrossRef PubMed.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef PubMed.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef.
- P. B. Zetterlund and S. b. Perrier, Macromolecules, 2011, 44, 1340–1346 CrossRef.
- M. Kemmere, M. Kuijpers, L. Jacobs and J. Keurentjes, Macromol. Symp., 2004, 206, 321–331 CrossRef.
- J. Phommalysack-Lovan, Y. Chu, C. Boyer and J. Xu, Chem. Commun., 2018, 54, 6591–6606 RSC.
- P. N. Kurek, A. J. Kloster, K. A. Weaver, R. Manahan, M. L. Allegrezza, N. Boyer, C. De Alwis Watuthanthrige, J. A. Reeves and D. Konkolewicz, Ind. Eng. Chem. Res., 2018, 57, 4203–4213 CrossRef.
- A. Wolpers and P. Vana, Macromolecules, 2014, 47, 954–963 CrossRef.
- Y. Nakamura, T. Arima, S. Tomita and S. Yamago, J. Am. Chem. Soc., 2012, 134, 5536–5539 CrossRef PubMed.
- C. C. Kathrein, W. K. Kipnusu, F. Kremer and A. Boeker, Macromolecules, 2015, 48, 3354–3359 CrossRef.
- D. Wei, Y. Xu, C. Liu, Y. Zhai, H. Chen, L. Bai, H. Yang, L. Yang, W. Wang and Y. Niu, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1265–1269 CrossRef.
- M. Tan, Y. Shi, Z. Fu and W. Yang, Polym. Chem., 2018, 9, 1082–1094 RSC.
- T. D. Nguyen, S. T. Vo, M. R. Islam, K. T. Lim, D.-V. N. Vo and L. G. Bach, J. Chem. Technol. Biotechnol., 2019, 94, 1416–1424 CrossRef.
- Z. Li, H. Tang, A. Feng and S. H. Thang, Prog. Chem., 2018, 30, 1097–1111 Search PubMed.
- F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 199–210 CrossRef PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- X. Pan, M. Fantin, F. Yuan and K. Matyjaszewski, Chem. Soc. Rev., 2018, 47, 5457–5490 RSC.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2015, 116, 1969–1992 CrossRef PubMed.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef PubMed.
- Y. Song, Y. Kim, Y. Noh, V. K. Singh, S. K. Behera, A. Abudulimu, K. Chung, R. Wannemacher, J. Gierschner, L. Lüer and M. S. Kwon, Macromolecules, 2019, 52, 5538–5545 CrossRef.
- S. Shanmugam, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55, 1036–1040 CrossRef.
- W. Ma, H. Chen, Y. Ma, C. Zhao and W. Yang, Macromol. Chem. Phys., 2014, 215, 1012–1021 CrossRef.
- J. Xu, K. Jung and C. Boyer, Macromolecules, 2014, 47, 4217–4229 CrossRef.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef PubMed.
- N. Corrigan, D. Rosli, J. W. J. Jones, J. Xu and C. Boyer, Macromolecules, 2016, 49, 6779–6789 CrossRef.
- Y. Huang, Y. Zhu and E. Egap, ACS Macro Lett., 2018, 7, 184–189 CrossRef.
- E. Kolvari, M. Zolfigol, N. Koukabi and B. Shirmardi-Shaghasemi, Chem. Pap., 2011, 65, 898–902 Search PubMed.
- S. V. Bhagwat, S. Jouen, D. C. Kundaliya, H. Singh, T. Jagadale, A. A. Athawale and S. B. Ogale, J. Nanosci. Nanotechnol., 2009, 9, 5823–5828 CrossRef PubMed.
- J. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chem. Sci., 2014, 5, 3568–3575 RSC.
- J. Jiang, G. Ye, Z. Wang, Y. Lu, J. Chen and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 12037–12042 CrossRef.
- S. Shanmugam, S. Xu, N. N. M. Adnan and C. Boyer, Macromolecules, 2018, 51, 779–790 CrossRef.
- K. Kołątaj, R. Ambroziak, M. Kędziora, J. Krajczewski and A. Kudelski, Appl. Surf. Sci., 2019, 470, 970–978 CrossRef.
- J. Wang, M. Rivero, A. Muñoz Bonilla, J. Sanchez-Marcos, W. Xue, G. Chen, W. Zhang and X. Zhu, ACS Macro Lett., 2016, 5, 1278–1282 CrossRef.
- L. Bartolome, M. Imran, K. G. Lee, A. Sangalang, J. K. Ahn and D. H. Kim, Green Chem., 2014, 16, 279–286 RSC.
- Y. F. Yang, F. Y. Meng, X. H. Li, N. N. Wu, Y. H. Deng, L. Y. Wei and P. Zeng, J. Nanosci. Nanotechnol., 2019, 19, 7517–7525 CrossRef PubMed.
- X. Li, J. L. Li, W. G. Huang, X. Z. Zhang, B. Zhang and T. Cai, Nanoscale, 2018, 10, 19254–19261 RSC.
- K. Li, P. Y. Dugas, M. Lansalot and E. Bourgeat-Lami, Macromolecules, 2016, 49, 7609–7624 CrossRef.
- Q. Xu, P. Pu, J. Zhao, C. Dong, C. Gao, Y. Chen, J. Chen, Y. Liu and H. Zhou, J. Phys. Chem. A, 2015, 3, 542–546 Search PubMed.
- Z. Huang, L. Feng, H. Ming, C. Li, T. Xu, C. Chen and X. Guo, J. Lumin., 2014, 151, 100–105 CrossRef.
- V. Strauss, J. T. Margraf, K. Dirian, Z. Syrgiannis, M. Prato, C. Wessendorf, A. Hirsch, T. Clark and D. M. Guldi, Angew. Chem., 2015, 127, 8410–8415 CrossRef.
- A. Cadranel, V. Strauss, J. T. Margraf, K. A. Winterfeld, C. Vogl, L. Dordevic, F. Arcudi, H. Hoelzel, N. Jux, M. Prato and D. M. Guldi, J. Am. Chem. Soc., 2018, 140, 904–907 CrossRef CAS PubMed.
- X. Wang, L. Cao, F. Lu, M. J. Meziani, H. Li, G. Qi, B. Zhou, B. A. Harruff, F. Kermarrec and Y. P. Sun, Chem. Commun., 2009, 3774–3776 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00401d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |