
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of surface oxidation on the electronic transport properties of phosphorene gas sensors: a computational study
Juan M. Marmolejo-Tejada a and
Andres Jaramillo-Botero*ba
a and
Andres Jaramillo-Botero*ba
aElectronics and Computer Science Division, Pontificia Universidad Javeriana, Cali 760031, Colombia
bChemistry and Chemical Engineering Division, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: ajaramil@caltech.edu
First published on 14th February 2020
Abstract
The potential for phosphorene-based devices has been compromised by the material's fast degradation under ambient conditions. Its tendency to fully oxidize under O2-rich and humid environments, leads to the loss of its appealing semiconducting properties. However, partially-oxidized phosphorene (po-phosphorene), has been demonstrated to remain stable over significantly longer periods of time, thereby enabling its use in sensing applications. Here, we present a computational study of po-phosphorene-based gas sensors, using the Density-Functional-based Tight Binding (DFTB) method. We show that DFTB accurately predicts the bandgap for the pristine material and po-phosphorene, the electronic transport properties of po-phosphorene at different surface oxygen concentrations, and the appropriate trends in Density-of-States (DOS) contributions caused by adsorbed gas molecules, to demonstrate its potential application in the development of gas sensors. Results are compared against the more traditional and expensive Density Functional Theory (DFT) method using generalized gradient approximation (GGA) exchange–correlation functionals, which significantly underestimates the material's bandgap.
1 Introduction
Sensor approaches based on two-dimensional (2D) heterostructured materials, such as graphene, phosphorene, transition metal dichalcogenides (TMDs) and others,1–3 in conjunction with big data analytics, are expected to enable the real-time detection of adsorbates through differentiable electronic signatures. This is due to their usually high electronic mobility, mechanical rigidity and surface-to-volume ratio, which bring important benefits to the development of nanosensors. Measuring targeted biological markers (biomarkers), such as primary and secondary metabolites, volatile organic compounds (VOCs), and other biomolecules in nano-molar (nM) or pico-molar (pM) concentrations4,5 will enable fast, reliable and inexpensive diagnosis and tracking of health in human, animal and plant organisms. The potential for non- or minimally-invasive sensor coupled to mobile platforms for real-time measurements, also offers a unique opportunity within the Internet of Things (IoT) paradigm.Here, we focus on phosphorene, as a two-dimensional material and allotrope of phosphorus made up of a single layer of black phosphorus. Black phosphorus is a layered material where P atoms covalently bond to 3 adjacent P atoms, forming a wrinkled honeycomb structure that stacks to other individual layers through van der Waals forces.6–8 A single layer is known as phosphorene and has a direct bandgap of ∼2 eV, that gradually decreases with increased stacking of layers down to ∼0.3 eV in the bulk.9–12 Phosphorene is susceptible to mechanical deformation and stacking order,13,14 yet it has very high carrier mobility, up to >50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1.15,16
000 cm2 V−1 s−1.15,16
Several authors have reported theoretical and experimental studies that show phosphorene's interaction with many small molecules, and their effect on its resistance and carrier density capabilities, enables the selective detection and measurement of such molecules.5,8,17–30 Recently published reviews associated to advances in the field of 2D materials for sensing applications, provide a clear picture of the opportunities and challenges offered by phosphorene.31–34 One of the main problems in its widespread use for sensing applications is its stability under ambient conditions. Phosphorene reacts with water vapor and oxygen assisted by visible light35 to degrade within the scope of hours. One way to control this degradation, is to partially oxidize phosphorene (po-phosphorene). Po-phosphorene has been shown to remain stable in air and moisture, preserving its electronic properties at room temperatures and above,36,37 and enabling field-effect transistor (FET) devices with sub-nM concentration capabilities for different analytes (in particular nitrogenated species5).
Here, we report on a systematic study of the effects of surface oxidation on the electronic transport properties of phosphorene-based gas sensors. The paper is organized as follows: Section 2 introduces the first-principles based computational methods and procedures used in Section 3 to predict the electronic transport properties of pristine and po-phosphorene with different physically-adsorbed molecules, and the sensing capabilities of a po-phosphorene FET sensor device under various oxygen concentrations. Section 4 summarizes the importance and applicability of our results.
2 Methods and procedures
To determine the electronic transport properties of pristine and functionalized phosphorene, from first-principles, we solve the non-relativistic Schrödinger equation38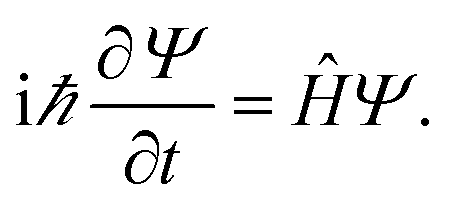 Particle wave functions in a periodic potential (Bloch waves) are used as solutions for band structure calculations, as Ψnk(r) = eikrunl(r), where ψ(r) is an eigenfunction of the system and is defined as the product of a plane wave envelope and a periodic Bloch function u(r); n is the band index and k the wave vector associated with the direction of motion of the electron in the crystal, which takes on values within the corresponding Brillouin zone (BZ). Consequently, the band structure is determined by the orbital energies εnk for each band n and momentum k, obtained with the Kohn–Sham approach to density functional theory (DFT).39
Particle wave functions in a periodic potential (Bloch waves) are used as solutions for band structure calculations, as Ψnk(r) = eikrunl(r), where ψ(r) is an eigenfunction of the system and is defined as the product of a plane wave envelope and a periodic Bloch function u(r); n is the band index and k the wave vector associated with the direction of motion of the electron in the crystal, which takes on values within the corresponding Brillouin zone (BZ). Consequently, the band structure is determined by the orbital energies εnk for each band n and momentum k, obtained with the Kohn–Sham approach to density functional theory (DFT).39
Although GW calculations and DFT calculations with hybrid exchange–correlation functionals provide a very accurate band structure prediction of semiconductor materials, these are computationally very expensive or not available in electronic transport packages capable of solving the non-equilibrium Green's functions (NEGF) machinery40,41 on two/three-probe systems. Although exchange–correlation functionals within the generalized gradient approximation (GGA) usually underestimate the bandgap of semiconductor materials, we used Perdew–Burke–Ernzerhof (PBE) functionals42 as reference. Therefore, geometry optimization and electronic properties calculations were carried out using linear combination of atomic orbitals (LCAO) basis sets, as implemented in the QuantumATK package,43–45 where the Kohn–Sham (KS) Hamiltonian is represented in the basis of the PseudoDojo norm-conserving pseudopotentials46 on all atoms and PBE parametrization with van der Waals correction,47 using a 75 hartree density mesh cut-off, and a 5 × 10−5 self-consistent field (SCF) convergence criteria. A 1 × 3 × 3 k-point mesh was used for the BZ sampling of a rectangular unit cell consisting of 48 P atoms, and additional O, N, C and Na atoms, as discussed in the following Section. 2 nm of vacuum was added to the directions normal to the transport direction, z, to avoid unrealistic interactions between atoms in neighboring periodic cells. Atoms were allowed to relax until forces converged below 0.05 ev Å−1, using the Limited-memory Broyden–Fletcher–Goldfarb–Shanno (LBFGS) method.48
To enable faster calculations on the relatively-large atomistic system models involved in this study, we used the Density-functional-based tight binding (DFTB) approximate methods. DFTB has been increasingly used in recent years, for predicting different phosphorene properties.30,49–54 Here, we use the third-order parameterization for organic and biological systems (3OB)55–57 Slater–Koster parameters to determine the electronic properties of interest, including band-structures and bandgaps. DFTB was coupled to the NEGF machinery, to determine the electronic transport characteristics for the different device configurations, involving pristine and po-phosphorene semiconducting channels in a three-probe, back-gated, field-effect configuration.
3 Results and discussion
3.1 Electronic transport properties of phosphorene with adsorbed molecules
We employed a 3 × 4 phosphorene surface unit cell as starting point, and characterized the effect on transport properties with different adsorbed organic molecules on it, including CH4, CO, CO2, HCN, HNC, NH3, NO, and NO2. Similar configurations have been reported in previous theoretical and experimental publications,19–23,29,30 to study the selective detection of such molecules. After minimizing each structure with DFT, we obtained the binding energy of each analyte to phosphorene from Eb = Ec − (Ep + Eanalyte), where Eb is the binding energy, Ec the complex's total energy, Ep the pristine phosphorene energy, and Eanalyte the single molecule energy. Then, we computed the system's fat bandstructure (FBS) and projected density of states (PDOS), to determine the contribution of each analyte on phosphorene's electronic properties. We also minimized the structure using the DFTB approximation, and performed FBS and PDOS calculations, to benchmark against DFT results.Fig. 1(a) compares the analyte's binding energy to phosphorene, using DFT, DFTB, and existing published data.17,21,23,25,26,29,30 As observed by Kou et al.,17 NO and NO2 molecules consistently have the largest binding energy to pristine phosphorene, implying it is more sensitive to these molecules. Fig. 2(a–i) depicts the changes in band structure as a function of analyte adsorption, and Fig. 3(a–i) shows the PDOS obtained for each system. NO and NO2 are the only molecules among the tested gas phase molecules, that exhibit a significant contribution to the system's electronic properties around the Fermi level. Furthermore, DFT and DFTB results are qualitatively consistent, albeit the evident differences in bandgap values (∼0.81 eV and ∼1.93 eV, respectively). Contributions from CH4, CO, CO2, HCN, HNC and NH3 molecules are a few eVs from the Fermi level, and have little effect on phosphorene's bandgap and semiconducting properties.
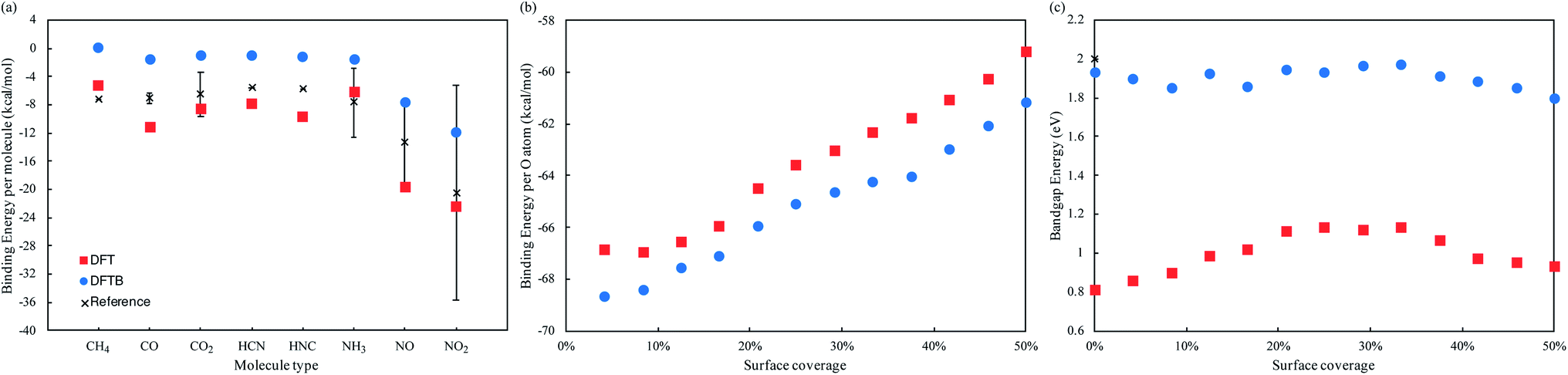 | ||
| Fig. 1 (a) Binding energy of CH4, CO, CO2, HCN, HNC, NH3, NO, and NO2 to phosphorene, using DFT and DFTB methods, compared to reference results in ref. 17, 21, 23, 25, 26, 29 and 30. (b) Binding energy per oxygen atom and (c) bandgap energy, as a function of adsorbed oxygen concentrations in the T1 position, using DFT and DFTB methods. | ||
 | ||
| Fig. 2 Fat bandstructure of (a) pristine phosphorene, and phosphorene with adsorbed (b) CH4, (c) CO, (d) CO2, (e) HCN, (f) HNC, (g) NH3, (h) NO, (i) NO2, (j) 4.2% O, (k) 50% O and (l) Na. Fat bands in blue show the contribution of the adsorbed molecule to the electronic structure of phosphorene. | ||
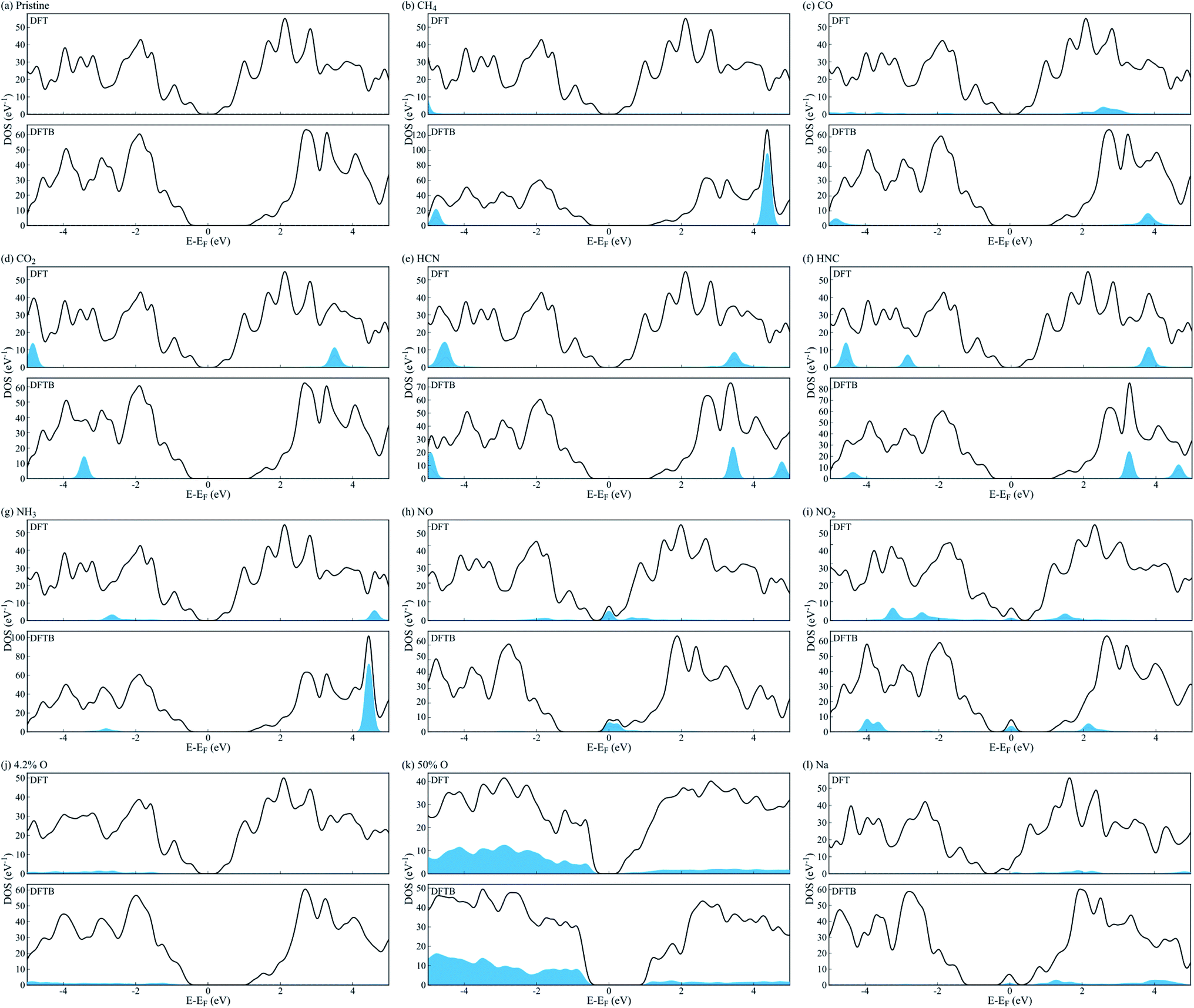 | ||
| Fig. 3 Projected density of states of (a) pristine phosphorene, and phosphorene with adsorbed (b) CH4, (c) CO, (d) CO2, (e) HCN, (f) HNC, (g) NH3, (h) NO, (i) NO2, (j) 4.3% O, (k) 50% O and (l) Na. Blue areas correspond to the contribution of the adsorbed molecule to phosphorene's density of states. | ||
Similar to n-type doping of phosphorene with Cu adatoms,58 other atomic dopants, such as Li, Na, Al and Cr, induce metallic behaviour on phosphorene,59 and may be useful for avoiding Schottky barriers when contacting phosphorene with conventional metal electrodes.60 This is observed in Fig. 2(l) and 3(l), where metalization of phosphorene is achieved via doping with Na atoms in the hollow position at a concentration of 5% (1 out of 20 available sites). We chose doping our electrodes with Na atoms in this study, because of the availability of Slater–Koster parameters to computationally characterize the electronic transport properties under a DFTB-NEGF formalism. Na-doping effectively metalizes phosphorene electrodes without compromising the electronic transport characterization of the proposed device, albeit it is not practical to realize via experiments. Cu doping metallization is favored experimentally,58 but Cu interaction parameters are not currently available for DFTB, hence our choice of Na.
3.2 Electronic transport properties of po-phosphorene as a function of oxygen concentration
We now consider po-phosphorene with moderate oxygen concentrations, to improve its stability under ambient conditions, and retain its sensing capabilities. As we had reported in ref. 5, oxygen adsorption in the dangling position (top T1 sites), corresponds to the most energetically favorable configuration for low oxygen concentrations. Hence, we started from the optimized pristine phosphorene surface and randomly added oxygen atoms on T1 sites, from 4.2% up to a 50% surface coverage.Fig. 1(b) compares the binding energy per oxygen atom for different adsorbed concentrations on T1 positions, using DFT and DFTB. We observe a reduced rate of binding past 25% surface oxygen concentration, which picks up again past 37.5%. Fig. 2(j, k) and 3(j, k) show the calculated FBS and PDOS for the lower and upper limit cases (4.2% and 50% oxygen concentration), where contributions to the system's electronic properties are a few eVs away from the Fermi level; thus, only slight alterations to the bandgap are observed, and semiconducting properties are preserved in all cases. This is summarized in Fig. 1(c), where bandgap estimations are compared as a function of different adsorbed oxygen concentrations in the T1 position, for both DFT and DFTB. As also pointed out in ref. 5 and 61, DFT calculations result in higher bandgap energies at low oxygen concentrations, while indirect bandgap energies on the transport direction z. DFTB calculations, on the other hand, show indirect bandgap energies with very little variations across the range of oxygen concentrations studied here. A fully oxidized phosphorene structure, i.e. a phosphorus pentoxide (P2O5) monolayer, results in a 11.62 eV direct bandgap (7.45 eV using hybrid functionals62).
We also calculated the binding energy of NO and NO2 molecules to po-phosphorene, finding that surface adsorption is energetically favorable, at the different adsorbed oxygen concentrations on T1 sites. Adsorption of NO/NO2 species on pristine phosphorene is energetically more favorable (by ∼8–12 kcal mol−1) when compared to adsorption onto available P sites of po-phosphorene, due to redistribution of surface charges caused by the oxygen population. In spite of this, we expect a po-phosphorene-based sensor is able of detecting NO, NO2 and other gases whose electronic contributions are localized near the Fermi level.
3.3 Device geometry and electronic transport properties as a function of oxygen and adsorbed molecule concentrations
Fig. 4 shows a schematic illustration of the proposed sensor architecture, were Na-doped phosphorene is used for the electrodes (metal-like behavior), and pristine or po-phosphorene for the semiconducting channel. The y direction is assumed to be infinitely periodic, x is finite, and the z transport length (L) is set to 6.5 nm, for a total of 120 T1 surface sites available. The po-phosphorene surface is partially oxidized with 8.3% and 16.7% oxygen concentration on T1 sites, leaving 110 and 100 available sites for analyte adsorption, respectively. A metallic back-gate lays under the semiconducting junction, separated by a dielectric region with a relative dielectric constant set to 3.9 (i.e. equivalent to SiO2).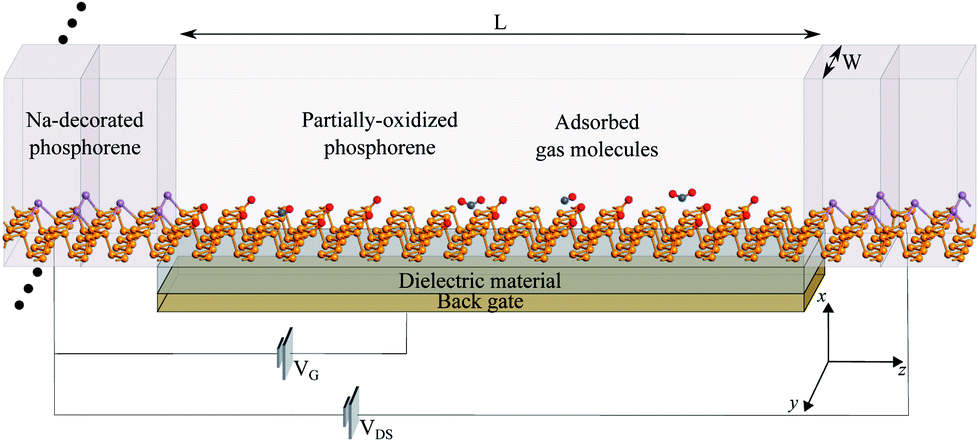 | ||
| Fig. 4 Isometric view of the po-phosphorene FET sensor. We set L = 6.5 nm and W = 1.345 nm. | ||
Fig. 5(a) shows conductance as a function of gate voltage, VG, with a maximum conductance in the order of tenths of μS. The proposed device is assumed to operate at room temperature 298 K ± 50 K, hence we confirmed that it exhibits minimal changes in thermionic conductance over the range 250–350 K. Fig. 5(b) shows the IV characteristics for the bare device (i.e. no adsorbed gas molecules) with on-state currents of a few tenths of μA, which demonstrates a feasible operation for different VG values. Fig. 5(c) shows the zero-bias projected (along the conduction direction) local device DOS (LDDOS), which corresponds to the number of allowed electron (or hole) states per volume at a given energy projected in real space, averaged over x and y, and plotted along z as a function of energy. A VG = −1.0 V was chosen to guarantee a fast on-off device transition, as corroborated from the bare IV characteristics in Fig. 5(b). The available states in the semiconducting junction are shifted along the energy axis when applying a gate voltage VG due to charge injection at the gate electrode.
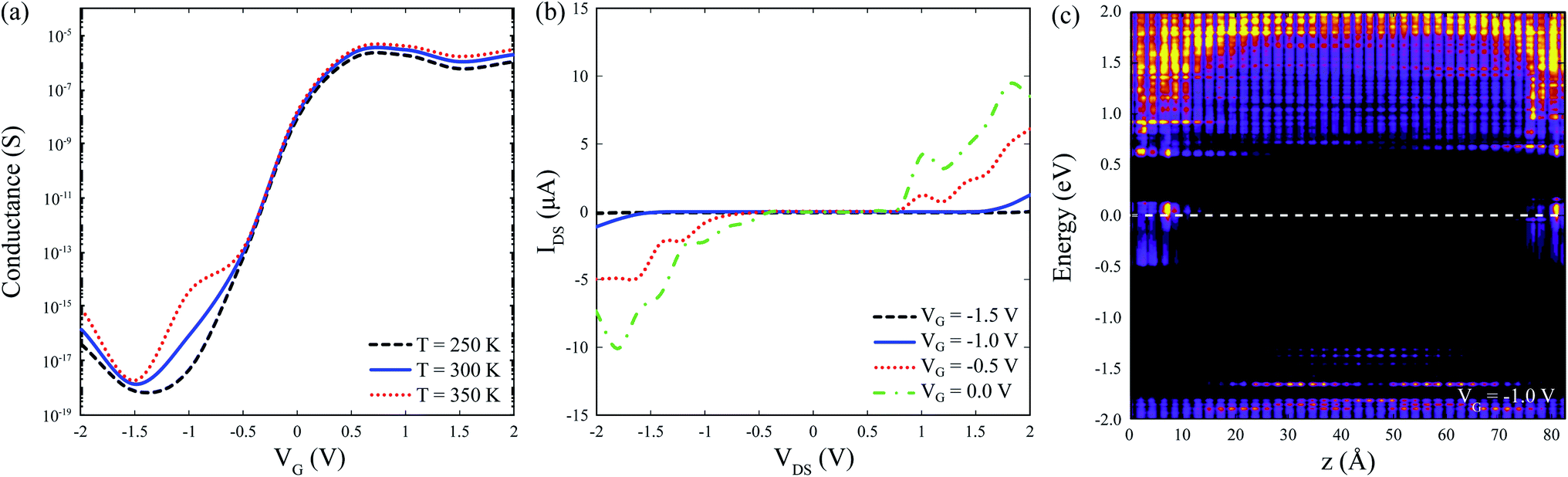 | ||
| Fig. 5 Electronic transport characteristics for the bare system. (a) Zero-bias conductance as a function of gate voltage, VG. (b) IV characteristics. (c) Zero-bias device density of states (DDOS) averaged over x and y, plotted along z as a function of energy at VG = −1.0 V. | ||
IV calculations on Fig. 6 show the device is capable of detecting single physisorbed NO and NO2 molecules on the channel, with differentiable electronic signatures at VG = −1 V and VDS = −2 V, disregarding surface oxidation. Although different peak current values are obtained for each oxidation level, NO and NO2 signals differ by a few μA, suggesting the need for a precise control of surface oxidation during device fabrication, and most likely a calibration step before usage. Notwithstanding, the device is capable of selectively detecting single NO and NO2 molecules.
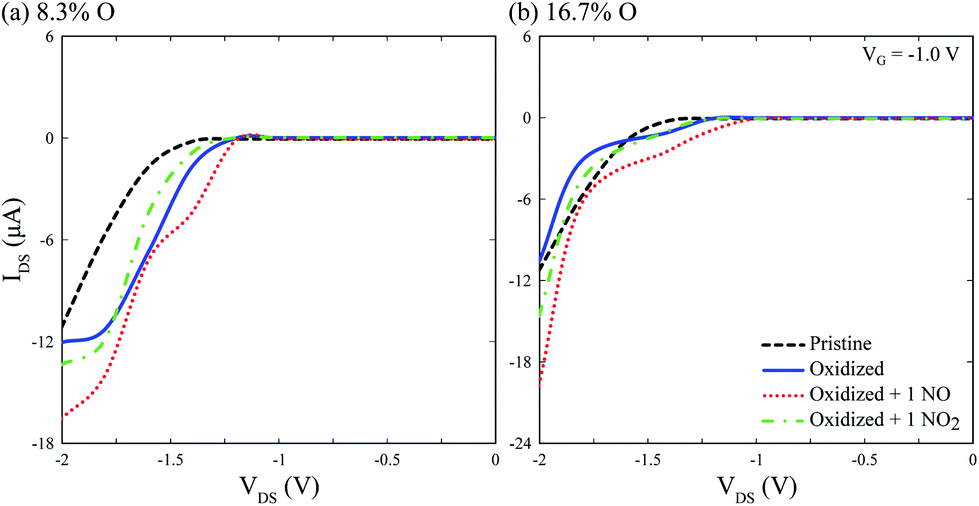 | ||
| Fig. 6 Current vs. voltage (IV) characteristics for the device at VG = −1 V with (a) 8.3% and (b) 16.7% surface oxidation, with and without physisorbed NO and NO2 at 0.87% concentration. For comparison, IV characteristics for the bare device are included (black dashed line). | ||
4 Conclusions
Our results demonstrate po-phosphorene can be practically used for gas sensing applications of nitrogen–oxygen moieties. Controlled oxidation of T1 sites on the phosphorene surface, up to 50% coverage, leads to preserved semiconducting properties, and sufficient binding sites for the potential detection of small organic molecules. Particularly, adsorbed molecules whose electronic contribution is located near the systems' Fermi level. We have shown that NO and NO2 gas molecules bind to po-phosphorene's surface with favorable energy, and that they can be selectively detected by means of a field-effect sensor based on po-phosphorene. Notwithstanding, the high level of control over surface oxidation needed to achieve chemical stability of phosphorene, our IV calculations show that a po-phosphorene based sensor device is capable of single molecule detection with differentiable electronic signature for NO-adsorbed and NO2-adsorbed molecules at minimum concentration for VG = −1 V and negative VDS. Then, we estimate a 19 nM L−1 limit of detection (LOD), assuming a capture gas chamber on top of the semiconducting channel with 8.775 μm2 surface area and 10 nm height.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. M. M.-T. would like to thank Branislav Nikolić for enabling computational resources within the Department of Physics and Astronomy at University of Delaware, and the Pontificia Universidad Javeriana, Cali, and Colciencias under the Postdoctoral Scholarship Program (811-C160I637000000881) for partial support. Authors acknowledge partial support from ICETEX, the Colombian Ministries of Education, and Industry/Tourism, within the Scientific Colombia Program OMICAS (Award 792-61187).Notes and references
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- S. Lebègue, T. Björkman, M. Klintenberg, R. M. Nieminen and O. Eriksson, Phys. Rev. X, 2013, 3, 031002 Search PubMed.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699 CrossRef CAS PubMed.
- A. Jaramillo-Botero and J. M. Marmolejo-Tejada, IEEE Sens. J., 2019, 19, 3975–3983 CAS.
- J. M. Marmolejo-Tejada and A. Jaramillo-Botero, Phys. Chem. Chem. Phys., 2019, 21, 19083–19091 RSC.
- A. Brown and S. Rundqvist, Acta Crystallogr., 1965, 19, 684–685 CrossRef CAS.
- L. Cartz, S. R. Srinivasa, R. J. Riedner, J. D. Jorgensen and T. G. Worlton, J. Chem. Phys., 1979, 71, 1718–1721 CrossRef CAS.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed.
- R. W. Keyes, Phys. Rev., 1953, 92, 580–584 CrossRef CAS.
- Y. Takao, H. Asahina and A. Morita, J. Phys. Soc. Jpn., 1981, 50, 3362–3369 CrossRef CAS.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed.
- J. Qiao, X. Kong, Z.-X. Hu, F. Yang and W. Ji, Nat. Commun., 2014, 5, 4475 CrossRef CAS PubMed.
- R. Fei and L. Yang, Nano Lett., 2014, 14, 2884–2889 CrossRef CAS PubMed.
- M. Wu, X. Qian and J. Li, Nano Lett., 2014, 14, 5350–5357 CrossRef CAS PubMed.
- Y. Akahama, S. Endo and S.-i. Narita, J. Phys. Soc. Jpn., 1983, 52, 2148–2155 CrossRef CAS.
- S. Narita, Y. Akahama, Y. Tsukiyama, K. Muro, S. Mori, S. Endo, M. Taniguchi, M. Seki, S. Suga, A. Mikuni and H. Kanzaki, Physica B+C, 1983, 117–118, 422–424 CrossRef CAS.
- L. Kou, T. Frauenheim and C. Chen, J. Phys. Chem. Lett., 2014, 5, 2675–2681 CrossRef CAS PubMed.
- H. Liu, Y. Du, Y. Deng and P. D. Ye, Chem. Soc. Rev., 2015, 44, 2732–2743 RSC.
- A. N. Abbas, B. Liu, L. Chen, Y. Ma, S. Cong, N. Aroonyadet, M. Kopf, T. Nilges and C. Zhou, ACS Nano, 2015, 9, 5618–5624 CrossRef CAS PubMed.
- M. Donarelli, L. Ottaviano, L. Giancaterini, G. Fioravanti, F. Perrozzi and C. Cantalini, 2D Materials, 2016, 3, 025002 CrossRef.
- Y. Cai, Q. Ke, G. Zhang and Y.-W. Zhang, J. Phys. Chem. C, 2015, 119, 3102–3110 CrossRef CAS.
- S. Cui, H. Pu, S. A. Wells, Z. Wen, S. Mao, J. Chang, M. C. Hersam and J. Chen, Nat. Commun., 2015, 6, 8632 CrossRef CAS PubMed.
- A. Srivastava, M. S. Khan, S. K. Gupta and R. Pandey, Appl. Surf. Sci., 2015, 356, 881–887 CrossRef CAS.
- V. V. Kulish, O. I. Malyi, C. Persson and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 992–1000 RSC.
- M. Lalitha, Y. Nataraj and S. Lakshmipathi, Appl. Surf. Sci., 2016, 377, 311–323 CrossRef CAS.
- Q. Yang, Y. Huang, R. Meng and X. Chen, 2016 17th International Conference on Electronic Packaging Technology, ICEPT, 2016, pp. 749–752 Search PubMed.
- N. Suvansinpan, F. Hussain, G. Zhang, C. H. Chiu, Y. Cai and Y.-W. Zhang, Nanotechnology, 2016, 27, 065708 CrossRef PubMed.
- Q. Yang, R. S. Meng, J. K. Jiang, Q. H. Liang, C. J. Tan, M. Cai, X. Sun, D. G. Yang, T. L. Ren and X. P. Chen, IEEE Electron Device Lett., 2016, 37, 660–662 CAS.
- V. Nagarajan and R. Chandiramouli, J. Mol. Graphics Modell., 2017, 75, 365–374 CrossRef CAS PubMed.
- J. Pang, Q. Yang, X. Ma, L. Wang, C. Tan, D. Xiong, H. Ye and X. Chen, Phys. Chem. Chem. Phys., 2017, 19, 30852–30860 RSC.
- V. Sorkin, Y. Cai, Z. Ong, G. Zhang and Y. W. Zhang, Crit. Rev. Solid State Mater. Sci., 2017, 42, 1–82 CrossRef CAS.
- M. Donarelli and L. Ottaviano, Sensors, 2018, 18(11), 3638 CrossRef PubMed.
- R. Irshad, K. Tahir, B. Li, Z. Sher, J. Ali and S. Nazir, J. Ind. Eng. Chem., 2018, 64, 60–69 CrossRef CAS.
- A. Yang, D. Wang, X. Wang, D. Zhang, N. Koratkar and M. Rong, Nano Today, 2018, 20, 13–32 CrossRef CAS.
- A. Favron, E. Gaufrès, F. Fossard, A.-L. Phaneuf-L'Heureux, N. Y.-W. Tang, P. L. Lévesque, A. Loiseau, R. Leonelli, S. Francoeur and R. Martel, Nat. Mater., 2015, 14, 826 CrossRef CAS PubMed.
- B. Tian, B. Tian, B. Smith, M. C. Scott, Q. Lei, R. Hua, Y. Tian and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4345–4350 CrossRef CAS PubMed.
- T. Nilges, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4311–4313 CrossRef CAS PubMed.
- E. Schrödinger, Ann. Phys., 1926, 384, 361–376 CrossRef.
- A. Jaramillo-Botero, J. Tahir-Kheli, P. von Allmen and W. A. Goddard III, in CRC handbook of Nanoscience, Engineering and Technology, ed. W. A. Goddard, D. Brenner, S. E. Lyshevski and G. J. Iafrate, 2012, pp. 935–982 Search PubMed.
- S. Datta, Superlattices Microstruct., 2000, 28, 253–278 CrossRef CAS.
- M. Brandbyge, J.-L. Mozos, P. Ordejón, J. Taylor and K. Stokbro, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 165401 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- QuantumATK version O-2019.03, Synopsys QuantumATK, https://www.synopsys.com/silicon/quantumatk.html Search PubMed.
- S. Smidstrup, D. Stradi, J. Wellendorff, P. A. Khomyakov, U. G. Vej-Hansen, M.-E. Lee, T. Ghosh, E. Jónsson, H. Jónsson and K. Stokbro, Phys. Rev. B, 2017, 96, 195309 CrossRef.
- S. Smidstrup, T. Markussen, P. Vancraeyveld, J. Wellendorff, J. Schneider, T. Gunst, B. Verstichel, D. Stradi, P. A. Khomyakov, U. G. Vej-Hansen, M.-E. Lee, S. T. Chill, F. Rasmussen, G. Penazzi, F. Corsetti, A. Ojanpera, K. Jensen, M. L. N. Palsgaard, U. Martinez, A. Blom, M. Brandbyge and K. Stokbro, J. Phys.: Condens. Matter, 2019, 32(1), 15901 CrossRef PubMed.
- M. van Setten, M. Giantomassi, E. Bousquet, M. Verstraete, D. Hamann, X. Gonze and G.-M. Rignanese, Comput. Phys. Commun., 2018, 226, 39–54 CrossRef CAS.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- D. C. Liu and J. Nocedal, Math. Program., 1989, 45, 503–528 CrossRef.
- V. Sorkin and Y. W. Zhang, Nanotechnology, 2015, 26, 235707 CrossRef CAS PubMed.
- S. Carmel, A. Pon, N. Meenakshisundaram, R. Ramesh and A. Bhattacharyya, Phys. Chem. Chem. Phys., 2018, 20, 14855–14863 RSC.
- A. Pihlajamäki, MSc thesis, University of Jyvaskyla, Finland, 2018.
- V. Sorkin and Y. W. Zhang, Nanotechnology, 2018, 29, 235707 CrossRef CAS PubMed.
- A. S. Carmel, A. Pon, R. Ramesh and A. Bhattacharryya, 2017 International conference on Microelectronic Devices, Circuits and Systems, ICMDCS, 2017, pp. 1–4 Search PubMed.
- T. A. Niehaus, S. T. A. G. Melissen, B. Aradi and S. M. V. Allaei, J. Phys.: Condens. Matter, 2019, 31, 395901 CrossRef CAS PubMed.
- M. Gaus, A. Goez and M. Elstner, J. Chem. Theory Comput., 2013, 9, 338–354 CrossRef CAS PubMed.
- M. Gaus, X. Lu, M. Elstner and Q. Cui, J. Chem. Theory Comput., 2014, 10, 1518–1537 CrossRef CAS PubMed.
- M. Kubillus, T. Kubar, M. Gaus, J. Rezac and M. Elstner, J. Chem. Theory Comput., 2015, 11, 332–342 CrossRef CAS PubMed.
- S. P. Koenig, R. A. Doganov, L. Seixas, A. Carvalho, J. Y. Tan, K. Watanabe, T. Taniguchi, N. Yakovlev, A. H. Castro Neto and B. Özyilmaz, Nano Lett., 2016, 16, 2145–2151 CrossRef CAS PubMed.
- T. Hu and J. Hong, J. Phys. Chem. C, 2015, 119, 8199–8207 CrossRef CAS.
- X. Zhang, Y. Pan, M. Ye, R. Quhe, Y. Wang, Y. Guo, H. Zhang, Y. Dan, Z. Song, J. Li, J. Yang, W. Guo and J. Lu, Nano Res., 2018, 11, 707–721 CrossRef CAS.
- G. Wang, R. Pandey and S. P. Karna, Nanoscale, 2015, 7, 524–531 RSC.
- A. Ziletti, A. Carvalho, P. E. Trevisanutto, D. K. Campbell, D. F. Coker and A. H. Castro Neto, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 085407 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |