
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic properties of 3d transition metal (Sc–Ni) doped plumbene
Daniel Hashemi * and
Hideo Iizuka
* and
Hideo Iizuka
Toyota Research Institute of North America, Toyota Motor North America, Ann Arbor, MI 48105, USA. E-mail: daniel.hashemi@toyota.com
First published on 14th February 2020
Abstract
Recently, a synthesized two-dimensional layer structured material, so-called “plumbene”, has attracted research interests because of its sizeable spin–orbit coupling. To study the potential of this material as a dilute magnetic semiconductor, we computationally investigate the structure, electronic, and magnetic properties of 3d transition metal (TM) doped plumbene using density functional theory (DFT). These calculations show that Ti, V, Cr, Mn, Fe, and Co-doped plumbene systems are magnetic while no magnetic solution was found for Sc and Ni. We also calculate the magnetic couplings between two TM impurities in the system with an impurity concentration of less than 2%. For V, Mn, Fe, Co-doped systems with short inter-impurity distances, we obtain a Curie temperature above room temperature using the mean-field approximation, indicating the potential for magnetic storage and spintronics applications.
I. Introduction
Two-dimensional (2D) materials, particularly those in group 14, have attracted extensive interest due to their potential in terms of the topological insulating properties,1–8 the chemical reactivity,9 the magnetic behavior,10,11 and the thermal properties.12 A recently synthesized 2D material, called plumbene13–17 has generated much attention because it has the largest spin–orbit interaction among all group 14 elements18 as the magnitude of spin–orbit coupling increases approximately as the fourth power of the effective nuclear charge. Moreover, rich physical characteristics are expected in plumbene due to strong spin–orbit coupling (SOC) as compared to graphene and phosphorene,18 i.e. huge host-induced magnetic anisotropy of impurities due to its strong SOC. In terms of mechanical properties, a recent molecular dynamics simulation has shown that plumbene is several times stronger than bulk lead.17 In terms of topological insulating properties, plumbene becomes a topological insulator with a large band gap (≈200 meV) through electron doping as shown by ab initio calculations.15 According to theoretical calculations, plumbene is also a good candidate for realizing the quantum spin Hall effect at room temperature (RT).19 Unexpected quantum spin Hall insulating properties were also reported for bilayer and decorated plumbene.20,21Over the past decades, there have been many studies regarding the fundamental science of dilute magnetic semiconductors (DMSs).22–26 It has been shown that the combination of 2D semiconductors and magnetic data storage could lead to 2D spintronics devices with a wide range of applications such as nonvolatile magnetoresistive memories, spin valve, spin-based transistors, and magnetically enhanced optoelectronics devices.27
The electronic and magnetic properties of 2D materials can be engineered with substitutional doping or adsorbing of TM elements, which can lead to DMSs in 2D semiconductors.28–34 The magnetic substitutional doping can induce the spin and valley polarization due to the spin–orbit and exchange interaction in transition-metal dichalcogenides (TMDs).35
In this paper, inspired by the recent synthesis of plumbene,13 we use 3d TM elements as magnetic impurities to show that it is possible to achieve magnetism in plumbene. First, we study the stability and electronic properties of TM defects in plumbene, and thereafter, the magnetic orders are acquired through the long-range interaction between the diluted TM-doped systems. The Curie temperature is also estimated using the mean-field approximation for the ferromagnetic (FM) cases.
II. Computational details
The calculations were performed within the framework of spin-polarized density functional theory (DFT) including SOC, using the Vienna ab initio simulation package (VASP).36,37 SOC has a huge impact on the electronic properties of plumbene in such a way that without taking it into account, the global band gap disappears.16 The frozen-core full-potential projector augmented-wave method (PAW) was used38 with the Perdew, Burke, and Ernzerhof (PBE)39 generalized gradient approximation (GGA). These functionals provide a reasonably good description of the magnetic states of TM nanostructures such as adatoms40,41 and nanowires.42–44The convergence with respect to the size of k-sampling, cutoff radius, and vacuum thickness was checked. These resulted in a k-point mesh of 2 × 2 × 1 for Density of States (DOS) of the TM-doped system, a plane-wave cutoff radius of 600 eV, and a vacuum distance between the periodic images of 15 Å. We used the conjugate-gradient algorithm for structural relaxation with a force convergence criterion of 0.01 eV Å−1.
The pristine plumbene structure was fully optimized. To obtain the fundamental band gap, the electronic band structures were calculated by the PBE functional, hybrid Heyd–Scuseria–Ernzerhof functionals (HSE03)45–47 and (HSE06).48 We used the standard values for the mixing parameter (0.25) and the range-separation parameters of 0.3 Å−1 and 0.2 Å−1 for HSE03 and HSE06 functionals, respectively. Charge transfer analysis between TM and plumbene was carried out using Bader formalism.49–51 The reciprocal space was sampled by a 6 × 6 × 1 k-points mesh in the Brillouin zone of the primitive unit cell. The electronic and magnetic properties of TM-doped systems were studied using the PBE-optimized structures. We used noncollinear spin polarization in a fully relativistic framework for the TM-doped plumbene systems to determine the easy magnetization axis and to calculate the magnetic interaction between two TM impurities. The structures, charge densities, and magnetization densities were visualized using VESTA code.52
III. Results and discussions
A. Structure and energetics
The calculated lattice constant of freestanding plumbene using PBE, HSE03, and HSE06 functionals, as shown on Table 1, are 4.93 Å, 4.88 Å, and 4.87 Å, respectively, which shows a good agreement between these methods. The calculated buckling heights obtained by PBE, HSE03, and HSE06 functionals are also 1.01 Å, 0.97 Å, and 0.97 Å, respectively.| System | PBE | HSE03 | HSE06 |
| Band gap (eV) | 0.43 | 0.50 | 0.58 |
| Lattice constant (Å) | 4.93 | 4.88 | 4.87 |
| h (Å) | 1.01 | 0.97 | 0.97 |
The calculated band gap energies are 0.43 eV, 0.50 eV, and 0.58 eV using PBE, HSE03, and HSE06 functionals, respectively. The electronic band structures obtained by these methods are displayed in Fig. 1(d). It is known that PBE is expected to underestimate the band gap energies53–55 which may be reflected in our calculations. HSE functionals are well-established and more accurate for describing electronic properties of 2D materials as compared to PBE, but they are computationally very expensive.56 We expect the experimental band gap energy to be higher than the calculated PBE band gap of 0.43 eV.
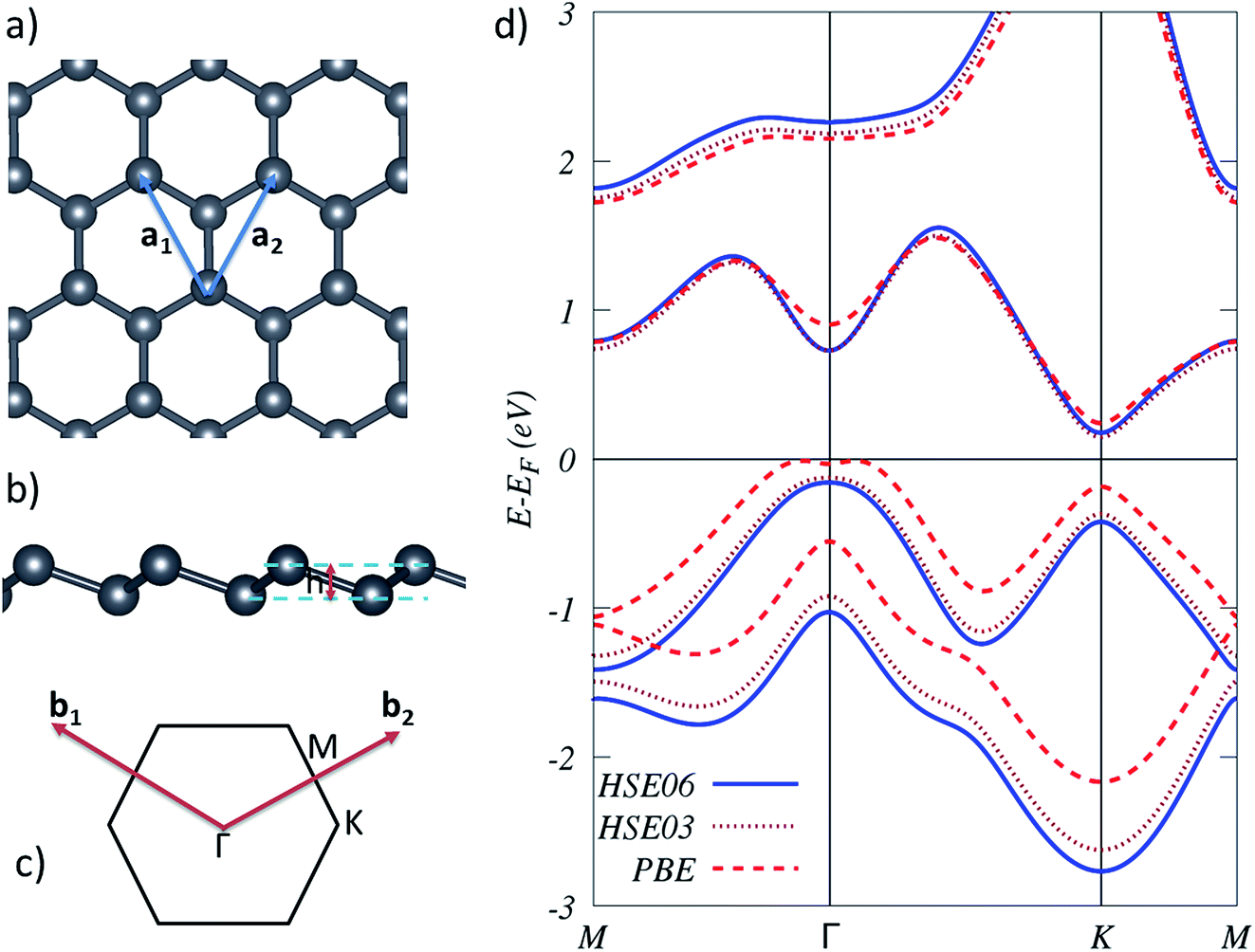 | ||
| Fig. 1 (a and b) Top and side views of buckled plumbene, respectively. The blue solid vectors indicate the primitive vectors. h is the vertical distance between the Pb planes. (c) First Brillouin zone with high-symmetry points. (d) Electronic band structures for plumbene calculated with the HSE06 (blue solid line), HSE03 (brown dotted line), and PBE (red dashed line) functionals. | ||
We checked the reliability of the PBE calculations and found consistent results across the PBE and the HSE methods in terms of magnetic configurations of the Fe-doped system. Due to the high computational workload and the reasonable agreement with the PBE results, we did not perform a similar study with the HSE approach.
To model the single impurity TM-doped systems, we chose a 4 × 2 supercell along the zigzag and armchair directions, respectively. Thus, the PBE lattice constants of a = 19.74 Å and b = 17.09 Å were used. This corresponds to 31 Pb atoms in a TM atom in the supercell. To calculate the magnetic exchange coupling between the two TM impurities, a 7 × 4 supercell was chosen with lattice constants of a = 34.54 Å and b = 34.18 Å, which corresponds to 110 Pb atoms and two TM atoms in this supercell. For structural relaxations of the TM-doped system in this supercell, we used only the Γ point sampling of the Brillouin zone. We first test the equilibrium configuration of a pristine plumbene sheet. The equilibrium structure for pristine plumbene is shown in Fig. 1(a and b). As obtained by PBE calculations, the equilibrium lattice constant, Pb–Pb bond length, and buckling height were estimated to be 4.93 Å, 3.02 Å, and 1.01 Å, respectively, which are consistent with previous studies.14–16
Table 2 lists the geometric and magnetic characters of the TM-substituted plumbene. Pb binding energy is also calculated as a reference system. The binding energy in our calculations is defined as follows:
| Eb = EPb−Vac + ETM − EPb−TM, | (1) |
| Dopant | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Pristine |
| Eb (eV) | 3.10 | 2.79 | 2.54 | 1.72 | 1.70 | 2.61 | 2.87 | 3.37 | 1.97 |
| TM–Pb (Å) | 2.92 | 2.80 | 2.79 | 2.85 | 3.02 | 2.69 | 2.53 | 2.50 | 3.02 |
| Easy axis | ![[x with combining circumflex]](https://www.rsc.org/images/entities/i_char_0078_0302.gif) |
|
ŷ | ẑ | ẑ | ẑ | |||
| |ms| (μB) | 1.32 | 2.79 | 3.98 | 4.10 | 2.95 | 1.52 | |||
| |mo| (μB) | 0.18 | 0.32 | 0.04 | 0.04 | 0.49 | 0.22 | |||
| TM charge transfer (e) | −1.09 | −0.81 | −0.50 | −0.36 | −0.37 | −0.09 | +0.04 | +0.20 | |
| Pb charge transfer (e) | +0.35 | +0.27 | +0.16 | +0.12 | +0.14 | +0.02 | −0.01 | −0.07 | |
| ΔE(z1) (meV) | 115 | −430 | −576 | 523 | 175 | ||||
| TC (K) | 445 | 2023 | 677 | ||||||
| ΔE(z2) (meV) | −83 | −125 | −98 | 128 | 91 | 12 | |||
| TC (K) | 495 | 352 | 46 | ||||||
| ΔE(z3) (meV) | 57 | 24 | 22 | 43 | −78 | −18 | |||
| TC (K) | 220 | 93 | 85 | 166 | |||||
| ΔE(z4) (meV) | −22 | −12 | 1 | −34 | 24 | 18 | |||
| TC (K) | 4 | 93 | 70 | ||||||
| ΔE(z5) (meV) | −1 | −7 | 7 | −5 | −15 | −16 | |||
| TC (K) | 27 | ||||||||
| ΔE(a1) (meV) | −45 | −125 | −78 | 101 | 77 | 17 | |||
| TC (K) | 391 | 298 | 66 | ||||||
| ΔE(a2) (meV) | −12 | 34 | −99 | −23 | −33 | −70 | |||
| TC (K) | 132 | ||||||||
| ΔE(a3) (meV) | 11 | −34 | −36 | −34 | 13 | 14 | |||
| TC (K) | 43 | 50 | 54 | ||||||
| ΔE(a4) (meV) | −21 | −47 | −41 | 12 | 18 | −5 | |||
| TC (K) | 46 | 70 | |||||||
| ΔE(a5) (meV) | −0.5 | −9 | −10 | 8 | 15 | 1 | |||
| TC (K) | 31 | 58 | 4 |
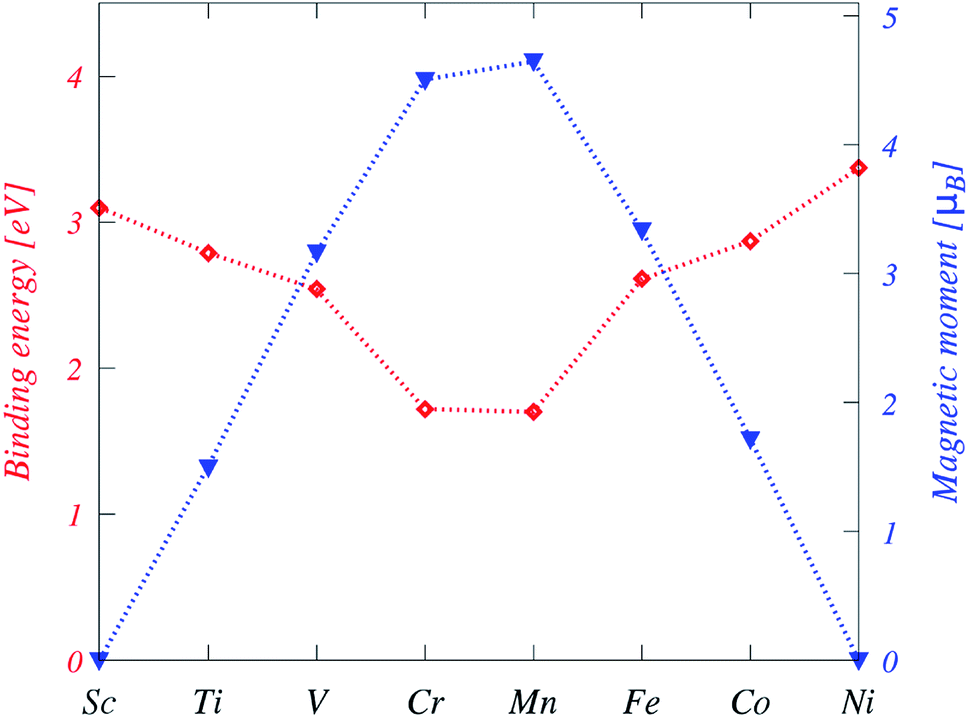 | ||
| Fig. 2 Binding energies (red) and magnetic moments (blue) of the TM-doped plumbene system. The dotted lines between symbols are a guide to the eye. | ||
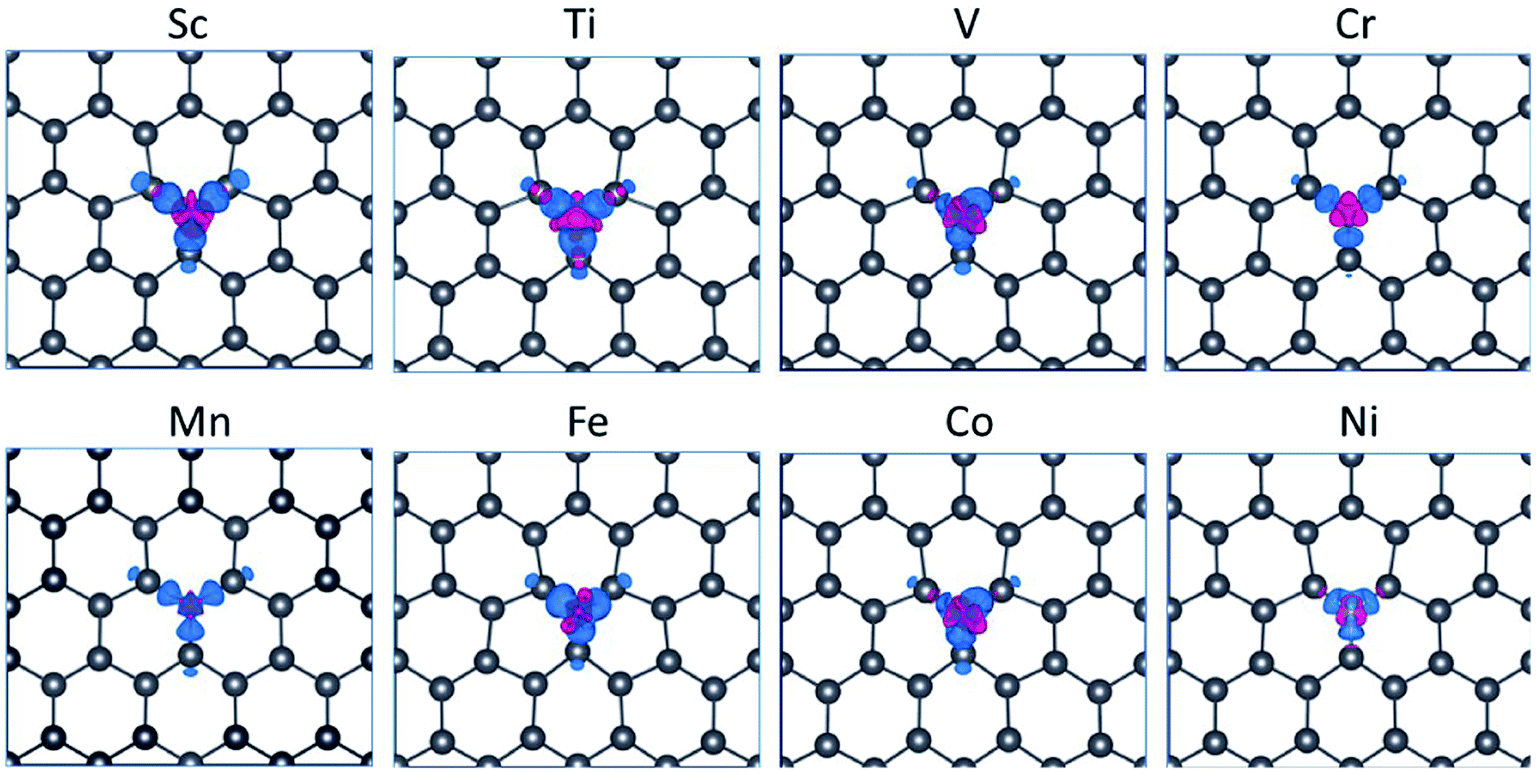 | ||
| Fig. 3 Charge density difference of plumbene doped by TM atoms with an isosurface value of 0.004e Å−3, where e is the elementary electron charge. Blue and red regions denote loss (depletion) and gain (accumulation) of charges, respectively. | ||
Additionally, to check the thermal stability of TM-doped systems, we performed ab initio molecular dynamics (MD) simulations at RT. As an example, we focused on the Mn case as it has the lowest binding energy. These simulations showed that the structure of Mn-doped system does not collapse throughout the MD simulation at 300 K, and this system can maintain its structural integrity at this temperature. Since this stability was checked for the Mn case, TM-doped systems with higher binding energies are expected to be even more stable.
The calculated band structures (Fig. 4) of 3.1% concentration single TM-doped systems show that there are impurity states that cross the Fermi level. This indicates the metallic nature of TM-substituted systems with the exception of Fe and Ni. The band gaps are calculated to be 0.03 eV for Fe and 0.19 eV for Ni, respectively.
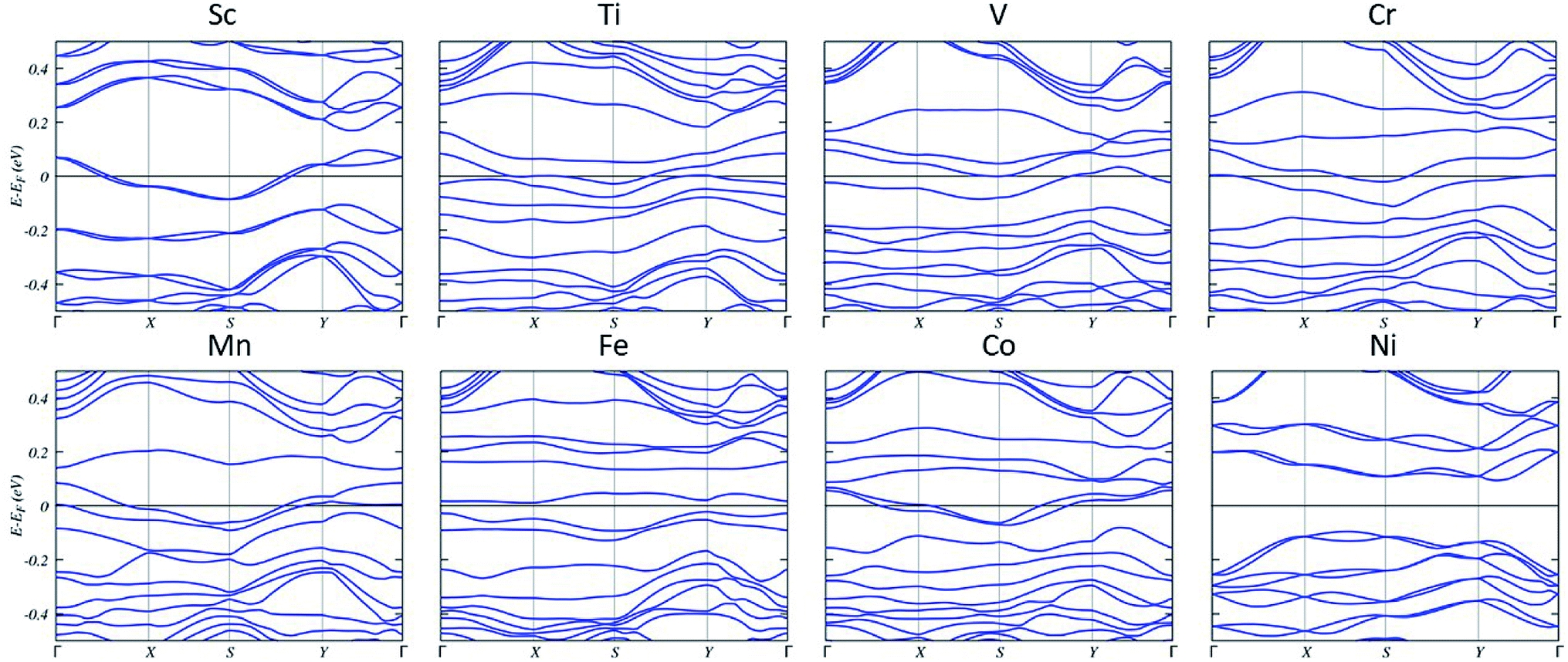 | ||
| Fig. 4 Band structures of TM-doped plumbene systems. The fermi energy is set at zero. | ||
The DOS and projected DOS (PDOS) of TM-doped systems (Fig. 5) show that the energy splitting between the TM 3d peaks is large, signifying that magnetism mainly originates from TM impurities. Several resonance peaks can be found in the DOS, which is caused by the strong hybridization between the 3d orbitals of TM and 3p orbitals of Pb.
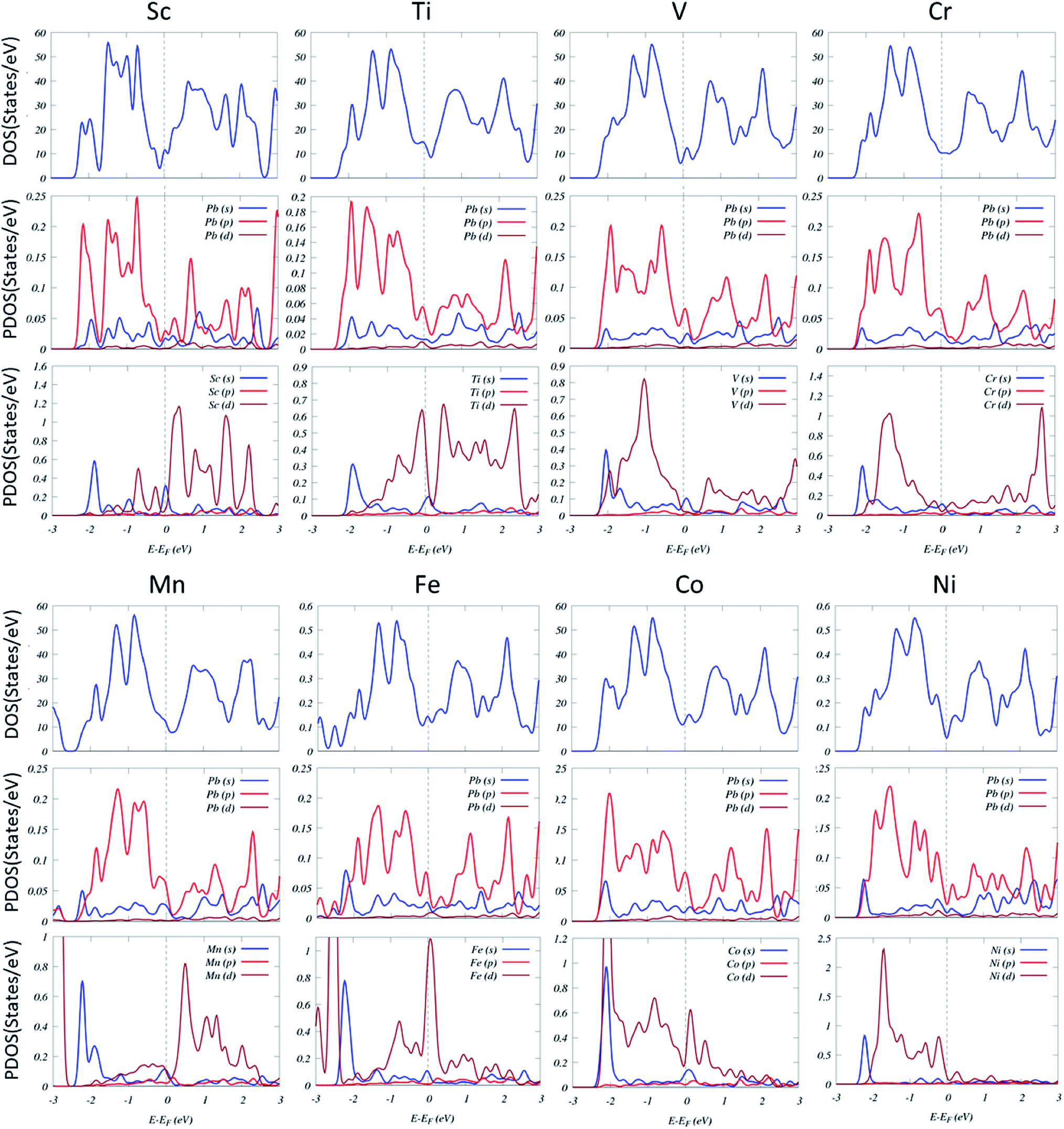 | ||
| Fig. 5 DOS and projected DOS of the TM-substituted systems. The Fermi level has been set to zero and indicated by a vertical gray dashed line. | ||
B. Magnetic properties
First-principles calculations suggest that doping with Ti, V, Cr, Mn, Fe, and Co results in magnetic states, while no magnetic solutions are found for Sc and Ni-doped plumbene systems. Particularly, the impurities from V to Co have magnetic states due to more valence electrons than Pb and an open shell structure. Isolated transition metal atoms have a large spin and orbital magnetic moments according to Hund's rules. However, electron delocalization and crystal field effects compete with intra-atomic Coulomb interactions leading to an overall reduction of spin moments as well as quenching of orbital moments in TM impurities dissolved in nonmagnetic hosts. The absolute values of spin magnetic moments are 1.32 μB, 2.79 μB, 3.98 μB, 4.10 μB, 2.95 μB, and 1.52 μB for Ti, V, Cr, Mn, Fe, and Co, respectively, as shown in Table 2. Unlike graphene, plumbene doped with TM impurities has a fractional magnetic moment due to a partially occupied 4s orbital, which is unoccupied for TM-substituted graphene. The orbital magnetic moments of the TM-doped systems are 0.18 μB for Ti, 0.32 μB for V, 0.04 μB for Cr, 0.04 μB for Mn, 0.49 μB for Fe and 0.22 μB for Co, which display a minimum value at the middle of the 3d series.The pd hybridization in TM-doped plumbene is characterized by long-range and anisotropic interactions between TM defects as listed on Table 2 and shown in Fig. 6. The TM impurity concentration is 1.79% in this case. The energy difference between parallel (E↑↑) and antiparallel spins (E↑↓) is exchange coupling. The spins are oriented along the corresponding preferential orientation (easy axis) in each case.
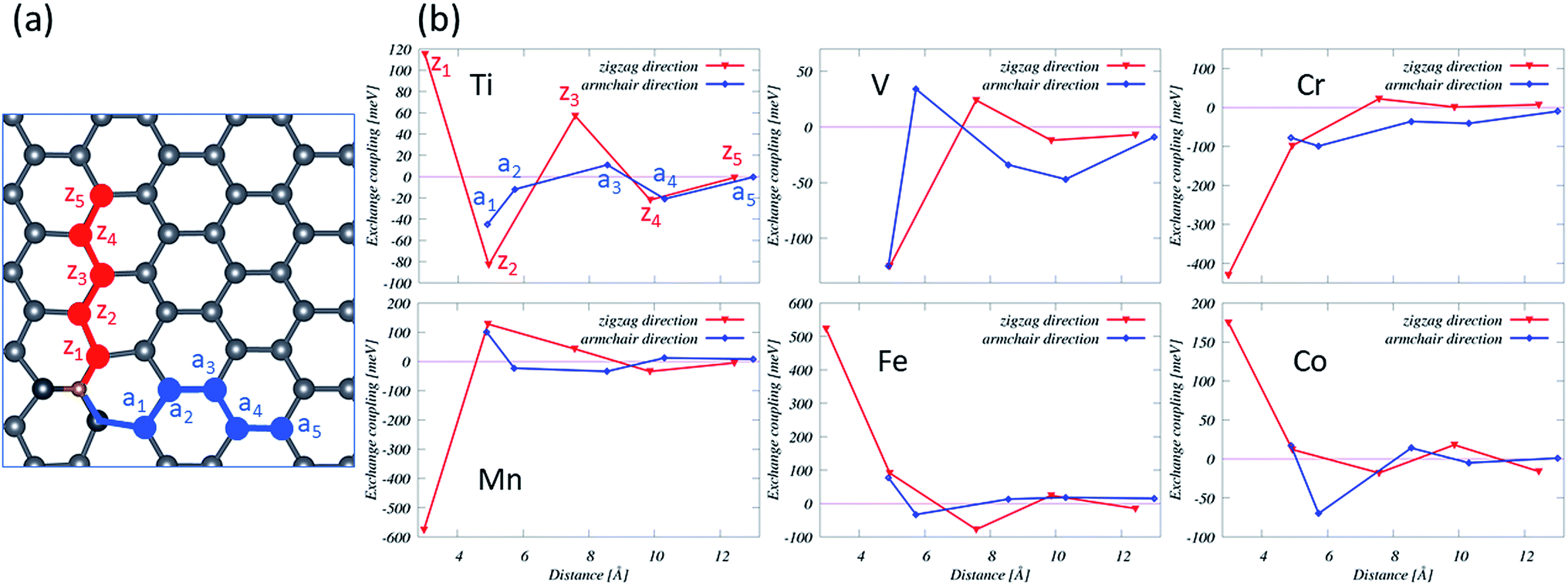 | ||
| Fig. 6 (a) Schematic representation of TM impurity (yellow atom) doped in plumbene in sites along the zigzag (zi) and the armchair (aj) directions. (b) Energy differences between antiparallel (E↓↑) and parallel (E↑↑) spins as a function of the distance. The solid lines between symbols are a guide to the eye. zi and aj are noted on the Ti plot as an example. | ||
For the nearest-neighbor interactions (z1), spins align parallel for Ti, Fe, and Co and align antiparallel for Mn, Cr with the exchange couplings being very strong for Cr (−430 eV), Mn (−576 eV), and Fe (523 eV). Magnetic configurations of V could not be stabilized in this case. For the next nearest neighbor interaction (z2 and a1), spins align parallel for Mn, Fe, and Co and align antiparallel for Ti, V, and Cr. For the third and higher-order nearest-neighbor interactions, the inter-impurity exchange couplings of the TM across the plumbene oscillate and decay with the inter-impurity separation. These couplings are not typically strong. These positive and negative exchange couplings as a function of the distance lead to ferromagnetic (FM) and antiferromagnetic (AF) orderings. We note that the same magnetic orderings were obtained using the HSE methods for a Fe-doped system with a relatively short distance between the Fe impurities.
We chose the Mn-substituted system as a typical example, as shown in Fig. 7. These magnetic orders are shown by spin density oriented along the +z (blue) and the −z (red) directions, respectively. The spin density suggests that the magnetism originates from the TM atom, and the spin density is slightly induced in the nearest Pb atoms. There is a strong interaction for short distance between the two Mn atoms, resulting in orbital reconstruction, which causes no isolated unpaired electrons.
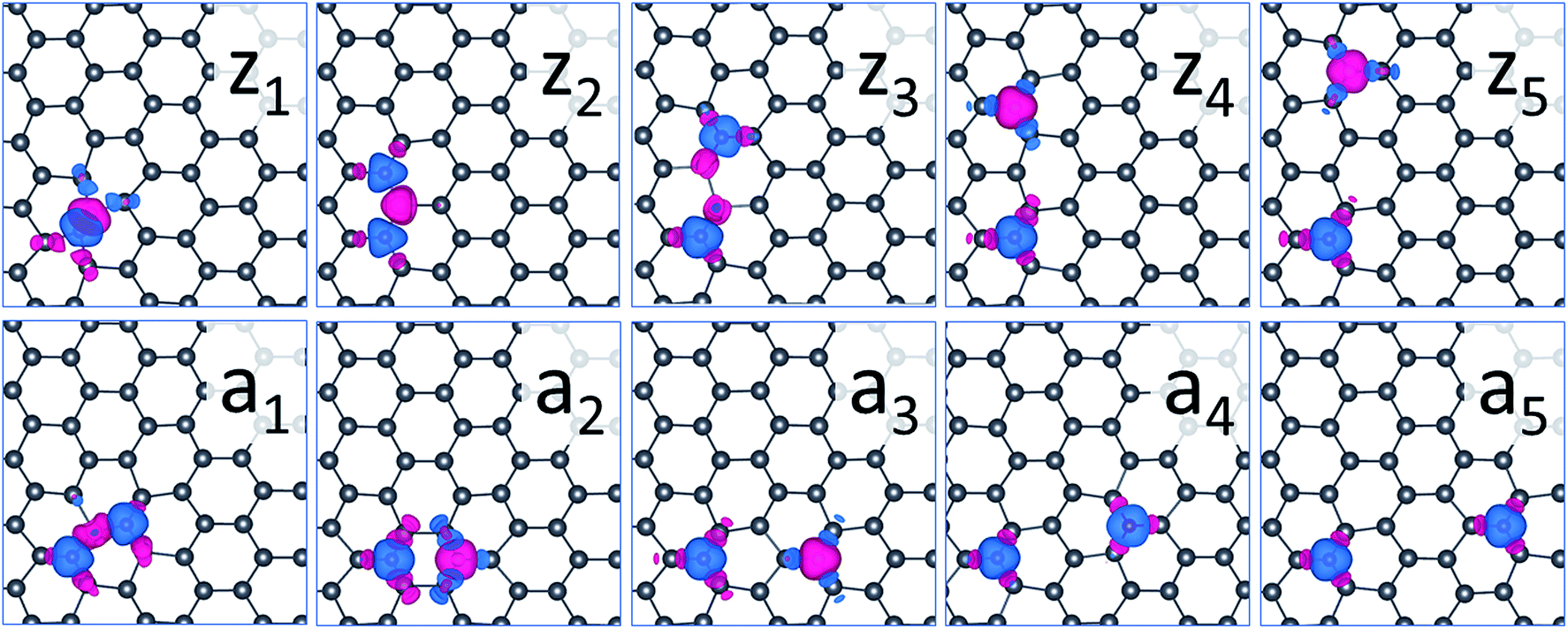 | ||
| Fig. 7 Magnetization density for ground state systems consisting two Mn impurities doped in plumbene at sites along the zigzag (zi) and the armchair (aj) directions. The blue and red colors represent spin (oriented along the z and −z directions) densities with an isosurface value of 0.003 μB Å−3, where μB is the Bohr magneton. | ||
It is worth noting that some metals, instead of substitutional doping, may tend to diffuse on 2D materials and form clusters.57 In this case, an ensemble of single domain clusters or nanoparticles becomes a paramagnet as each independent particle has an effective magnetic moment which eventually leads to superparamagnetism.58 This superparamagnet system behaves as an ensemble of non-interacting huge magnetic moments with no effective magnetic anisotropy.59
C. Transition temperature
We can estimate the Curie temperature (TC) in the mean-field approximation (MFA) via
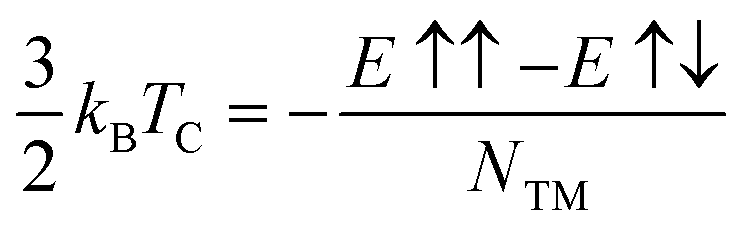 | (2) |
As listed on Table 2, the TC above RT are estimated to be about 445 K for Ti (z1), 495 K for Mn (z2), 391 K for Mn (a1), 2023 K for Fe (z1), 352 K for Fe (z2), 677 K for Co (z1), respectively. These high TCs can be traced back to the high values of the exchange couplings for these systems.
To confirm the MFA results above, the TC of Fe (z2) was further determined using Monte Carlo (MC) simulations as it has the lowest TC among the calculated TCs above RT. These simulations are used to evaluate the stability of 2D magnetic semiconducting60–62 or conducting43,63 systems.
TC is primarily determined by the strength of the exchange coupling and influenced by the magnetic anisotropy energy (MAE). The latter is not taken into account by the MFA, but it is by the MC. In agreement with the Mermin–Wagner theorem,64 we observed no magnetic ordering for a vanishing MAE. Introducing the MAE stabilizes the moments against thermal fluctuation and, thus, leads to a TC.
The TC was determined to be 300 K using the magnetic susceptibility with an MAE of 5.0 meV. The MFA overestimates the TC by about 15%. Based on this calculation, a similar overestimation of TC may be assumed for other TM-doped systems. This assumption does not decrease the TCs mentioned above to lower than RT. Our preliminary non-collinear DFT calculations suggest a colossal MAE for the TM-doped plumbene systems, which are significantly larger than 5.0 meV. An increase of the MAE will raise the TC. Therefore even higher TCs are expected for these systems.
IV. Conclusions
Calculations within the DFT framework suggest that with a proper choice of TM doped in plumbene, we can control its electronic and magnetic properties. The spin polarization is found in Ti, V, Cr, Mn, Fe, and Co-doped systems. The long-range and anisotropic interactions between two TM impurities in plumbene with an impurity concentration of 1.79% were calculated. The Curie temperature above RT was estimated for Ti, Mn, Fe, and Co-doped systems, demonstrating the potential for spintronics applications.Conflicts of interest
There are no conflicts to declare.References
- S. Cahangirov, M. Topsakal, E. Aktürk, H. Şahin and S. Ciraci, Two- and one-dimensional honeycomb structures of silicon and germanium, Phys. Rev. Lett., 2009, 102, 236804 CrossRef CAS PubMed.
- C.-C. Liu, W. Feng and Y. Yao, Quantum spin hall effect in silicene and two-dimensional germanium, Phys. Rev. Lett., 2011, 107, 076802 CrossRef PubMed.
- C.-C. Liu, H. Jiang and Y. Yao, Low-energy effective hamiltonian involving spin-orbit coupling in silicene and two-dimensional germanium and tin, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 195430 CrossRef.
- P. Tang, P. Chen, W. Cao, H. Huang, S. Cahangirov, L. Xian, Y. Xu, S.-C. Zhang, W. Duan and A. Rubio, Stable two-dimensional dumbbell stanene: A quantum spin hall insulator, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 121408 CrossRef.
- Y. Ren, Z. Qiao and Q. Niu, Topological phases in two-dimensional materials: a review, Rep. Prog. Phys., 2016, 79, 066501 CrossRef PubMed.
- S.-S. Li, W.-X. Ji, S.-J. Hu, C.-W. Zhang and S.-S. Yan, Effect of amidogen functionalization on quantum spin hall effect in Bi/Sb(111) films, ACS Appl. Mater. Interfaces, 2017, 9, 41443–41453 CrossRef CAS PubMed.
- Y.-P. Wang, W.-X. Ji, C.-W. Zhang, P. Li, S.-F. Zhang, P.-J. Wang, S.-S. Li and S.-S. Yan, Two-dimensional arsenene oxide: A realistic large-gap quantum spin hall insulator, Appl. Phys. Lett., 2017, 110, 213101 CrossRef.
- R.-W. Zhang, C.-W. Zhang, W.-X. Ji, P. Li, P.-J. Wang, S.-S. Li and S.-S. Yan, Silicon-based chalcogenide: Unexpected quantum spin hall insulator with sizable band gap, Appl. Phys. Lett., 2016, 109, 182109 CrossRef.
- Q. H. Wang, Z. Jin, K. K. Kim, A. J. Hilmer, G. L. C. Paulus, C.-J. Shih, M.-H. Ham, J. D. Sanchez-Yamagishi, K. Watanabe, T. Taniguchi, J. Kong, P. Jarillo-Herrero and M. S. Strano, Understanding and controlling the substrate effect on graphene electron-transfer chemistry via reactivity imprint lithography, Nat. Chem., 2012, 4, 724–732 CrossRef CAS PubMed.
- M. Ali, X. Pi, Y. Liu and D. Yang, Electronic and magnetic properties of graphene, silicene and germanene with varying vacancy concentration, AIP Adv., 2017, 7, 045308 CrossRef.
- O. V. Yazyev, Emergence of magnetism in graphene materials and nanostructures, Rep. Prog. Phys., 2010, 73, 056501 CrossRef.
- J. H. Seol, I. Jo, A. L. Moore, L. Lindsay, Z. H. Aitken, M. T. Pettes, X. Li, Z. Yao, R. Huang, D. Broido, N. Mingo, R. S. Ruoff and L. Shi, Two-dimensional phonon transport in supported graphene, Science, 2010, 328, 213–216 CrossRef CAS PubMed.
- J. Yuhara, B. He, N. Matsunami, M. Nakatake and G. Le Lay, Graphene's latest cousin: Plumbene epitaxial growth on a “nano watercube”, Adv. Mater., 2019, 31, 1901017 CrossRef PubMed.
- D. K. Das and S. K. Singh, Plumbene: A new 2d-material resembling graphene, in Advances in Industrial and Production Engineering, Springer, Singapore, 2019, pp. 193–197 Search PubMed.
- X.-L. Yu, L. Huang and J. Wu, From a normal insulator to a topological insulator in plumbene, Phys. Rev. B, 2017, 95, 125113 CrossRef.
- Y. Li, J. Zhang, B. Zhao, Y. Xue and Z. Yang, Constructive coupling effect of topological states and topological phase transitions in plumbene, Phys. Rev. B, 2019, 99, 195402 CrossRef CAS.
- D. K. Das, J. Sarkar and S. Singh, Effect of sample size, temperature and strain velocity on mechanical properties of plumbene by tensile loading along longitudinal direction: A molecular dynamics study, Comput. Mater. Sci., 2018, 151, 196–203 CrossRef CAS.
- F. Herman, C. D. Kuglin, K. F. Cuff and R. L. Kortum, Relativistic corrections to the band structure of tetrahedrally bonded semiconductors, Phys. Rev. Lett., 1963, 11, 541–545 CrossRef.
- X.-L. Yu and J. Wu, Evolution of the topological properties of two-dimensional group iva materials and device design, Phys. Chem. Chem. Phys., 2018, 20, 2296–2307 RSC.
- H. Zhao, C.-W. Zhang, W.-X. Ji, R.-W. Zhang, S.-S. Li, S.-S. Yan, B.-M. Zhang, P. Li and P.-J. Wang, Unexpected giant-gap quantum spin hall insulator in chemically decorated plumbene monolayer, Sci. Rep., 2016, 6, 20152 CrossRef CAS PubMed.
- L. Zhang, H. Zhao, W. X. Ji, C. W. Zhang, P. Li and P. J. Wang, Discovery of a new quantum spin hall phase in bilayer plumbene, Chem. Phys. Lett., 2018, 712, 78–82 CrossRef CAS.
- T. Dietl, H. Ohno, F. Matsukura, J. Cibert and D. Ferrand, Zener model description of ferromagnetism in zinc-blende magnetic semiconductors, Science, 2000, 287, 1019–1022 CrossRef CAS PubMed.
- H. Ohno, D. Chiba, F. Matsukura, T. Omiya, E. Abe, T. Dietl, Y. Ohno and K. Ohtani, Electric-field control of ferromagnetism, Nature, 2000, 408, 944–946 CrossRef CAS PubMed.
- K. Sato, L. Bergqvist, J. Kudrnovský, P. H. Dederichs, O. Eriksson, I. Turek, B. Sanyal, G. Bouzerar, H. Katayama-Yoshida, V. A. Dinh, T. Fukushima, H. Kizaki and R. Zeller, First-principles theory of dilute magnetic semiconductors, Rev. Mod. Phys., 2010, 82, 1633–1690 CrossRef CAS.
- T. Dietl, A ten-year perspective on dilute magnetic semiconductors and oxides, Nat. Mater., 2010, 9, 965 CrossRef CAS PubMed , review article.
- J. M. D. Coey, M. Venkatesan and C. B. Fitzgerald, Donor impurity band exchange indilute ferromagnetic oxides, Nat. Mater., 2005, 4, 173–179 CrossRef CAS PubMed.
- I. Žutić, J. Fabian and S. Das Sarma, Spintronics: Fundamentals and applications, Rev. Mod. Phys., 2004, 76, 323–410 CrossRef.
- A. Hashmi and J. Hong, Transition metal doped phosphorene: First-principles study, J. Phys. Chem. C, 2015, 119, 9198–9204 CrossRef CAS.
- T. Hu and J. Hong, First-principles study of metal adatom adsorption on black phosphorene, J. Phys. Chem. C, 2015, 119, 8199–8207 CrossRef CAS.
- Y. C. Cheng, Z. Y. Zhu, W. B. Mi, Z. B. Guo and U. Schwingenschlögl, Prediction of two-dimensional diluted magnetic semiconductors: Doped monolayer MoS2 systems, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 100401 CrossRef.
- L. Seixas, A. Carvalho and A. H. Castro Neto, Atomically thin dilute magnetism in co-doped phosphorene, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 155138 CrossRef.
- J. He, K. Wu, R. Sa, Q. Li and Y. Wei, Magnetic properties of nonmetal atoms absorbed MoS2 monolayers, Appl. Phys. Lett., 2010, 96, 082504 CrossRef.
- M. Sun, W. Tang, Q. Ren, Y. Zhao, S. Wang, J. Yu, Y. Du and Y. Hao, Electronic and magnetic behaviors of graphene with 5d series transition metal atom substitutions: A first-principles study, Phys. E, 2016, 80, 142–148 CrossRef CAS.
- Y. Luo, S. Wang, S. Li, Z. Sun, J. Yu, W. Tang and M. Sun, Transition metal doped puckered arsenene: Magnetic properties and potential as a catalyst, Phys. E, 2019, 108, 153–159 CrossRef CAS.
- Y. C. Cheng, Q. Y. Zhang and U. Schwingenschlögl, Valley polarization in magnetically doped single-layer transition-metal dichalcogenides, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 155429 CrossRef.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for open-shell transition metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- I. G. Rau, S. Baumann, S. Rusponi, F. Donati, S. Stepanow, L. Gragnaniello, J. Dreiser, C. Piamonteze, F. Nolting, S. Gangopadhyay, O. R. Albertini, R. M. Macfarlane, C. P. Lutz, B. A. Jones, P. Gambardella, A. J. Heinrich and H. Brune, Reaching the magnetic anisotropy limit of a 3d metal atom, Science, 2014, 344, 988–992 CrossRef CAS PubMed.
- C. F. Hirjibehedin, C.-Y. Lin, A. F. Otte, M. Ternes, C. P. Lutz, B. A. Jones and A. J. Heinrich, Large magnetic anisotropy of a single atomic spin embedded in a surface molecular network, Science, 2007, 317, 1199–1203 CrossRef CAS PubMed.
- H. Hashemi, W. Hergert and V. S. Stepanyuk, Magnetic states of M-Fe wires (M = Sc–Ni) on vicinal Cu(111) from first principles, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 104418 CrossRef.
- H. Hashemi, G. Fischer, W. Hergert and V. S. Stepanyuk, Magnetic properties of 3d transition metal wires on vicinal Cu(111) surfaces at finite temperature, J. Appl. Phys., 2010, 107, 09E311 CrossRef.
- H. Hashemi, A. Bregman, H. S. Nabi and J. Kieffer, Magnetic anisotropy energies of M-Fe wires (M = V-Co) on vicinal Cu(111), RSC Adv., 2016, 6, 108948–108954 RSC.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Hybrid functionals based on a screened coulomb potential, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Erratum: “hybrid functionals based on a screened coulomb potential” [J. Chem. Phys. 118, 8207 (2003)], J. Chem. Phys., 2006, 124, 219906 CrossRef.
- J. Heyd and G. E. Scuseria, Efficient hybrid density functional calculations in solids: Assessment of the Heyd–Scuseria–Ernzerhof screened coulomb hybrid functional, J. Chem. Phys., 2004, 121, 1187–1192 CrossRef CAS PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the exchange screening parameter on the performance of screened hybrid functionals, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, A grid-based bader analysis algorithm without lattice bias, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, Improved grid-based algorithm for bader charge allocation, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- K. Momma and F. Izumi, VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- J. P. Perdew and M. Levy, Physical content of the exact Kohn-Sham orbital energies: Band gaps and derivative discontinuities, Phys. Rev. Lett., 1983, 51, 1884–1887 CrossRef CAS.
- L. J. Sham and M. Schlüter, Density-functional theory of the energy gap, Phys. Rev. Lett., 1983, 51, 1888–1891 CrossRef.
- P. Mori-Sánchez, A. J. Cohen and W. Yang, Localization and delocalization errors in density functional theory and implications for band-gap prediction, Phys. Rev. Lett., 2008, 100, 146401 CrossRef PubMed.
- K. Choudhary, I. Kalish, R. Beams and F. Tavazza, High-throughput identification and characterization of two-dimensional materials using density functional theory, Sci. Rep., 2017, 7, 5179 CrossRef PubMed.
- A. Maiti and A. Ricca, Metal–nanotube interactions – binding energies and wetting properties, Chem. Phys. Lett., 2004, 395, 7–11 CrossRef CAS.
- W. C. Elmore, The magnetization of ferromagnetic colloids, Phys. Rev., 1938, 54, 1092–1095 CrossRef CAS.
- Magnetism goes nano: electron correlations, spin transport, molecular magnetism, ed. S. Blügel, T. Brückel and C. M. Schneider, Forschungszentrum, Zentralbibliothek, Jülich, 2005, vol. 26 Search PubMed.
- Y.-P. Wang, W.-X. Ji, C.-W. Zhang, P. Li, P.-J. Wang, B. Kong, S.-S. Li, S.-S. Yan and K. Liang, Discovery of intrinsic quantum anomalous hall effect in organic mn-dca lattice, Appl. Phys. Lett., 2017, 110, 233107 CrossRef.
- M.-H. Zhang, C.-W. Zhang, P.-J. Wang and S.-S. Li, Prediction of high-temperature chern insulator with half-metallic edge states in asymmetry-functionalized stanene, Nanoscale, 2018, 10, 20226–20233 RSC.
- S.-J. Zhang, C.-W. Zhang, S.-F. Zhang, W.-X. Ji, P. Li, P.-J. Wang, S.-S. Li and S.-S. Yan, Intrinsic dirac half-metal and quantum anomalous hall phase in a hexagonal metal-oxide lattice, Phys. Rev. B, 2017, 96, 205433 CrossRef.
- D. Hashemi, M. J. Waters, W. Hergert, J. Kieffer and V. S. Stepanyuk, Substrate-controlled magnetism: Fe nanowires on vicinal cu surfaces, Nanomaterials, 2020, 10, 159 CrossRef CAS PubMed.
- N. D. Mermin and H. Wagner, Absence of ferromagnetism or antiferromagnetism in one- or two-dimensional isotropic heisenberg models, Phys. Rev. Lett., 1966, 17, 1133–1136 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |