DOI:
10.1039/D0RA00411A
(Review Article)
RSC Adv., 2020,
10, 15461-15492
Advances in the chemical and biological diversity of heterocyclic systems incorporating pyrimido[1,6-a]pyrimidine and pyrimido[1,6-c]pyrimidine scaffolds
Received
14th January 2020
, Accepted 20th March 2020
First published on 20th April 2020
Abstract
Heterocycles incorporating a pyrimidopyrimidine scaffold have aroused great interest from researchers in the field of medical chemistry because of their privileged biological activities; they are used as anti-bacterial, antiviral, anti-tumor, anti-allergic, antihypertensive, anticancer, and hepatoprotective agents. Therefore, the present study aims to investigate the chemistry of heterocycles incorporating pyrimido[1,6-a]pyrimidine and pyrimido[1,6-c]pyrimidine skeletons and their biological characteristics. The main sections discuss (1) the synthetic routes to obtain substituted pyrimidopyrimidines, pyrimido[1,6-a]pyrimidin-diones, pyrimidoquinazolines, tricyclic, tetracyclic, and binary systems; (2) the reactivity of the substituents attached to the pyrimidopyrimidine skeleton, including thione and amide groups, nucleophilic substitutions, condensations, ring transformations, and coordination chemistry; (3) compounds of this class of heterocycles containing a significant characteristic scaffold and possessing a wide range of biological characteristics.
 Khaled M. Elattar | Dr Khaled M. Elattar was born in Menyet Sammanoud, Aga, El-dakahlia, Egypt, in 1979. He received his BSc in 2002 from the Faculty of Science, Mansoura University, Egypt and his MSc in 2006 from the Faculty of Science, Benha University, Benha, Egypt. He obtained his PhD in organic chemistry in 2011 from the Faculty of Science, Mansoura University, Egypt (PhD supervisors: Prof. A. A. Fadda and Prof. A. S. Fouda). He was a Lecturer in Organic Chemistry at the Faculty of Education of Sirte University, Sirte, Libya from 2012 to 2015. He is a member of the Egyptian Chemical Society. He is a reviewer for some international scientific journals. His main research interests are in the field of organic synthesis, the synthesis of heterocyclic compounds of pharmaceutical interest and medicinal chemistry. He joined the editorial board of OA Journal – Pharmaceutics in 2018. http://oa.enpress-publisher.com/index.php/Pha/about/editorialTeam. |
 Başak Doğru Mert | Başak Doğru Mert was born in 1982 in Adana, Turkey. She received her BSc in 2004 from the Chemistry Department of Science and Letters Faculty, Çukurova University, Turkey, and she obtained her MSc in 2007 and her PhD in 2012 in physical chemistry from the Science and Letters Faculty of Çukurova University (PhD thesis advisor: Prof. B. Yazıcı). She was a researcher at Çukurova University from 2006 to 2017 and at Mardin Artuklu University from 2017 to 2018. She has worked as a researcher at Adana Alparslan Türkeş Science and Technology University, Department of Energy Systems Engineering, Adana, Turkey since 2018. She is a member of the Corrosion Association of Turkey and the Hydrogen Technologies Association. Her research interests are corrosion and energy. |
 M. Monier | Dr Mohammed Monier Mohammed Ibrahim received his BSc. in chemistry (2000) as well as MSc. in organic chemistry (2005) and PhD in Organic and Polymer Chemistry (2011) from the Faculty of Science, Mansoura University, Egypt. His main research fields are bioengineering and applied chemistry. His main research interests include the uses of natural and synthetic polymers in biomedical and environmental applications. Dr Monier is currently an Associate Professor at Mansoura University, Faculty of Science, Chemistry Department, Egypt (Tenure-Permanent) and at Taibah University, Chemistry Department, Yanbu, KSA (Contracted). He was a demonstrator (2000–2005), assistant lecturer (2005–2009), research assistant (2009–2011), and assistant professor (2011–2014) in the Chemistry Department, Drexel University, Philadelphia, PA, USA. He was a Fulbright Visiting Scholar at the Chemistry Department, Drexel University, Philadelphia PA, USA, in 2015. |
 Ahmed El-Mekabaty | Dr Ahmed El-Mekabaty Mohamed was born in 1982 in Salamoon El-Komash, Mansoura, Egypt. He received his BSc in 2003 from the Faculty of Science, Mansoura University, Mansoura, Egypt and his MSc in 2007 (with a thesis on the synthesis of new heterocyclic compounds of pharmaceutical interest) from the same university. He obtained his PhD in Organic and Petroleum Chemistry in 2011 from the Faculty of Science, Mansoura University, Egypt (PhD thesis advisor: Prof. O. M. O. Habib, Mansoura University). He is an Associate Professor of Organic and Petroleum Chemistry at the Faculty of Science, Mansoura University, Egypt. He is a member of the American Chemical Society. He is a reviewer for some international journals. His research focuses on the synthesis of heterocyclic compounds of pharmaceutical interest and evaluating their efficiency as oxidation and corrosion inhibitors for lubricating oils. |
1. Introduction
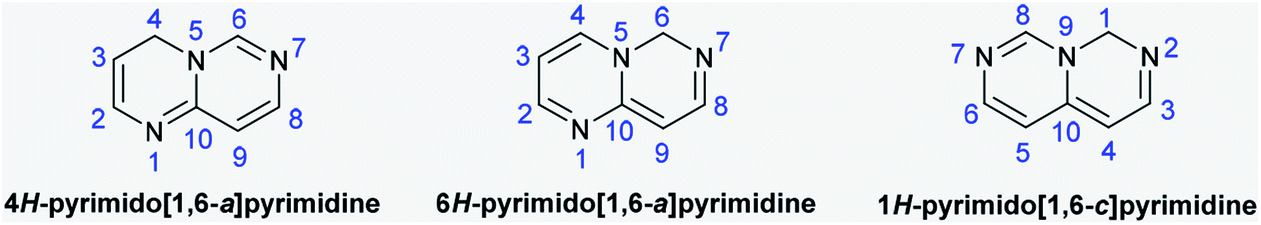
Due to their various substantial biological characteristics, pyrimidopyrimidine compounds have recently established a notable place in the work of researchers in the field of medicinal chemistry.1 Pyrimido[4,5-d]pyrimidines are used as cancer cell growth inhibitors,2 antioxidants,3 agents to reduce dihydrofolic acid to tetrahydrofolic acid,4 antidiabetics,5 angiogenesis inhibitors,6 resistance modification agents,7 Mycobacterium tuberculosis,8 hypertension,9 and allergy symptom treatments,10 antibacterial11 and antiviral agents,12 antitumor agents that inhibit monocarboxylate transporters (MCTs),13–15 anti-inflammatory16 and hepatoprotective agents,17 and tyrosine kinase inhibitors.18 The pyrimidopyrimidines of this class comprise 4H-pyrimido[1,6-a]pyrimidine, 6H-pyrimido[1,6-a]-pyrimidine, and 1H-pyrimido[1,6-c]pyrimidine (Fig. 1) and their fused benzene derivatives. Accordingly, the analogs pyrimido[1,2-c]pyrimidines are a class of 4H-pyrimido[1,6-a]pyrimidines. Sirakanyan et al.19 reported the synthesis of a pentacyclic system, pyranopyridofuro[2,3-e]pyrimido[1,2-c]pyrimidine, in 71% yield by the chlorination of the desired pyrimidine-aminoalcohol with phosphorus oxychloride and subsequent intramolecular cyclization under reflux conditions. Moreover, Vivek's group20 prepared a series of tricyclic systems, ethyl 8-substituted-10-(methylthio)-4-oxo-4,8-dihydropyrazolo[4,3-e]pyrimido[1,2-c]pyrimidine-3-carboxylates, by the reactions of 1-substituted-3-(methylthio)-1H-pyrazolo-[3,4-d]pyrimidin-4-amines with diethyl 2-(ethoxymethylene)malonate under either solvent-free microwave irradiation (81–85%) or thermal conditions (51–62%) in diphenyl ether. Tanarro and Gutschow21 reported the synthesis of 10-benzyl-3,4,9,10,11,12-hexahydro-2H-pyrido[4′,3′:4,5]thieno[3,2-e]-pyrimido[1,2-c]pyrimidine from ethyl 2-amino-6-benzyl-4,5,6,7-tetrahydro-thieno[2,3-c]pyridine-3-carboxylate and evaluated its activity as an inhibitor for acetyl-cholinesterase from EeAChE, hAChE, and hBChE.
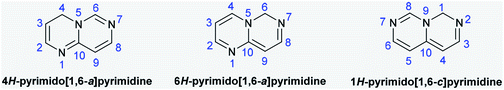 |
| | Fig. 1 Structures and atomic numbering of the 4H-, 6H-pyrimido[1,6-a]pyrimidine and 1H-pyrimido[1,6-c]pyrimidine skeletons. | |
4H-Pyrimido[1,6-a]pyrimidines are rarely reported despite the fact they provide an exceptional ring structure and multiple substitution designs, polarities, and H-bonding proficiencies. In view of this, 4H-pyrimido[1,6-a]pyrimidin-4-ones were obtained by the condensation of 4-aminopyrimidine with Meldrum's acid22 or ethyl acetoacetate.23 Kitagawa's group24 reported the synthesis of 2,3,4,7-tetrahydro-6H-pyrimido[1,6-a]pyrimidin-6-one (50% yield) and its 8-methyl analog (15% yield) as cyclization products by the treatment of both 2,4-dichloro-pyrimidine and 2,4-dichloro-6-methylpyrimidine with 3-aminopropan-1-ol and subsequent treatment with thionyl chloride in tetrahydrofuran; an additional product was formed (62% yield) due to the chlorination of the 8-methyl analog at C9 of the pyrimidopyrimidinone ring. Lulle et al.25 conveyed the synthesis of 1-substituted-1,2,3,4-tetrahydro-6H-pyrimido[1,6-a]pyrimidine-6,8(7H)-diones by the amination of 1-(3-bromopropyl)-pyrimidine-2,4(1H,3H)-dione with different amines, such as ammonia, propylamine, butylamine, and benzylamine, and subsequent intramolecular cyclization. Previously, methyl 2-benzoylamino-3-dimethylaminopropenoate served as a synthon for the preparation of N-(6,8-dihydroxy-4-oxo-4H-pyrimido[1,6-a]pyrimidin-3-yl)benzamide from heterocyclic α-amino compounds in acetic acid.26 A series of 1,4,9b-triazaphenalenes as types of pyridopyrimido[1,6-a]pyrimidines were synthesized by the thermal condensation reactions of 3-substituted-cyclopropane-1,1,2,2-tetracarbonitriles in 1,2-dichlorobenzene at reflux temperatures, and their potential as materials for electronic applications was studied.27 In addition, the conformers of perhydro-3a,6a,9a-triazaphenalene were studied by stereoelectronic stabilization, gauche interactions, DFT calculations, and spectral data.28
A book chapter by Vasvári-Debreczy et al. described the chemistry of bicyclic heterocyclic systems of the type [6 + 6] containing a nitrogen atom at the ring junction.29a One of the heterocyclic compounds investigated in this chapter incorporated a pyrimido[1,6-a]pyrimidine scaffold. In this chapter, the researchers discussed the synthesis of pyrimido[1,6-a]pyrimidine-6-thione, pyrimido[1,6-a]quinazolinone, pyrimido[1,2-c]quinazoline, pyrimido[6,1-b]quinazolinone, and pyrimido-[5,6,1-ij]quinazolinone heterocyclic systems. Hermecz and Vasvári-Debreczy wrote a book chapter on 6-6 bicyclic systems with a ring junction nitrogen atom, in which the synthesis of 2H-pyrimido[1,6-a]pyrimidines was briefly discussed.29b
On the other hand, Elattar et al. reviewed the chemistry of pyrimido[1,2-a]pyrimidines,30 pyrimido[4,5-b]pyrimidines, and pyrimido[5,4-b]pyrimidines.31 The pyrimido[1,2-a]-pyrimidines30 were synthesized from the reaction of guanidine with ethyl 2-methyl-3-oxopropanoate, unsaturated ketones or unsaturated nitriles. Acid treatment of dihydro-4H-pyrimido[1,2-a]pyrimidines led to the formation of pyrimidopyrimidinium salts. In addition, pyrimido[1,2-a]pyrimidines were synthesized from amino-pyrimidines by multicomponent reactions with aryl aldehydes and barbituric or thiobarbituric acids, substituted-dihydro-3H-pyrazol-3-ones or active nitriles. The reactions of β-ketoesters with amino-pyrimidines yielded the anticipated ring systems; also, the reactions of alkynones, diesters or enaminoesters with aminopyrimidines yielded the same target heterocycles. In another route, pyrimido[2,1-b]quinazolinones are a type of pyrimido[1,2-a]pyrimidines which are prepared by ring transformation of 6-amino-3,4-dihydro-2H-pyrimido[1,2-c]quinazolines; moreover, thienyl-pyrimido[1,2-a]pyrimidines are analgesic and antimicrobial agents. Also, the salts of these compounds are inhibitors of human platelet aggregation, and the tricyclic systems are potent gastroprotective agents. 3,8-diaryl-4H-pyrimido[1,2-a]pyrimidinone has antagonistic effects on melanin-concentrating hormone receptor and pyrimido[1,2-a]quinazolinones are protein kinase inhibitors. Additionally, dipyridamole, a type of pyrimido[5,4-d]pyrimidine,31 is an anticoagulant agent and cAMP-phosphodiesterase platelet inhibitor; it also decreases pulmonary hypertension.32 The present study aims to explore and investigate the chemistry of pyrimido[1,6-a]pyrimidines and pyrimido[1,6-c]pyrimidines as important classes of heterocycles and to inspect their diverse biological activities. This is considered to be a complementary study to our previous studies in this field,33–37 which are considered to be an addition to the field of medicinal chemistry.
2. Synthetic methods
2.1. Synthesis of substituted pyrimidopyrimidines
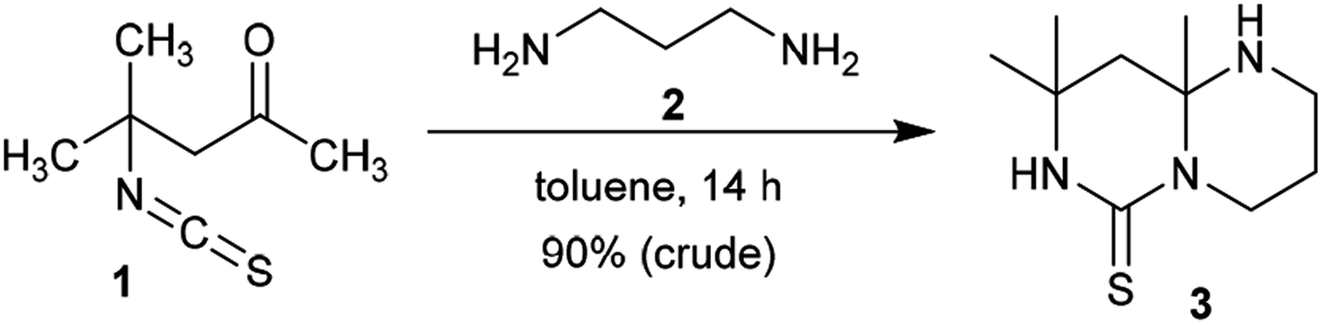
2.1.1. Synthesis from acyclic reactants. In light of observations, 4-isothiocyanato-4-methylpentan-2-one (1) was reacted with propane-1,3-diamine (2) in a molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to afford the anticipated product, pyrimidopyrimidine-thione 3, in excellent yield. The product was formed based on the molar ratio of the reactants. The functional group of the substituent in position 1 was ring-closed to afford binuclear junction 3. The condensed heterocycle 3 was formed by preliminary condensation of the amino function of the diamine with the ketonic carbonyl function of 4-isothiocyanato-4-methylpentan-2-one (1), followed by a subsequent intramolecular nucleophilic attack of the terminal amino function at the C
:1 to afford the anticipated product, pyrimidopyrimidine-thione 3, in excellent yield. The product was formed based on the molar ratio of the reactants. The functional group of the substituent in position 1 was ring-closed to afford binuclear junction 3. The condensed heterocycle 3 was formed by preliminary condensation of the amino function of the diamine with the ketonic carbonyl function of 4-isothiocyanato-4-methylpentan-2-one (1), followed by a subsequent intramolecular nucleophilic attack of the terminal amino function at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S function of the isothiocyanate fragment (Scheme 1).38
S function of the isothiocyanate fragment (Scheme 1).38
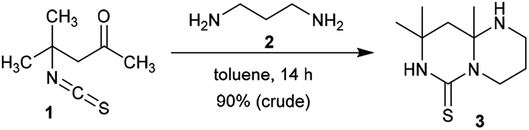 |
| | Scheme 1 Synthesis of 8,8,9a-trimethyloctahydro-6H-pyrimidopyrimidine-thione. | |
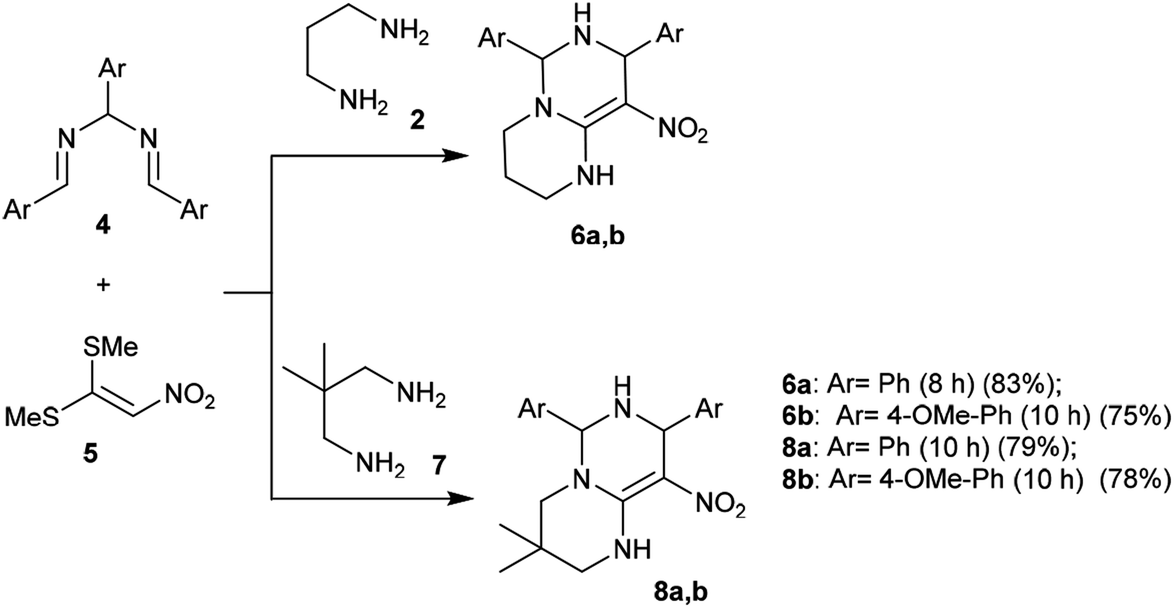
A proficient synthetic route was reported by Alizadeh et al.39 through a multicomponent procedure. Subsequently, the reactions of (2-nitroethene-1,1-diyl)bis(methylsulfane) (5) with propane-1,3-diamine (2) or 2,2-dimethylpropane-1,3-diamine (7) followed by reaction with N,N′-(arylmethylene)bis(1-arylmethanimine) (4) at reflux temperature yielded the desired diaryl-hexahydro-2H-pyrimidopyrimidines 6a, 6b, 8a, and 8b, respectively, with yields ranging from 75%–83% (Scheme 2). This procedure offers an alternative technique for application in drug discovery.39
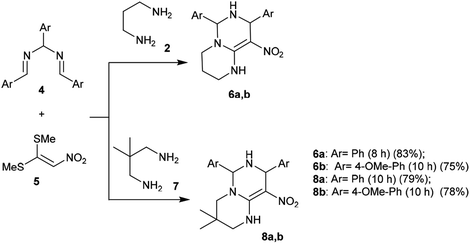 |
| | Scheme 2 Synthesis of hexahydro-2H-pyrimido[1,6-a]-pyrimidines. | |
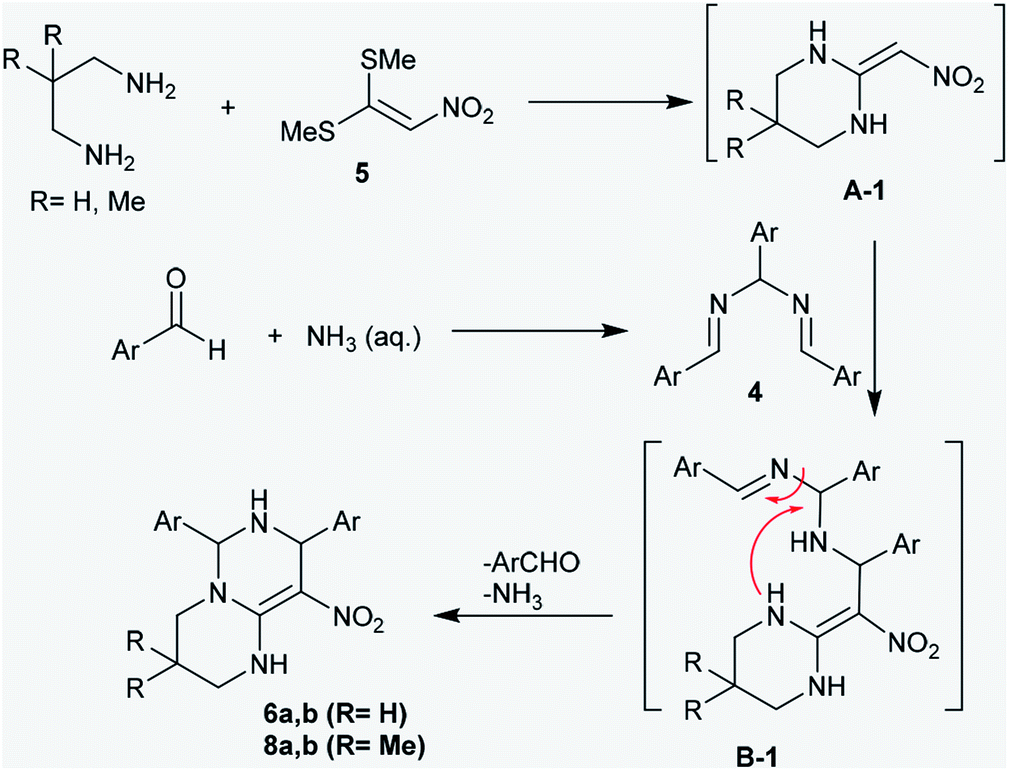
The proposed mechanism of these reactions was demonstrated through nucleophilic substitution of the amino groups of acyclic diamines with methyl-mercaptan groups to generate the first pyrimidine ring (intermediate A-1). Next, the addition of the intermediate A-1 to N,N′-(arylmethylene)bis(1-aryl-methanimine) (4) generated the intermediate B-1, which underwent successive cyclization to afford the products 6 and 8 through the elimination of aryl aldehyde and ammonia molecules by a hydrolysis step. The general mechanism involved the cycloaddition of diamines to bis-methylsulfane 5, attack of the generated intermediate at the CN bond of compound 4, and subsequent intramolecular cyclization to construct another pyrimidine ring (Scheme 3).39
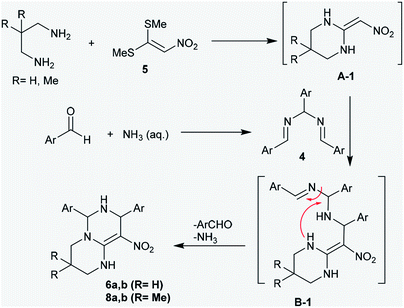 |
| | Scheme 3 The proposed mechanism for the ring construction of pyrimidopyrimidines 6 and 8. | |
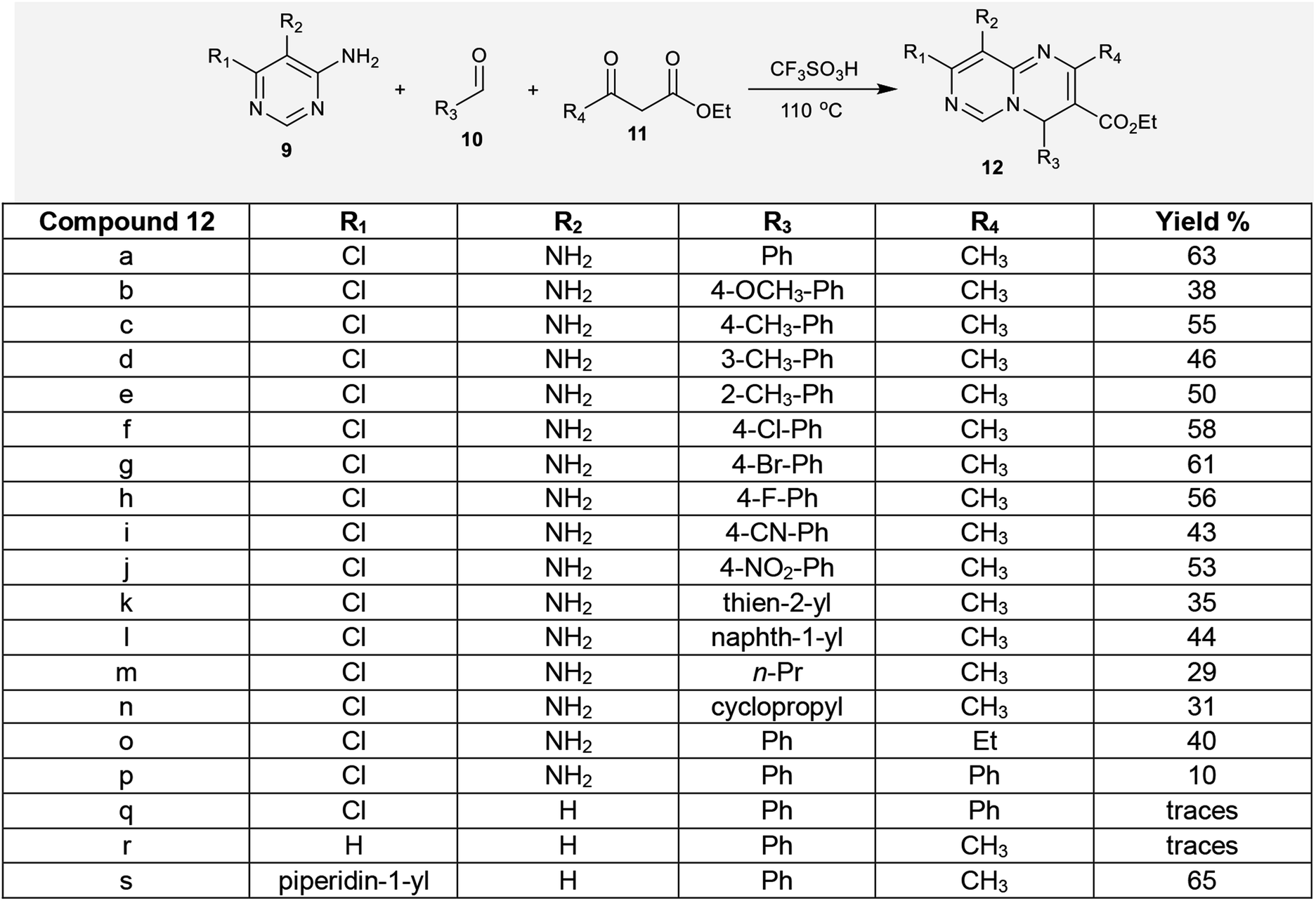
2.1.2. Multicomponent synthesis. Multicomponent reactions were employed for the synthesis of ethyl 2,4,8,9-tetrasubstituted-4H-pyrimido[1,6-a]pyrimidine-3-carboxylates through a facile synthetic route. In this route, one-pot three-component reactions of aminopyrimidines 9 and aldehydes 10 with β-ketoesters 11 yielded the respective ethyl carboxylates 12. The compounds contain an sp3 carbon at the C4 position, which diminishes the aromaticity of the 6–6 bicyclic system. Additionally, the chlorine atom and amino group (R1 = Cl, R2 = NH2) increase the diversity of structures 12a–p. The reactions of 10a with 11a and 9a to prepare ethyl carboxylate 12a were carried out using different acids, such as hydrochloric acid, sulfuric acid, and trifluoromethane–sulfonic acid, in different equivalent amounts using acetonitrile as a solvent and by solvent-free reactions. As a result, better product yields were achieved under solvent-free conditions with trifluoromethanesulfonic acid (0.5 equiv.) by heating at 110 °C. In the first step, the aldehydes 10 were condensed with the respective β-ketoesters 11 to form the arylidene intermediates, which reacted with the aminopyrimidines to afford the desired ethyl carboxylates 12 (Scheme 4).40
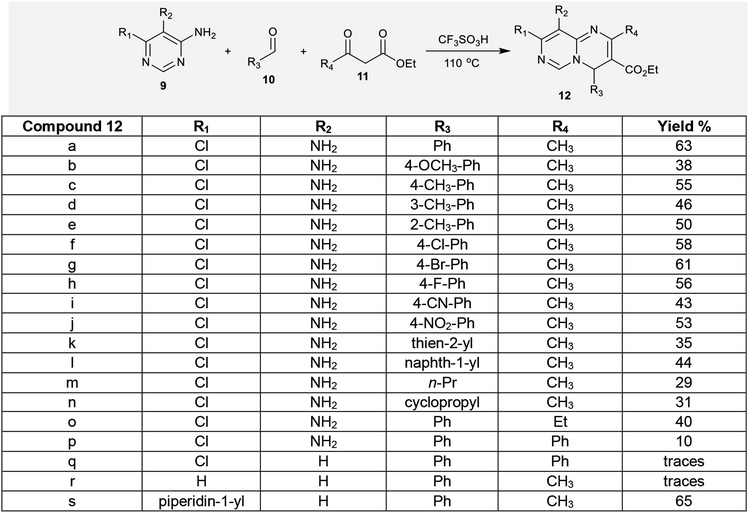 |
| | Scheme 4 Multicomponent synthesis of ethyl 2,4,8,9-tetrasubstituted-4H-pyrimido[1,6-a]pyrimidine-3-carboxylates. | |
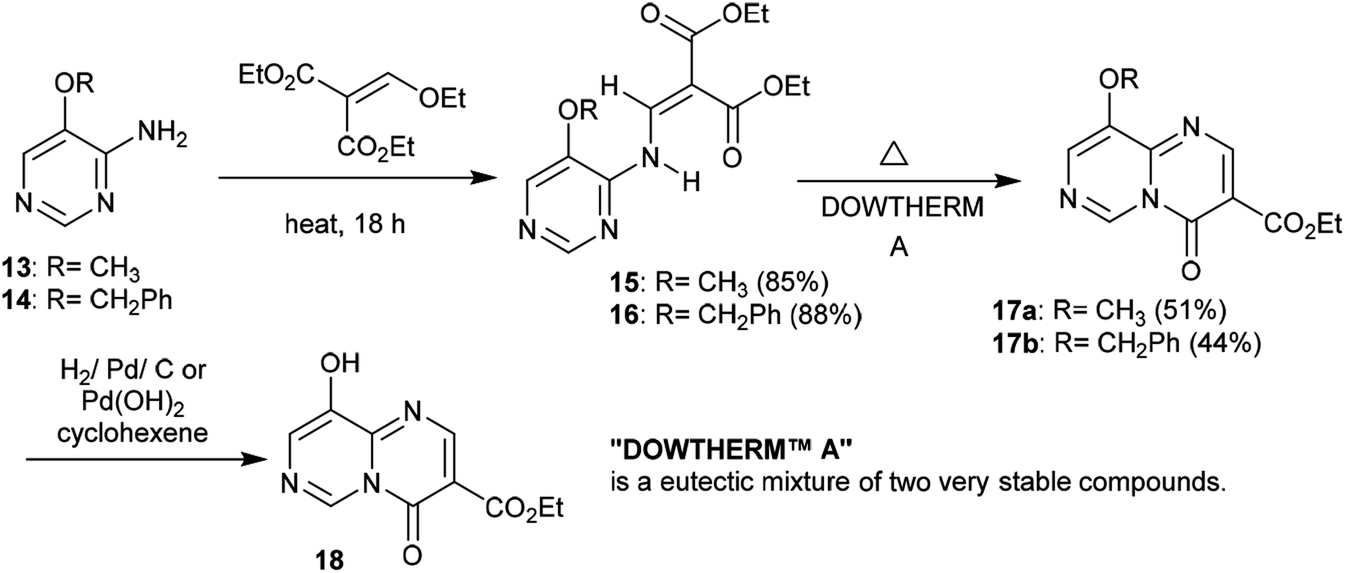
2.1.3. Synthesis from 4-aminopyrimidines. Heating 5-(benzyloxy)pyrimidin-4-amine (13) and 5-methoxypyrimidin-4-amine (14) with diethyl 2-(ethoxymethylene)malonate yielded the respective enamines 15 and 16, respectively, in excellent yields (88% and 85%) after elimination of an ethanol molecule. Cyclization of the enamines 15 and 16 was accomplished by heating with Dowtherm reagent to afford ethyl carboxylates 17a and 17b in moderate yields. The cyclization step proceeded through an initial rearrangement involving a1,3 H shift followed by an intramolecular nucleophilic attack of the formed imine at the carbonyl ester group and subsequent elimination of an ethanol molecule. Hydrogenolysis of pyrimido[1,6-a]pyrimidine 17b with catalytic palladium over carbon or palladium hydroxide produced the desired ethyl carboxylate 18 through the cleavage of O–C bonds with the elimination of a benzyl alcohol molecule (Scheme 5).22
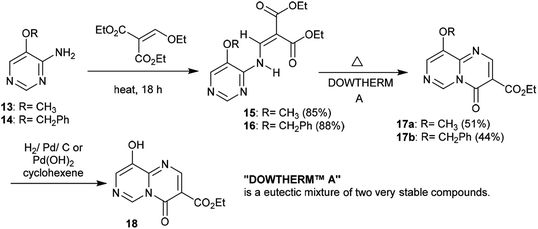 |
| | Scheme 5 Synthesis of the ethyl carboxylate of pyrimidopyrimidine 18. | |
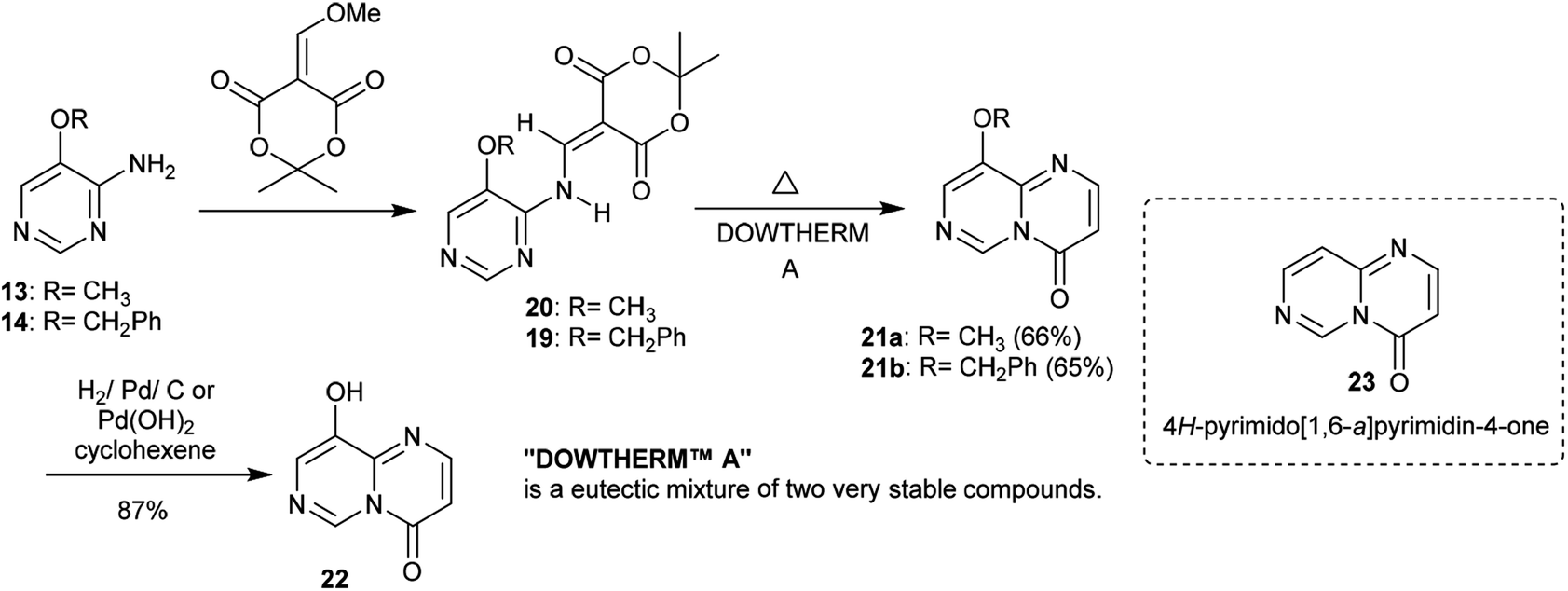
Similarly, the condensation of 4-alkoxy-4-aminopyrimidines 13 and 14 with Meldrum's acid derivative afforded, after thermal cyclization, the enaminoester intermediates 19 and 20 with the formation of 9-alkoxy-1-pyrimido[1,6-a]pyrimidines 21a and 21b substituted by a 3-ethoxycarbonyl group in the first issue. The hydrogenolysis of the anticipated benzyl ether afforded 9-hydroxy-4H-pyrimido[1,6-a]pyrimidin-4-one 22 as a heterocyclic phenol and the analog ketone 23 (Scheme 6).22
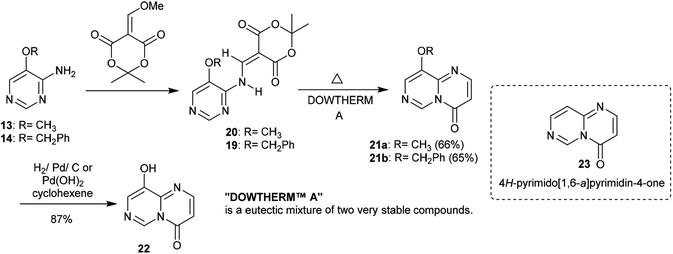 |
| | Scheme 6 Synthesis of 9-hydroxy-4H-pyrimido[1,6-a]pyrimidin-4-one. | |
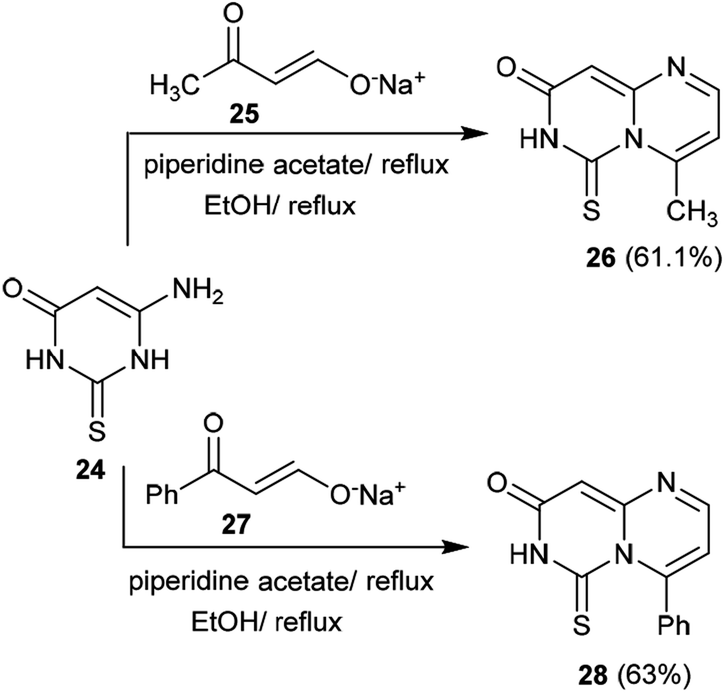
The previous procedure was applied for acyclic ketones such as sodium oxobutenolate 25 and sodium propenolate 27. Therefore, 6-aminothiouracil (24) was subsequently reacted with sodium salts 25 and 27 in piperidine acetate at their reflux temperatures to afford the desired 4-substituted-pyrimidopyrimidinones 26 and 28, respectively. The proposed mechanistic route adopted to obtain these compounds proceeded according to the same mechanism proposed for the formation of cycloalkyl[e]pyrimido[1,6-a]pyrimidin-3(7H)-ones (Scheme 7).41
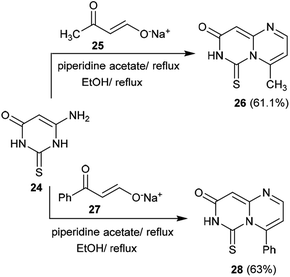 |
| | Scheme 7 Synthesis of 4-substituted-6-thioxo-6,7-dihydro-8H-pyrimido[1,6-a]pyrimidin-8-ones. | |
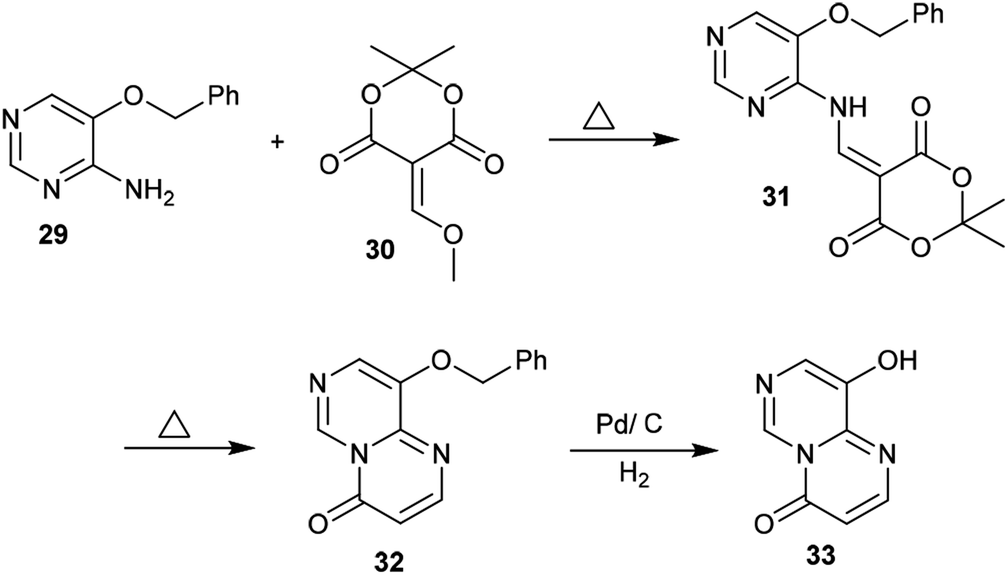
Condensation of Meldrum's acid with methyl formate catalyzed by zinc chloride provided a facile in situ preparation of its methoxy-methylene derivative 30. Consequently, 5-(benzyloxy)pyrimidin-4-amine (29) was reacted with a derivative of Meldrum's acid (30) to afford the pyrimidinyl-dioxane-dione 31 in a yield of 68%. The respective pyrimidopyrimidinone 32 was generated by heating compound 31 to release carbon dioxide and acetone molecules. Hydrogenolysis of 32 using a palladium over carbon system led to removal of the protecting benzyl group to afford 9-hydroxy-4H-pyrimido[1,6-a]pyrimidin-4-one (33) (Scheme 8).42
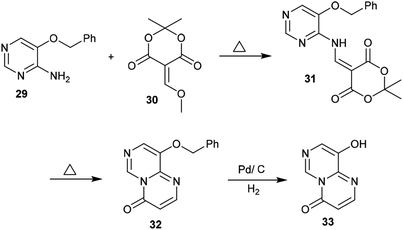 |
| | Scheme 8 Synthesis of 9-hydroxy-4H-pyrimido[1,6-a]pyrimidin-4-one. | |
2.2. Synthesis of pyrimido[1,6-a]pyrimidin-diones
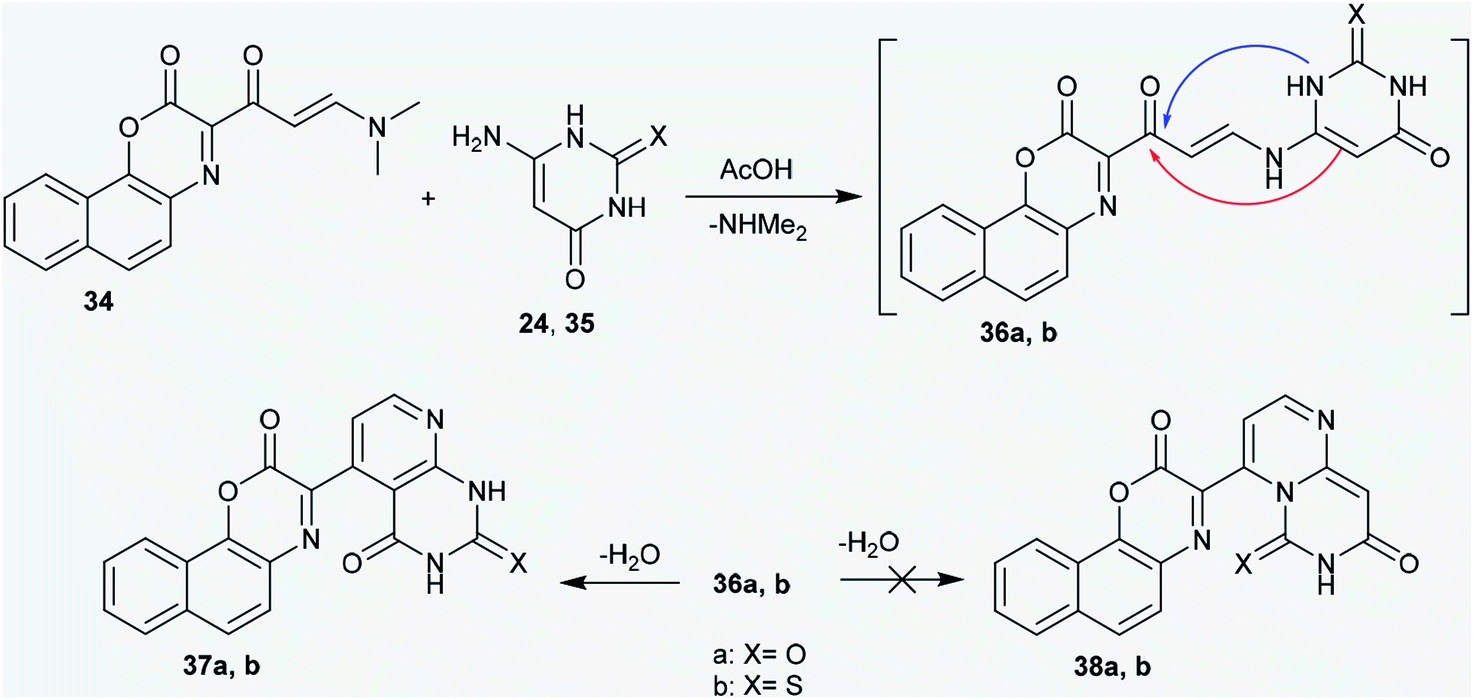
2.2.1. Ring annulation attempts. The enaminone derivative of 2H-naphtho1,4 oxazinone (34) was reacted with 6-amino-2-thioxo-2,3-dihydro-pyrimidin-4(1H)-one (24) and 6-aminopyrimidine-2,4(1H,3H)-dione (35), respectively,43 by heating in acetic acid to afford the respective 2-thioxo(oxo)-4-oxo-pyrido[2,3-d]pyrimidines 37a and b, respectively, instead of the formation of 6H-pyrimido[1,6-a]pyrimidines 38a and b. However, the reactions failed to afford the anticipated dihydro-6H-pyrimido[1,6-a]pyrimidines 38a and b. A Michael addition-type reaction was proposed for the detection of the formed products (37a,b), in which the exocyclic amino group of the amino-pyrimidine attacks the active CC double bond of enaminone 34; this resulted in the elimination of a dimethylamine molecule and the generation of the non-isolable intermediates 36a and b. The endocyclic imino group of aminopyrimidine is known to be a stronger nucleophile;44,45 however, it has high steric hindrance.46 In this route, the cyclization of the intermediates (36a,b) into the desired pyridopyrimidines (37a,b) or pyrimidopyrimidines (38a,b) could be achieved. The addition of the exocyclic CC to the carbonyl ketone took place rather than the intramolecular nucleophilic addition of the NH group of the pyrimidinone ring at the carbonyl ketone. However, the formation of the pyridopyrimidines (37a,b) was accomplished based on the spectroscopic data of the isolated products (Scheme 9).47
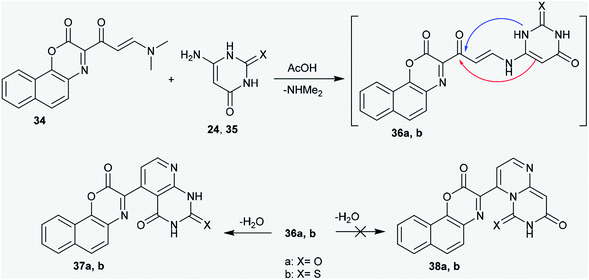 |
| | Scheme 9 Synthesis of 2-thioxo(oxo)-4-oxo-pyrido[2,3-d]pyrimidines. | |
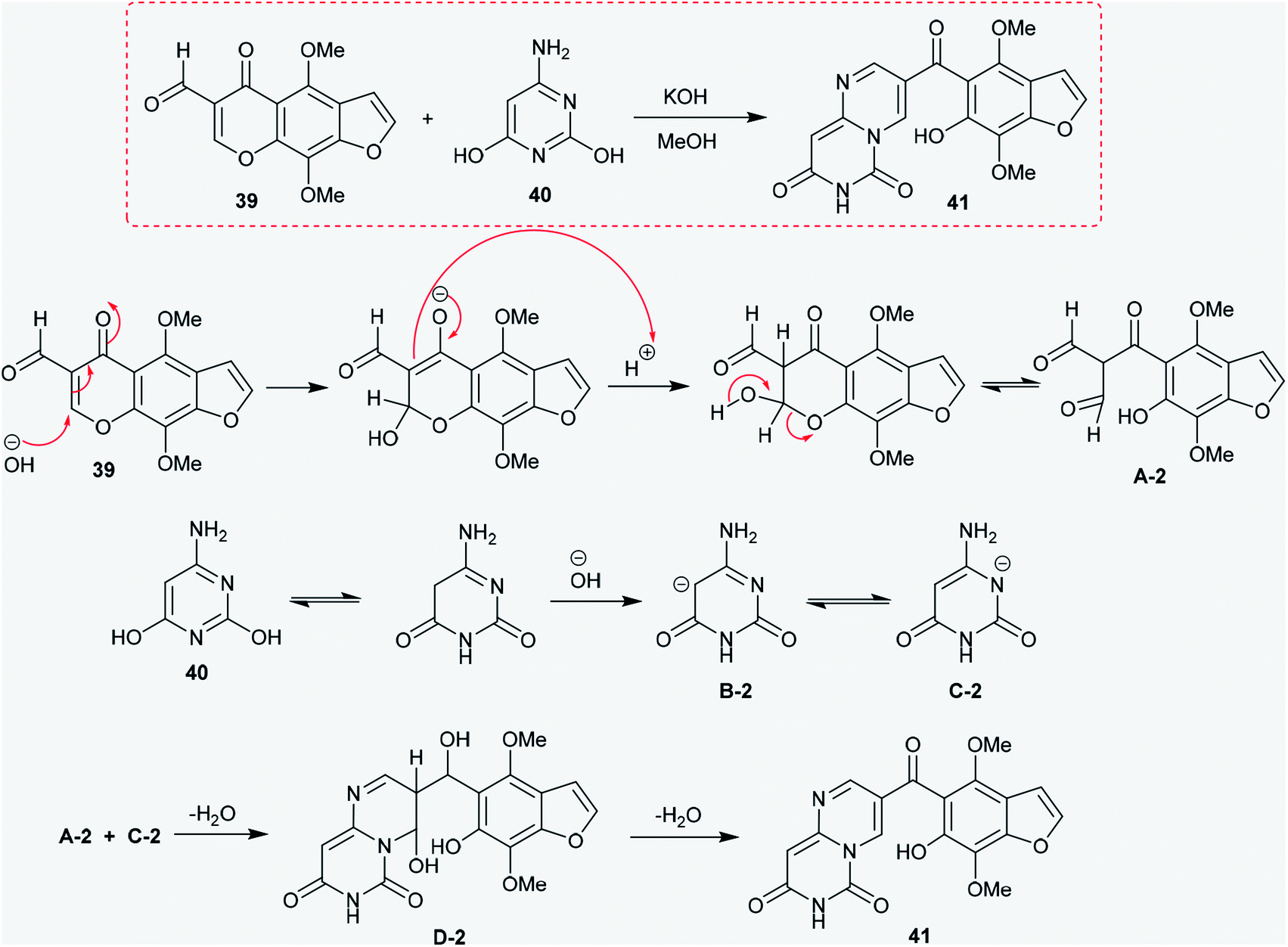
2.2.2. Synthesis from 6-aminopyrimidine-2,4-diol. Moreover, the hetryl aldehyde 39 was reacted with 6-aminopyrimidine-2,4-diol (40) in an alcoholic solution of potassium hydroxide at reflux temperature to afford 3-hetryl-carbonyl-pyrimidopyrimidine-dione 41. The mechanism of this reaction is proposed in Scheme 10. An initial nucleophilic attack of the hydroxide ion generated from potassium hydroxide took place at C2 of the chromen-4-one ring, followed by protonation and tautomerization of the carbonyl group at C4. Then, pyranone ring cleavage was accomplished to generate the intermediate A-2. In another route, 6-aminopyrimidine-2,4-diol (40) was rearranged to the keto-form with generation of the anion by abstraction of the proton at C3 by the action of hydroxide ion with the formation of the intermediate B-2, which was tautomerized to the intermediate C-2. The interaction of the intermediates A-2 and C-2 yielded the respective pyrimido[1,6-a]pyrimidine-dione (41) through nucleophilic attack of the anion carbon at the carbonyl group of the intermediate A-1, followed by the cyclocondensation step.48
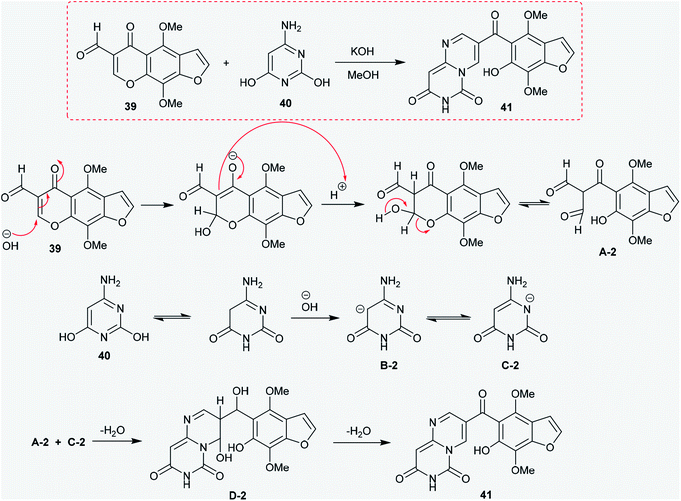 |
| | Scheme 10 Synthesis of 3-(aroyl)-6H-pyrimido[1,6-a]pyrimidine-6,8(7H)-dione. | |
2.3. Synthesis of pyrimidoquinazolines
Quinazolinone analogs were reported to have a comprehensive diversity of biological activities and have been widely used and applied in various medical and pharmaceutical fields;49 they are applied as antioxidant,50 antihyperlipidemic,51 antiviral,52 antitumor,53 analgesic, anti-inflammatory,54 anticonvulsant,55 antihypertensive,56 and antimicrobial agents.57
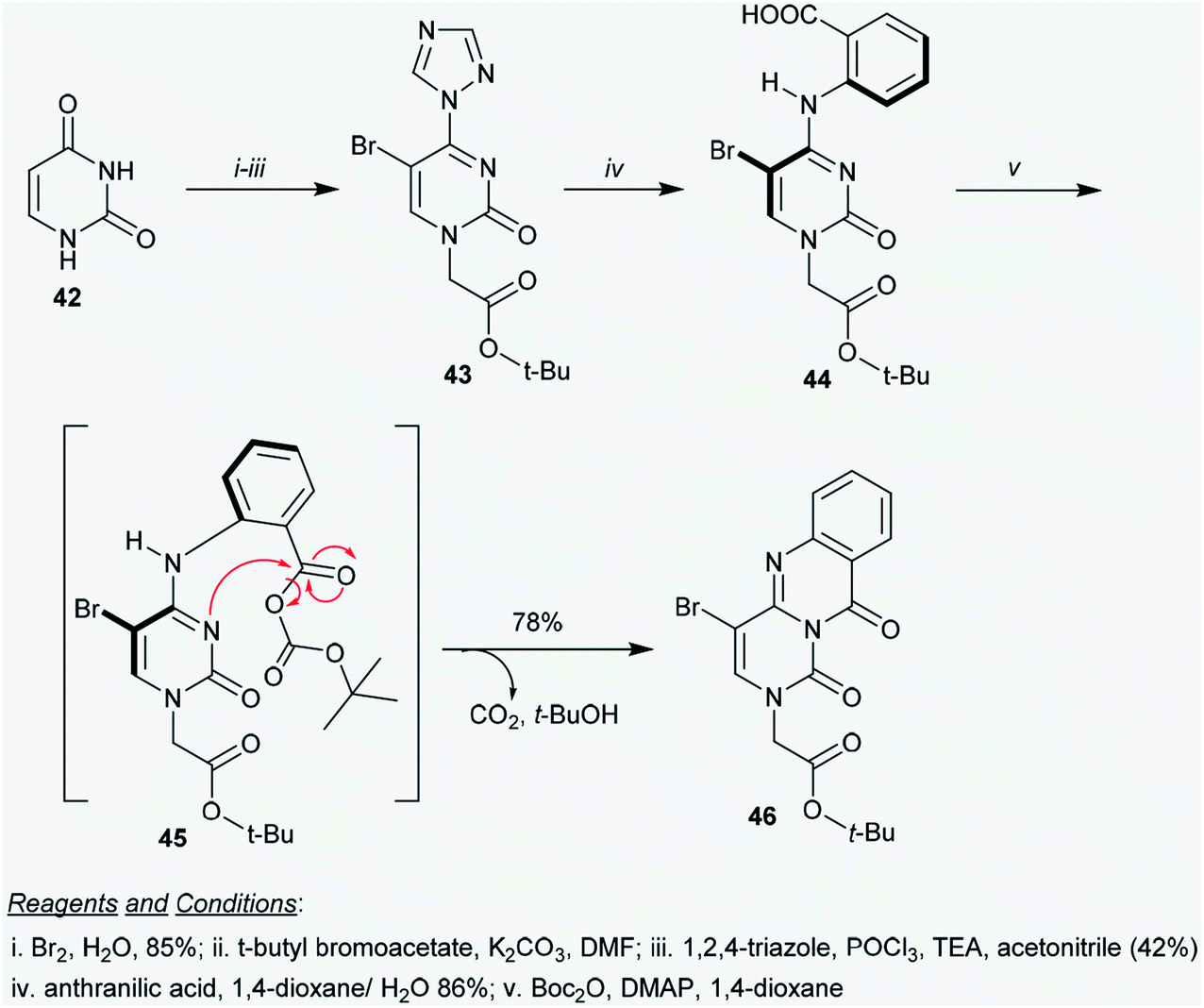
2.3.1. Synthesis from pyrimidine-2,4(1H,3H)-dione. Suchy and Hudson58 reported a simple multi-step synthetic route for the construction of tert-butyl 2-(4-bromo-1,10-dioxo-1H-pyrimido[6,1-b]quinazolin-2(10H)-yl)acetate (46) via an elegant procedure. This process represents the first synthesis of a heterocyclic compound incorporating a 1,10-dioxo-1H-pyrimido[6,1-b]quinazoline core. Thus, tert-butyl acetate 43 was obtained in moderate yield by bromination of uracil (42)59 in the first step, followed by treatment with t-butyl bromoacetate in DMF containing K2CO3, treatment with phosphorus oxychloride, and subsequent reaction with 1,2,4-triazole in acetonitrile catalyzed by TEA. Heating the respective 1H-1,2,4-triazolyl-pyrimidine 43 with anthranilic acid in 1,4-dioxane yielded the aryl cytosine 44. The reaction proceeded in the presence of 10% water to facilitate the nucleophilic substitution and in the absence of base. The aromatic nucleophilic substitution of 43 with anthranilic acid progressed according to the method reported by Pedroso et al.60 Product 43 was obtained after recrystallization in 86% yield. In the last step, tert-butyl 2-(4-bromo-1,10-dioxo-1H-pyrimido[6,1-b]quinazolin-2(10H)-yl)acetate (46) was synthesized by treatment of aryl cytosine 44 with Boc anhydride or di-tert-butyl pyrocarbonate (Boc2O) in the presence of 4-dimethyl-aminopyridine (DMAP) as a base. The product 46 was formed in 78% yield through in situ generation of the intermediate 45, which followed an intramolecular decarboxylative coupling.61 The intramolecular nucleophilic attack of the nitrogen atom at position 3 of the cytosine ring on the carbonyl group of the carboxylate fragment proceeded with the elimination of tert-butanol and carbon dioxide molecules (Scheme 11).
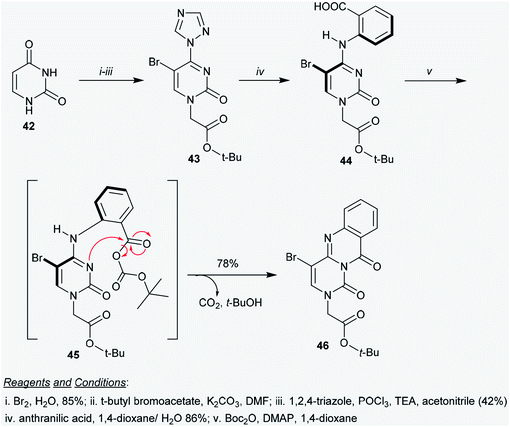 |
| | Scheme 11 Synthesis of tert-butyl 2-(4-bromo-1,10-dioxo-1H-pyrimido[6,1-b]quinazolin-2(10H)-yl)acetate. | |
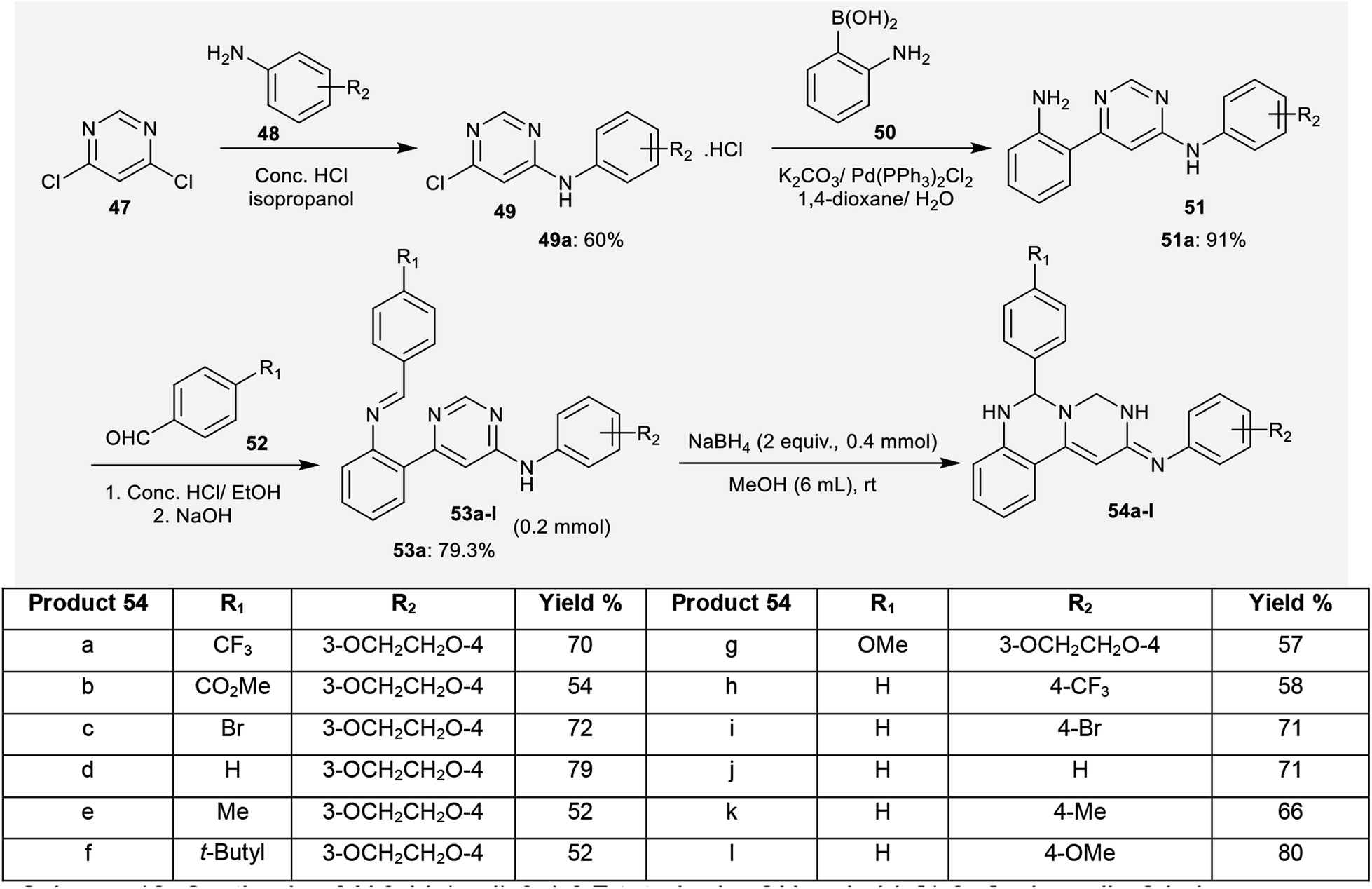
2.3.2. Synthesis from 4,6-dichloropyrimidine. Proficient multi-step synthetic routes have been investigated by Li et al.62 for the construction of N,6-bis(aryl)-3,4,6,7-tetrahydro-2H-pyrimido[1,6-c]quinazolin-2-imines 54a–l in good yields (52–80%). Therefore, 4,6-dichloropyrimidine (47) reacted with aryl amines 3 in isopropanol catalyzed by hydrochloric acid to afford 6-chloro-N-aryl-pyrimidin-4-amines (49).63 Palladium-catalyzed Suzuki–Miyaura cross-coupling reactions of 49 with boronic acid 50 yielded the desired N,6-diaryl-pyrimidin-4-amines (51). Subsequently, condensation of 51 with 4-substituted-benzaldehydes (52) afforded the Schiff bases 53. Cyclization of compounds 53 to the desired pyrimidoquinazolines 54 was accomplished by sodium borohydride treatment in methanol at room temperature. In this sequence, different conditions were applied for these reactions, in which the best yields (53a, 70%) of the reactions were obtained using sodium borohydride as an additive in methanol as a solvent at room temperature (Scheme 12).62
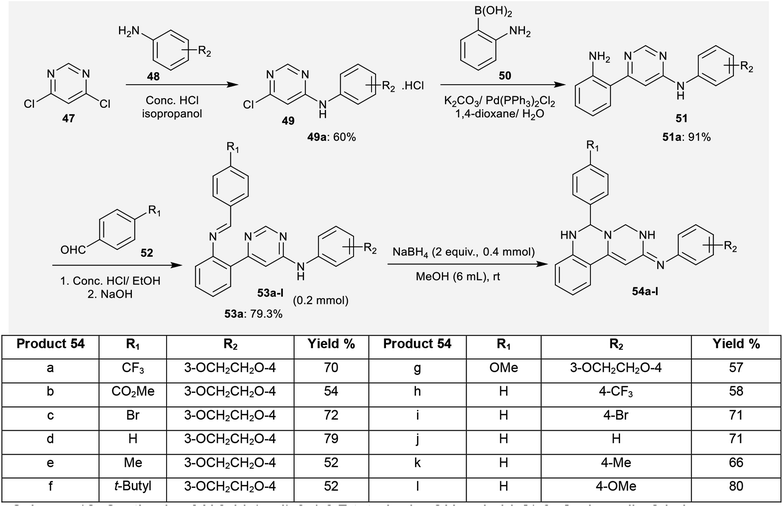 |
| | Scheme 12 Synthesis of N,6-bis(aryl)-3,4,6,7-tetrahydro-2H-pyrimido[1,6-c]quinazolin-2-imines. | |
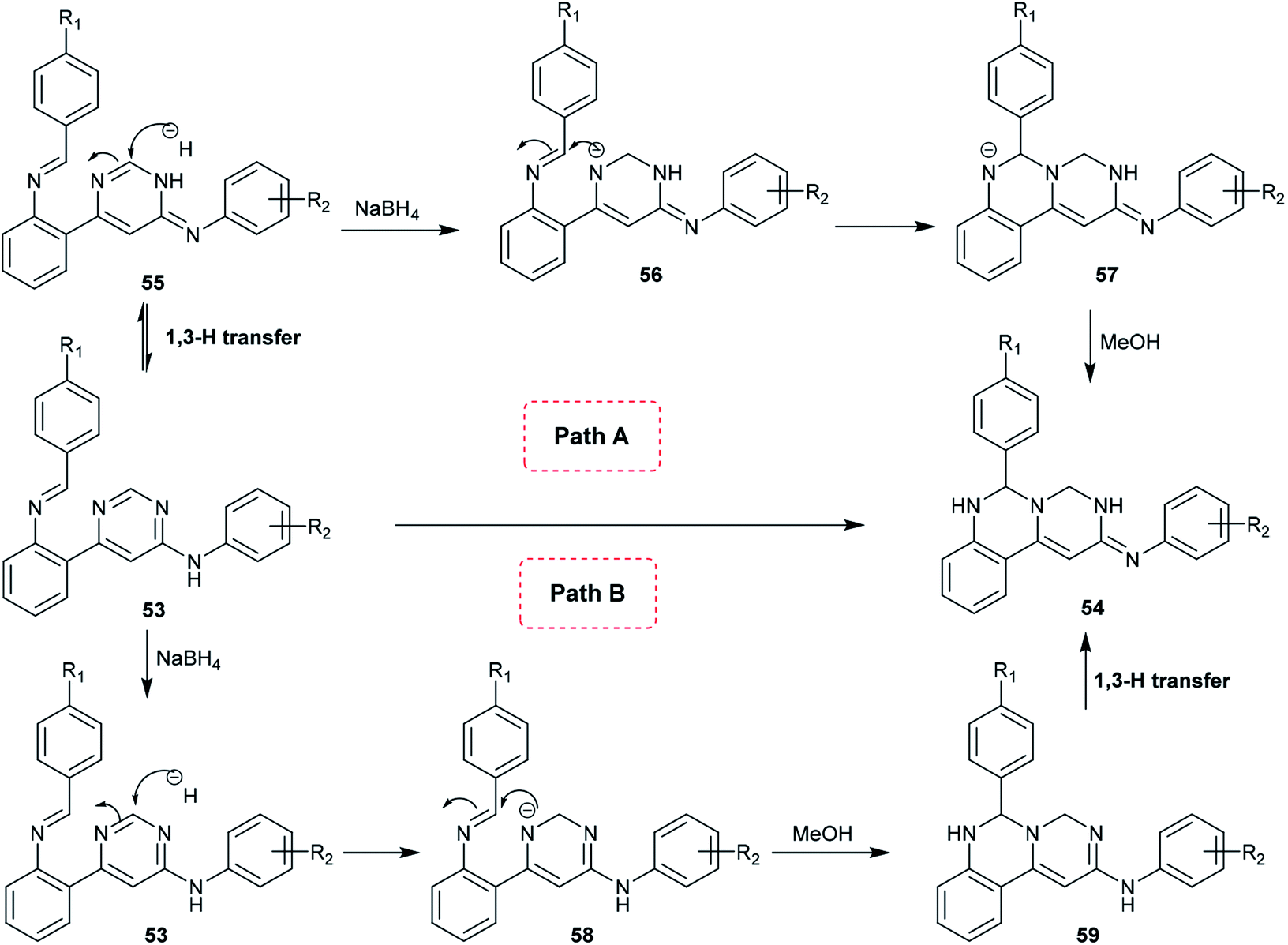
A proposed mechanism for the synthesis of pyrimidoquinazolines 54 is presented in Scheme 13. Accordingly, there are two possible pathways (A and B) for the cyclization step of compounds 53 to afford the pyrimido[1,6-c]quinazolines (54). In route A,1,3 proton transfer generates the tautomer 55. The carbon at position 2 of the pyrimidine ring is attacked by the hydrogen anion produced from sodium borohydride to form the intermediate 56. The nitrogen anion of 56 enables intramolecular cyclization by attacking the CN bond to form intermediate 57. Alcoholysis of the intermediate 57 with methanol yields the target products 54. In route B, reductive dearomatization of 53 occurs first by the action of sodium borohydride; then, the generated nitrogen anion of the intermediate 58 undergoes alcoholysis to form the intermediate 59, and finally,1,3 proton transfer yields the products 54.62
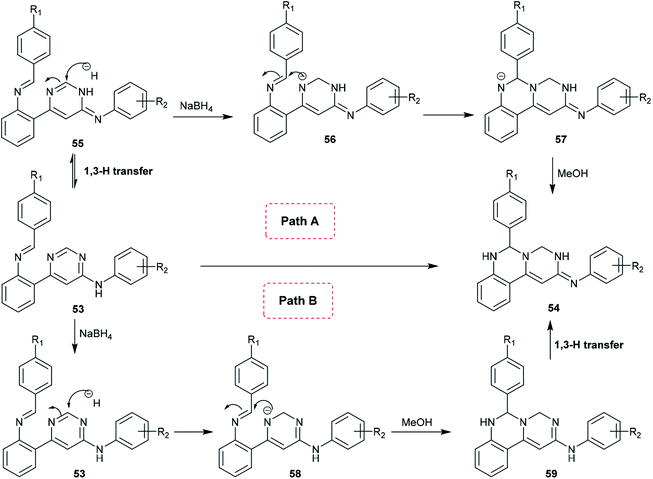 |
| | Scheme 13 Proposed mechanism for the synthesis of 2H-pyrimidoquinazolines 54. | |
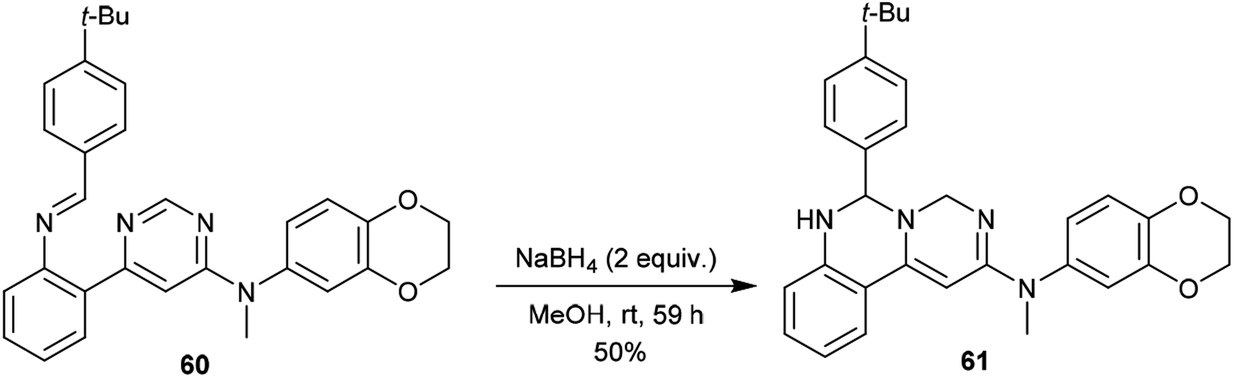
In the same way, pyrimido[1,6-c]quinazoline (61) was synthesized with a yield of 50% by treatment of N-methyl-pyrimidine 60 with NaBH4/MeOH at room temperature. Product 61 was obtained by following pathway B without the step of1,3 proton transfer (Scheme 14).62
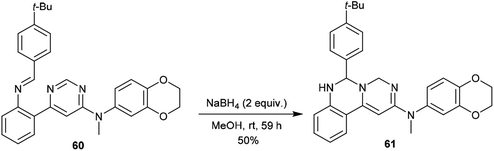 |
| | Scheme 14 Synthesis of N-methyl-6,7-dihydro-4H-pyrimido[1,6-c]quinazoline. | |
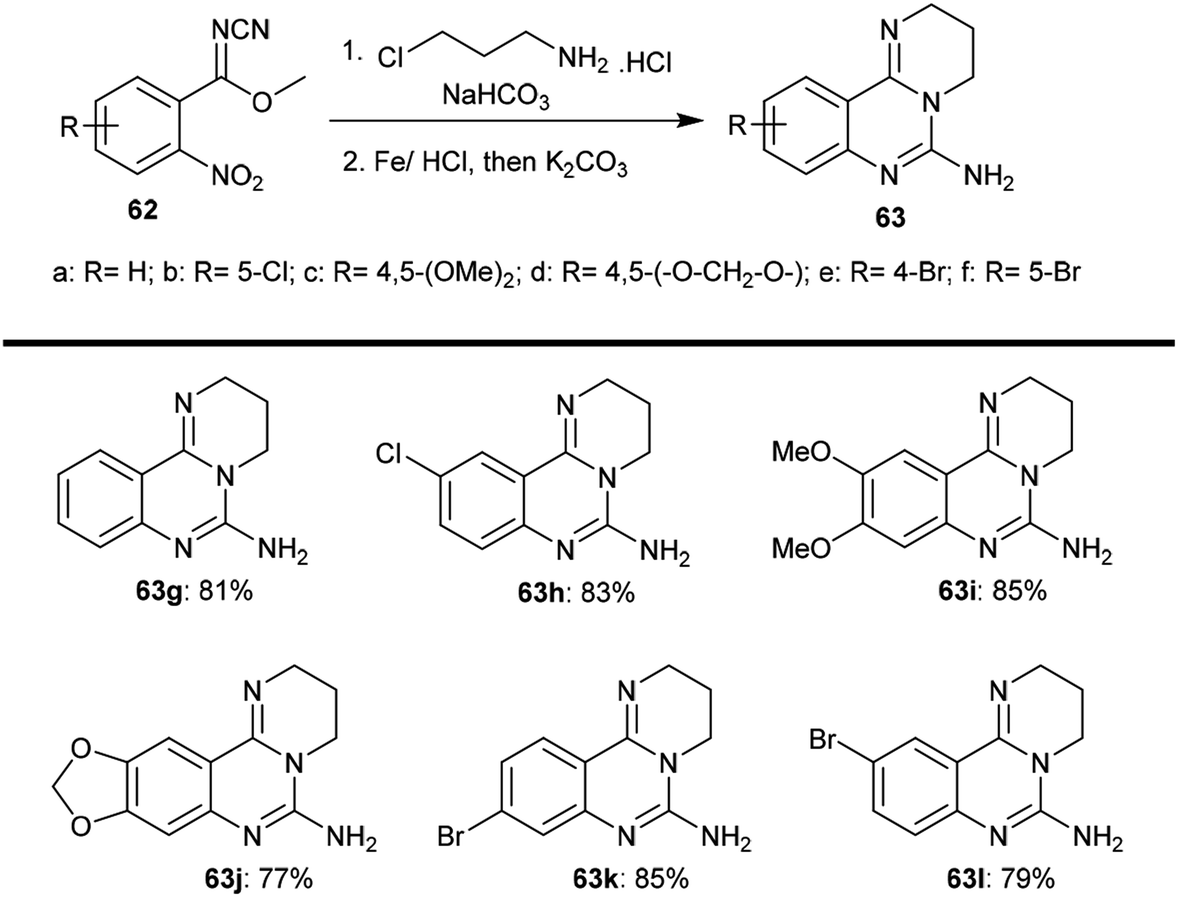
2.3.3. Synthesis from methyl N-cyano-2-nitrobenzimidates. Yin et al.64 employed a method of tandem condensation for the synthesis of substituted-6-amino-3,4-dihydro-2H-pyrimido[1,2-c]quinazolines (63) with excellent yields through a two-step synthesis. The products 63 were obtained from the reaction of methyl benzimidates 62 each with 3-chloropropan-1-amine hydrochloride catalyzed by sodium bicarbonate and subsequent reductive cyclization by treatment with an Fe/HCl system. In the first step, two equivalents of chloroalkyl-amines (hydrochloride and sodium bicarbonate) were used; meanwhile, four equivalents of iron and excess potassium carbonate were used in the next step, which involved processing by refluxing the reactants in an Fe–HCl system for three hours and subsequent addition of potassium carbonate with refluxing for an additional six hours. The procedure involved a one-pot sequence of intramolecular N-alkylation (Scheme 15).
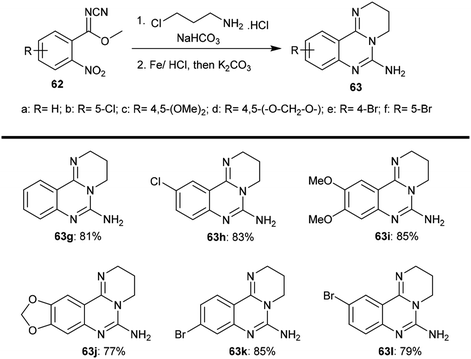 |
| | Scheme 15 Synthesis of 3,4-dihydro-2H-pyrimido[1,2-c]quinazolines. | |
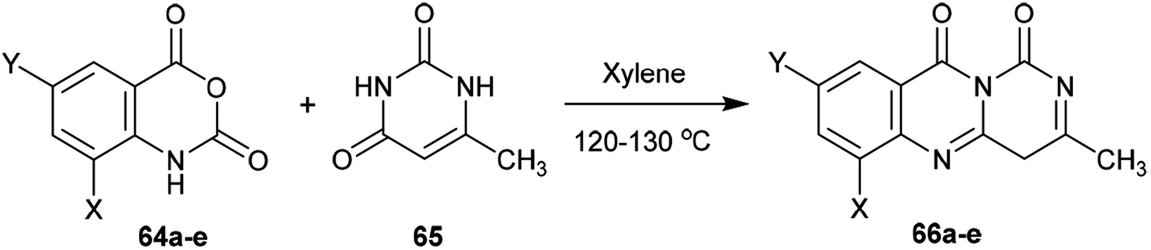
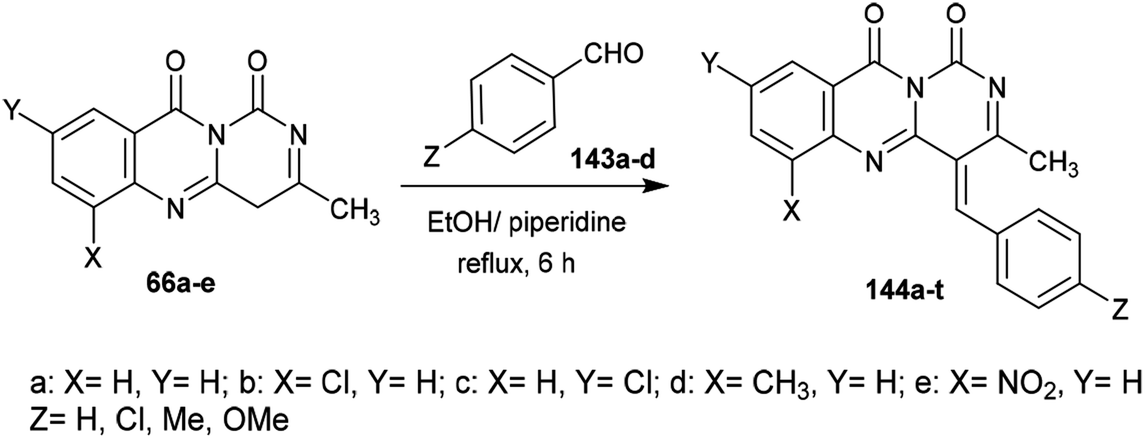
2.3.4. Synthesis from pyrimidine-dione. Heating of substituted isotonic anhydrides (64a–e) with 6-methylpyrimidine-2,4(1H,3H)-dione (65) in xylene afforded the respective 1H-pyrimido[6,1-b]quinazoline-1,10(4H)-diones 66a–e, respectively. The products were formed through ring-opening of the respective anhydride with condensation and subsequent ring closure (Scheme 16).65
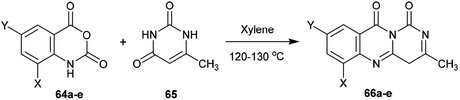 |
| | Scheme 16 Synthesis of 3-methyl-1H-pyrimido[6,1-b]quinazoline-1,10(4H)-diones. | |
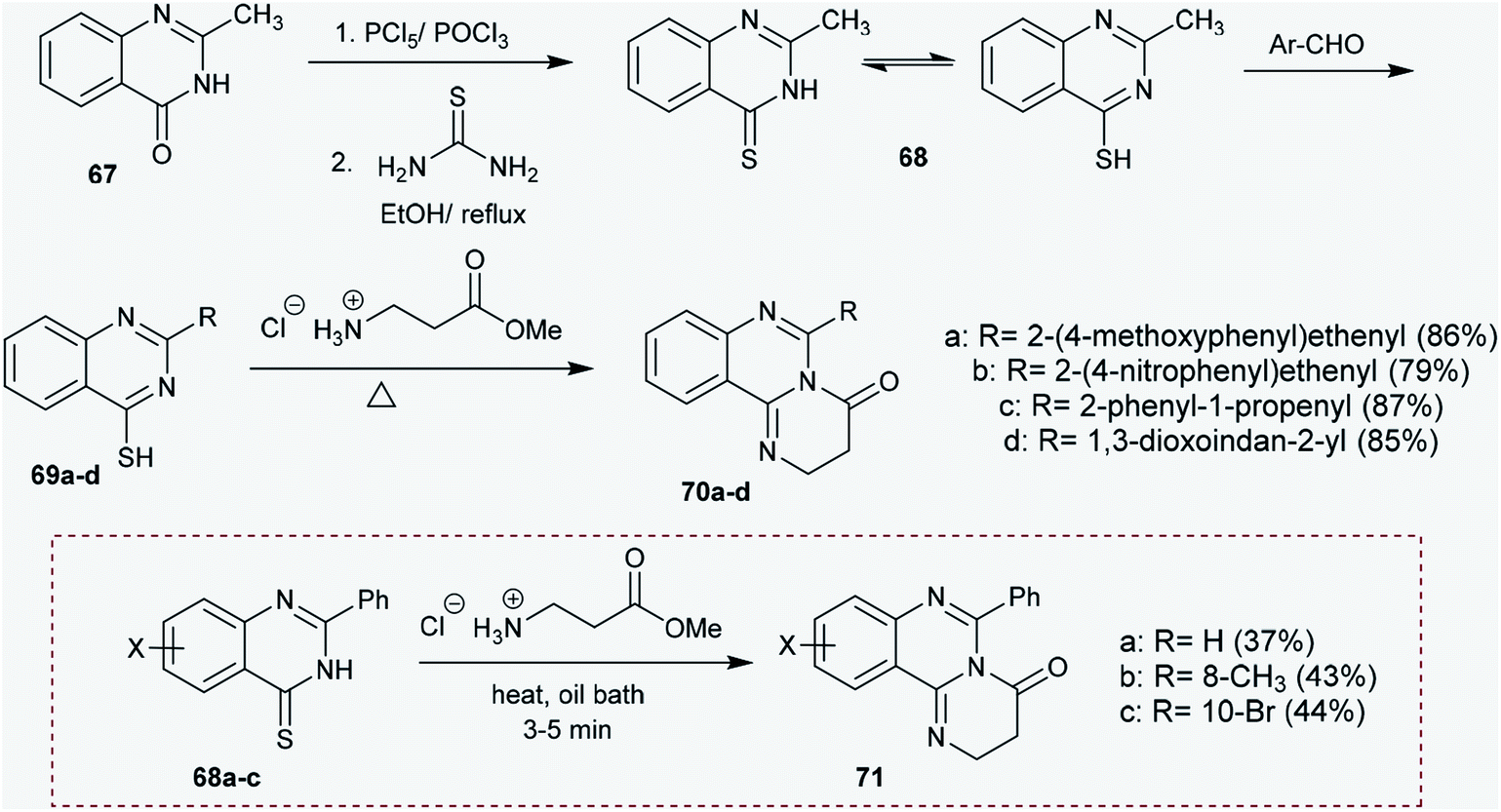
2.3.5. Synthesis from quinazolinones. Treatment of 2-methylquinazolin-4(3H)-one (67) with phosphorus pentasulfide and phosphorus oxychloride followed by reaction with thiourea in ethanol at reflux temperature yielded 2-methylquinazoline-4-thiol (68). The respective 2-substituted-quinazoline-4-thiol derivatives (69a–d) were synthesized by reactions of 68 with aryl aldehydes. Solvent-free reactions of 69a–d with esters of amino acids catalyzed by TEA at their reflux temperatures yielded the corresponding 6-substituted-2,3-dihydro-4H-pyrimidoquinazolinones 70a–d in respectable yields (79–87%). The use of solvents and basic catalysts in the previous reactions reduces the yield of the products. The products 70a–d were obtained by reactions involving nucleophilic substitution and thermal cyclization processes.66 Otherwise, Špirková and Stankovský67 reported the synthesis of pyrimidoquinazolinones 71 by heating 2-phenyl-quinazoline-4(3H)-thiones (68a–c) with equimolar amounts of methyl esters of glycine, α-alanine or β-alanine in an oil bath in the absence of basic catalyst (Scheme 17).
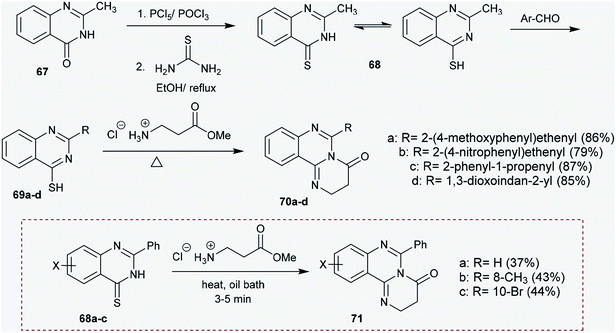 |
| | Scheme 17 Synthesis of 6-substituted-2,3-dihydro-4H-pyrimido[1,2-c]quinazolin-4-ones. | |
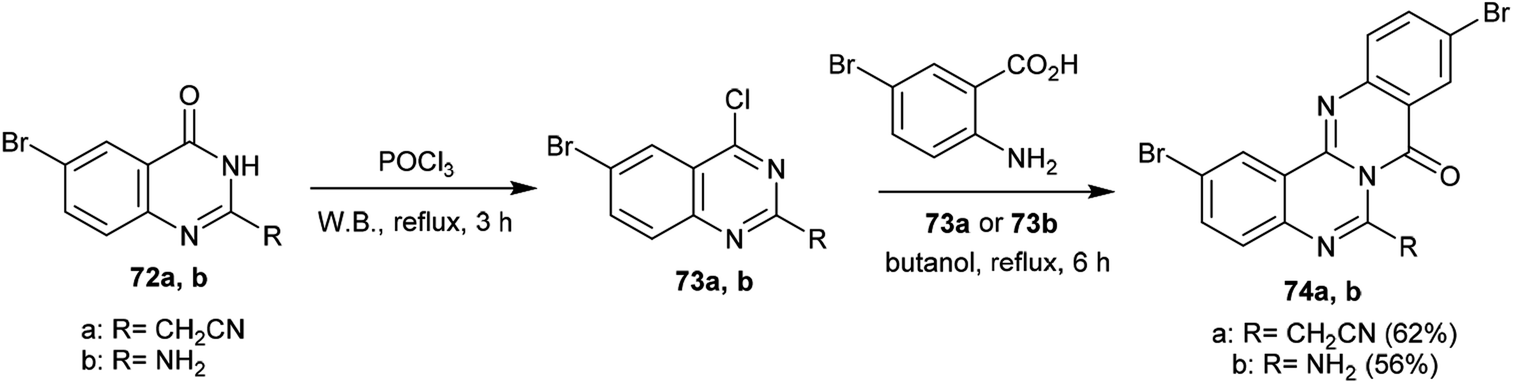
Congruently, chlorination of 6-bromo-2-substituted-quinazolin-4(3H)-ones 72a,b with phosphorus oxychloride yielded the corresponding chlorinated products 73a,b, respectively, in which the chlorination occurred at C4 of the quinazolin-4(3H)-one ring. Accordingly, cyclization step reactions of 73a and 73b with 2-amino-5-bromobenzoic acid, respectively, were performed by heating in butanol at its reflux temperature to afford 2,10-dibromo-6-substituted-8H-quinazolinoquinazolinones 74a and 74b. The products were acquired by nucleophilic attack of the amino group at C4 of the quinazoline ring with chlorine atom substitution. Subsequent1,3 H transfer and intramolecular cyclocondensation yielded the respective products 74a and 74b (Scheme 18).68
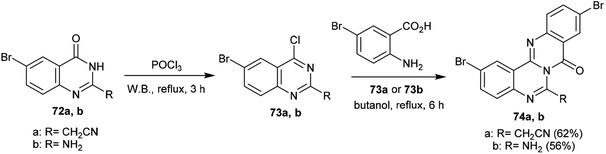 |
| | Scheme 18 Synthesis of 2,10-dibromo-6-substituted-8H-quinazolinoquinazolinones 74. | |
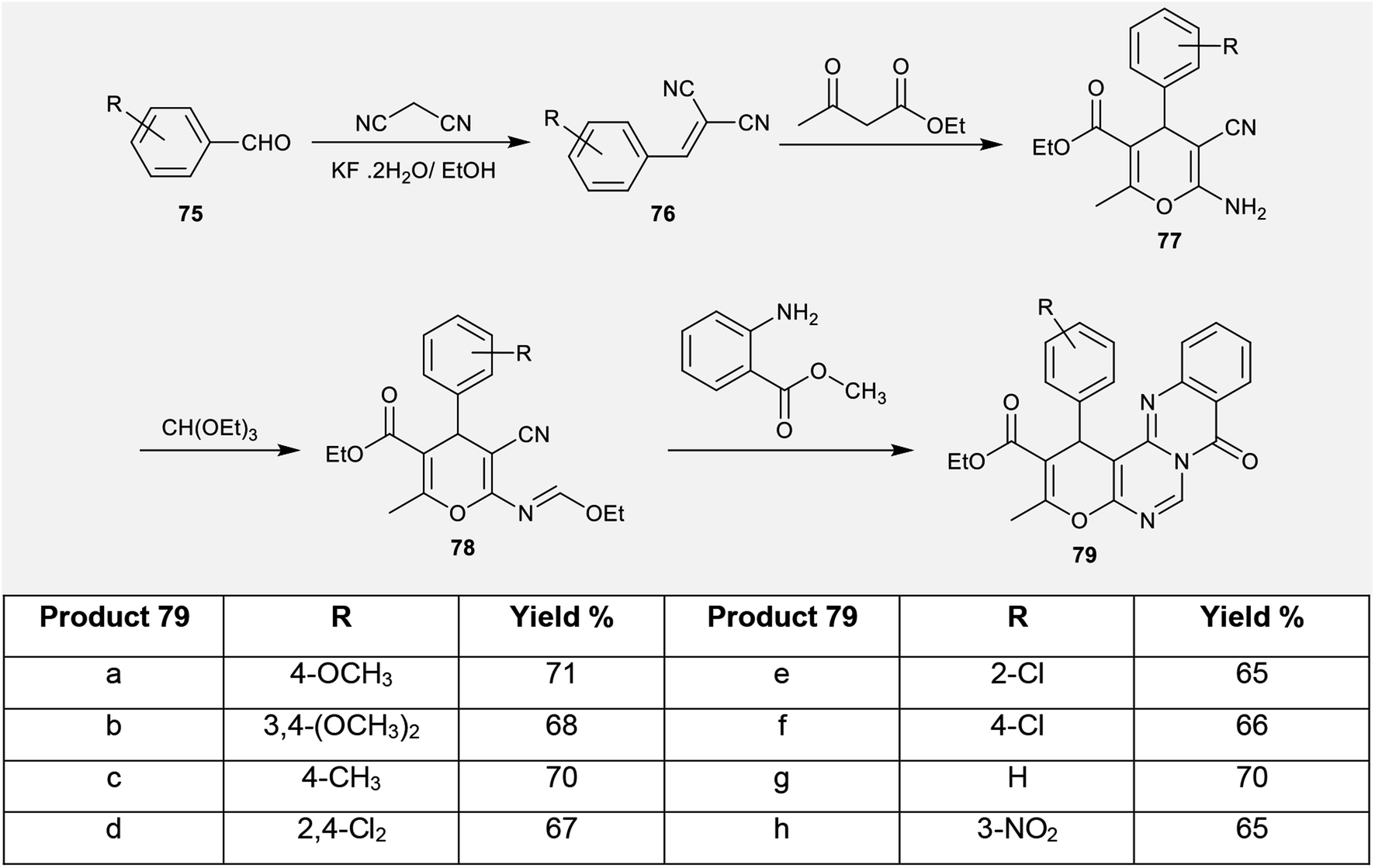
2.3.6. Synthesis from aryl aldehydes. An unpretentious method was conveyed for the synthesis of ethyl pyrano-pyrimido[6,1-b]quinazoline-2-carboxylates (79) by Shi et al.69 through multi-step reactions. Consequently, condensation of aryl aldehydes 75 with malononitrile yielded the arylidene malononitriles 76 according to the procedure reported by Venkateswarlu et al.70 Arylidenes 76 were reacted with ethyl 3-oxobutanoate to afford the desired ethyl carboxylates 77. Refluxing of ethyl carboxylates 77 with triethyl orthoformate in acetic anhydride yielded the ethoxymethylene-amino analogs 78. The cyclization of 78 with methyl 2-aminobenzoate in acetic acid at reflux temperature yielded the corresponding ethyl pyrano-pyrimido[6,1-b]quinazoline-2-carboxylates (79) with yields ranging from 65% to 71%. The cyclization step followed an initial nucleophilic attack of the arylamino function of methyl 2-aminobenzoate at the ethoxymethylene carbon with the elimination of an ethanol molecule and subsequent intramolecular nucleophilic attack at the nitrile function to generate an imino group, which attacked the ester group to afford the target products 79 (Scheme 19).69
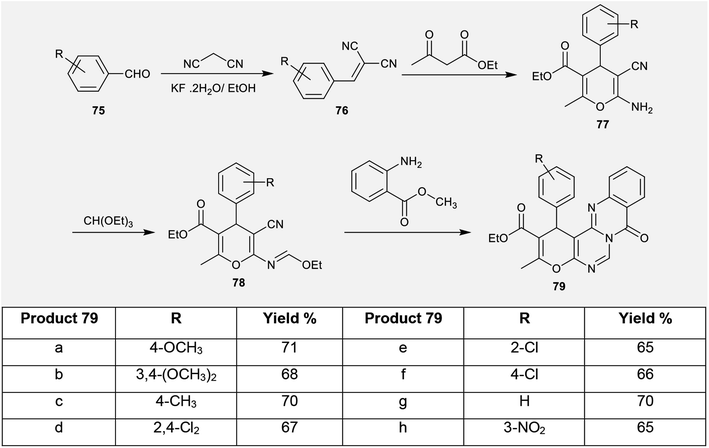 |
| | Scheme 19 Synthesis of ethyl 8-oxo-1H,8H-pyrano-pyrimido[6,1-b]quinazoline-2-carboxylate. | |
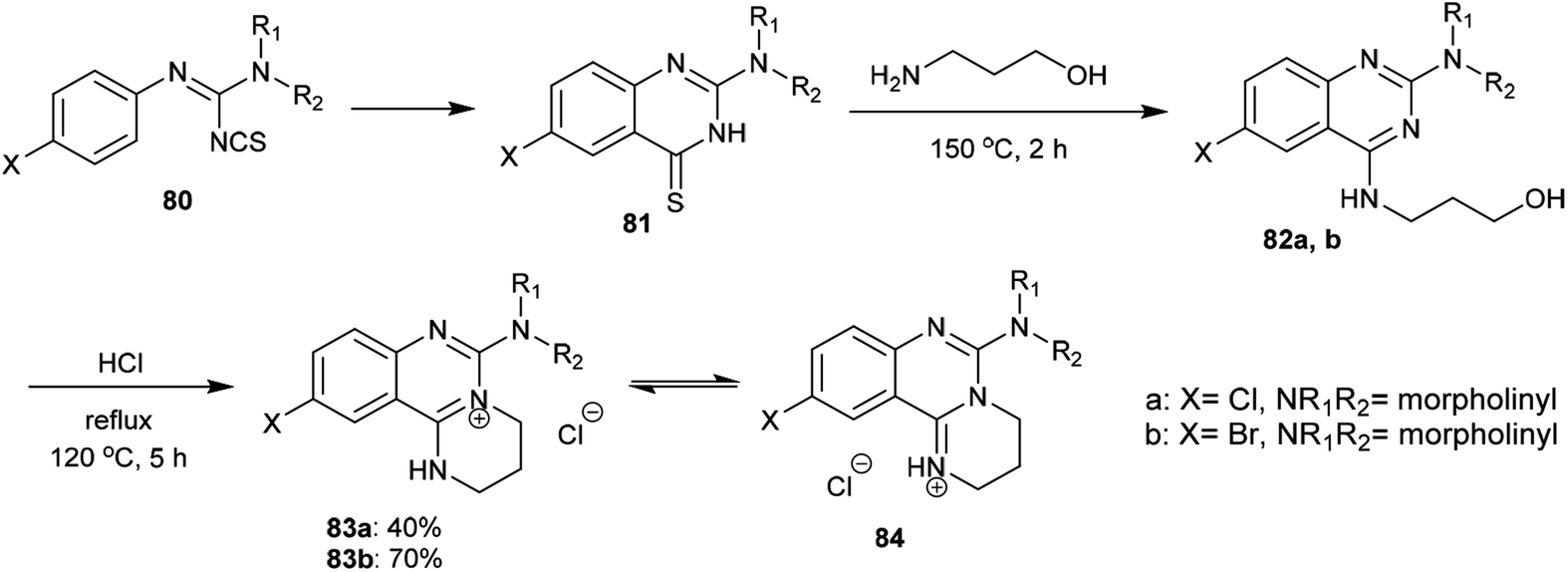
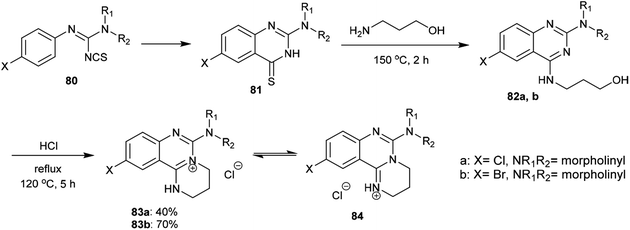
2.3.7. Synthesis of quaternary salts. A proficient procedure for the synthesis of tetrahydropyrimido[1,2-c]quinazolin-5-ium salts (84) was conveyed by Stankovský and Filip.71 Accordingly, heating of amidinoyl isothiocyanates (80) in benzene yielded the corresponding 2-(disubstituted-amino)-6-substituted-quinazoline-4(3H)-thiones (81) in quantitative yields. The substitution of the SH group “generated from enolization of the carbothioamide group” with the terminal amino group of 3-aminopropan-1-ol was achieved by heating the reactants to afford the respective 4-(3-hydroxypropylamino)quinazolines 82a,b. Subsequent heating of 82a,b with hydrochloric acid yielded the corresponding pyrimidoquinazolinium chlorides 83a,b, respectively. The structures of 83a,b have two other possible tautomeric structures, identified as pyrimidoquinazolinium chlorides 84 (Scheme 20).
 |
| | Scheme 20 Synthesis of pyrimidoquinazolinium chlorides 84. | |
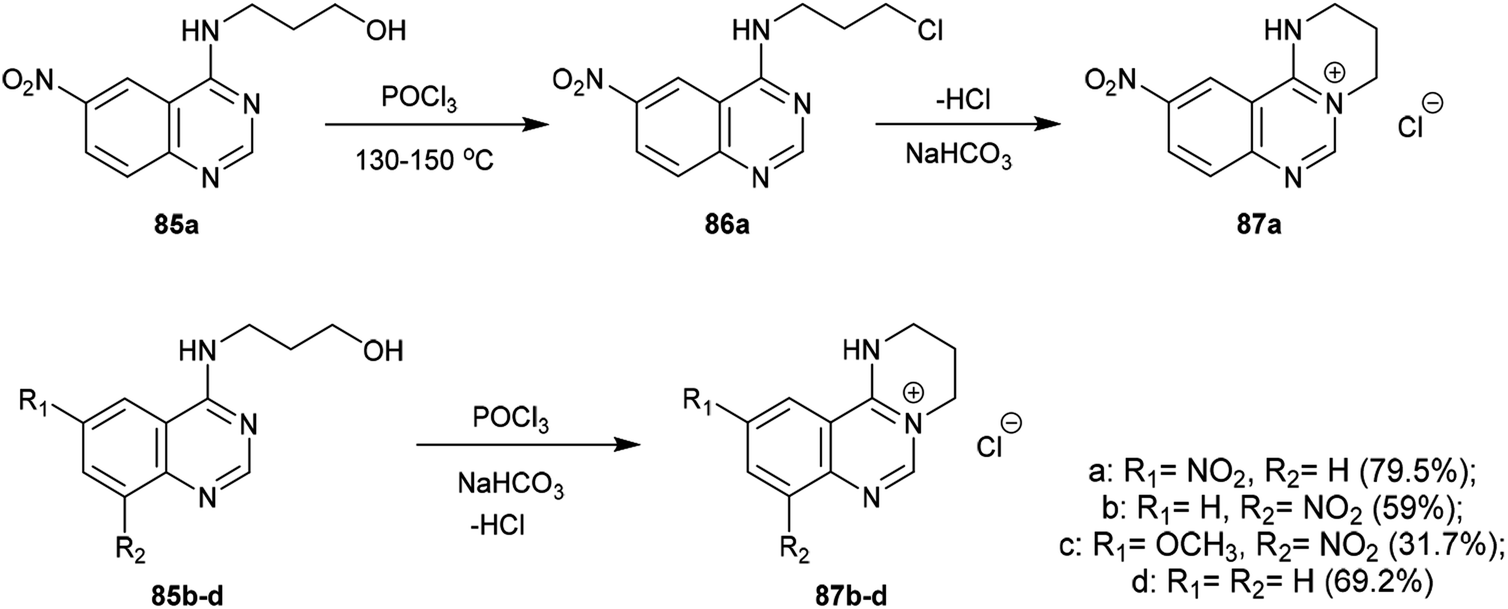
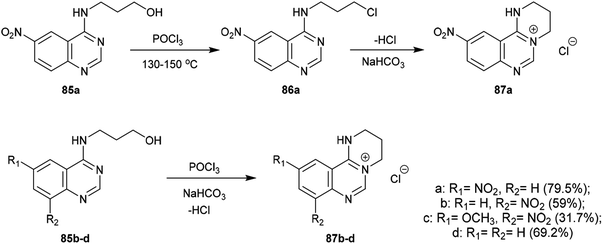
An efficacious method for the synthesis of quaternary salts was conveyed by Yoshikawa and Shitago72 involving intramolecular cyclization reactions of the anticipated 4-amino-hydroxy-propane-6,8-disubstituted-quinazolines 86. As a result, the treatment of 4-amino-6,8-disubstituted-quinazolines 85a–d with phosphorus oxychloride at reflux temperature yielded the desired chlorinated products 86, which were cyclized to the tetrahydropyrimido[1,2-c]quinazolin-5-ium chlorides 87a–d by treatment with sodium bicarbonate. The mechanism of this cyclization followed an initial1,3 proton transfer of the proton of the imine group followed by elimination of the HCl molecule (Scheme 21).
 |
| | Scheme 21 Synthesis of pyrimidoquinazolinium chlorides. | |
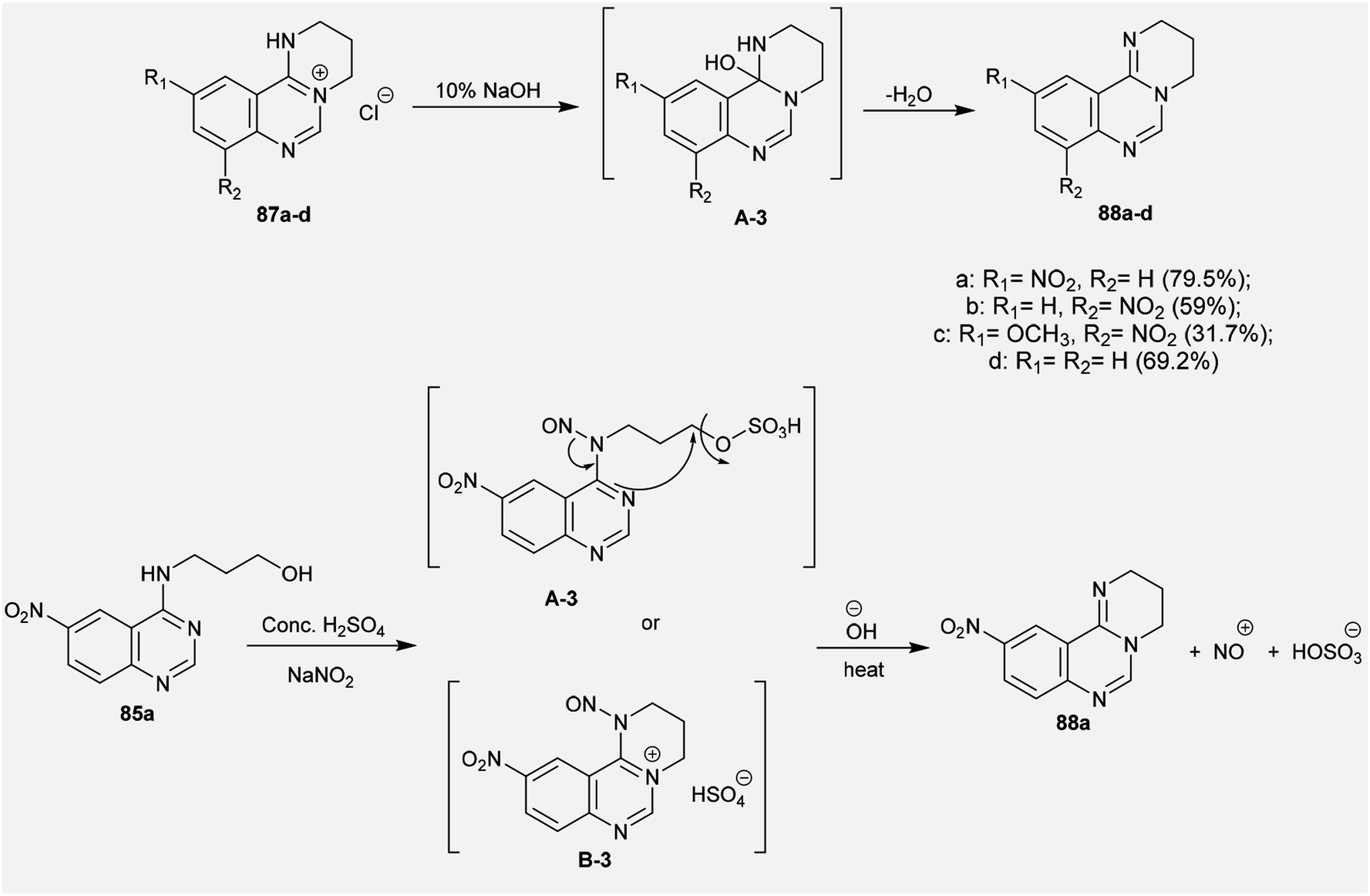
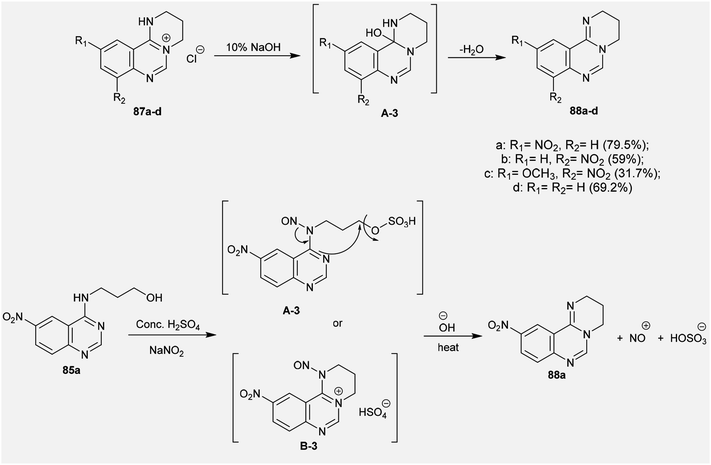
2.3.8. Synthesis from quaternary salts. Treatment of tetrahydropyrimido[1,2-c]quinazolin-5-ium chlorides 87a–d with 10% sodium hydroxide solution yielded the free bases of pyrimido[1,2-c]quinazolines 88a–d in moderate to good yields (31.7–79.5%). The products were formed by the dehydration of the generated intermediate A-3. On the other hand, treatment of 3-((6-nitroquinazolin-4-yl)amino)propan-1-ol (85a) with conc. sulfuric acid and sodium nitrite and subsequent heating with sodium hydroxide solution yielded pyrimidoquinazoline 88a through two possible mechanistic routes. Nitrosation of the imine group with nitrosyl sulfate and sulfonation of the terminal hydroxy group activated the cyclization by abstraction of the nitroso group and subsequent substitution of the hydrogen sulfate group of the intermediate A-3. Alternatively, the cyclization step proceeded with the elimination of a hydrogen sulfate group to generate the tetrahydropyrimido[1,2-c]quinazolin-5-ium hydrogen sulfate, intermediate B-3 (Scheme 22).72
 |
| | Scheme 22 Synthesis of 8,10-disubstituted-3,4-dihydro-2H-pyrimidoquinazolines. | |
2.4. Synthesis of tricyclic systems
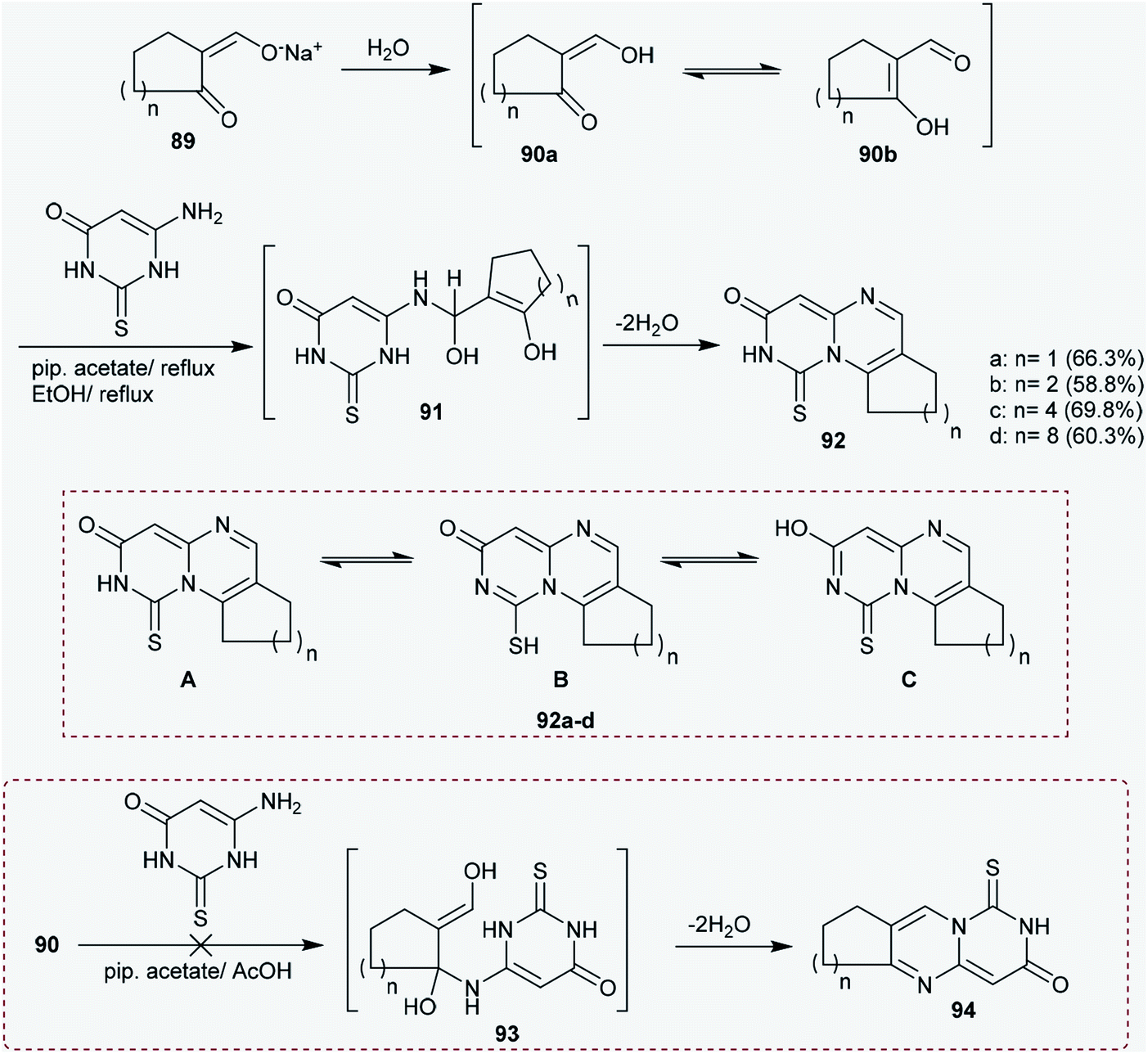
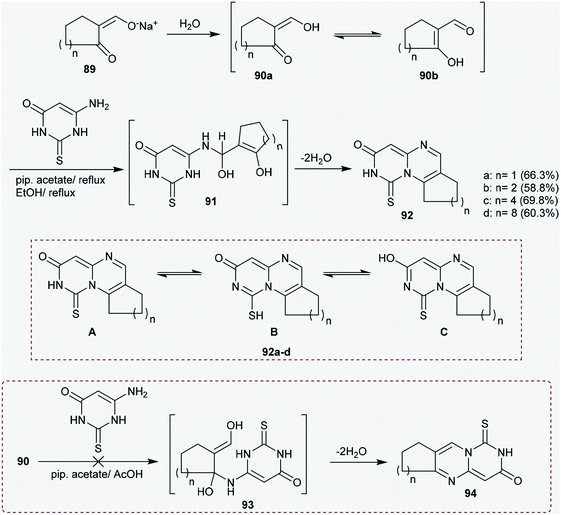
2.4.1. Synthesis of cycloalka-pyrimido[1,6-a]pyrimidinones. From the literature viewpoint, the construction of the pyrimido[4,5-d]pyrimidine heterocyclic system has been achieved through multistep synthetic routes.73,74 Moreover, Hussein and coworkers41,75 reported an unpretentious method for the construction of 1-thioxo-1,2,8,9-tetrahydro-cycloalkyl-pyrimido-pyrimidinones 92a–d. Therefore, condensation of the sodium salts of formyl cyclic ketones (89) with 6-aminothiouracil in piperidine acetate/acetic acid systems at reflux temperature afforded the cyclocondensation products 92a–d in noteworthy yields. The reactions progressed by nucleophilic addition of the amino group of 6-aminothiouracil to the aldehydic carbonyl group of 90b (in situ generated from the hydrolysis of sodium salts 89 with water) in piperidine acetate to generate the intermediates 91. The tricyclic systems, cycloalkyl-pyrimidopyrimidinones 92a–d, have three possible tautomeric structures, A–C. The nitrogen atom at position 2 of the pyrimidopyrimidine ring has a hydrogen atom, which enables the enolization of the amidic carbonyl group at position 3 of the ring, and thione-thiol tautomerization at position 1 forms the possible structures A–C.41 Intramolecular cyclocondensation of the intermediates 91 yielded the respective pyrimidopyrimidines 92a–d instead of generating the planned products 9476–78 (Scheme 23).
 |
| | Scheme 23 Synthesis of 1-thioxo-1,2,8,9-tetrahydro-cycloalkyl-pyrimidopyrimidinones. | |
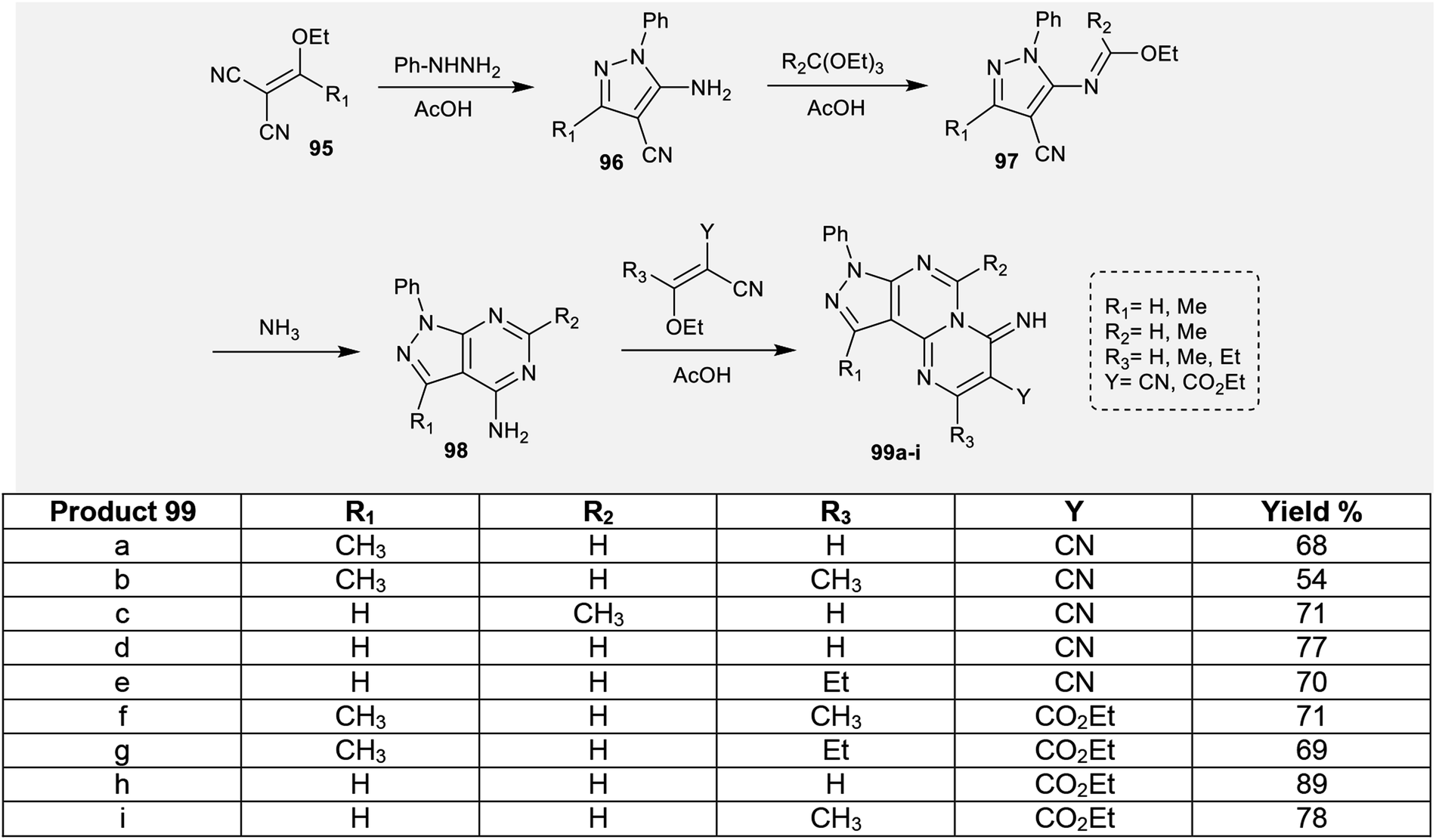
2.4.2. Synthesis of pyrazolo-pyrimido[1,2-c]pyrimidines. To construct the pyrimidopyrimidine ring, an efficient multi-step synthetic route was applied by Karoui et al.79 by succeeding acid-catalyzed condensation reactions. Consequently, the reactions of 2-(substituted-(ethoxy)methylene)malononitriles 95 with phenylhydrazine yielded the respective 4-cyano-5-amino-3-substituted-1-phenyl-1H-pyrazoles 96. The imino group of phenyl-hydrazine performed a nucleophilic attack at the nitrile function of malononitriles 95. Subsequent treatment of pyrazoles 96 with triethylorthoformates in acetic acid as a catalyst at reflux temperature yielded the desired ethyl carbonoimidates 97.80,81 The ethyl carbonoimidates 97 were further treated with ammonia in ethanol containing a catalytic amount of acetic acid at reflux temperature to afford the respective 3,6-disubstituted-4-amino-1-phenyl-1H-pyrazolopyrimidines 98.82 Afterwards, pyrazolopyrimidines 98 were reacted with 2,3-disubstituted-3-ethoxyacrylonitriles in ethanol catalyzed by acetic acid to afford the anticipated tricyclic iminopyrazolo[4,3-e]pyrimido[1,2-c]pyrimidines 99 (Scheme 24).79
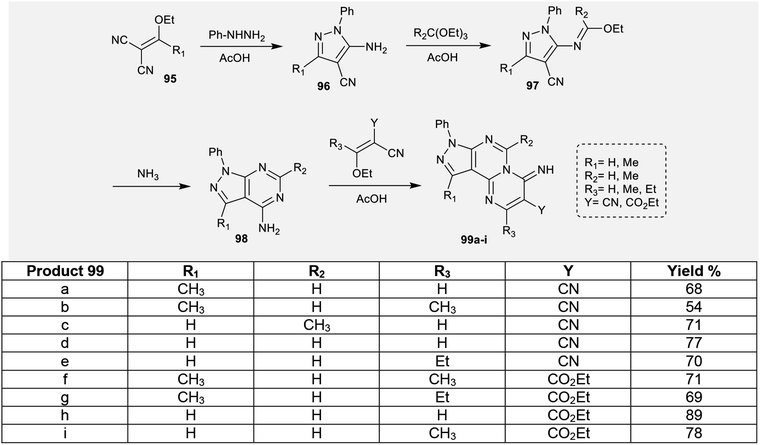 |
| | Scheme 24 Synthesis of 2,3,6,10-tetrasubstituted-8-phenylpyrazolopyrimidopyrimidin-4(8H)-imines. | |
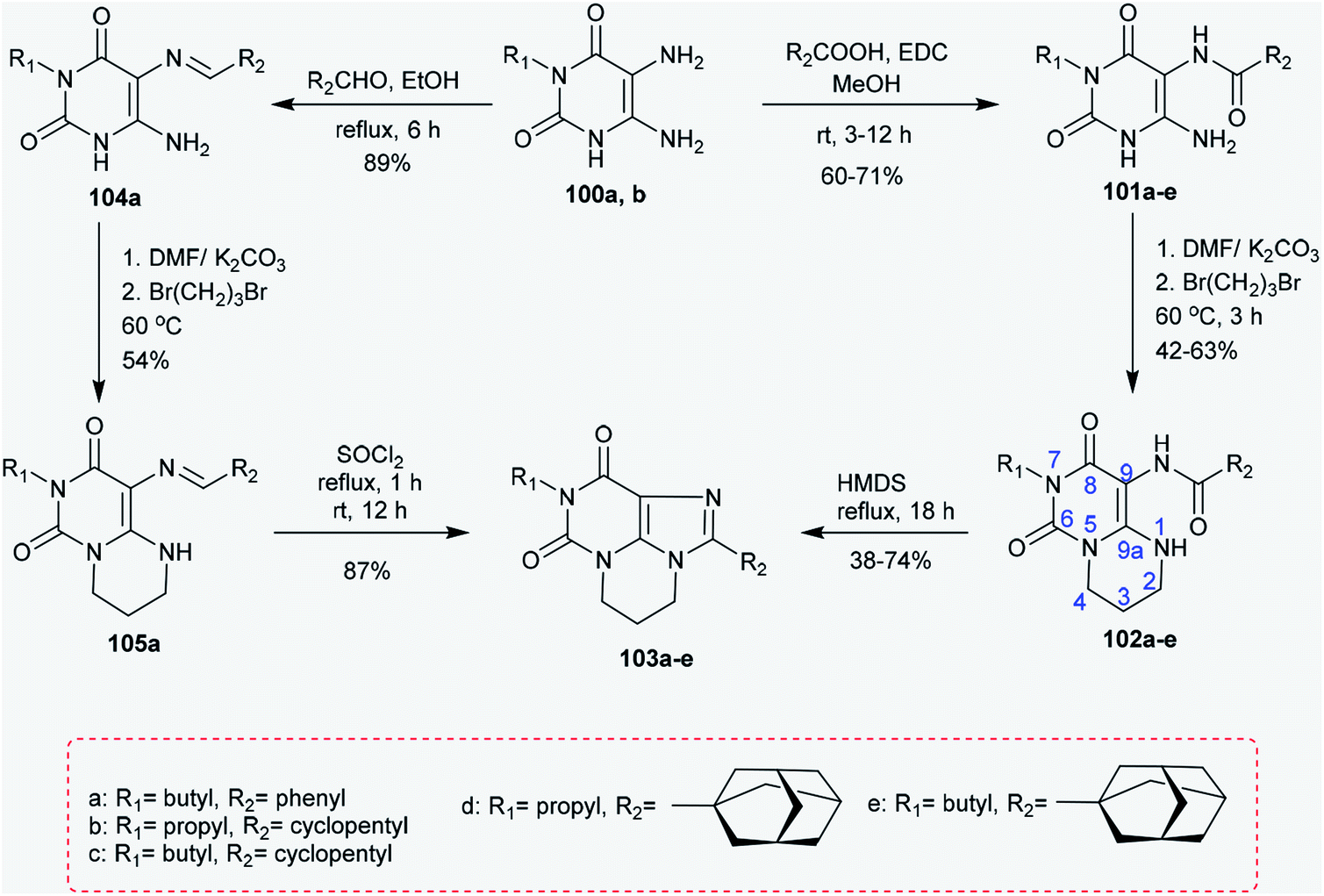
2.4.3. Synthesis of pyrimidopurine-diones. Treatment of 5,6-diamino-3-substituted-pyrimidine-2,4(1H,3H)-diones 100a and 100b each with cycloalkyl carboxylic acids or benzoic acid in methanol catalyzed by EDC yielded the corresponding amides 101a–e.83 Alternatively, the amides 101a–e reacted with 1,3-dibromopropane in DMF/K2CO3 to afford pyrimidopyrimidindiones 102a–e, respectively, with yields of 42–63%. The high reactivity of 1,3-dibromopropane allowed the reaction between the nitrogen atom at position 1 and the amino group attached to position 6 of the uracil. Further refluxing of 102a–e with HMDS at reflux temperature for 18 h yielded the tricyclic systems 103a–e, respectively, with yields of 38–74%. An alternative route for the synthesis of a xanthine tricyclic system was reported through the preparation of compound 103a. Therefore, the reaction of 100a with benzaldehyde in ethanol at reflux temperature yielded compound 104a.84 The bicyclic 105a was synthesized in a yield of 54% by the reaction of 1,3-dibromopropane with 104a in a DMF/K2CO3 system. Ring closure of compound 105a was accomplished by an oxidative step by reaction with thionyl chloride to yield xanthine product 103a in 87% yield (Scheme 25).85 The substituents of 2-(3-noradamantyl)-, such as 2-(adamantan-1-yl)-9-propyl-5,6-dihydro-4H,8H-pyrimidopurine-dione (102), have been verified to be selective antagonists for adenosine A1 receptor.86,87
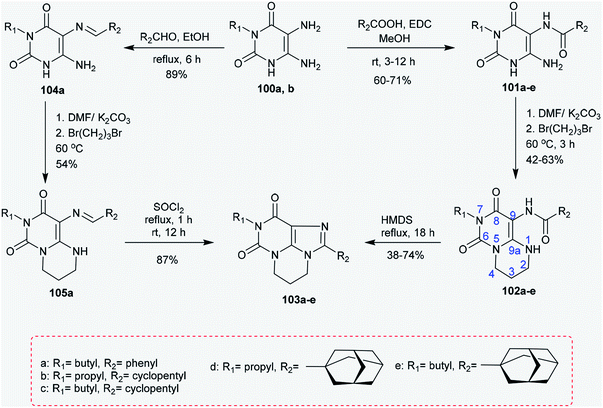 |
| | Scheme 25 Synthesis of pyrimidopurine-diones. | |
2.5. Synthesis of tetracyclic systems
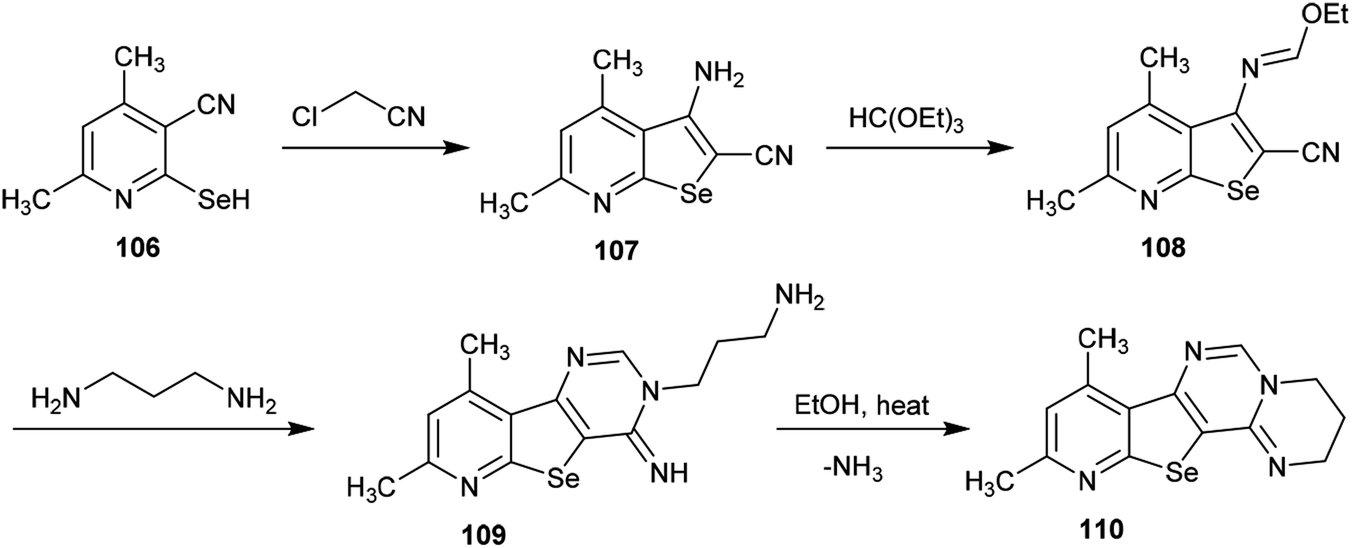
A simple multi-step synthetic method was reported by Abdel-Hafez et al.88 for the preparation of a tetracyclic system incorporating the pyrimido[1,2-c]pyrimidine scaffold. Thus, 3-amino-4,6-dimethyl-selenopheno[2,3-b]pyridine-2-carbonitrile (107) was prepared by reaction of 2-hydroseleno-4,6-dimethyl-nicotinonitrile (106) with 2-chloroacetonitrile.89 The reaction of selenopheno[2,3-b]pyridine 107 with triethyl orthoformate yielded the respective ethyl formimidate 108, which was treated with propane-1,3-diamine in 1,4-dioxane at room temperature to afford the tricyclic pyrido-selenopheno[3,2-d]pyrimidine 109. The intramolecular cyclization of compound 109 to the target product 110 was accomplished by heating in ethanol. In this cyclization, the terminal amino group of the propyl chain acts as a nucleophile which attacks the CNH at position 4 of the pyrimidine ring, resulting in the elimination of an ammonia molecule (Scheme 26).88
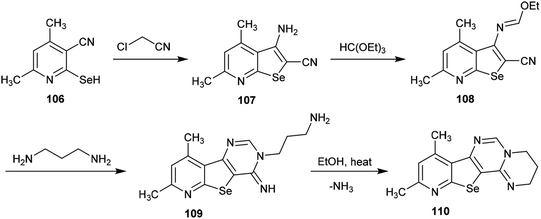 |
| | Scheme 26 Synthesis of pyrido-selenopheno[2,3-e]pyrimidopyrimidine. | |
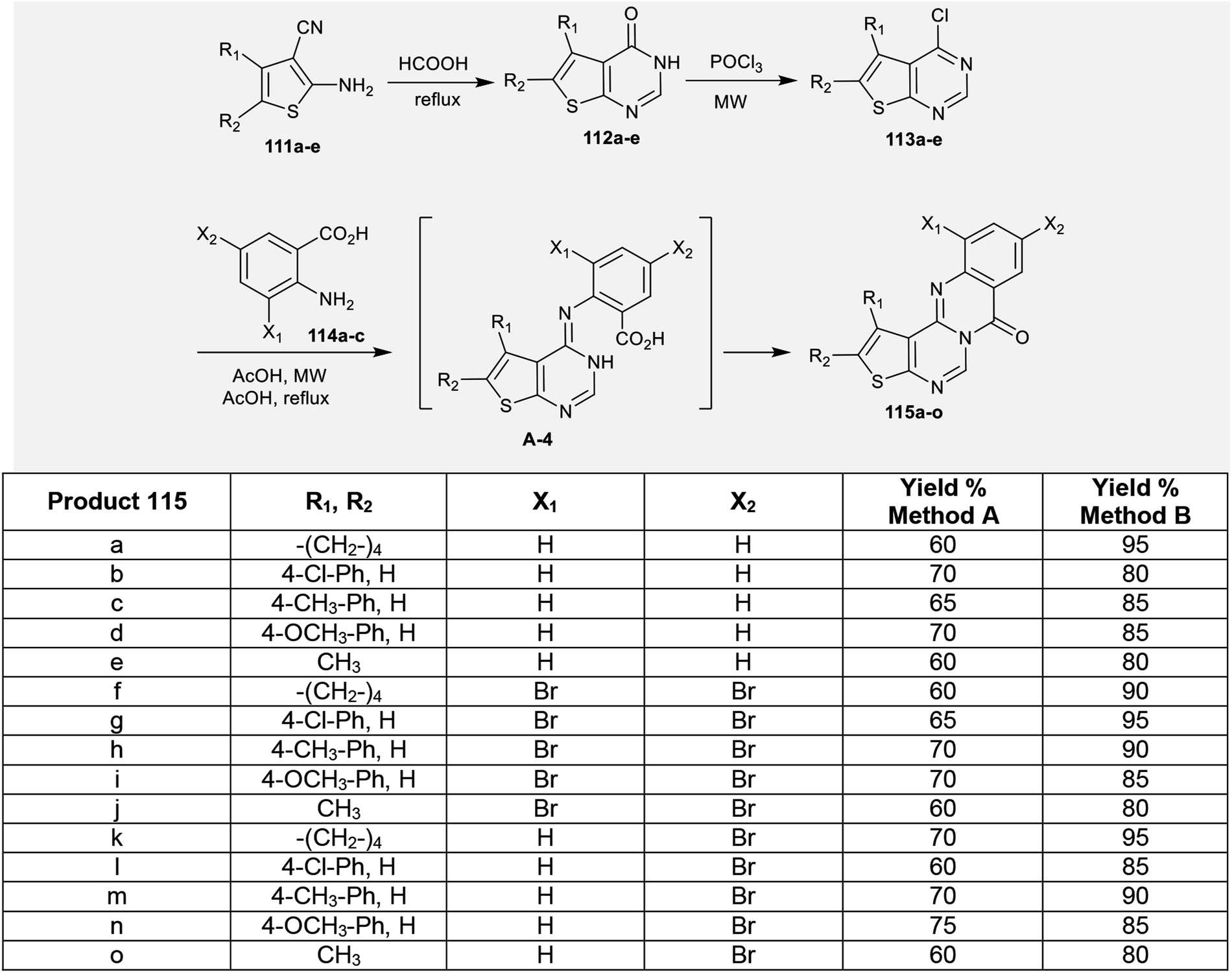
The reaction of enaminonitriles of thiophenes 111a–e with formic acid at reflux temperature yielded thienopyrimidinones 112a–e, respectively.90 Further treatment of 112a–e with phosphorus oxychloride under microwave irradiation conditions yielded the anticipated chlorinated thienopyrimidines 113a–e. A series of 7H-thienopyrimidoquinazolinones 115a–o was synthesized by Niementowski reactions91 of 3a–e with anthranilic acids 114a–c under microwave irradiation or reflux92–95 conditions. The products 115a–o were formed through nucleophilic attack of the amino group of the anthranilic acids at C4 of thieno[2,3-d]pyrimidines 113a–e, followed by1,3 proton transfer and subsequent intramolecular cyclocondensation (Scheme 27).96
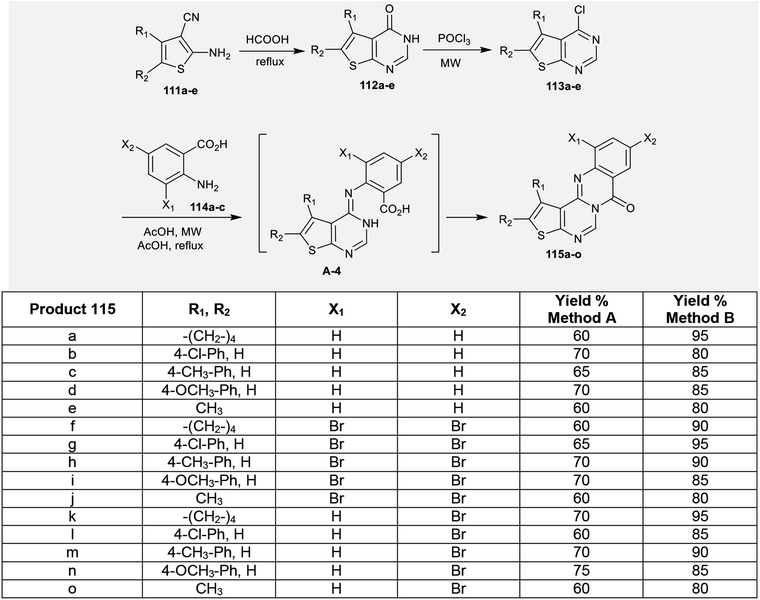 |
| | Scheme 27 Synthesis of 7H-thienopyrimidoquinazolinones 115. | |
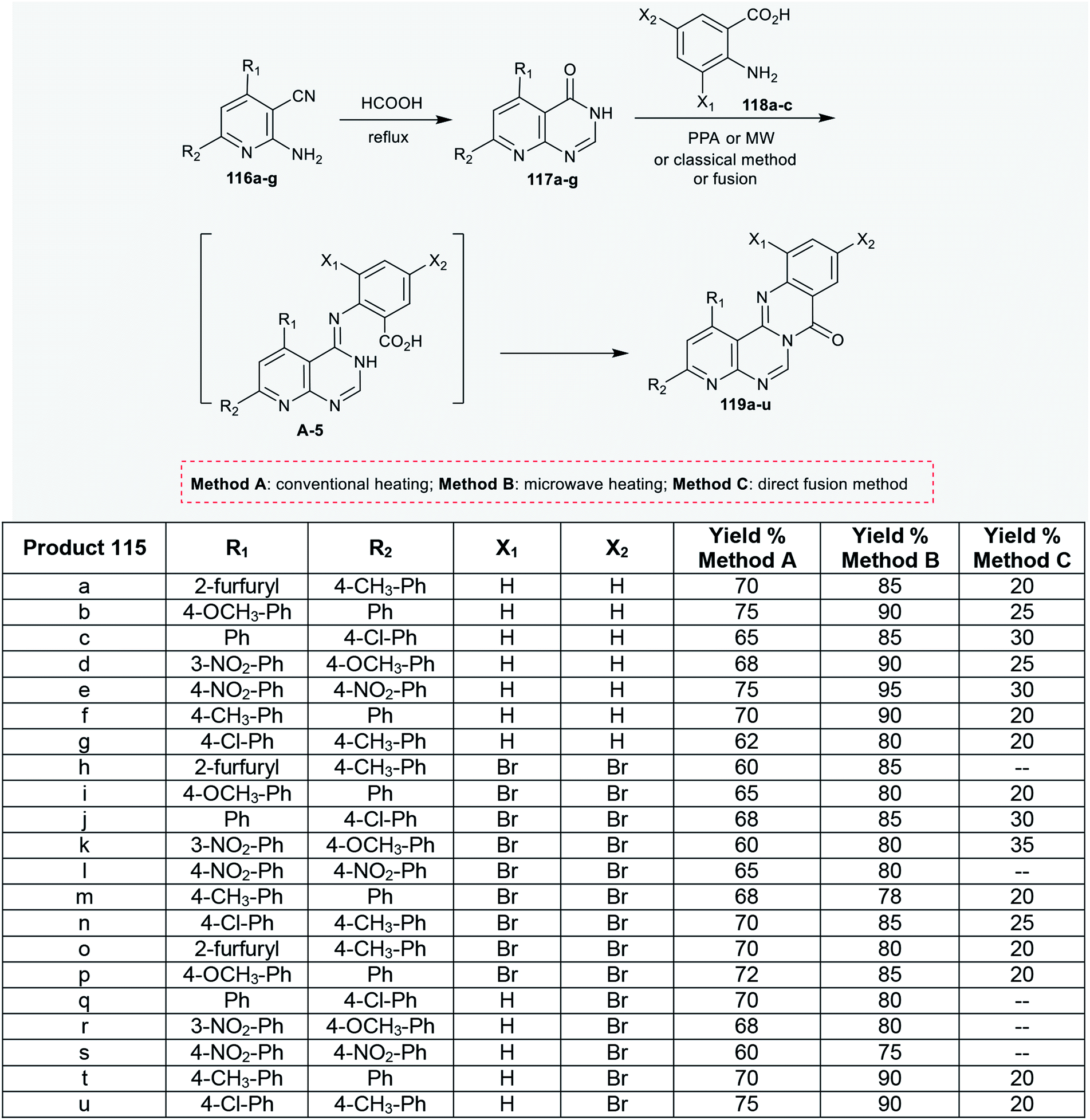
As well, pyridopyrimidinones 117a–g were synthesized by refluxing 2-amino-nicotinonitriles 116a–g in formic acid. Niementowski condensation of 117a–g with anthranilic acids 118a–c under microwave irradiation conditions, by refluxing in the presence of polyphosphoric acid, or by the direct fusion method yielded the anticipated 8H-pyridopyrimidoquinazolinones 119a–u. The products 119a–u were formed through initial condensation between the amino group of the anthranilic acid and the carbonyl group of the pyrimidine ring and a subsequent intramolecular cyclocondensation step of the generated intermediate A-5 (Scheme 28).92
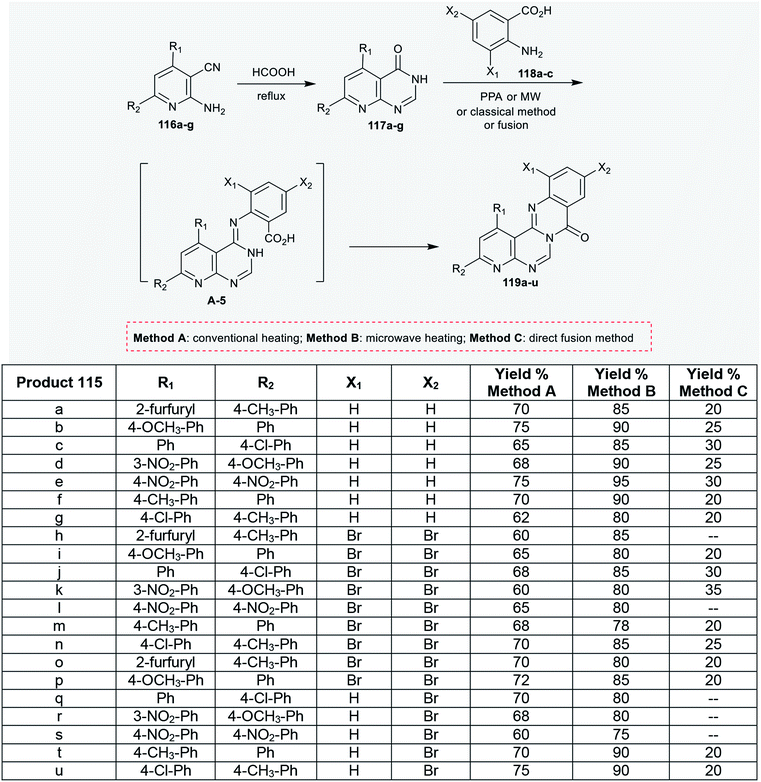 |
| | Scheme 28 Synthesis of 8H-pyridopyrimidoquinazolinones 119. | |
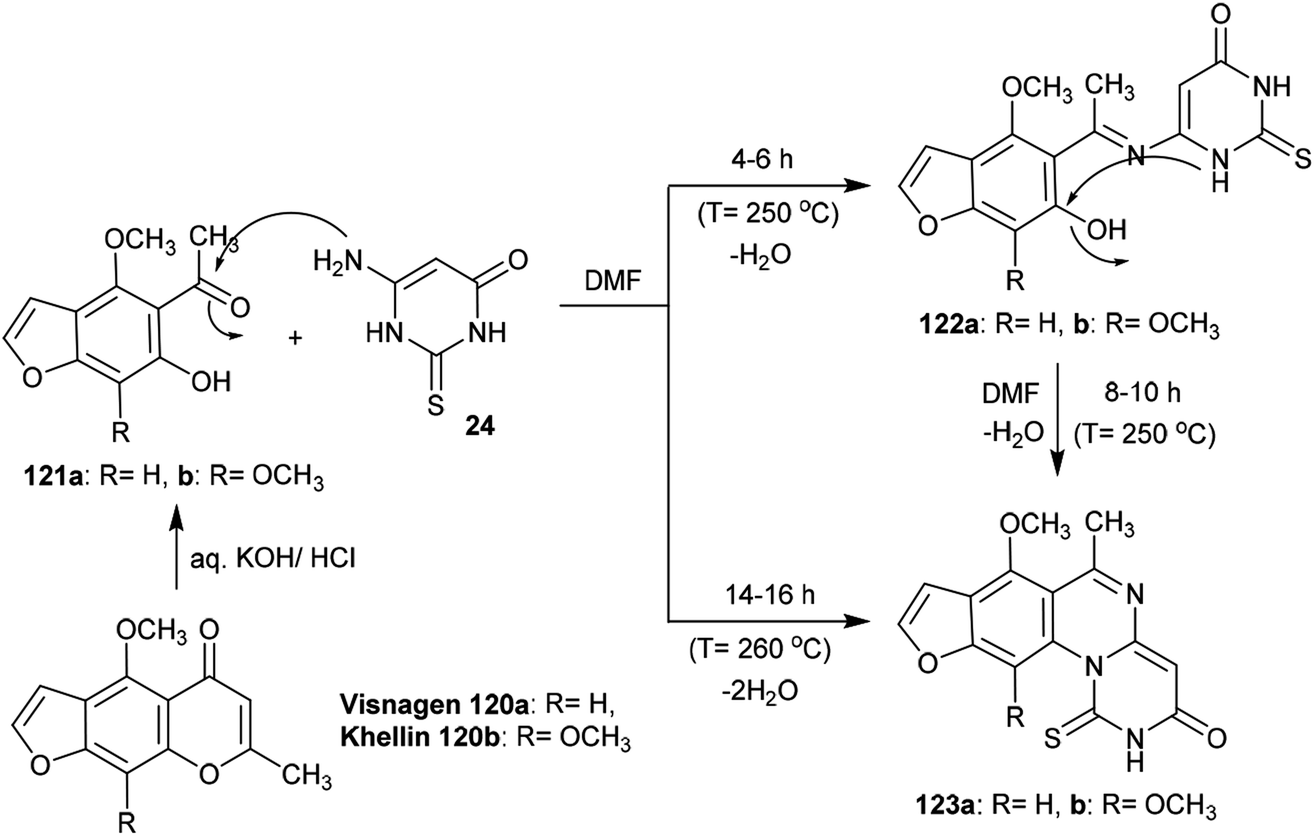
Treatment of Visnagen 120a (R = H) and Khellin 120b (R = OCH3), respectively, in an aqueous solution of potassium hydroxide generated the respective 5-acetyl-6-hydroxy-4-methoxybenzofuran derivatives 121a and 121b, which were condensed with dihydropyrimidinone 24 in DMF at reflux temperature to yield 3H-furopyrimidoquinazolinones 123a and 123b. The reactions of benzofuran derivatives 121a and 121b with dihydropyrimidinone 24 produced compounds 122a and 122b after a short time; these were condensed in DMF to afford the target compounds 123a and 123b, respectively. In the first step, nucleophilic attack of the amino group of dihydropyrimidinone 24 took place at the acetyl carbonyl group of benzofuran derivatives 121a and 121b, and subsequent intramolecular cyclocondensation afforded compounds 123a and 123b97 (Scheme 29).
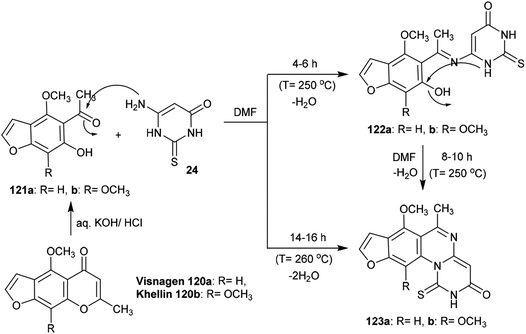 |
| | Scheme 29 Synthesis of furopyrimidoquinazolinones. | |
2.6. Synthesis of binary heterocycles
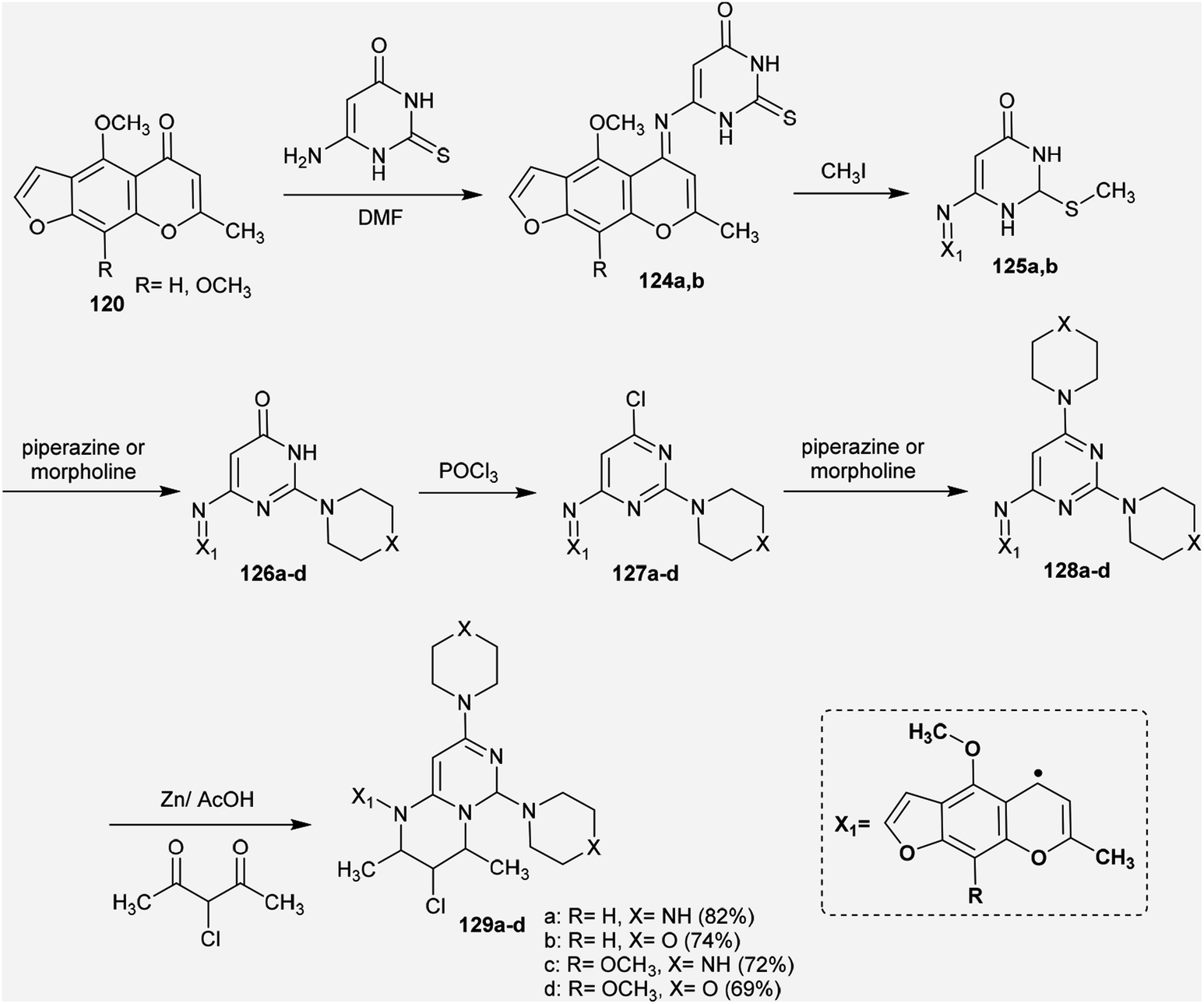
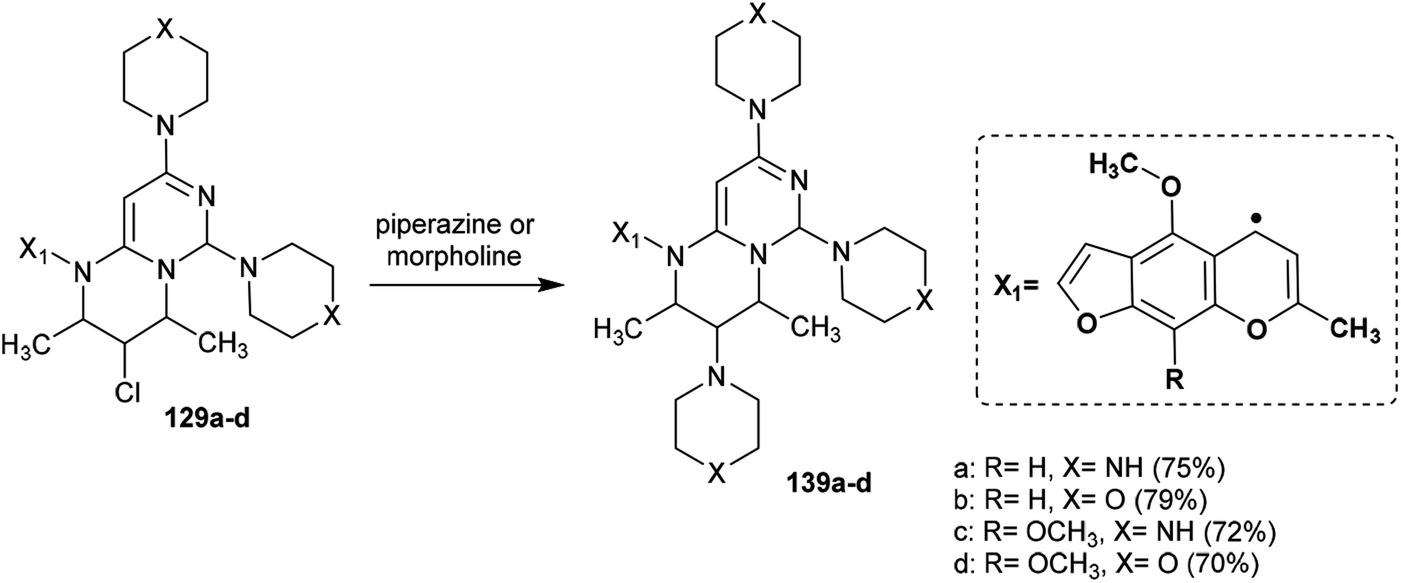
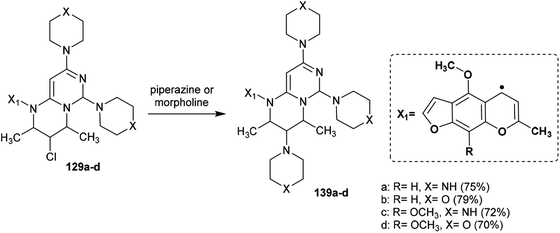
Abu-Hashem and Youssef98 reported a proficient procedure for the synthesis of 2H-pyrimido[1,6-a]pyrimidines 129a–d through multi-step reactions. Firstly, condensation reactions of Visnagen and Khellin 120 in refluxing DMF with 6-amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one afforded the Schiff bases 124a and b, respectively. Succeeding methylation of the thione group of 124a and b with methyl iodide afforded the corresponding methylmercaptan analogs 125a and b, which followed nucleophilic substitution by reactions with piperazine and morpholine to yield a series of pyrimidinones 126a–d. Chlorination of the ketone group at C4 of the pyrimidine ring of 126a–d was accomplished by reaction with phosphorus oxychloride in dry 1,4-dioxane to afford the anticipated chlorinated products 127a–d. Consequent halogen substitution of the formed products 127a–d was achieved by reactions with piperazine and morpholine through nucleophilic attack of the cyclic amine at the chlorine atom at C4 of the pyrimidine ring to afford the trisubstituted pyrimidines 128a–d, respectively. Lastly, the cyclization step of 128a–d proceeded by cyclocondensation with 3-chloropentane-2,4-dione in acetic acid containing zinc dust to afford the corresponding 2H-pyrimidopyrimidines 129a–d in good yields (69–82%) (Scheme 30).
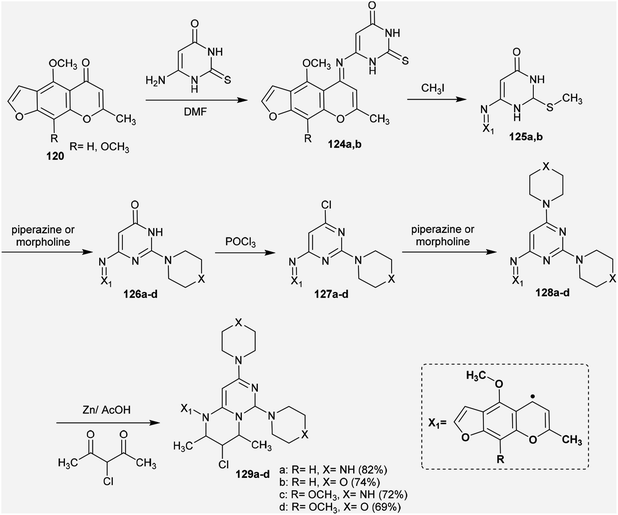 |
| | Scheme 30 Synthesis of tetrahydro-2H-pyrimidopyrimidines. | |
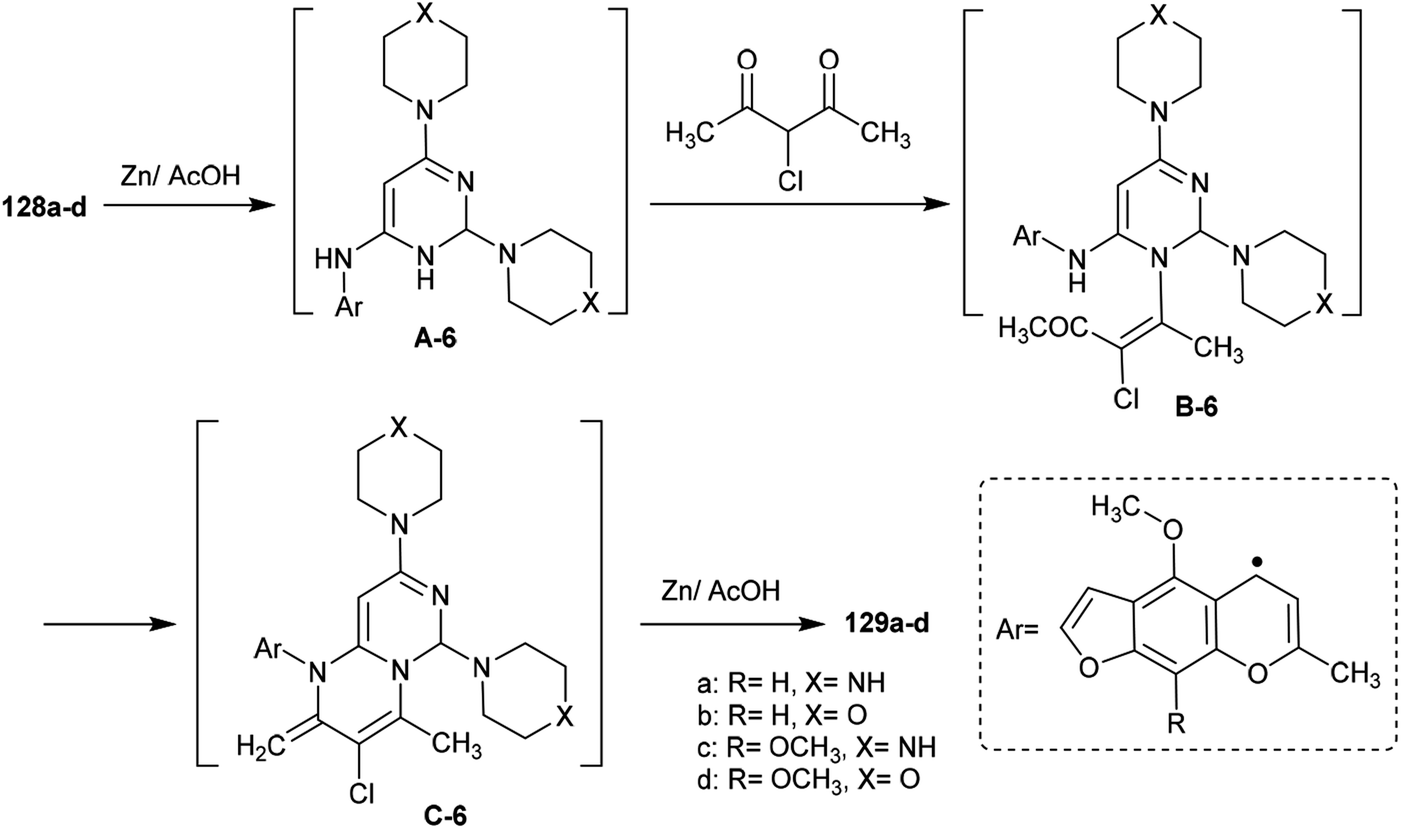
The mechanism of the cyclization of the trisubstituted pyrimidines 128a–d to the respective tetrahydro-2H-pyrimido[1,6-a]pyrimidines 129a–d is illustrated in Scheme 31. The initial step in the reactions of pyrimidines 128a–d with 3-chloropentane-2,4-dione is the reduction of the pyrimidine ring and the substituted imine at C6 of the ring with Zn in acetic acid to generate the intermediate A-6. Condensation of the intermediate A-6 with 3-chloropentane-2,4-dione, which was tautomerized to the enol form, produced the intermediate B-6; subsequent intramolecular cyclization after rearrangement of another ketonic carbon to the enol form and condensation with the imine group at C6 of the pyrimidine ring generated the intermediate C-6. Subsequent reduction of the intermediate C-6 with zinc yielded a series of pyrimidopyrimidines 129a–d.98
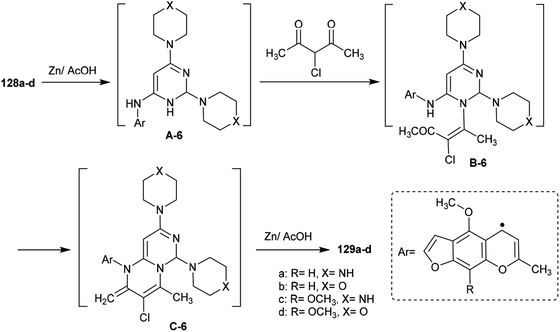 |
| | Scheme 31 The proposed ring construction of the pyrimidopyrimidines. | |
3. Reactions
3.1. Reactivity of the thione group
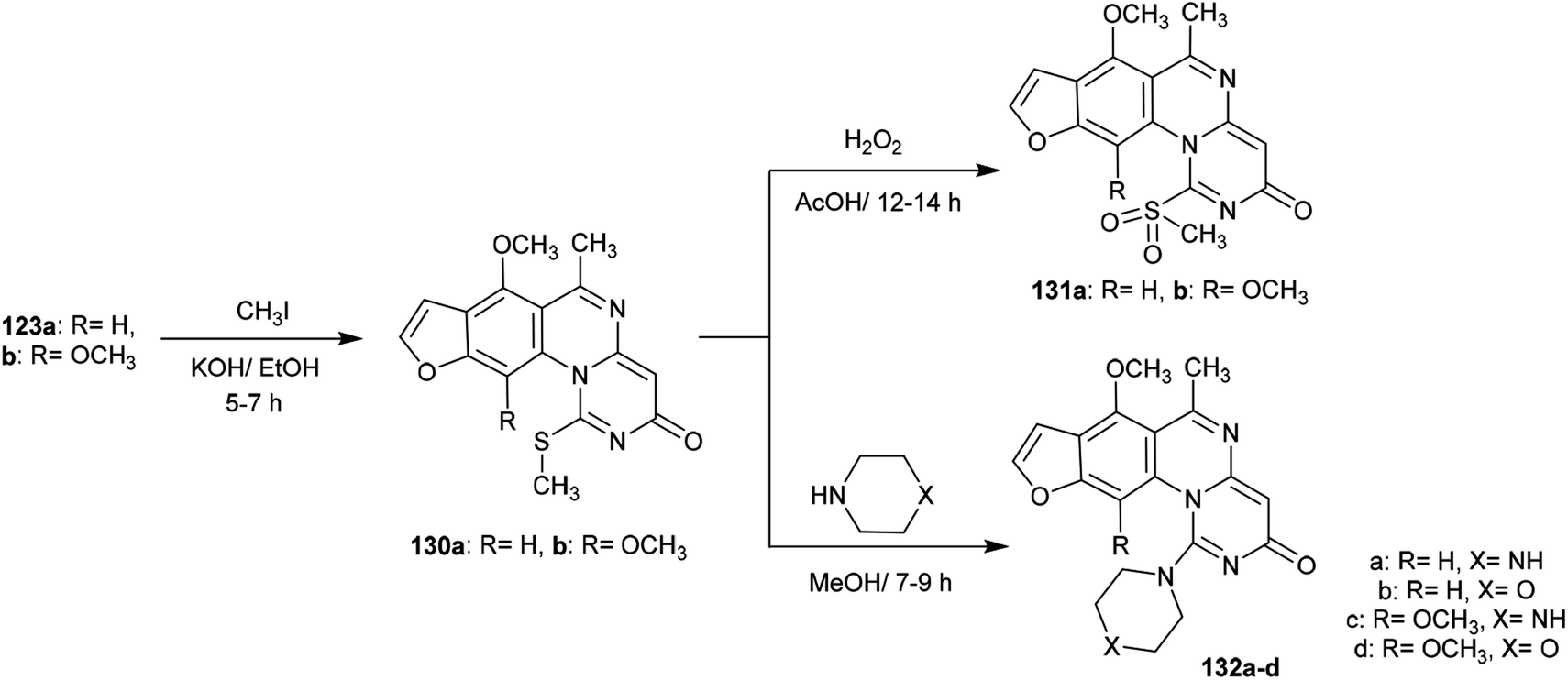
Methylation of the thione groups of 3H-furopyrimidoquinazolinones 123a and 123b with methyl iodide in alcoholic potassium hydroxide solution yielded the methylmercaptan derivatives 130a and 130b, respectively. Oxidation of 130a and 130b with hydrogen peroxide yielded the (methylsulfonyl)-3H-furo-pyrimido[1,6-a]quinazolin-3-ones 131a, and 131b. Alternatively, treatment of 130a and 130b with cyclic amines such as piperazine or morpholine afforded the respective amine-linked furo-pyrimido-quinazolinones 132a–d through nucleophilic substitution reactions in which SCH3 acted as a good leaving group97 (Scheme 32).
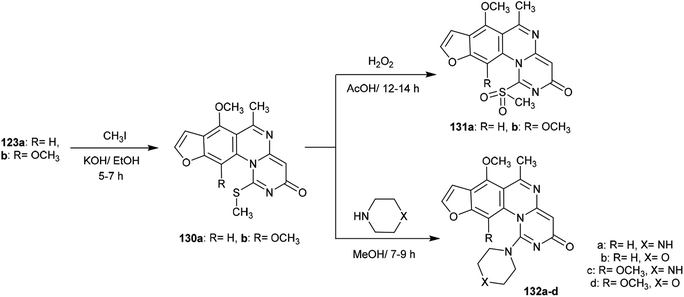 |
| | Scheme 32 Oxidation and amination of furopyrimidoquinazolinones. | |
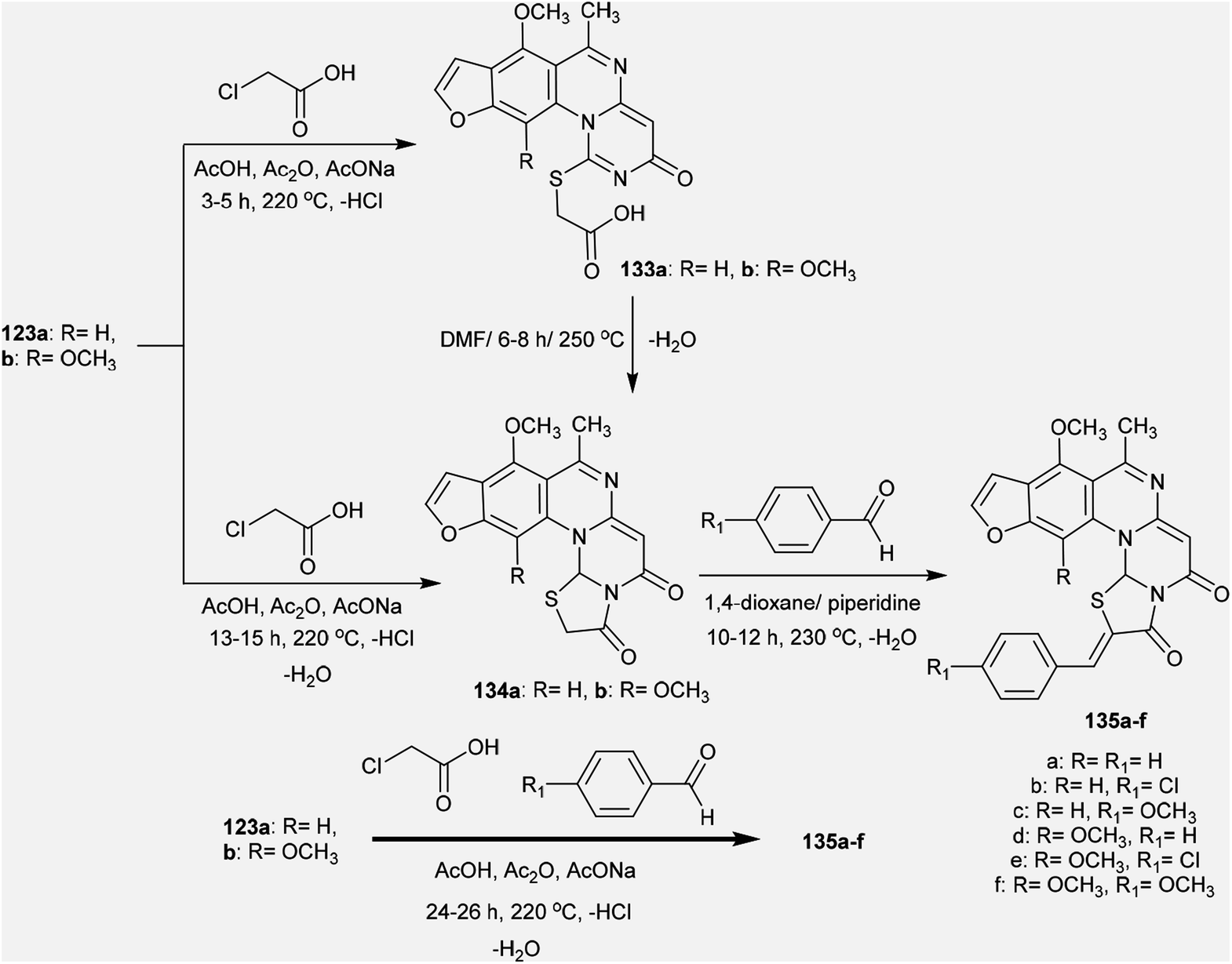
The reactivity of 3H-furopyrimidoquinazolinones 123a and 123b was investigated by Abu-Hashem97 through reactions with α-halo-acids. Thus, heating of compounds 123a and 123b with 2-chloroacetic acid in AcOH/Ac2O catalyzed by sodium acetate yielded furo-thiazolo-pyrimido[1,6-a]quinazoline-3,5-diones 134a and 134b, respectively; these were obtained from the same reactions through the formation of compounds 133a and 133b and subsequent condensation. The formation of compounds 134a,b is not discussed because the mechanism of reduction of the CN bond of the pyrimidinone ring to complete the cyclization step is unclear. Additionally, condensation of compounds 134a and 134b with aryl aldehydes in 1,4-dioxane containing piperidine yielded a series of arylidenes 135a–f. One-pot reactions of compounds 123a and 123b with 2-chloroacetic acid and aryl aldehydes in AcOH/Ac2O catalyzed by sodium acetate directly afforded the products 135a–f (Scheme 33).
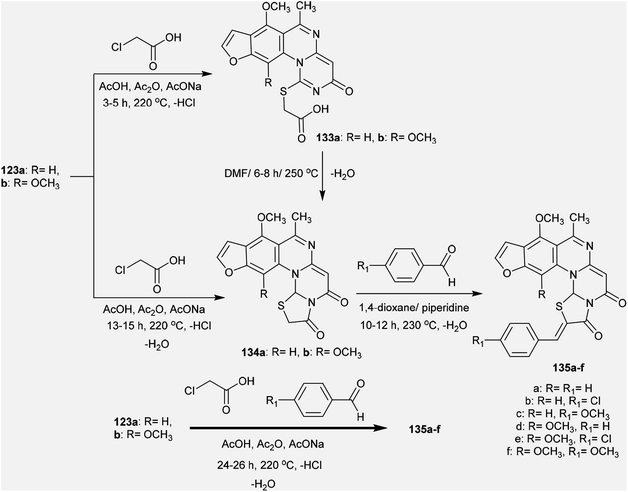 |
| | Scheme 33 Synthesis of arylidene of furothiazolo-pyrimido[1,6-a]quinazoline-3,5-diones. | |
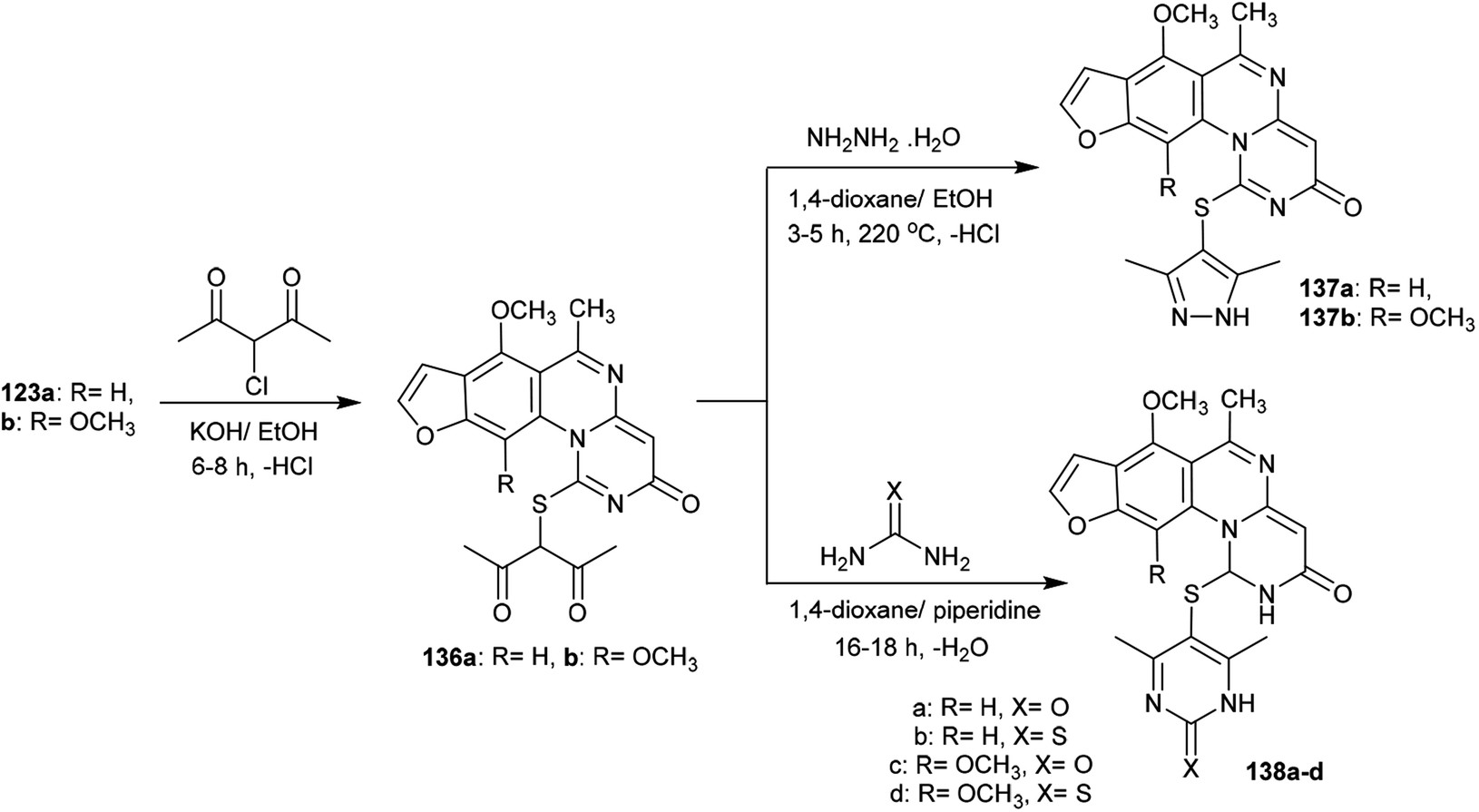
The alksylation process of 3H-furopyrimidoquinazolinones 123a and 123b was achieved by treatment of alcoholic potassium hydroxide solutions of the reactants with 3-chloropentane-2,4-dione to afford the desired thiopentane-2,4-diones 136a and 136b, respectively. The alkylation of the thione groups of 123a and 123b was accomplished after the tautomerization of the thione groups to the thiol forms. Compounds 136a and 136b were reacted with hydrazine hydrate, urea and thiourea in 1,4-dioxane containing piperidine at reflux temperature to yield the binary heterocycles 137a, 137b, and 138a–d, respectively. The reactions are cyclocondensations of 1,3-diketones with 1,2- or 1,3-binucleophiles97 (Scheme 34).
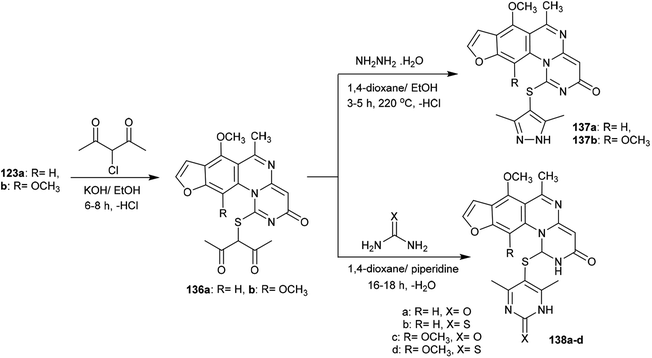 |
| | Scheme 34 Synthesis of hetrylthio-3H-furopyrimidoquinazolinones. | |
3.2. Reactivity of the amide group
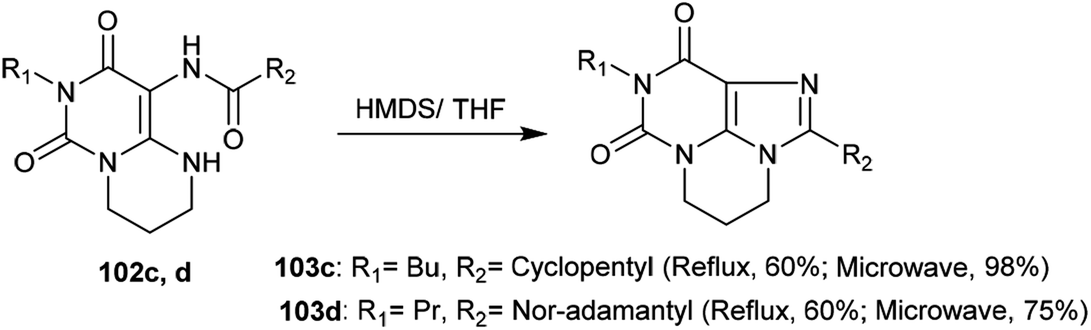
More recently, the tricyclic systems 103c and 103d were also prepared under microwave irradiation conditions to improve the yield and shorten the reaction time. The reaction of 102d to prepare 103d failed to give a good yield due to the low solubility of the reactant. By dissolving compound 102d in THF and applying microwave irradiation conditions (20 min at 100 W) with the addition of HMDS with careful stirring, compound 103d was obtained in 75% yield. On the other side, pyrimidopurine-dione 103c was synthesized under microwave irradiation conditions from the reaction of pyrimido[1,6-a]pyrimidine 102c with HMDS in 98% yield under the previously described conditions (100 W, 140 °C, 20 min) (Scheme 35).99
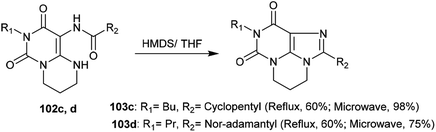 |
| | Scheme 35 Synthesis of pyrimidopurine-diones. | |
3.3. Nucleophilic substitution reactions
Substitution of the chlorine atom at C3 of tetrahydro-pyrimidopyrimidines 129a–d with nucleophiles such as piperazine and morpholine was achieved in boiling methanol to afford the respective 1-aryl-2,4-dimethyl-3,6,8-tri(piperidinyl) and (morpholinyl)-pyrimidopyrimidines 139a–d (Scheme 36).98
 |
| | Scheme 36 Synthesis of 1-aryl-3,6,8-trisubstituted-2H-pyrimidopyrimidines. | |
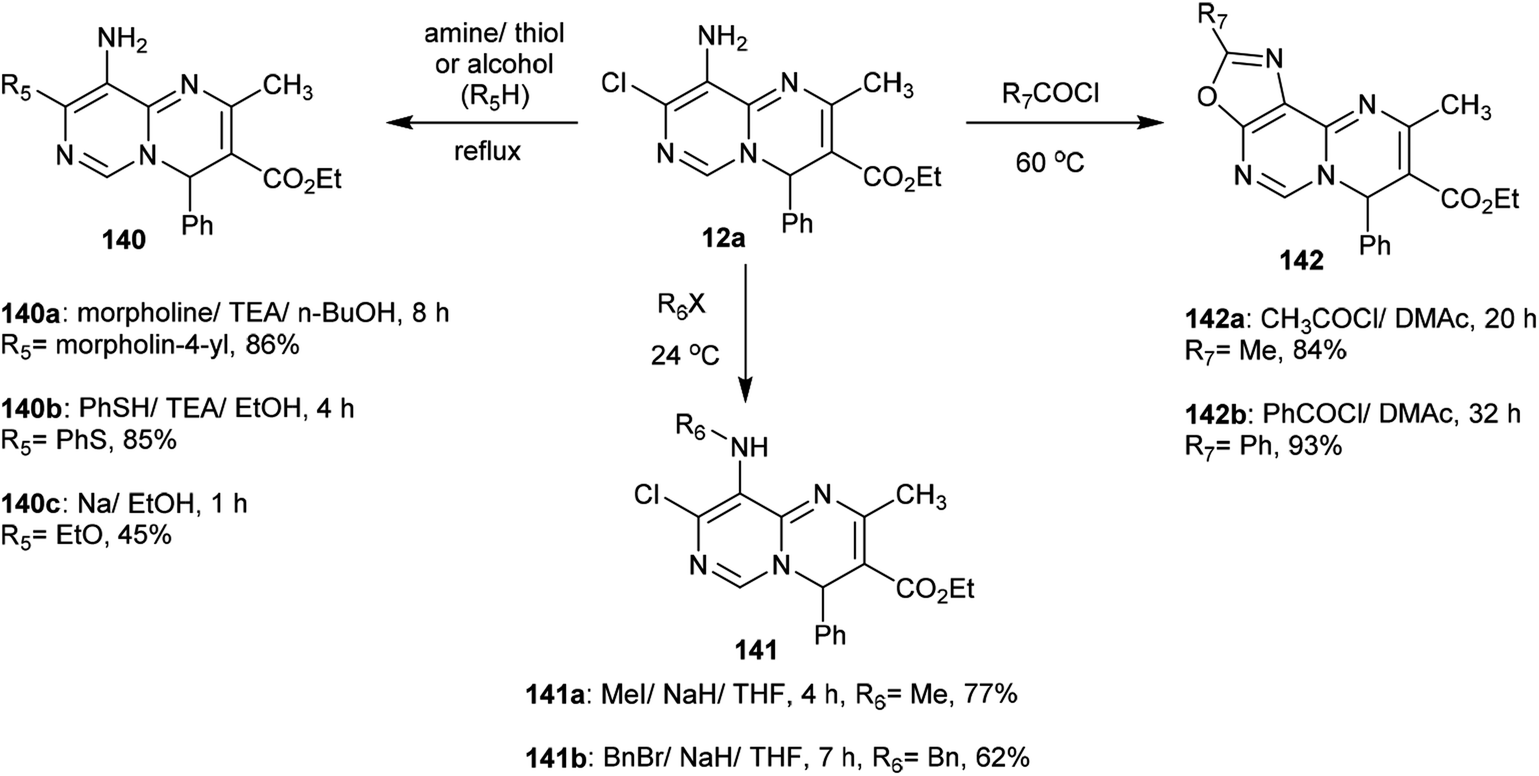
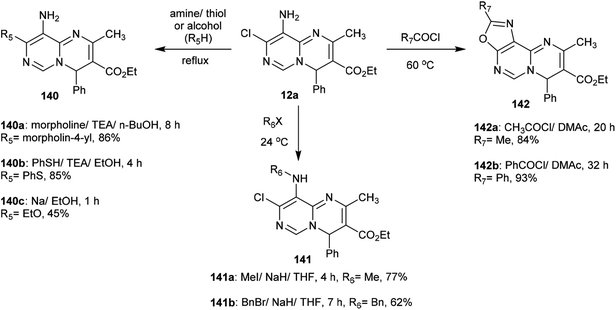
The reactivity of ethyl carboxylate 12a was verified by reactions with different electrophiles and nucleophiles. Consequently, ethyl carboxylate 12a was reacted with morpholine, benzenethiol, and sodium ethoxide solutions to afford the anticipated ethyl carboxylates 140a, 140b, and 140c, respectively. In another route, the chlorine atom at C8 of compound 12a was substituted by reactions with methyl iodide and benzyl bromide to afford the products 141a and 141b in 77% and 62% yield, respectively. Additionally, reactions of 12a with acetyl chloride and benzoyl chloride led to the generation of tricyclic systems, oxazolopyrimidopyrimidines 142a and 142b, in 84% and 93% yield, respectively (Scheme 37). The chlorine atom and amino group linked to the basic skeleton of 4H-pyrimidopyrimidine provide potential for future studies.40
 |
| | Scheme 37 Reactivity of ethyl 4H-pyrimidopyrimidine-carboxylate. | |
3.4. Condensation reactions
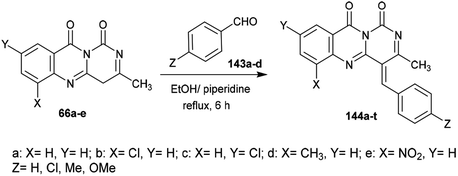
Condensation of 1H-pyrimido[6,1-b]quinazoline-1,10(4H)-diones 66a–e with aryl aldehydes 143a–d in ethanol containing piperidine at reflux temperature yielded the respective arylidenes of 3-methyl-1H-pyrimido[6,1-b]quinazoline-1,10(4H)-diones 144a–t (Scheme 38).65
 |
| | Scheme 38 Synthesis of arylidenes of 3-methyl-1H-pyrimido[6,1-b]quinazoline-1,10(4H)-diones. | |
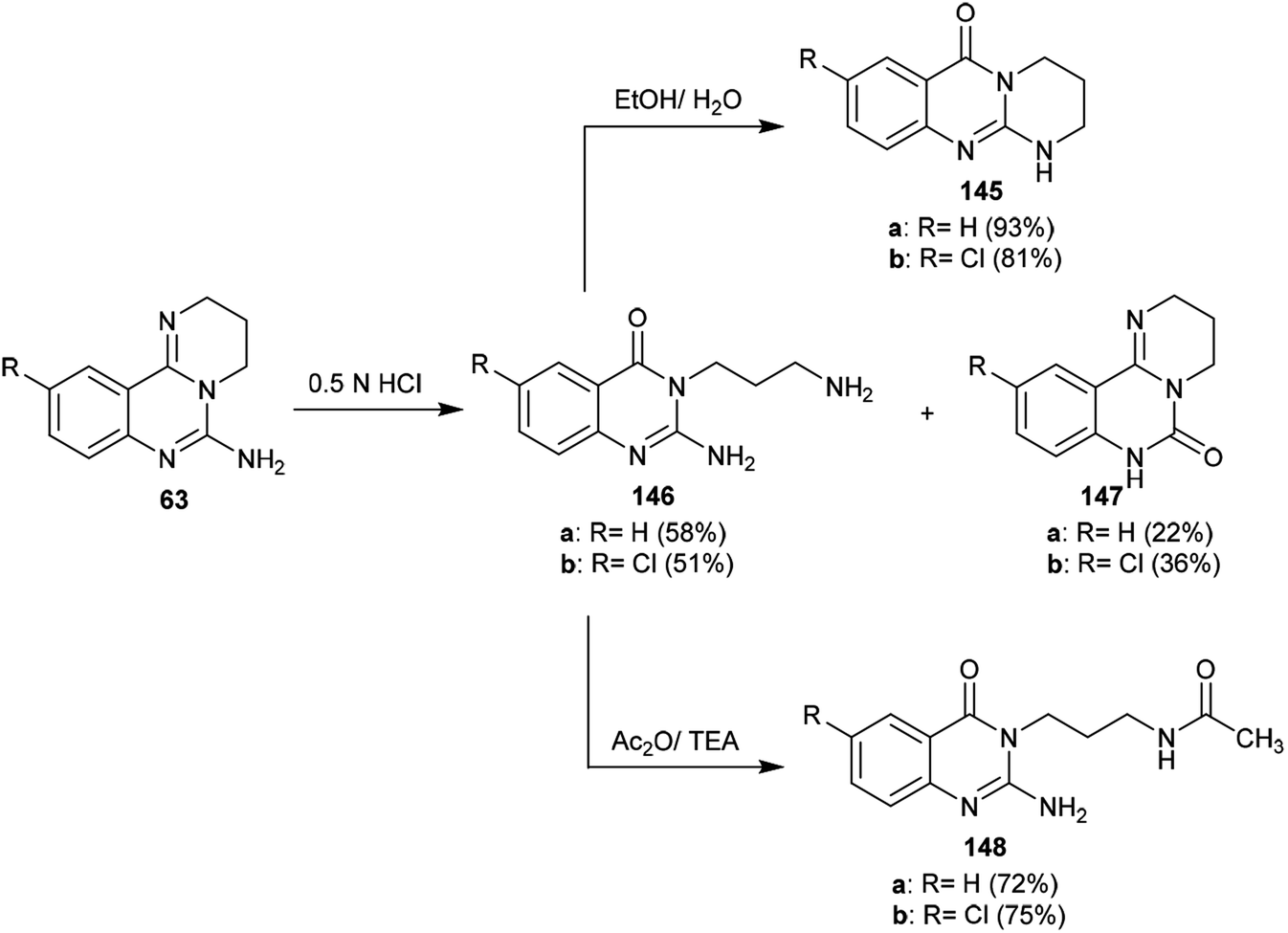
3.5. Ring transformations
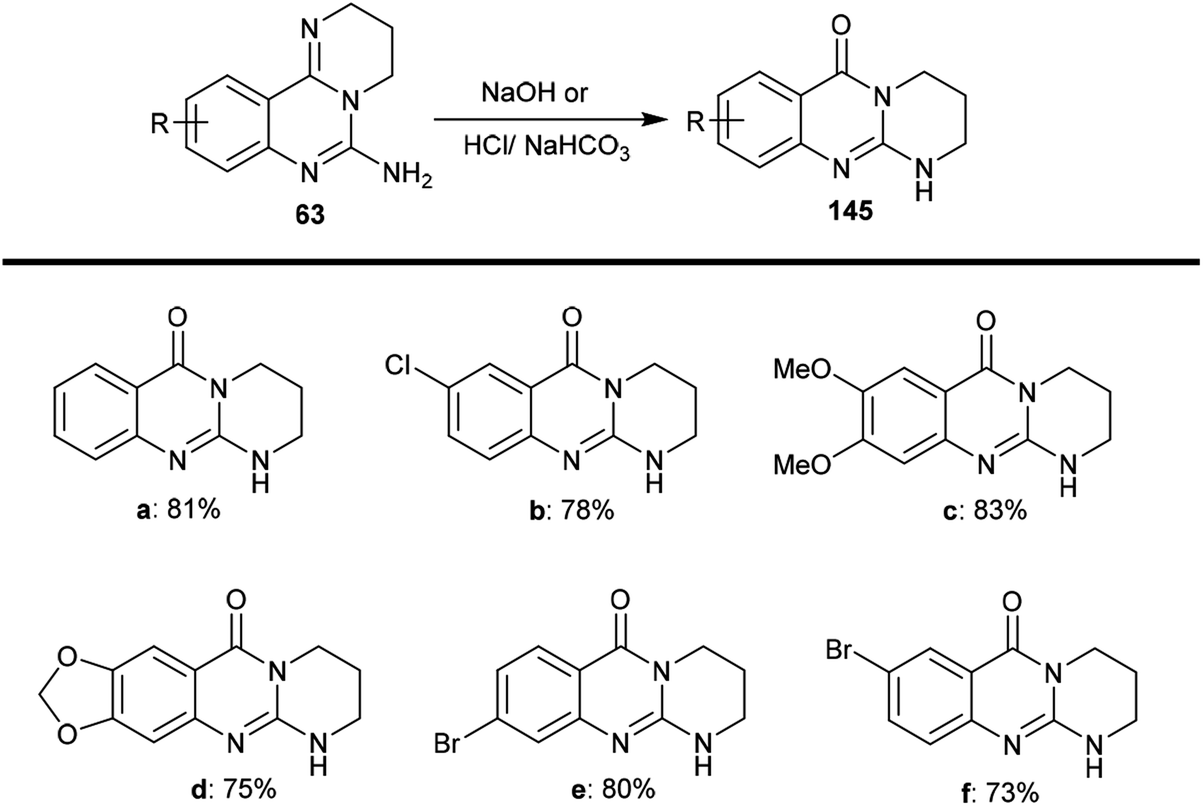
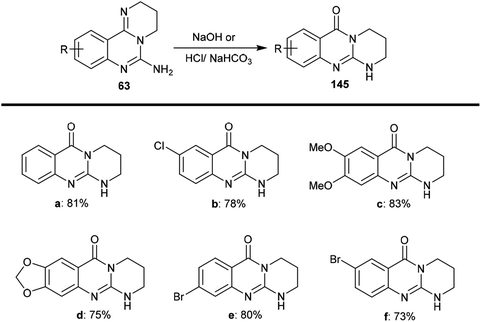
The selective acidic or basic hydrolysis of substituted-6-amino-3,4-dihydro-2H-pyrimido[1,2-c]quinazolines (63) furnished the analogue 1,2,3,4-tetrahydro-6H-pyrimido[2,1-b]quinazolin-6-ones (145). The processes involved ring-opening/ring-closing, in which the cleavage of the pyrimidine ring was accomplished in the presence of sodium hydrate and subsequent refluxing in a mixture of ethanol/water to achieve the intramolecular cyclization. Compounds 145 were obtained with excellent yields ranging from 73% to 83% (Scheme 39).64
 |
| | Scheme 39 Synthesis of pyrimidoquinazolinones. | |
The sequence of the tentative mechanistic routes of ring-opening/closure for the transformation of pyrimidoquinazolines 63 with the formation of by-products was reported. Therefore, the hydrolysis of 63 in acid medium resulted in cleavage of the pyrimidine ring with the formation of quinazolinones 146, along with the unexpected formation of pyrimidoquinazolinones 147 as side products. In fact, the intermediates which were formed after the ring-opening process were produced in the hydrochloride form, which hindered the cyclization in the next step. Neutralization of the formed hydrochloride of the anticipated aminoquinazolinone in a refluxing mixture of EtOH/H2O yielded the tricyclic pyrimido-quinazolinones 145 through simple intramolecular cyclization. Moreover, acetylation of 146 with acetic anhydride in the presence of triethylamine yielded the monoacetylated products 148 in good yields. The basic hydrolysis of the 2H-pyrimidine series provides an exceptional and selective route which is superior to acid hydrolysis (Scheme 40).64
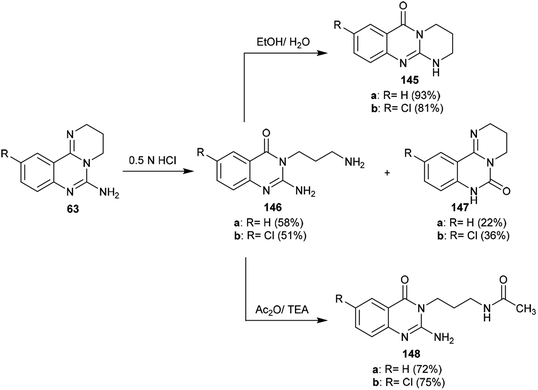 |
| | Scheme 40 Study of the ring-cleavage sequence. | |
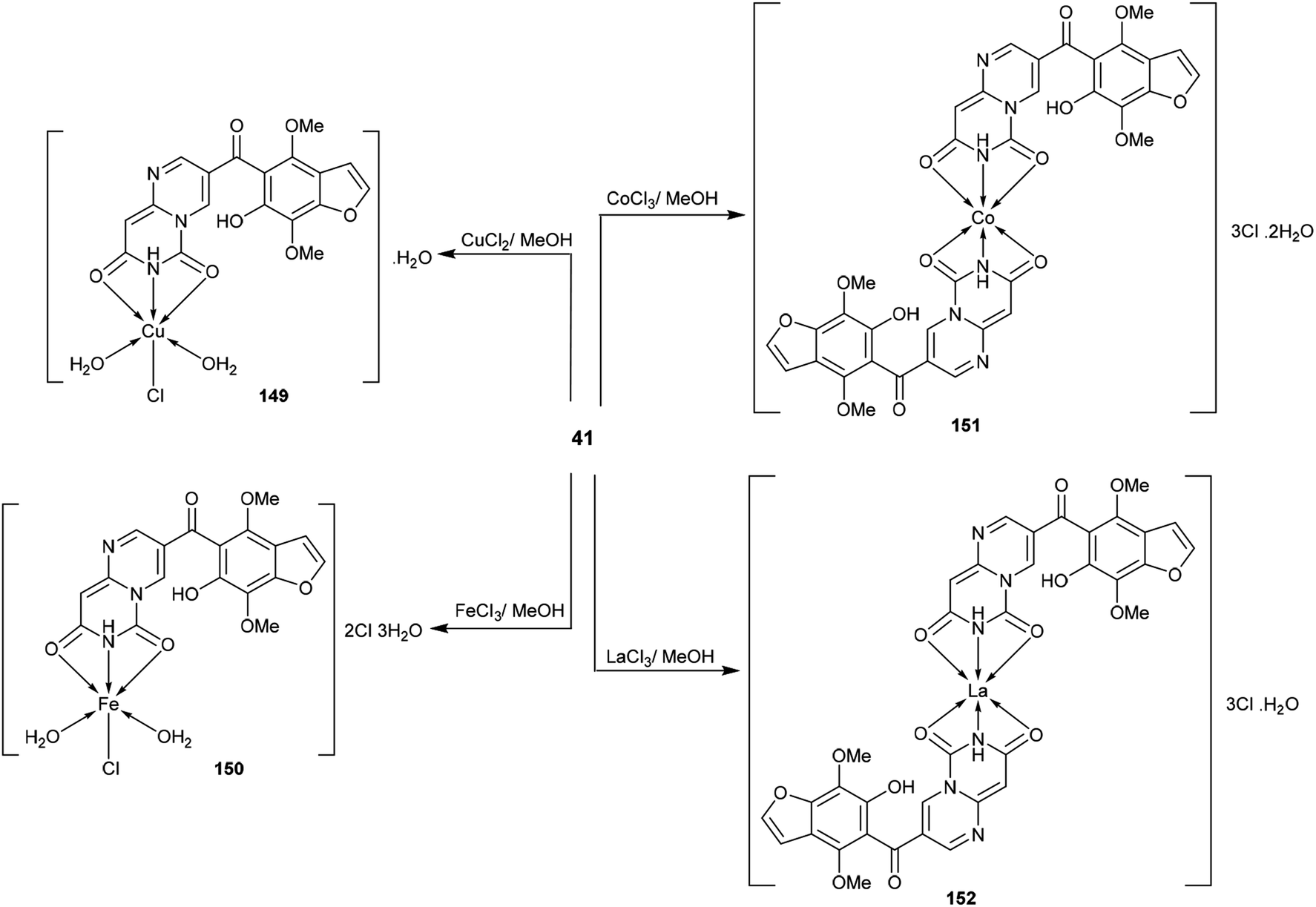
3.6. Coordination chemistry
The respective pyrimido[1,6-a]pyrimidine-dione (41) was applied as a tridentate ligand with two oxygen atoms and one nitrogen atom, which can donate electrons to the empty d-orbital of the metal to form three coordination bonds. Therefore, compound 41 tends to form 1:1 or 1:2 (metal:ligand) metal complexes 149–152 (Scheme 41)48 by reaction with metal ions such as copper chloride, ferric chloride, cobalt chloride, and lanthanum chloride in methanol at reflux temperature. The metal complexes 149–152 were synthesized by applying the method reported by Merchán et al.100
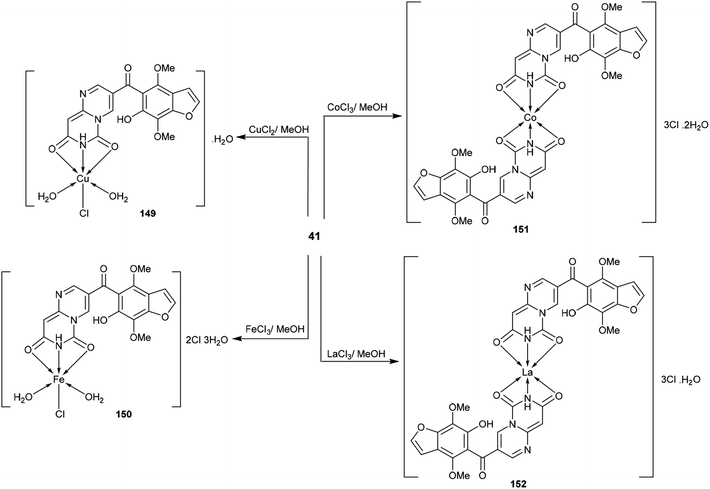 |
| | Scheme 41 Synthesis of pyrimido[1,6-a]pyrimidine-transition metal complexes. | |
4. Biological Activities
4.1. Cytotoxic Activity
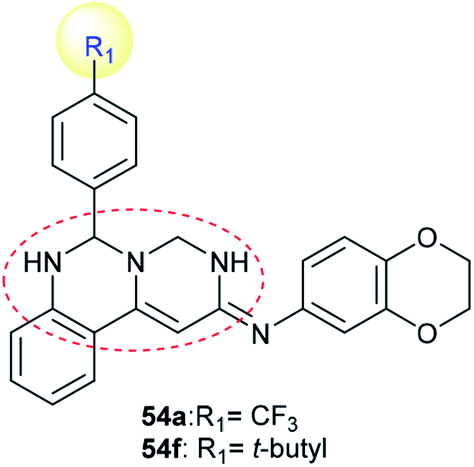
Compounds 54a and 54f (Scheme 12) were assessed as anticancer agents against ZR-75-30, HCT116, and A549 cancer cells using the MTT assay. The results revealed that both compounds displayed potent activities against the inspected cell lines. Generally, compound 54f revealed potent activity against ZR-75-30 cancer cells, with IC50 = 0.71 μM, HCT116 cancer cells, with IC50 = 1.1 μM, and A549 cancer cells, with IC50 = 2.2 μM. Pyrimido[1,6-c]quinazoline 54f was used as a guide compound for further drug development for cancer treatment. The structure–activity relationships revealed that incorporation of the pyrimido[1,6-c]quinazoline scaffold is essential for potent cytotoxic results; additionally, the incorporation of a tert-butyl substituent at C4′ of the phenyl ring at C6 of the pyrimido[1,6-c]quinazoline core enhanced the activity (Fig. 2).62
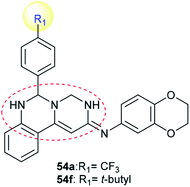 |
| | Fig. 2 Structure of potent cytotoxic agents and tyrosine kinase inhibitors. | |
Moreover, the cytotoxic activity was evaluated for pyrimido[1,6-a]pyrimidine-dione (41) and its metal complexes 149–152 (Schemes 10 and 41) against the hepatocyte cell line by the MTT-based cell viability assay. The results indicated that Cu-complex 149 displayed potent cytotoxicity, with CC50 = 93 μM relative to VX-950 (CC50 = 90 μM) against the tested cell line. In general, the formation of complexes is essential for potent cytotoxic results; Fe-complex 150, Co-complex 151, and La-complex 152 showed activities of CC50 = 330, 154, and 213 μM, respectively. Compound 5 showed the lowest cytotoxic activity, with CC50 = 341 μM. The Cu-, Fe-, Co-, and La- complexes 149–152 could reduce cell viability by 50%.48
4.2. Antimicrobial activity
The antimicrobial activity of 6-substituted-2,3-dihydro-4H-pyrimido[1,2-c]quinazolin-4-ones (70a–d) (Scheme 17) was investigated against B. cereus, B. subtilis, and E. coli as examples of Gram +ve and −ve bacterial strains with A. niger as a fungal strain and methaqualone as an antibiotic standard using the disc diffusion technique. Compounds 70a (MIC = 0.75 × 10−3 mmol mL−1) and 70b (MIC = 0.36 × 10−3 mmol mL−1) revealed moderate activities relative to the antibiotic results; however, compounds 70c (MIC = 0.79 × 10−3 mmol mL−1) and 70d (MIC value = 0.14 × 10−2 mmol mL−1) showed weak activity against B. cereus. Compound 70b showed good activity against B. subtilis and E. coli. Meanwhile, compounds 70b and 70c displayed reasonable activities against Aspergillus niger.66
The antimicrobial activity of quinazolinoquinazolinone 74b (Scheme 18) was inspected against S. aureus, B. subtilis, P. aeruginosa, E. coli, A. fumigatus, G. candidum, C. albicans, and S. racemosum. The compound revealed potent results against S. aureus and B. subtilis, with inhibition zones of 18.2 and 17.3 mm relative to the results of the standard drugs Penicillin G and Streptomycin. In addition, compound 74b showed moderate activities against the E. coli bacterial strain and fungal strains, i.e. A. fumigatus, G. candidum, and C. albicans; meanwhile, this compound was found to be inactive against P. aeruginosa and S. racemosum. The MIC (μg mL−1) was investigated for compound 74b against different bacterial and fungal strains, in which the MIC was found to be 78 μg mL−1 against the S. aureus bacterial microorganism and G. candidum fungal strain.68
The MICs of the anticipated cycloalkyl-pyrimidopyrimidinones 92a–d and pyrimido[1,6-a]pyrimidinone 28 (Schemes 7 and 23) were evaluated against S. aureus and B. subtilis as Gram-positive bacterial strains, E. coli and P. aeruginosa as Gram-negative bacterial strains, and C. albicans as a fungus. The compounds were prepared with serial dilution at concentrations of 25, 50, 100, 200, and 400 μg mL−1. Ciprofloxacin and Triflucan were used as antibiotic standards. The compounds showed moderate MIC activities against the tested microbial strains. The MIC inhibits the growth of visible colonies, by which S. aureus was found to be the most resistant microorganism. The MIC was found to be ≥ 1600 μg mL−1 against S. aureus for all the inspected compounds and ≥ 800 μg mL−1 against the other microorganisms.75
The antibacterial activities of compounds 123a,b, 130a,b, 131a,b, 132a–d, 133a,b, 134a,b, 135a–f, 136a,b, 137a,b, and 138a–d (Schemes 29 and 32–34) were evaluated against S. aureus, S. pyogenes, E. coli, and K. pneumoniae using cefotaxime sodium as antibiotic standard. Compounds 135a–f, 137a,b, and 138a–d revealed potent activities against the inspected bacterial strains. The remaining compounds displayed moderate activities. On the other hand, two series of compounds, 135a–f and 138a–d, have potent antifungal activities against A. Niger, A. Alternata, C. Lunata, and C. Albicans fungal strains, with MICs of 2–9 μmol mL−1 and 7–12 μmol mL−1, respectively. The SAR studies verified that potent antimicrobial results were achieved by the incorporation of alkyl groups such as methyl, electron-donating groups such as thioxo, hydroxyl, methoxy, amino, methylsulfonyl, and chlorine atom, heterocyclic cores such as thiazole, pyrazole, pyrimidine, and quinazoline, and cyclic amines such as piperazine and morpholine rings.97
The antibacterial activity of the desired arylidenes of 3-methyl-1H-pyrimido[6,1-b]quinazoline-1,10(4H)-diones 144a–t was evaluated using the agar cup technique against E. coli and S. typhi Gram-negative strains. The results showed that the compounds have more potent activity against E. coli than against the strain of S. typhi. In addition, compounds 144e (X = H, Y = NO2, Z = H), g (X = Cl, Y = H, Z = Cl), h (X = H, Y = Cl, Z = Cl), j (X = H, Y = NO2, Z = Cl), o (X = H, Y = NO2, Z = CH3), and t (X = H, Y = NO2, Z = OCH3) demonstrated potent activity relative to the standard antibiotic penicillin (24 mm). The antifungal activity was investigated by poison plate assays101 against A. niger and P. chrysogenum fungal strains. No growth of the fungal strains was noted for compounds 144b, c, e, g, h, j, l, m, o, q, r, or t (Scheme 38).65
4.3. Antihyperglycemic and antihyperlipidemic potentials
Antihyperglycemic and antihyperlipidemic tests were carried out for compound 92b using albino mice and pregnant female albino rats. The test was performed using six groups of eight male albino mice; one group served as a control, and the other groups were administered the inspected compound by gastric tube in regularly increasing doses (200, 400, 600, 800 and 1000 mg per kg b.w.). Accordingly, pyrimidoquinazolinone 92b (Scheme 23) was investigated as an anti-hyperglycemic, antihyperlipidemic and antioxidant agent using a sublethal dose of 10 mg per kg b.w. per day for three weeks; it was found to be a potent agent, showing good results in neonatal streptozotocin-induced (n-STZ) diabetic male and female albino rats. The potent results can be attributed to the insulinogenic action and extrapancreatic effects in addition to the improving action on the antioxidant defense system.75
4.4. Antidiabetic and antioxidant activities
The antidiabetic and antioxidant effects were assessed for pyrimidoquinazolinone 92b (Scheme 23); the results indicated that the compound displayed potent antihyperglycemic and antihyperlipidemic potentials in n5-STZ-induced type 2 diabetic male and female rats. The effects are related to the insulinogenic action and extrapancreatic properties in addition to the improving action on the antioxidant defense system. However, additional clinical research is necessary to estimate the safety and effectiveness of the investigated compound in diabetic human beings.102
4.5. Gastroprotective agents
In this type, pyrazolopyrimidopyrimidines 99a, 99b, 99f, and 99g (Scheme 24) were evaluated as gastroprotective agents against gastric ulcer induced by HCl/ethanol solution. The samples were tested at two different concentrations (50 and 100 mg kg−1). The results demonstrated that the tricyclic pyrazolo-pyrimido-pyrimidine 99f at a concentration of 100 mg kg−1 showed remarkably higher inhibition of gastric lesions (91.42%) relative to the results obtained by cimetidine (74.03% at 100 mg kg−1), which was utilized as a reference drug. The structure–activity relationships (SARs) specified that the replacement of hydrogen atom with methyl substituents is necessary for potent gastroprotective results in addition to the presence of the pyrazolopyrimidopyrimidine skeleton, which improves the biological results.79
4.6. Anticonvulsant activity
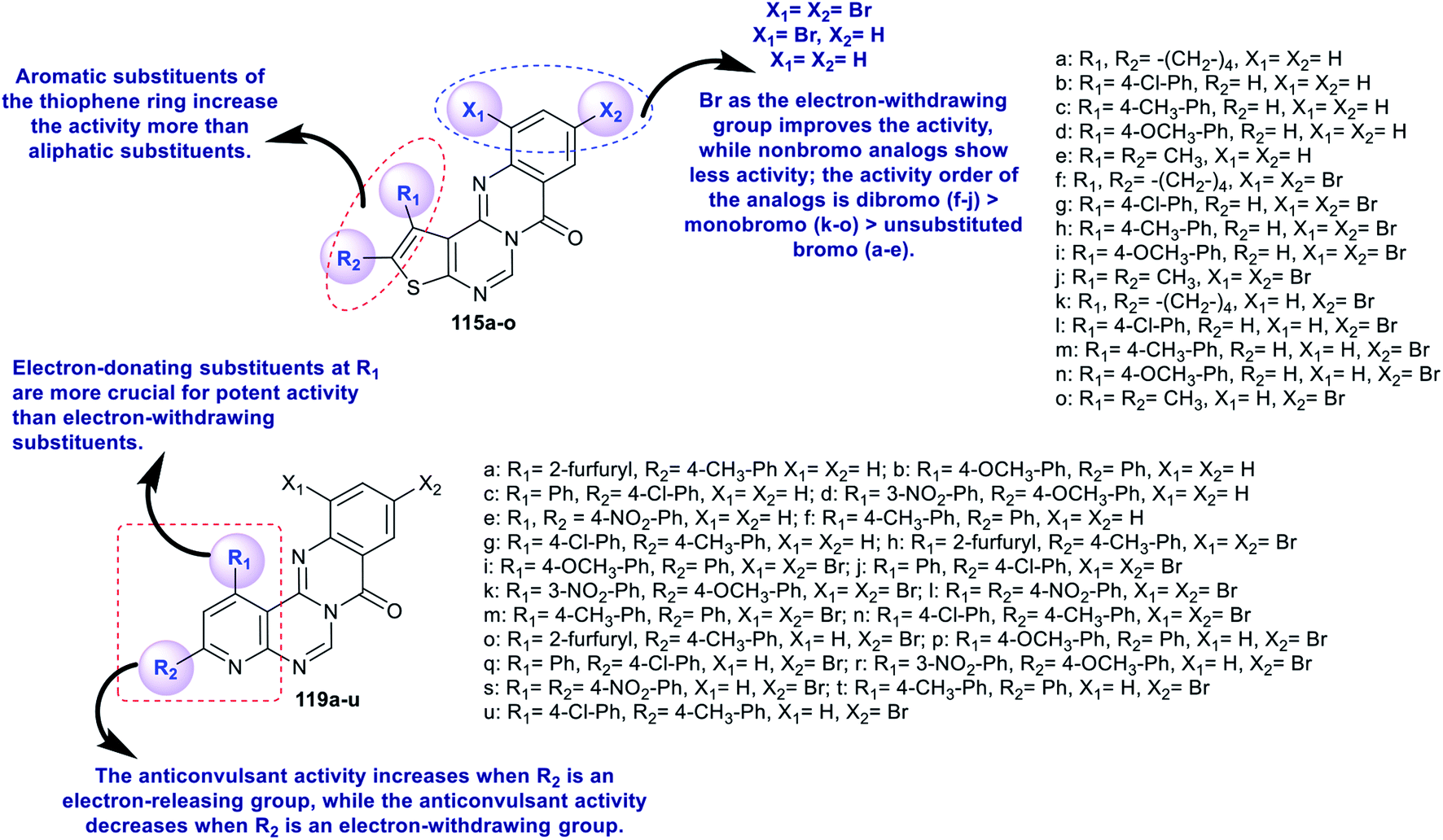
The advanced detection and progress of new chemical agents for the remediation of epilepsy is based on the use of predictable animal models, whereby MES and scPTZ-induced seizure models in mice are known as reference standards in the primary periods of testing. Two series of thienopyrimido-quinazolinones 115a–o and pyridopyrimidoquinazolinones 119a–u (Schemes 27 and 28 and Fig. 3)92,96 were assessed as anticonvulsant agents (anti-MES and anti-scPTZ). The compounds showed potent activities. The neurotoxicity was evaluated through a Rotarod procedure. All tested intraperitoneal compounds were administered in different doses ranging from 15 to 175 mg per kg b.w., medium toxic dose values (DT50) and post-summer protection. The substituents attached to the heterocyclic and aromatic benzene rings have high impacts on the biological characteristics, in which the groups responsible for the penetration across the blood–brain barrier arise from modulating the lipophilicity. The protection index (PI) values indicate the relationship between lipophilicity and toxicity. Compounds 119j and 119n revealed potent activities. More potent activity against seizures induced by scPTZ than against seizures induced by MES was noted for compounds 115a–o. Additionally, compounds 119a–u revealed greater decreases in activity against seizure induced by scPTZ than against seizure induced by MES. The structures and SARs of the anticonvulsant activity of the thieno- and pyrido-fused pyrimidoquinazolinones 115a–o and 119a–u are specified in Fig. 3.103,104
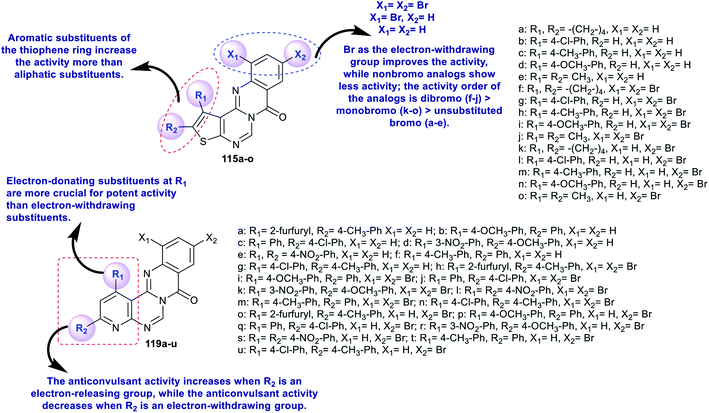 |
| | Fig. 3 Structures and SARs of the potent anticonvulsant agents. | |
4.7. Anti-inflammatory activity
The respective pyrazolopyrimidopyrimidines 99a, 99b, 99f, and 99g (Scheme 24) were evaluated as anti-inflammatory agents using carrageenan-induced rat paw edema assessments. The results presented that the most potent analogs are compounds 99a, 99b, and 99f (50-100 mg kg−1, i.p.) relative to the results of the reference drug, acetylsalicylic-lysine (300 mg kg−1, i.p.). The most potent anti-inflammatory analog is 99f. The percent inhibition for compound 99f ranged from 60.02% to 82.83% three hours after injection of the carrageenan and reduction of edema. The nature of the substituents of pyrazolopyrimido-pyrimidines affected the obtained results of the anti-inflammatory activities of the inspected compounds. Consequently, replacement of the hydrogen atom at position 5 with methyl or ethyl groups and replacement of the nitrile group with an ester function is essential for potent anti-inflammatory activity.79
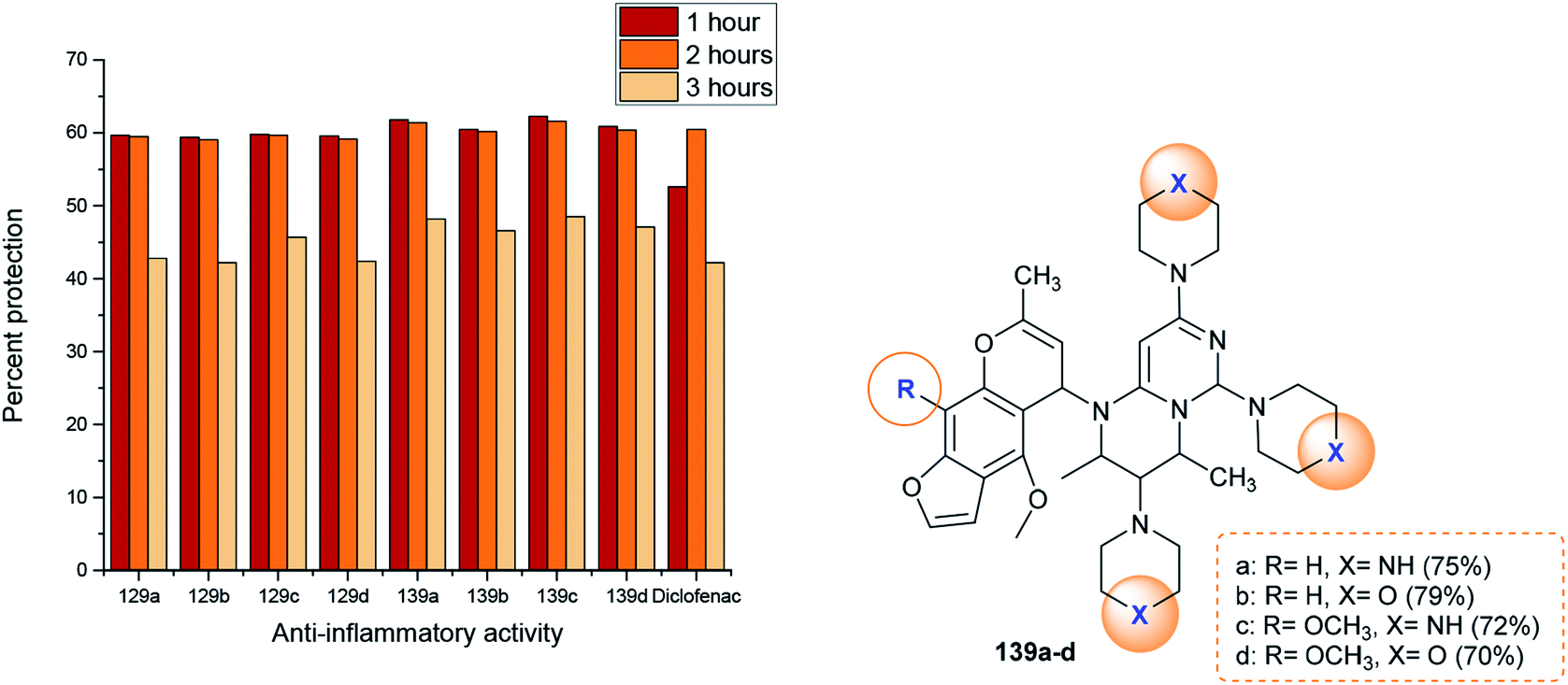
The anti-inflammatory activities of pyrimidopyrimidines 129 and 139a–d (Scheme 36 and Fig. 4) were evaluated by a carrageenan-induced paw edema assay using diclofenac sodium as a standard drug. The results obviously indicated that most of the compounds have potent activities relative to those obtained by diclofenac sodium after 1–3 hours. Compounds 129 and 139a–d inhibited carrageenan-induced paw edema at 59.4–62.3% (after 1 hour), 59.1–61.6% (after 2 hours), and 42.2–48.5% (after 3 hours). As a result, pyrimido[1,6-a]pyrimidines 139a–d are generally more potent than compounds 129a–d. The substitution of a chlorine atom at C3 of pyrimido[1,6-a]pyrimidines 129a–d with cyclic amines is crucial for potent anti-inflammatory activity. In addition, the order of the activities of compounds 139a–d was noted to be 139c > 139a > 139d > 139b. Therefore, the presence of methoxy substituents (compound 139c) instead of unsubstituted analogs is necessary for potent anti-inflammatory activity. Moreover, piperazine substituents are better than morpholine substituents for potent activity.98
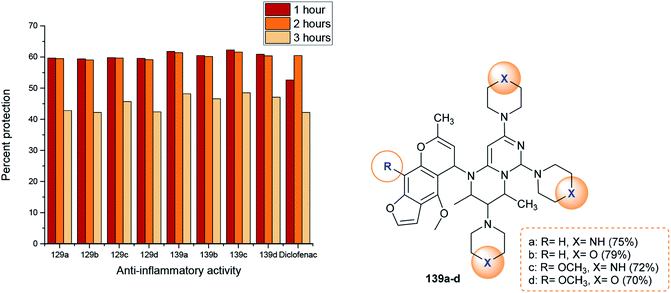 |
| | Fig. 4 Comparison of the anti-inflammatory activities of 2H-pyrimido[1,6-a]pyrimidines. | |
4.8. Analgesic activity
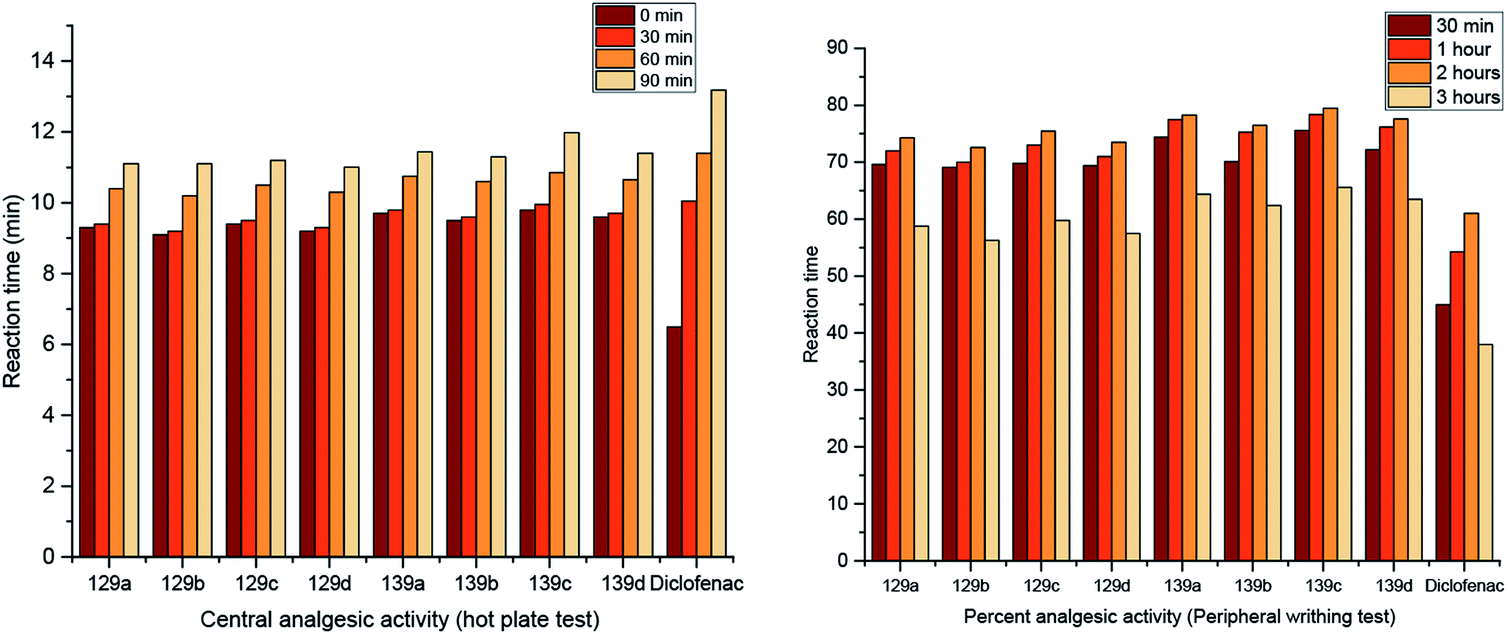
Two series of pyrimidopyrimidines, 129 and 139a–d (Scheme 36 and Fig. 5), were evaluated as analgesic agents by the hot-plate (central analgesic activity) and acetic acid-induced writhing tests. The compounds showed potent activities using both methods compared to the results obtained for diclofenac sodium. Hence, pyrimidopyrimidine 139c displayed higher analgesic activity than diclofenac sodium. The SARs indicated that the incorporation of methoxy substituents at C9 of the furochromenyl core is essential for potent activity. The efficiency of the tested analgesic agents increases with time and subsequently decreases after three hours. Frequently, pyrimidopyrimidines 139a–d are more potent agents than pyrimidopyrimidines 129a–d. These results can be explained by the action of the chemical structures. Accordingly, the substitution of the chlorine atom at C3 with the piperazine ring of the pyrimidopyrimidine core is essential for potent results of analgesic activity.98
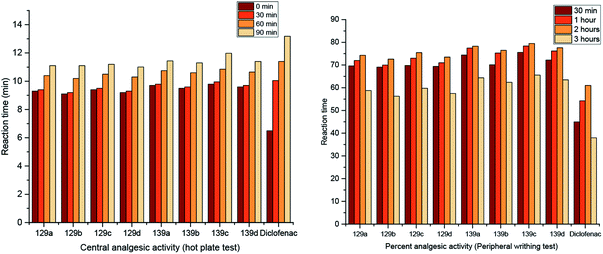 |
| | Fig. 5 Comparison of the analgesic activity results using hot plate and peripheral writhing tests. | |
4.9. Antiviral activity
The pyrimidopyrimidine-dione 41 and its metal complexes 149–152 (Schemes 10 and 41) were evaluated as HIV agents using Atevirdine as an antibiotic standard. The compounds displayed good activities relative to the antibiotic standard. Compound 41 presented the most potent activity, with IC50 = 40.31 μM; meanwhile the metal complexes 149–152 revealed IC50 values ranging from 78.67 to 96.18 μM. The tested compounds generally indicated respectable HIV-1 RT inhibitory activity but showed lower activities compared to Atevirdine. On the other hand, compound 41 and its metal complexes 149–152 presented potent HCV NS3-4A protease inhibitory activities, with IC50 = 0.388, 0.445, 0.987, 0.456, and 0.765 μM, respectively, relative to the results of the standard VX-950 (IC50 = 0.20 μM). The complexation step generally improves the activity against HCV NS3-4A protease; however, in this case, the complexation step of compound 41 decreases the activities of the formed metal complexes to a lower degree.48
4.10. Kinase inhibitory activity
Pyrimido[1,6-c]quinazoline 54f (Scheme 12) was evaluated as an inhibitor for protein tyrosine kinases such as FLT3, INSR, and VEGFR-2, with potent results. The IC50 values ranged from 0.94 to 0.86 and 1.0 μM against FLT3, INSR and VEGFR-2 protein tyrosine kinases, respectively. In addition, pyrimido[1,6-c]quinazoline 54a showed no activity against any of the tested kinases.62 Also, INSR and VEGFR-2 are closely related to breast cancer.105,106
4.11. Affinity for ET(A) receptors
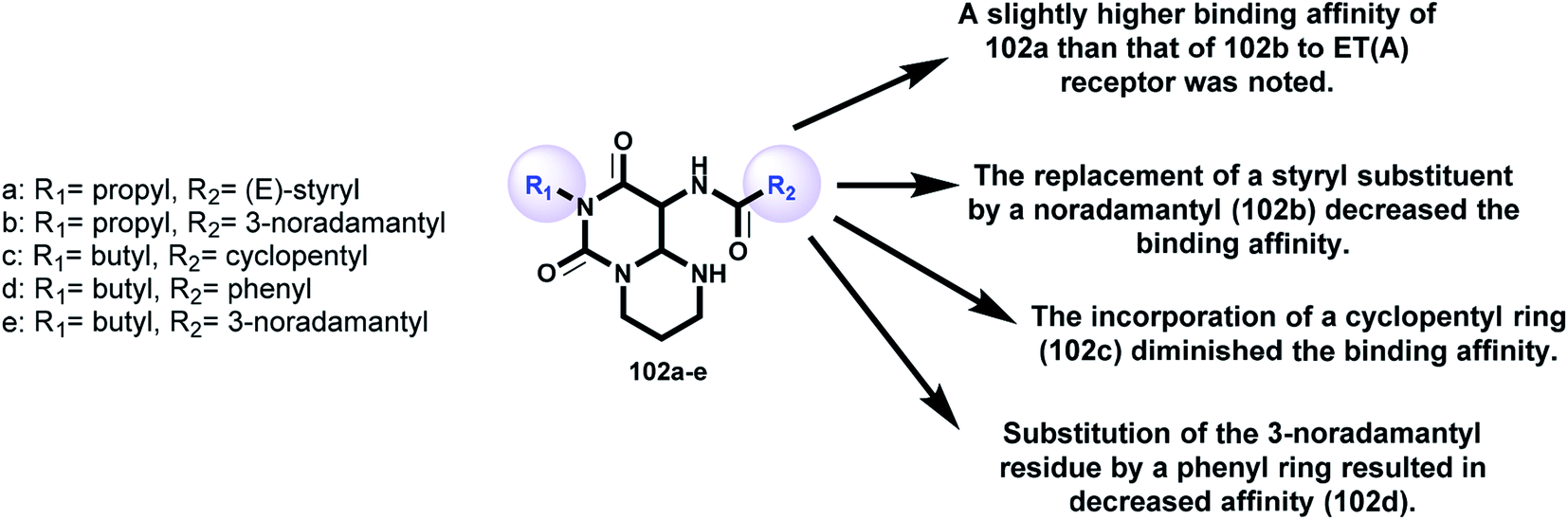
Griessmeier and Müller107 demonstrated the affinities of a series of substituted 6H-pyrimidopyrimidine-diones 102 (Scheme 25 and Fig. 6) as receptors for endothelin ET(A)-selective antagonists against human astrocytoma 1321N1 cell lines by applying [3H]BQ-123. The affinity was found to be moderate, with [3H]BQ-123 binding inhibition in the range of 30–40% at a definite concentration. The radioligand binding inhibition was determined to be 10 μmol l−1 [3H]BQ-123. The inhibition of radioligand binding indicated that compound 102a displayed moderate affinity to ET(A) receptors in human astrocytoma 1321N1 cells, with values of 33% for 102a, 20% for 102b, 2% for 102c, 14% for 102d, and 35% for 102e (Fig. 6) against the human astrocytoma 1321N1 cell line. The expression level of ET(A) receptors for the prepared membranes of the cell line was found to be high (Bmax = 128 fmol mg−1 protein). The K(D) value for [3H]BQ-123 (KD = 2.29 nmol l−1) was equivalent to that previously investigated in the human neuroblastoma SK-N-MC cell line. The rank order of a series of agonists and antagonists clarified the individual labeling of ET(A) receptors on the studied cell line “astrocytoma 1321N1” by [3H]BQ-123. Astrocytoma 1321N1 cells are useful for screening potential ET(A) ligands in radioligand binding assessments with [3H]BQ-123; these cells express high levels of ET(A) receptors.
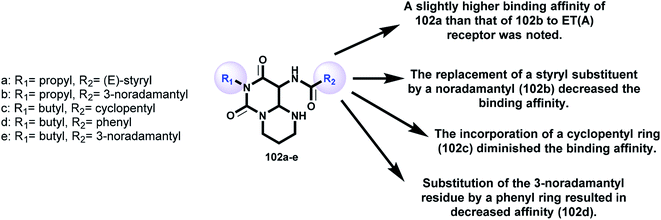 |
| | Fig. 6 SARs of pyrimidopyrimidine-diones as bioactive molecules. | |
5. Concluding remarks
The present survey provides a description of the chemistry and biological importance of heterocycles incorporating pyrimido[1,6-a]pyrimidine and pyrimido[1,6-c]pyrimidine scaffolds. The pyrimido-pyrimidines of this class demonstrated valuable and diverse biological properties. The investigated compounds were synthesized through the formation of three C–N bonds by the reaction of acyclic reactants such as 1,3-diamine with 4-isothiocyanato-4-methylpentan-2-one. In another route, one-pot multicomponent reactions of aminopyrimidines, aldehydes, and β-ketoesters yielded the substituted analogs. Pyrimidopyrimidines were also synthesized from the reaction of aminopyrimidines with unsaturated esters, unsaturated ketones or Meldrum's acid. In addition, pyrimidine-2,4(1H,3H)-diones were synthesized from uracil, and 4,6-dichloropyrimidine served as a reactive synthon for the synthesis of pyrimido[1,6-c]quinazolines. Also, the chlorination of 6-bromo-2-substituted-quinazolinones and subsequent reaction with 2-amino-5-bromobenzoic acid yielded the respective quinazolino-quinazolinones. Cyclization of the respective 4-(3-hydroxy-propyl-amino)-quinazolines in hydrochloric acid produced the quaternary salts. In another route, pyrimidopurine-diones were prepared from 5,6-diamino-3-substituted-pyrimidine-diones by amidation or condensation with acids and cyclization with 1,3-dibromopropane. Moreover, tri-, tetracyclic, and binary heterocycles were synthesized. These studies reported the reactivity of the substituents, nucleophilic substitution reactions, condensations, ring transformations, and complex formation. The different synthetic methods investigated for the construction of the pyrimidopyrimidine ring provide various heterocycles that are useful in different areas of chemistry, such as drug design. Consequently, the compounds are potent cytotoxic, antimicrobial, antihyperglycemic, antihyperlipidemic, antidiabetic, antioxidant, gastroprotective, anticonvulsant, anti-inflammatory, analgesic, and antiviral agents and act as inhibitors of protein tyrosine kinases. Preferred structures can bind to several high-affinity targets, affording novel biologically active agents. Thus, pyrimido[1,6-a]pyrimidines and pyrimido[1,6-c]pyrimidines represent substantial characteristic scaffolds that possess a wide range of biological characteristics.
6. Literature overview
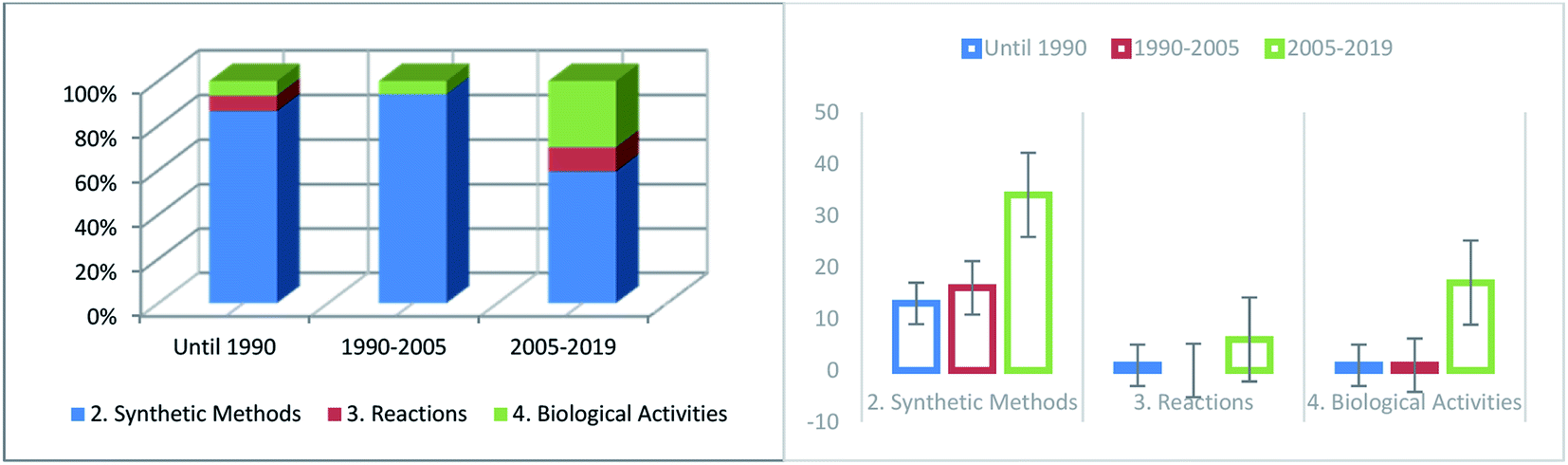
Since Yani and coworkers24 reported the synthesis of 2,3,4,7-tetrahydro-6H-pyrimido[1,6-a]pyrimidin-6-ones, several scientific studies have been published regarding the synthesis of this category of heterocyclic compounds. Several surveys have demonstrated the frequency with which published work relating to the diverse sections of this paper has increased throughout the preceding period (Fig. 7). During this period (before 1990), much interest was aroused in the preparation of compounds with the same biological behavior as pyrimidopyrimidine isomers [4,5-d] and [5,4-d]. The numbers of synthetic methods (Section 2) and reactions (Section 3) have remarkably increased in recent years owing to the significant and valuable biological activities of these compounds. The biological applications (Section 4) have increased in recent years, although several types of biological characteristics have not been yet applied.
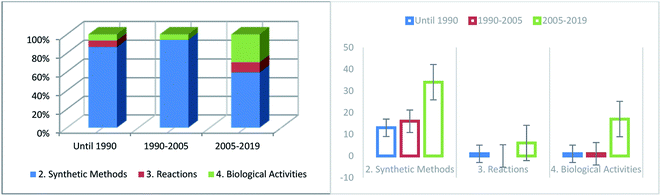 |
| | Fig. 7 Comparison of the number of reports related to the different sections of this paper in the last decade. | |
7. Future perspectives
The diverse biological activities which have been attributed to pyrimidopyrimidines and pyridopyrimidine analogs should be applied to this class of compounds depending on the nature of the substituents attached to the basic ring skeleton. Therefore, the pyrimido[1,6-a]pyrimidine-6,8(7H)-dione moiety provides a possible ligand for the formation of complexes, which are potent cytotoxic agents; further evaluation of their cytotoxic and antiviral activities should be a research focus. Compounds incorporating the pyrimido[1,2-c]quinazolinone scaffold are potent antimicrobial agents; this provides further areas of study for the synthesis of a series of these compounds with the same potency. The cycloalkyl[e]pyrimidopyrimidinone compounds have potent antihyperglycemic, antihyperlipidemic, antidiabetic, and antioxidant activities; the tricyclic system of pyrimidopyrimidine fused with a cycloalkane ring provides efficient bioactive components. Furthermore, tricyclic pyrazolo-pyrimido[1,2-c]pyrimidin-4(8H)-imines display potent gastroprotective activities. In addition, thieno-pyrimidoquinazolinones 115a–o and pyrido-pyrimidoquinazolinones 119a–u are potent anticonvulsant agents; this provides a route to prepare biologically active compounds with diverse activities and to achieve potent results. The attachment of cyclic amines and binary heterocycles to pyrimido[1,6-a]pyrimidine skeletons is an efficient synthetic route to prepare compounds with anti-inflammatory and analgesic activities. The compounds of pyrimidopyrimidines and pyridopyrimidines are potent inhibitors of protein kinase; therefore, 2-imino-pyrimidoquinazolines are potent inhibitors of protein tyrosine kinases.
Abbreviations
| EeAChE | Electrophorus electricus acetylcholinesterase |
| hAChE | Human acetyl-cholinesterase |
| hBChE | Human butyryl-cholinesterase |
| CF3SO3H | Trifluoromethanesulfonic acid |
| equiv. | Equivalent |
| DOWTHERM™A | A eutectic mixture of two very stable compounds |
| Pd(OH)2 | Palladium hydroxide |
| K2CO3 | Potassium carbonate |
| DMF | N,N-Dimethylformamide |
| POCl3 | Phosphorus oxychloride |
| TEA | Triethylamine |
| Boc2O | Boc anhydride or di-tert-butyl pyrocarbonate |
| NaBH4 | Sodium borohydride |
| SOCl2 | Thionyl chloride |
| HC(OEt)3 | Triethyl orthoformate |
| HCOOH | Formic acid |
| PPA | Polyphosphoric acid |
| MW | Microwave |
| Zn | Zinc |
| NaH | Sodium hydride |
| THF | Tetrahydrofuran |
| MeI | Methyl iodide |
| ZR-75-30 | Human breast cancer cells |
| HCT116 | Human cancer colon cells |
| A549 | Human lung cancer cells |
| IC50 | Inhibitive concentration at 50% of the original concentration |
| MIC | Minimum inhibitory concentration |
| EDC | N′-(3-Dimethyl-aminopropyl)-N-ethylcarbodiimide-hydrochloride |
| HMDS | 1,1,1,3,3,3-Hexamethyldisilazane |
| MES | Maximum electroshock-induced seizures |
| scPTZ | Subcutaneous pentylenetetrazole |
| PI | Protection index |
Gram-positive bacteria
| S. aureus | Staphylococcus aureus |
| B. subtilis | Bacillus subtilis |
| B. cereus | Bacillus cereus |
| S. pyogenes | Streptococcus pyogenes |
Gram-negative bacteria
| P. aeruginosa | Pseudomonas aeruginosa |
| E. coli | Escherichia coli |
| K. pneumoniae | Klebsiella pneumoniae |
| S. typhi | Salmonella typhi |
Fungal strains
| A. fumigatus | Aspergillus fumigatus |
| A. niger | Aspergillus niger |
| G. candidum | Geotrichum candidum |
| C. albicans | Candida albicans |
| S. racemosum | Syncephalastrum racemosum |
| A. alternata | Alternaria alternata |
| C. lunata | Curvularia lunata |
| P. chrysogenum | Penicillium chrysogenum |
| CC50 | 50% cytotoxic concentration |
Conflicts of interest
The authors declare no conflict of interest.
References
- R. Abdel-Rahman and K. El-Mahdy, Heterocycles, 2012, 85(10), 2391–2414, DOI:10.3987/REV-12-745.
- F. F. Solca, A. Baum, E. Langkopf, G. Dahmann, K.-H. Heider, F. Himmelsbach and J. C. A. van Meel, J. Pharmacol. Exp. Ther., 2004, 311, 502–509, DOI:10.1124/jpet.104.069138.
- J. P. De la Cruz, T. Carrasco, G. Ortega and F. S. De la Cuesta, Lipids, 1992, 27(3), 192–194 CrossRef CAS PubMed.
- M. G. Gebauer, C. Mc Kinlay and J. E. Gready, Eur. J. Med. Chem., 2003, 38(7–8), 719–728 CrossRef CAS PubMed.
- Y. Fang, J. Xu, Z. Li, Z. Yang, L. Xiong, Y. Jin, Q. Wang, S. Xie, W. Zhu and S. Chang, Bioorg. Med. Chem., 2018, 26(14), 4080–4087 CrossRef CAS PubMed.
- M. Simcox, B. Higgins, L. McDermott, T. Nevins, K. Kolinsky, M. Smith, H. Yang, J. Li, Y. Chen and K. Luk, Eur. J. Cancer Suppl., 2004, 2(8), 59 CrossRef.
- H. C. Barlow, K. J. Bowman, N. J. Curtin, A. H. Calvert, B. T. Golding, B. Huang, P. J. Loughlin, D. R. Newell, P. G. Smith and R. J. Griffin, Bioorg. Med. Chem. Lett., 2000, 10(6), 585–589 CrossRef CAS PubMed.
- A. H. Bacelar, M. A. Carvalho and M. F. Proença, Eur. J. Med. Chem., 2010, 45(7), 3234–3239 CrossRef CAS PubMed.
- P. Raddatz and R. Bergmann, Ger. Pat. 360731, 1988Chem. Abstr. 109, 54786.
- N. Kitamura and A. Onishi, Eur. Pat. 163599, 1984Chem. Abstr. 104, 186439.
-
(a) P. Sharma, N. Rane and P. Pandey, Arch. Pharm., 2006, 339(10), 572–578, DOI:10.1002/ardp.200600067;
(b) P. Sharma, N. Rane and V. K. Gurram, Bioorg. Med. Chem. Lett., 2004, 14, 4185–4190, DOI:10.1016/j.bmcl.2004.06.014.
-
(a) S. P. Iadonato, K. Bedard, M. W. Imanaka and K. W. Fowler, WO 2013049352 A2 20130404. PCT Int. Appl. 2013;
(b) A. C. Krueger, D. L. Madigan, D. W. Beno, D. A. Betebenner, R. Carrick, B. E. Green, W. He, D. Liu, C. J. Maring, K. F. McDaniel, H. Mo, A. Molla, C. E. Motter, T. J. Pilot-Matias, M. D. Tufano and D. J. Kempf, Bioorg. Med. Chem. Lett., 2012, 22, 2212–2215, DOI:10.1016/j.bmcl.2012.01.096.
-
(a) Y. Tong, T. D. Penning, A. S. Florjancic, J. Miyashiro and K. W. Woods, US. Pat. Appl. 0220572, 2012;
(b) S. M. El-Moghazy, D. A. Ibrahim, N. M. Abdelgawad, N. A. H. Farag and A. S. El-Khouly, Sci. Pharm., 2011, 79(3), 429–447 CrossRef CAS PubMed;
(c) T. J. Delia, M. Baumann and A. Bunker, Heterocycles, 1993, 35, 1397–1410 CrossRef CAS.
- J. R. Doherty, C. Yang, K. Scott, M. D. Cameron, M. Fallahi, W. Li, M. A. Hall, A. L. Amelio, J. K. Mishra, F. Li, M. Tortosa, H. M. Genau, R. J. Rounbehler, L. Yungi, C. V. Dang, K. G. Kumar, A. A. Butler, T. D. Bannister, A. T. Hooper, K. Unsal-Kacmaz, W. R. Roush and J. L. Cleveland, Cancer Res., 2014, 74(3), 908–920, DOI:10.1158/0008-5472.
- H. Wang, C. Yang, J. R. Doherty, W. R. Roush, J. L. Cleveland and T. D. Bannister, J. Med. Chem., 2014, 57, 7317–7324, DOI:10.1021/jm500640x.
- M. W. Martin, J. Newcomb, J. J. Nunes, C. Boucher, L. Chai, L. F. Epstein, T. Faust, S. Flores, P. Gallant, A. Gore, Y. Gu, F. Hsieh, X. Huang, J. L. Kim, S. Middleton, K. Morgenstern, A. Oliveirados-Santos, V. F. Patel, D. Powers, P. Rose, Y. Tudor, S. M. Turci, A. A. Welcher, D. Zack, H. L. Zhao, L. Zhu, X. T. Zhu, C. Ghiron, M. Ermann and D. Johnston, J. Med. Chem., 2008, 51(6), 1637–1648 CrossRef CAS PubMed.
- V. J. Ram, A. Goel, S. Sarkhel and P. R. Maulik, Bioorg. Med. Chem., 2002, 10, 1275–1280 CrossRef CAS PubMed.
- G. W. Rewcastle, A. J. Bridges, D. W. Fry, J. R. Rubin and W. A. Denny, J. Med. Chem., 1997, 40(12), 1820–1826 CrossRef CAS PubMed.
- S. N. Sirakanyan, D. Spinelli, A. Geronikaki, E. K. Hakobyan, H. Sahakyan, E. Arabyan, H. Zakaryan, L. E. Nersesyan, A. S. Aharonyan, I. S. Danielyan, R. E. Muradyan and A. A. Hovakimyan, Molecules, 2019, 24(21), E3952, DOI:10.3390/molecules24213952.
- D. S. Rina, M. S. Nirmal and C. R. Vivek, Asian J. Appl. Chem. Res., 2018, 1(1), 1–9 Search PubMed.
- C. M. G. Tanarro and M. Gutschow, J. Enzyme Inhib. Med. Chem., 2011, 26(3), 350–358, DOI:10.3109/14756366.2010.504674.
- F. Dennin, D. Blondeau and H. Sliwa, J. Heterocycl. Chem., 1990, 27(7), 1963–1967, DOI:10.1002/jhet.5570270721.
- A. Vasudevan, F. Mavandadi, L. Chen and A. Gangjee, J. Org. Chem., 1999, 64, 634–638, DOI:10.1021/jo9713870.
- M. Yani, S. Takeda, T. Baba and K. Kitagawa, Yakugaku Zasshi, 1974, 94(12), 1503–1514, DOI:10.1248/yakushi1947.94.12_1503.
- I. Zh. Lulle, R. A. Paégle, I. B. Mazheika and M. Yu. Lidak, Chem. Heterocycl. Compd., 1983, 19(4), 439–446, DOI:10.1007/BF00516219.
- B. Stanovnik, H. V. De Bovenkamp, J. Svete, A. Hvala, I. Simonič and M. Tišler, J. Heterocycl. Chem., 1990, 27(2), 359–361 CrossRef CAS.
- I. E. Kuznetsov, D. K. Susarova, L. A. Frolova, A. S. Peregudov, A. F. Shestakov, S. I. Troyanov, K. J. Stevenson and P. A. Troshin, Chem. Commun., 2017, 53, 4830–4833, 10.1039/c6cc10179h.
- C. Chapuis, H. Hagemann, W. Fieber, R. Brauchli and J.-Y. de Saint Laumer, J. Phys. Org. Chem., 2009, 22, 282–288, DOI:10.1002/poc.1465.
-
(a) L. Vasvári-debreczy, I. Hermecz and P. Mátyus, Bicyclic 6-6 Systems with One Ring Junction Nitrogen Atom: Two Extra Heteroatoms 1:1, Compr. Heterocycl. Chem. II, 1996, 8, 633–706, DOI:10.1016/B978-008096518-5.00192-1;
(b) I. Hermecz and L. Vasvári-Debreczy, Bicyclic 6-6 Systems with One Bridgehead (Ring Junction) Nitrogen Atom: Two Extra Heteroatoms 1:1, Compr. Heterocycl. Chem. II, 2008, 12, 257–320, DOI:10.1016/B978-008044992-0.01104-4.
- M. Monier, D. Abdel-Latif, A. El-Mekabaty and K. M. Elattar, Mini-Rev. Org. Chem., 2019, 16, 1–23, DOI:10.2174/1389557519666190925161145.
- M. Monier, D. Abdel-Latif, A. El-Mekabaty and K. M. Elattar, RSC Adv., 2019, 9, 30835–30867, 10.1039/C9RA05687D.
- D. D. Ivy, J. P. Kinsella, J. W. Ziegler and S. H. Abman, J. Thorac. Cardiovasc. Surg., 1998, 115(4), 875–882 CrossRef CAS PubMed.
- K. M. Elattar and B. D. Mert, RSC Adv., 2016, 6, 71827–71851, 10.1039/C6RA12364C.
- M. Monier, D. Abdel-Latif, A. El-Mekabaty, B. D. Mert and K. M. Elattar, Curr. Org. Synth., 2019, 16(6), 812–854, DOI:10.2174/1570179416666190704113647.
- K. M. Elattar, R. Rabie and M. M. Hammouda, Synth. Commun., 2016, 46, 1477–1498 CrossRef CAS.
- K. M. Elattar, R. Rabie and M. M. Hammouda, Monatsh. Chem., 2017, 148, 601–627, DOI:10.1007/s00706-016-1852-1.
- A. A. Fadda, S. A. El-Hadidy and K. M. Elattar, Synth. Commun., 2015, 45(24), 2765–2801, DOI:10.1080/00397911.2015.1089577.
- G. Zigeuner, W.-B. Lintschinger, A. Fuchsgruber and K. Kollmann, Monatsh. Chem., 1976, 107, 171–181 CrossRef CAS.
- A. Alizadeh, J. Mokhtari and M. Ahmadi, Tetrahedron, 2012, 68, 319–322 CrossRef CAS.
- J. Xiang, H. Li, K. Yang, L. Yi, Y. Xu, Q. Dang and X. Bai, Mol. Diversity, 2012, 16, 173–181, DOI:10.1007/s11030-011-9345-y.
- A. M. Hussein, J. Heterocycl. Chem., 2012, 49(2), 446–451 CrossRef CAS.
- F. Delnnin, D. Bulndeau and H. Sliwa, Tetrahedron Lett., 1989, 30(12), 1529–1530 CrossRef.
- H. M. Hassneen and T. A. Abdallah, Molecules, 2003, 8(3), 333–341, DOI:10.3390/80300333.
- M. H. Elnagdi, E. A. Abdel-All and G. E. H. Elgemeie, Heterocycles, 1985, 23(12), 3121–3153, DOI:10.3987/R-1985-12-3121.
- S. M. Sherief and A. M. Hussein, Monatsh. Chem., 1997, 128(6–7), 687–696, DOI:10.1007/BF00807600.
- K. C. Joshi, Y. N. Pathak and U. Grage, J. Heterocycl. Chem., 1979, 16(6), 1141–1145, DOI:10.1002/jhet.5570160611.
- I. H. El Azab and K. M. Khaled, Russ. J. Bioorg. Chem., 2015, 41(4), 421–436 CrossRef CAS.
- S. A. Galal, A. S. Abd El-All, K. H. Hegab, A. A. Magd-El-Din, N. S. Youssef and H. I. El-Diwani, Eur. J. Med. Chem., 2010, 45, 3035–3046 CrossRef CAS PubMed.
- P. Kaure, R. Kaur and K. Kaur, J. Global Pharma Technol., 2009, 2(4), 35–39 Search PubMed.
-
(a) M. A. Al-Omar, A. S. El-Azab, S. G. Abdel Hamide and H. A. El-Obeid, J. Saudi Chem. Soc., 2005, 10, 113–128 Search PubMed;
(b) M. A. Al-Fayez, A. M. Aleisa and M. A. Al-Omar, J. Biol. Sci., 2007, 7, 532–538, DOI:10.3923/jbs.2007.532.538.
- F. M. Refaie, A. Y. Esmat, S. M. A. Gawad, A. M. Ibrahim and M. A. Mohamed, Lipids Health Dis., 2005, 4, 22, DOI:10.1186/1476-511X-4-22.
- D. Murugesan, S. Periyaswamy, D. Erik and K. S. Seshaiah, Biol. Pharm. Bull., 2003, 26(9), 1278–1282 CrossRef PubMed.
- M. Al-Obaid, S. G. Abdel-Hameid, H. A. El-Kashef, A. A. M. Abdel-Aziz, A. S. El-Azab, H. A. Al-Khamees and H. I. El-Subbagh, Eur. J. Med. Chem., 2009, 44, 2379–2391 CrossRef PubMed.
- M. S. Hamed, M. M. Kamel, M. M. Kassem, N. Emad, M. S. Nofal and F. M. Ahmed, Acta Pol. Pharm., 2010, 67(2), 159–171 Search PubMed.
- V. B. Shashikant, J. D. Bhavana, C. D. Sudarshan, T. G. Suraj, T. R. Vankekesh, V. K. Chetan and P. S. Anik et, J. Pharmacol. Exp. Ther., 2008, 326(2), 604–613, DOI:10.1124/jpet.
- R. Kaur, M. Bansal and B. Kaur, Chem. Sci. J., 2011, CSJ-18 Search PubMed.
- P. S. N. Reddy, M. Vasantha and V. D. Reddy Rasayan, J. Chem., 2010, 3(4), 635–640 CAS.
- M. Suchy and R. H. E. Hudson, J. Org. Chem., 2014, 79, 3336–3347, DOI:10.1021/jo402873e.
- S. Y. Wang, J. Org. Chem., 1959, 24, 11–13 CrossRef CAS.
- C. Ausín, J. A. Ortega, J. Robles, A. Grandas and E. Pedroso, Org. Lett., 2002, 4, 4073–4075 CrossRef PubMed.
- L. J. Gooβen, G. Deng and L. M. Levy, Science, 2006, 313, 662–664 CrossRef PubMed.
- W. Li, X. Zhou, Y. Chen, S. Guo, F. Ba, W. Tian, C. Yang, M. Wang, Y. Liu, Y. Song, J. Zhu, Y. Zhou, F. Zhou, H. Guo and C. Zheng, Tetrahedron, 2016, 72(23), 3185–3192 CrossRef CAS.
- C. G. Hartung, A. C. Backes, B. Felber, A. Missio and A. Philipp, Tetrahedron, 2006, 62(43), 10055–10064, DOI:10.1016/j.tet.2006.08.065.
- P. Yin, N. Liu, Y.-X. Deng, Y. Chen, Y. Deng and L. He, J. Org. Chem., 2012, 77, 2649–2658, DOI:10.1021/jo2023697.
- R. R. Vidule, Indian J. Appl. Res., 2013, 3(8), 74–77 CrossRef.
- A. A. F. Wasfy, Phosphorus, Sulfur Silicon Relat. Elem., 2002, 177, 1349–1358 CrossRef CAS.
- K. Špirková and Š. Stankovský, Collect. Czech. Chem. Commun., 1996, 61, 957–961, DOI:10.1135/cccc19960957.
- N. F. Abdel-Ghaffar, Nat. Sci., 2011, 9(7), 190–201 Search PubMed.
- Q.-Z. Shi, Y.-N. Cao, S.-B. Ma, G.-X. Wang, G.-F. Han and Z. Xing, J. Chem. Res., 2016, 40, 767–771 CrossRef CAS.
- S. Venkateswarlu, M. Satyanarayana, P. Ravikiran and V. Siddaiah, J. Heterocycl. Chem., 2013, 50(5), 1089–1093, DOI:10.1002/jhet.1603.
- Š. Stankovský and A. Filip, Chem. Zvesti, 1984, 38(5), 677–685 Search PubMed.
- T. Yoshikawa and K. Shitago, Yakugaku Zasshi, 1974, 94(4), 417–423, DOI:10.1248/yakushi1947.94.4_417.
-
(a) K. Hirota, Y. Kitade, H. Sajiki and Y. Maki, Synthesis, 1984, 1984(7), 589–590, DOI:10.1055/s-1984-30901;
(b) K. Hirota, Y. Kitade, M. Sajika and Y. Maki, J. Chem. Soc., Perkin Trans. 1, 1990, 123–128, 10.1039/P19900000123.
- S. K. Srivastava, W. Haq and P. M. S. Chauhan, Bioorg. Med. Chem. Lett., 1999, 9(7), 965–966, DOI:10.1016/S0960-894X(99)00128-6.
- A. M. Hussein, J. Saudi Chem. Soc., 2010, 14, 61–68 CrossRef CAS.
- G. H. Elgemeie and H. A. Ali, Synth. Commun., 2002, 32(2), 253–264, DOI:10.1081/SCC-120002010.
- S. A. Ahmed, A. M. Hussein, W. G. M. Hozayen, A. H. H. El-Ghandour and A. O. Abdelhamid, J. Heterocycl. Chem., 2007, 44(4), 803–810 CrossRef CAS.
- A. M. Hussein and O. M. Ahmed, Bioorg. Med. Chem., 2010, 18(7), 2639–2644, DOI:10.1016/j.bmc.2010.02.028.
- A. Karoui, F. Allouche, M. Deghrigue, A. Agrebi, A. Bouraoui and F. Chabchoub, Med. Chem. Res., 2014, 23, 1591–1598, DOI:10.1007/s00044-013-0742-x.
- S. Gupta, L. M. Rodrigues, A. P. Esteves, A. M. F. Oliveira-Campos, M. S. Jose Nascimento, N. Nazareth, H. Cidade, M. P. Neves, E. Fernandes, M. Pinto, N. M. F. S. A. Cerqueira and B. Natercia, Eur. J. Med. Chem., 2008, 43, 771–780, DOI:10.1016/j.ejmech.2007.06.002.
- F. Allouche, F. Chabchoub, F. Carta and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2013, 28, 343–349, DOI:10.3109/14756366.2012.720573.
- M. Bakavoli, G. Bagherzadeh, M. Vaseghifar, A. Shiria, M. Pordel, M. Mashreghi, P. Pordeli and M. Araghi, Eur. J. Med. Chem., 2010, 45, 647–650, DOI:10.1016/j.ejmech.2009.10.051.
- C. E. Mueller, D. Shi, M. Manning Jr. and J. W. Daly, J. Med. Chem., 1993, 36(22), 3341–3349, DOI:10.1021/jm00074a015.
- A. M. Hayallah, J. Sandoval-Ramírez, U. Reith, U. Schobert, B. Preiss, B. Schumacher, J. W. Daly and C. E. Müller, J. Med. Chem., 2002, 45(7), 1500–1510, DOI:10.1021/jm011049y.
- S. Weyler, A. M. Hayallah and C. E. Muller, Tetrahedron, 2003, 59, 47–54, DOI:10.1016/S0040-4020(02)01485-0.
- S. Weyler, F. Fülle, M. Diekmann, B. Schumacher, S. Hinz, K. N. Klotz and C. E. Müller, ChemMedChem, 2006, 1(8), 891–902, DOI:10.1002/cmdc.200600066.
- O. M. Abo-Salem, A. M. Hayallah, A. Bilkei-Gorzo, B. Filipek, A. Zimmer and C. E. Müller, J. Pharmacol. Exp. Ther., 2004, 308(1), 358–366, DOI:10.1124/jpet.103.056036.
- S. H. Abdel-Hafez, R. A. Ahmed, M.
A. Abdel-Azim and K. M. Hassan, J. Chem. Res., 2009, 2009, 56–59 CrossRef.
- V. D. Dyachenko and V. P. Litvinov, Russ. Chem. Rev., 1997, 66(11), 923–951, DOI:10.1070/RC1997v066n11ABEH000323.
- P. R. Murumkar, M-Pharm, thesis submitted to North Maharastra University, Jalgaon, 2004.
- S. Von Niementowski, J. Prakt. Chem., 1895, 51, 564–572 CrossRef.
- S. S. Laddha and S. P. Bhatnagar, ARKIVOC, 2007,(xvi), 1–11, DOI:10.3998/ark.5550190.0008.g01.
- A. M. Abdel-Fattah, A. S. Aly, F. A. Gad, N. A. Hassan and A. B. A. El-Gazzar, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 163, 1–27 CrossRef CAS.
- R. O. Bora, I. S. Rathod, S. S. Toshniwal and M. Farooqui, Int. J. Chem. Sci., 2005, 3(3), 469–474 CAS.
- A. S. Aly, A. B. A. El-Gazzar and H. A. R. Hussein, Phosphorus, Sulfur Silicon Relat. Elem., 2007, 182, 35–56, DOI:10.1080/10426500600865293.
- S. S. Laddha and S. P. Bhatnagar, ARKIVOC, 2008,(xvii), 212–220, DOI:10.3998/ark.5550190.0009.h20.
- A. A. Abu-Hashem, Molecules, 2018, 23, 2793–2812, DOI:10.3390/molecules23112793.
- A. Abu-Hashem and M. M. Youssef, Molecules, 2011, 16, 1956–1972, DOI:10.3390/molecules16031956.
- J. C. Burbiel, J. Hockemeyer and C. E. Müller, Beilstein J. Org. Chem., 2006, 2(1), 20–25, DOI:10.1186/1860-5397-2-20.
- J. Garín, E. Meléndez, F. L. Merchán, D. Ortiz and T. Tejero, Synthesis, 1987, 1987(4), 368–370, DOI:10.1055/s-1987-27946.
- R. J. Cruickshank; P. Duguid; R. R. Swain J. Med. Microbiol., Vol. 1, New York: Churchill Livingstone Publishers, 1998 Search PubMed.
- O. M. Ahmed, A. M. Hussein and R. R. Ahmed, Med. Chem., 2012, 2(1), 020–028, DOI:10.4172/2161-0444.1000108.
- S. S Laddha and S. P Bhatnagar, Future Med. Chem., 2010, 2(4), 565–573, DOI:10.4155/FMC.10.16.
- S. S. Laddha, S. P. Bhatnagar, Novel Fused quinazolinones: Further studies on the anticonvulsant activity of 1,2,9,11 tetrasubstituted-7H-thieno[2′,3′:4,5]pyrimido[6,1-b]-quinazolin-7-one and 1,3,10,12-tetra-substituted-8H-pyrido[2′,3′:4,5]pyrimido[6,1-b]quinazolin-8-one. 13rd International Electronic Conference on Synthetic Organic Chemistry (ECSOC-13), November, 2009, pp. 1–30 Search PubMed.
- M. Shibuya, J. Biochem., 2013, 153(1), 13–19, DOI:10.1093/jb/mvs136.
- S. Guo, L. S. Colbert, M. Fuller, Y. Zhang and R. R. Gonzalez-Perez, Biochim. Biophys. Acta, 2010, 1806(1), 108–121, DOI:10.1016/j.bbcan.2010.04.004.
- K. J. Griessmeier and C. E. Müller, Pharmacology, 2005, 74, 51–56, DOI:10.1159/000083983.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Başak Doğru Mert
b,
M. Monier
a and
Ahmed El-Mekabaty
*a,
Başak Doğru Mert
b,
M. Monier
a and
Ahmed El-Mekabaty