
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanically tough and highly stretchable poly(acrylic acid) hydrogel cross-linked by 2D graphene oxide
Stephen Don Sarkar ,
Md. Mosfeq Uddin,
Chanchal Kumar Roy,
Md. Jahangir Hossen,
Majharul Islam Sujan and
Md. Shafiul Azam*
,
Md. Mosfeq Uddin,
Chanchal Kumar Roy,
Md. Jahangir Hossen,
Majharul Islam Sujan and
Md. Shafiul Azam*
Department of Chemistry, Bangladesh University of Engineering and Technology (BUET), Dhaka-1000, Bangladesh. E-mail: mdshafiulazam@chem.buet.ac.bd; azam@ualberta.ca
First published on 17th March 2020
Abstract
The mechanical performances of hydrogels are greatly influenced by the functionality of cross-linkers and their covalent and non-covalent interactions with the polymer chains. Conventional chemical cross-linkers fail to meet the demand of large toughness and high extensibility for their immediate applications as artificial tissues like ligaments, blood vessels, and cardiac muscles in human or animal bodies. Herein, we synthesized a new graphene oxide-based two-dimensional (2D) cross-linker (GOBC) and exploited the functionality of the cross-linker for the enhancement of toughness and stretchability of a poly(acrylic acid) (PAA) hydrogel. The 2D nanosheets of GO were modified in such a way that they could provide multifunctional sites for both physical and chemical bonding with the polymer chains. Carboxylic acid groups at the surfaces of the GO sheets were coupled with the acrylate functional groups for covalent cross-linking, while the other oxygen-containing functional groups are responsible for physical cross-linking with polymers. The GOBC had been successfully incorporated into the PAA hydrogel and the mechanical properties of the GOBC cross-linked PAA hydrogel (PAA-GOBC) were investigated at various compositions of cross-linker. Seven times enhancement in both toughness and elongation at break has been achieved without compromising on the modulus for the as-synthesized PAA-GOBC compared to the conventional N,N′-methylenebis(acrylamide) (MBA) cross-linked PAA hydrogel. This facile and efficient way of GO modification is expected to lead the development of a high-performance nanocomposite for cutting-edge applications in biomedical engineering.
1. Introduction
Hydrogels are three-dimensional cross-linked porous matrices of hydrophilic polymers with extraordinary capability of accommodating a large volume of water. The ‘soft and wet’ nature of hydrogels along with the unique combination of stimuli-responsiveness and biocompatibility have made them very attractive for manifold applications in tissue engineering,1,2 drug delivery,3 sensing technology,4 biochemical separation5,6 and catalysis.7,8 3D printed artificial organs have recently been successfully produced from hydrogel materials.9 Gong et al. developed a new kind of hydrogel that can spontaneously adhere to defective bones generating the potential for use in the treatment of joints and soft tissues.10 Although notable success has been achieved in the preparation, modification, and development of hydrogels in recent years, one of the key obstacles that still remains is the inadequate or improper combination of mechanical properties of hydrogels. For instance, hydrogels with low toughness are frequently associated with very low stretchability causing a big challenge for the hydrogels to be useful in biomedical engineering applications.The mechanical performances of hydrogels are greatly influenced by the activity of cross-linkers. Conventional chemical cross-linkers like N,N′-methylenebis(acrylamide) (MBA) forms covalent bonds with polymer chains and maintain the integrity of hydrogels. However, it leads to the improvement in toughness and modulus compromising on the extensibility. Covalent cross-linkers restrict the movement of polymer chains in a definite zone for finite extensibility.11 Recently, it has been considered that the inclusion of physical cross-linking may lead to high toughness with high extensibility.12 The physical cross-linking provides weak frictional resistance in polymer chain movement and ensure continuous energy dissipation during deformation. Molecular ionic interaction, hydrogen bonding, host–guest interaction, etc. have been used for physical cross-linking. Polymeric dispersive interaction of the surface of nanomaterials is an alternative over the conventional molecular physical cross-linking in hydrogels. Extraordinary reinforcing ability to improve the mechanical behavior of hydrogels has been achieved by the formation of nanocomposites using nanoclay,13 silica nanoparticles,14,15 carbon nanotubes16 etc. but they are still lagging from a desired mechanical performance which can lead to practical applications. The major drawbacks include random bonding of polymer and cross-linker which incurs non-uniform stress distribution. Moreover, the nanocomposites are very susceptible to environmental factors like temperature, pressure, and solvents, etc. putting forward lots of scope for further improvement.
Graphene oxide (GO) has attracted great attention as a promising filler material to enhance different functional properties of nanocomposite hydrogels.7,17–25 The GO nanosheets offer extremely large 2D surface area containing a good abundance of polar functional groups (carboxylic, hydroxyl and epoxide groups) on its basal planes and edges.26 Due to the presence of different polar functional groups the surface of GO can readily interact with different kinds of hydrophilic polymers through dispersive interactions like hydrogen bonding, dipole–dipole interaction, ionic interaction, etc. In addition to the 2D surface area, GO can offer a good sliding zone with frictional interaction for the unidirectional movement of the polymer during elongation. This can effectively incur good toughness with high extensibility in hydrogels.24 Moreover, the inherent excellent mechanical strength and easy preparation method of GO have made them very attractive. A good number of research works are available for the efficient incorporation of GO into the hydrogel to obtain enhanced mechanical properties.24,25,27,28 The most common strategy followed for the incorporation of GO into the nanocomposite hydrogels has been the in situ polymerization of water-soluble monomers containing traditional covalent cross-linkers (like MBA). However, this strategy often achieved inadequate improvement in mechanical properties such as tensile strength and stretchability of the hydrogels. For instance, Ye et al. reported the fabrication of PAA-MBA-GO nanocomposite hydrogel with such kind of strategy and found elongation at break only four times greater than that of neat PAA.24 The physical interactions between polymer chains and GO nanosheets could not offer very large energy dissipation restricting its function mainly as supporting or reinforcing material. Alternate strategies are therefore required to satisfy the requirement of even larger toughness and longer elongation.
Herein, we propose a modification of GO with both physical and chemical anchoring sites for polymeric interaction. The synergistic role of chemical and physical cross-linking in a single platform of cross-linker may lead to the improvement of mechanical performances.29 Functionalization of GO sheets with the acrylic groups leads to a special graphene oxide-based cross-linker (GOBC) capable of forming covalent and physical cross-linking with polymers, simultaneously. The as-synthesized GOBC cross-linkers have been incorporated into the conventional polymeric gel of PAA, and the mechanical properties and swelling behavior of the hydrogels have been investigated with various degrees of cross-linker concentrations. This new strategy of GO modification paves the way for the development of high-performance nanocomposites required for cutting-edge applications in biomedical engineering, particularly for the generation of artificial tissues.
2. Experimental
2.1 Chemicals and materials
High-purity graphite flakes (assay 99% carbon basis, −325 mesh particle size), hydrogen peroxide (H2O2), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), 2-aminoethyl methacrylate hydrochloride (AEM), N,N′-methylenebis(acrylamide) (MBA), acrylic acid (AA), and cellulose membrane dialysis tubing (avg. diam. 16 mm, MWCO 14 kDa) were purchased from Sigma Aldrich. Sulfuric acid (H2SO4), nitric acid (HNO3), sodium nitrate (NaNO3), potassium permanganate (KMnO4), hydrochloric acid (HCl), sodium hydroxide (NaOH), chloroacetic acid (ClCH2COOH) and potassium persulfate (KPS) were procured from Merck, Germany. All the chemicals were analytical reagent grade and used as received. Aqueous solutions of all the experiments were prepared using deionized (DI) water.2.2 Preparation of GO nanosheet
Graphene oxide nanosheet was prepared from graphite flakes according to the modified Hummers' method.30–32 At first, graphite flakes (2 g) was transferred into a mixture of concentrated H2SO4 (12 mL) and HNO3 (8 mL). It was heated at 80 °C for 8 h. After cooling at room temperature, 500 mL of DI water was added to dilute the mixture and left overnight. The mixture was filtered and the product washed with DI water to remove acid residue. The oxidized graphite powder was then dried overnight at room temperature. The obtained dry powder (1 g) was then treated in a round-bottomed flask with a mixture of NaNO3 (1.03 g) and concentrated H2SO4 (62 g). Under continuous stirring, 4.5 g of KMnO4 was slowly added and the temperature of the mixture was kept below 20 °C. The reaction was terminated by the addition of DI water after 2 days. The filtered of the reaction mixture was washed with 1 M HCl and subsequently with DI water. Then the mixture was dialyzed against DI water for one week to remove ionic species with a semipermeable membrane tube having a molecular weight cut-off of 14 kDa. The final product was obtained by drying in a vacuum oven at 60 °C.2.3 Preparation of carboxylated graphene oxide (CGO)
Carboxylated GO (CGO) was prepared by converting hydroxyl (–OH) and epoxide (C–O–C) groups into carboxylic acid (–COOH) through the treatment of GO surface with chloroacetic acid under a strong basic condition.33,34 10 mL of GO suspension (2 mg mL−1) was sonicated for 1 h to obtain a stable dispersion. Subsequently, 1.2 g of NaOH and 1.0 g of ClCH2COOH were added to the GO suspension and sonicated for another 2 h. The resulting solution was neutralized by HCl solution (1 M) and dialyzed against DI water at room temperature for 3 days to remove the unreacted chemicals and impurities. The resulted CGO solution was vacuum dried overnight at 60 °C.2.4 Synthesis of GOBC
GOBC was synthesized by following the EDC coupling reaction.35 15 mg of the prepared CGO was sonicated in 15 mL of DI water for 1 h. 35 mg of EDC and 30 mg of NHS were added to the suspension and stirred for 2 h at 0 °C. Finally, 300 mg of AEM was added to the mixture and stirred at room temperature overnight. The unreacted chemicals and reagents were removed from the mixture by dialysis against DI water for 3 days. The obtained GOBC solution was used for cross-linking of acrylic acid polymer chains for the synthesis of 2D cross-linked PAA composite hydrogels.2.5 Fabrication of PAA composite hydrogel
Different composition of 2D cross-linked PAA hydrogels were formed by the free radical polymerization reaction of AA with GOBC in presence of KPS initiator. 2 mL of AA monomer (density = 1.008 g mL−1) was added to 1 mL of DI water in a glass tube and sonicated for 30 min. A specific volume of GOBC solution and 10 mg of KPS were mixed in 1 mL of DI water in a vial and sonicated for 30 min. The mixture of cross-linker and initiator was transferred to AA solution under vigorous stirring. The total mixture was poured into a glass mold consisting of two parallel glass plates separated by 2 mm Teflon spacer. The sample was heated at 58 °C for 4 h in an inert atmosphere for polymerization. After completing the polymerization, prepared gels were taken off the glass plates and stored for further studies of mechanical properties and swelling behavior. PAA hydrogels containing MBA and GO were also prepared following the same procedure for comparison.2.6 Characterization
The Fourier transform infrared (FTIR) spectra of GO, CGO and GOBC were recorded in the range of 4000–400 cm−1 by preparing KBr pellet with samples in a Shimadzu FTIR-8400 instrument. UV-Vis absorption spectra were carried out in a UV/visible spectrophotometer (Shimadzu-1800). Thermogravimetric analyses (TGA) and differential scanning calorimetry (DSC) were performed using a NETZSCH STA 449F3 instrument. The samples were heated at a temperature increase rate of 10 °C min−1 from room temperature to 800 °C under the nitrogen atmosphere. The surface morphology of prepared cross-linkers and hydrogels was studied using field emission scanning electron microscope (FESEM, JSM-7600F, Tokyo, Japan) at a voltage of 10.0 kV. The samples for FESEM were completely freeze-dried before performing the experiment. X-ray diffraction (XRD) patterns of freeze-dried samples were recorded by an X-ray diffractometer (PANalytical Empyrean) using a Cu X-ray source (wavelength: Kα1 = 1.540598 Å and Kα2 = 1.544426 Å) in the 2θ range of 10° to 80°.2.7 Mechanical tests
All PAA composite hydrogels were cut into pieces of rectangular shape with 10 mm length, 4 mm width and 2 mm thickness for tensile measurements. The tensile measurements were conducted in a universal testing machine (TestResources, 100P250-12 System) at a crosshead speed of 300 mm min−1. The stress was calculated following the equation σ = F/A, where F is the recorded load and A is the cross-section area of the specimen. The strain of the samples was measured from the ratio of elongation of sample length (Δl) with that of initial length (l0), ε = Δl/l0. The Young's modulus was calculated from the initial linear region of the stress–strain curves. The toughness of each specimen was calculated by integrating the area underneath the stress–strain curve before fracture.2.8 Swelling ratio measurements
Small pieces of the as-prepared hydrogel of approximately similar weight were immersed in a large amount of DI water. The gel was freely swelled at room temperature to reach the swelling equilibrium. The water of gel samples was continuously changed at 12 h duration. The swelling ratio of the samples was calculated using the following equation:| SR = (Wt − Wd)/Wd |
3. Results and discussion
3.1 Synthesis and characterization of GO, CGO, GOBC
The main focus of this research was to fabricate a GO-based 2D cross-linker with the multifunctional cross-linking ability and investigate the influence of the cross-linker on the mechanical properties of PAA hydrogels. Accordingly, GO was prepared from graphite flakes via a modified Hummers' method. GO was converted into CGO via a reaction with chloroacetic acid in basic medium resulting in an increased number of –COOH groups on GO surface. To introduce vinyl groups over the surface, the carboxylic acid groups of CGO were functionalized using 2-aminoethyl methacrylate hydrochloride. The free-radical polymerization reaction was employed to prepare PAA composite hydrogels by cross-linking PAA polymer chains by GOBC in presence of KPS initiator. The schematic diagram of the stepwise preparation of PAA composite hydrogel is presented in Fig. 1.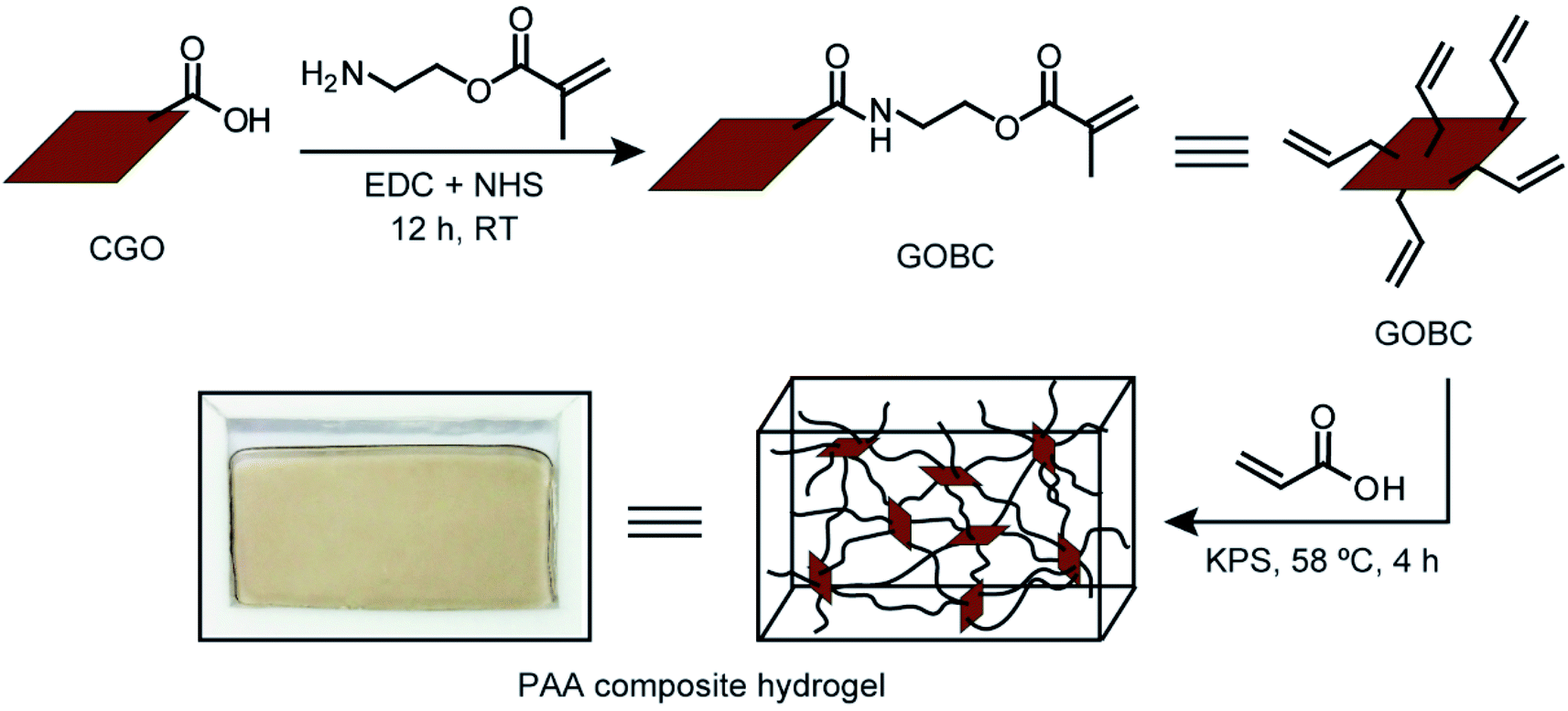 | ||
| Fig. 1 Schematic diagram of the preparation of GOBC cross-linked PAA composite hydrogel. | ||
The conversion of GO into GOBC was investigated by UV-Vis absorption spectroscopy. Absorption spectra of GO, CGO and GOBC were obtained from a dispersed solution of an equal concentration of each material and compared in Fig. 2a. The changes that appeared in the spectra are related to the modification of the GO matrix. As shown in Fig. 2a, the main characteristic peak for GO dispersion at 230 nm was attributed to the π–π* transitions of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and the shoulder at ∼300 nm was referred to the n–π* transitions of CO bonds. The overall feature is similar to the spectrum of GO synthesized using the conventional Hummers' method.36,37 For CGO dispersion, the main absorption peak at 230 nm remained unaltered; however, the peak at 300 nm disappeared. Moreover, CGO demonstrated much higher absorbance compared with GO dispersion. These observations are indicative of the activation of GO surface by converting the hydroxyl groups to carboxylic acid moieties under the basic medium.36,38,39 The red-shift of the peak from 230 nm to 250 nm for GOBC corresponds to the n–π* transitions occurred due to the formation of C–N bonds for amino functionalization of CGO.40,41
C bonds and the shoulder at ∼300 nm was referred to the n–π* transitions of CO bonds. The overall feature is similar to the spectrum of GO synthesized using the conventional Hummers' method.36,37 For CGO dispersion, the main absorption peak at 230 nm remained unaltered; however, the peak at 300 nm disappeared. Moreover, CGO demonstrated much higher absorbance compared with GO dispersion. These observations are indicative of the activation of GO surface by converting the hydroxyl groups to carboxylic acid moieties under the basic medium.36,38,39 The red-shift of the peak from 230 nm to 250 nm for GOBC corresponds to the n–π* transitions occurred due to the formation of C–N bonds for amino functionalization of CGO.40,41
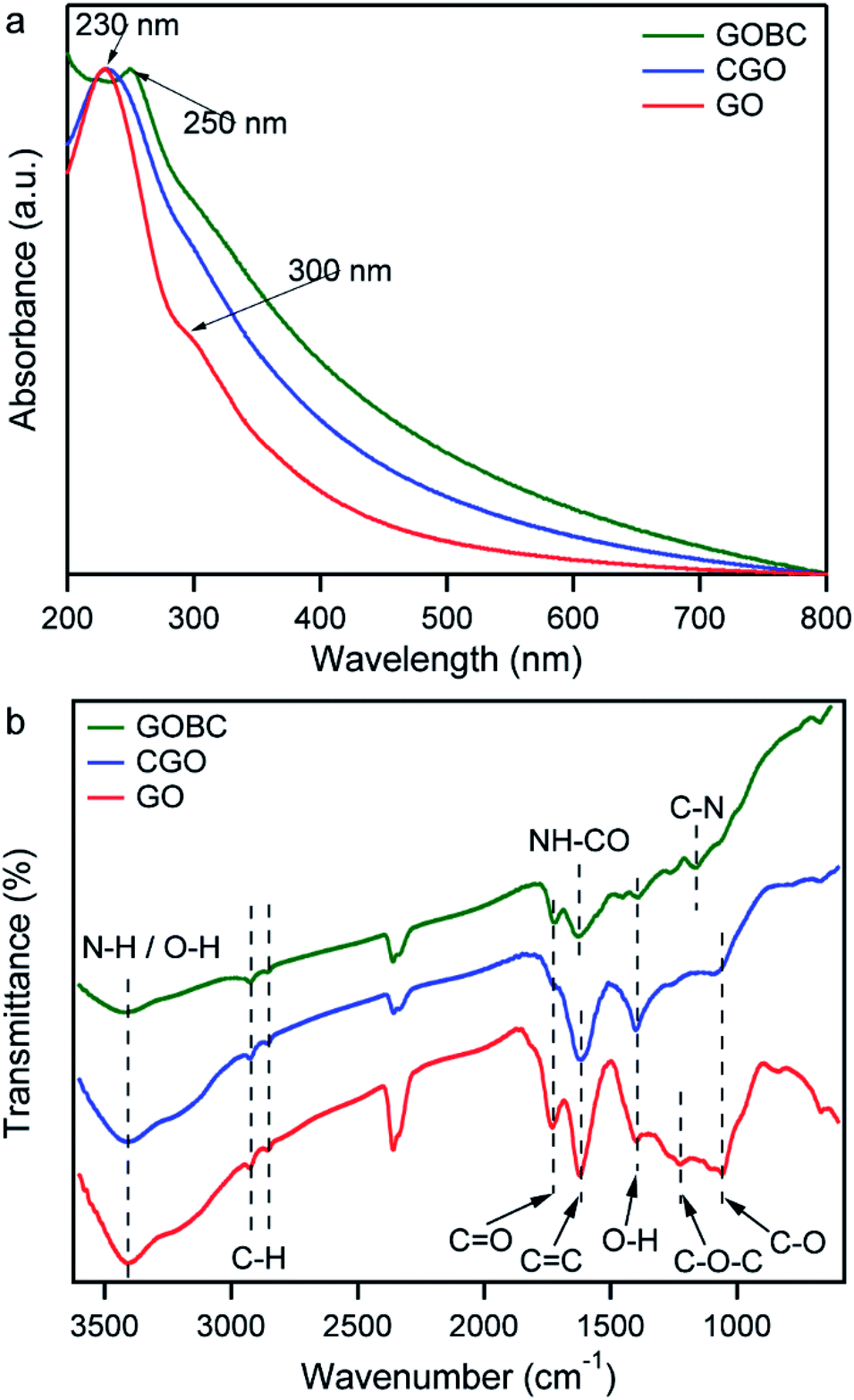 | ||
| Fig. 2 UV-Vis spectra (a), and FT-IR spectra (b) of GO (red), CGO (blue), and GOBC (green). | ||
The modifications of GO to CGO and GOBC were further investigated by the FT-IR spectra analysis. Fig. 2b displays the FT-IR spectra of GO, CGO, and GOBC providing strong evidence about the oxygen-containing functional groups. For GO, a broad peak at 3416 cm−1 and peak at 1732 cm−1 indicated the presence of O–H groups and CO bond of carboxylic acid groups, respectively. In-plane O–H bending mode appeared at 1397 cm−1 and the skeletal vibrations of aromatic CC were observed at 1620 cm−1. The peak at 1226 cm−1 was attributed to the presence of the C–O–C bond of epoxy groups. The stretching vibrations of alkoxy C–O occurred at 1055 cm−1.42–44 The spectral band information strongly validated the successful conversion of graphene oxide from graphite. The spectral differences between CGO and GO indicated the successful carboxylation of GO by the treatment of chloroacetic acid. Two major changes were observed in the spectrum of CGO. One was the reduction of intensity of the characteristic vibration band of C–O–C (1226 cm−1) and another was broadening of the absorption band of CO groups at 1732 cm−1.33,34,44 The shifting of the broad peak of O–H stretching vibration to lower wavenumber 3393 cm−1 indicated the formation of a large number of carboxyl groups on GO surface. The appearance of the peaks at 3408 cm−1, 1731 cm−1 and 1161 cm−1 in the spectrum of GOBC, were attributed to the stretching vibrations of N–H, CO, and C–N bonds, respectively, of methacrylate groups.30,41,42,45 The appearance of a new peak at 1652 cm−1 corresponded to stretching vibrations of NH–CO bond, which was formed by the coupling reaction of CGO with the AEM.30
TGA and DSC measurements were performed to investigate the thermal stability of GO, CGO, and GOBC. The obtained thermograms are presented in Fig. 3. Small weight losses below 100 °C for all samples were attributed to the evaporation of adhered moisture or some organic molecules over the surface of these materials. The major weight loss on the TGA curve of GO after 200 °C was associated with the thermal decomposition and rearrangements of oxygen-containing functional groups, which is supported by the DSC measurement showing a marked exothermic pick at 218 °C (Fig. 3a).34,46,47 The steady weight loss of GO in the range 300–800 °C might have been caused by the slow decomposition of the other functional groups. The thermal behavior of GO has been consistent with the previous reports.34,48 The decomposition temperature of oxygen-containing groups on TGA was shifted to a high temperature due to the conversion of GO into CGO (Fig. 3b).34 When the DSC curves of GO and CGO were compared, transformation of the sharp peak of 218 °C into broad peak yet again indicated the carboxylate surface functionalization of GO.34,49 Importantly, significant level of shifting of the decomposition temperature and gradual weight loss over a large range of temperature in the TGA curve of GOBC demonstrated high percentage of conversion of CGO to GOBC (Fig. 3c). DSC thermogram also supported the stability of GOBC over a wide range of temperatures. The enhancement of the thermal stability of GOBC was due to the successful attachment of large functional groups over the CGO surface.42,48
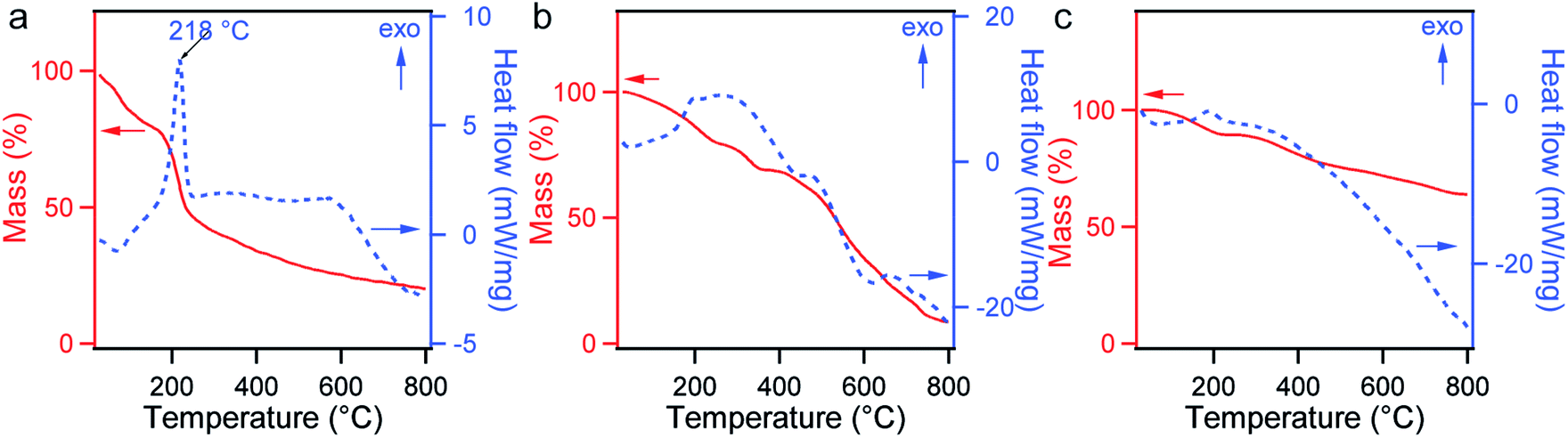 | ||
| Fig. 3 TGA (solid red lines) and DSC (dashed blue lines) curves of GO (a), CGO (b), and GOBC (c). | ||
The surface morphology of cross-linkers plays an important role in their functionalities while interacting with the polymer chains. The surface morphologies of GO, CGO, and GOBC were observed by FESEM and the microscopic images are presented in Fig. 4. The surface of GO had an irregular lamellar structure and maintained a layer to layer distance (Fig. 4a). This happened for the exfoliation of the sheet-like structure of graphite during the conversion into GO. The layer by layer structure suggested the intercalation of oxide functional groups (hydroxyl, carboxyl, and epoxide) which were covalently bonded to graphite surface. It is expected that each layer was connected with other closely due to the strong dipole–dipole interactions.34,50 The rough surface and irregular wrinkles of CGO (Fig. 4b) might have resulted during the carboxylation of GO.33 The lamellar morphology of GO was almost disappeared from the CGO in the event of exfoliation and sonication. Extensively exfoliated multi-layered porous structures were clearly visible for GOBC in Fig. 4c. The amino-functionalized graphene layers were irregularly folded to each other and formed tangled patches.35,42
 | ||
| Fig. 4 FESEM images of GO (a), CGO (b), and GOBC (c). | ||
3.2 Characterization of PAA-GOBC composite hydrogels
The in situ polymerization technique was employed to prepared PAA composite hydrogel because this technique enabled control over both the polymer architecture and the structure of the composite. To investigate the effect of GOBC as cross-linker on the mechanical behavior hydrogels, different PAA hydrogels with various concentrations of GOBC were prepared. FTIR spectrum of PAA-GOBC-0.05 nanocomposite hydrogel is shown in Fig. 5a. The peak at 3453 cm−1 was assigned to the O–H groups and a weak shoulder at 2924 cm−1 was attributed to C–H stretching vibrations.51 The peak at 1654 cm−1 corresponded to the CO stretching vibrations of acrylate units.52 The peak at 1380 cm−1 was due to the C–OH bond of carboxylic acid and the presence of C–N bonds of GOBC was observed at 1101 cm−1.42 XRD patterns of neat PAA and PAA-GOBC-0.05 are presented in Fig. 5b. The broad 2θ peak of neat PAA ranging from 15° to 30° corresponded to the well-known amorphous feature of cross-linked polymeric structure.52 Very similar XRD patterns of PAA-GOBC-0.05 and neat PAA supported the amorphous nature of both hydrogels. The absence of any notable change in the XRD patterns of PAA-GOBC-0.05 from neat PAA also demonstrated the uniform distribution of GOBC in the PAA matrix without any agglomeration, which is consistent with previous reports.52,53 The porosity of PAA-GOBC-0.05 hydrogel was investigated by FESEM analysis of the freeze-dried PAA-GOBC-0.05 hydrogel after swelling in DI water for 24 h. Sponge-like interconnected micropores were clearly evident (Fig. 5c) which is attributed to the removal of water molecules during freeze-drying.24 Most of the pore diameters were less than 100 μm and homogeneously distributed. A higher magnification image of the hydrogel exhibited a rod-shaped polymeric backbone, which might have resulted from the significant level of 2D surface cross-linking of GOBC (Fig. 5d). The functional groups present on the GOBC surface made the PAA polymer chains capable of forming strong physical and chemical interactions, which are responsible for its enhanced mechanical properties. Moreover, no visible level of aggregation was observed on the SEM images suggesting that the physical and chemical cross-linking formed by the GOBC with PAA was uniform.52
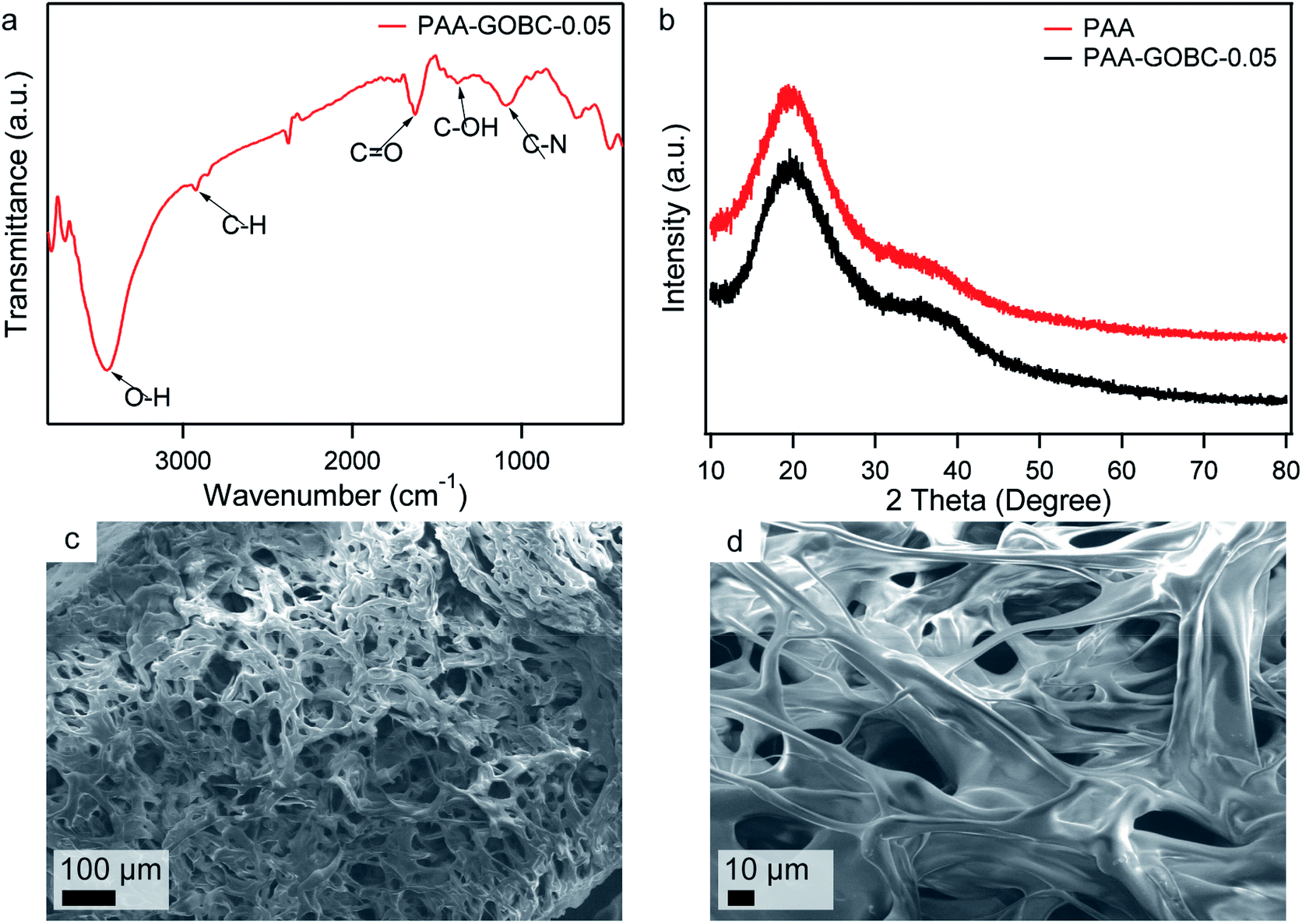 | ||
| Fig. 5 FTIR of freeze-dried PAA-GOBC-0.05 hydrogel (a), XRD patterns of neat PAA and PAA-GOBC-0.05 hydrogels (b), FESEM images of freeze-dried PAA-GOBC-0.05 hydrogel after swelling in DI water for 24 hours with 100× (c) and 500× (d) magnifications. | ||
3.3 Mechanical behavior of PAA composite hydrogels
The prepared PAA-GOBC hydrogels exhibited excellent tensile properties under external forces. The nanocomposite hydrogels possessed a high level of deformation (around 19 times) with remarkable flexibility without breaking as shown in Fig. 6b. The prepared hydrogel was also tough and ductile that it can be knotted (Fig. 6c) and was still highly stretchable even with that knot (Fig. 6d) indicating the presence of different types of physical and chemical bonds formed by GOBC with PAA polymer networks.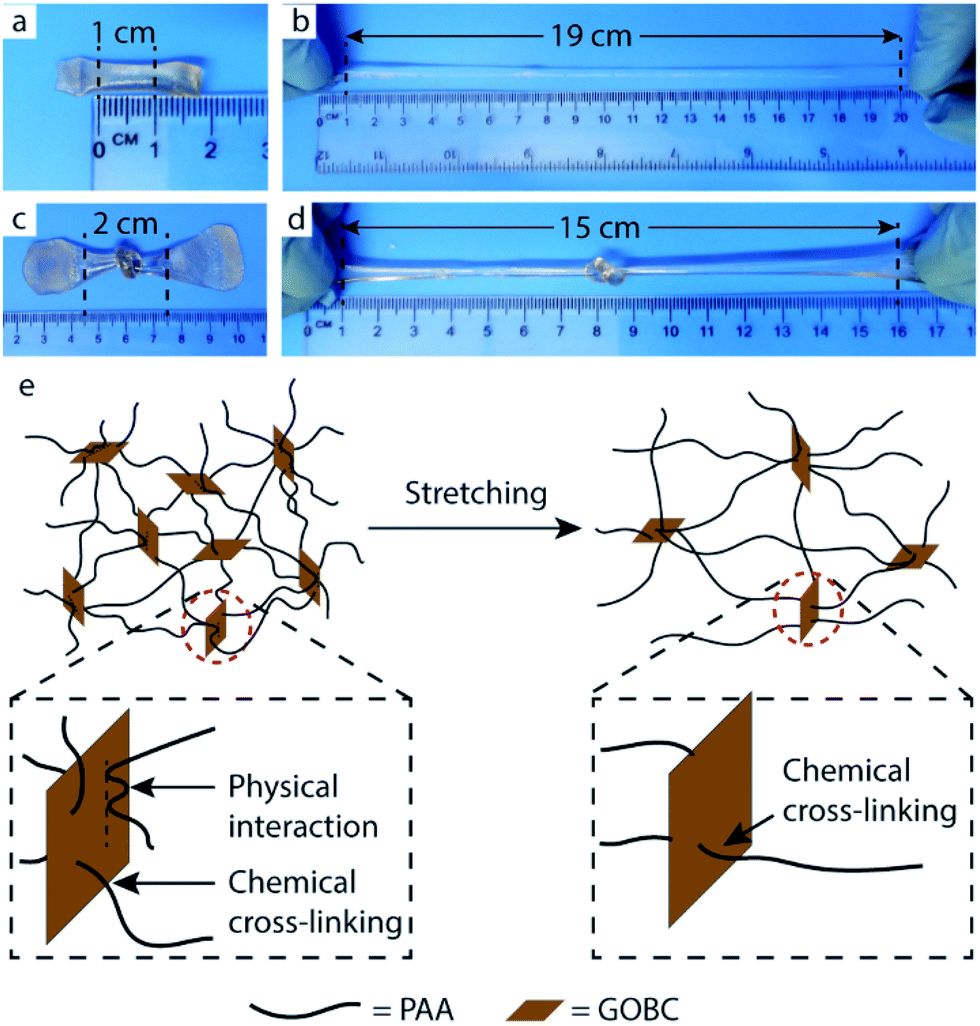 | ||
| Fig. 6 Photographs of high stretchability PAA-GOBC hydrogels to withstand 19 folds stretching (a and b), and 15 folds stretching from a knotted state (c and d), a schematic illustration of cross-linked polymer chains forming randomly entangled conformation, that dissipates deformation stress by chain disentanglement and desorption (e). | ||
Systematic tensile measurements were conducted to investigate the mechanical performance of PAA hydrogels influenced by the presence of GOBC cross-linker. Neat PAA, PAA-MBA, and GO reinforced PAA (PAA-GO) hydrogels were also prepared for comparison. Fig. 7a and b show the comparative stress–strain behavior of PAA hydrogels prepared with different types of cross-linkers and fillers. The measured mechanical parameters were determined from the stress–strain curves presented in Fig. 7c and d and summarized in Table 1. Compared to neat PAA hydrogel, all nanocomposite hydrogels (PAA-GO-0.05, PAA-MBA-0.05, and PAA-GOBC-0.05) exhibited greater Young's modulus, tensile strength, and toughness. Obviously, the PAA-GOBC hydrogels demonstrated exceptionally high elongation. PAA-GO-0.05 hydrogel showed significant improvement in elongation at break compared to neat PAA and PAA-MBA-0.05 with moderate enhancement in Young's modulus and tensile strength. This was due to the formation of physical and mechanical adhesive bonding offered by the chemical structure of GO. GO has a tendency to mechanically lock different kinds of polymers for its size and 2D shape, which is beneficial for the creation of load-bearing networks within the host polymer matrix.54 The large elongation can be explained by the fact that the network of PAA-GO-0.05 hydrogel was composed of soft and flexible PAA polymer chains impregnated with rigid GO sheets, which can ensure frictional energy dissipation during deformation.24 The physical interactions of GO and PAA chains include van der Waals interaction, ion-dipole, electrostatic interaction, etc. which had made the composites capable of this kind of sliding friction. Conversely, PAA-MBA-0.05 demonstrated a good tensile strength with very short elongation before breaking. This kind of hydrogels is elastic in nature due to the presence of large amount of covalent cross-linking and offer very small contribution to viscoelastic energy dissipation. Covalent cross-linking led to restricted elongation under tensile deformation.24 In the case of PAA-GOBC-0.05 hydrogel, exceptional high elongation along with large toughness had been obtained. GOBC cross-linker had been designed in such a way that it was capable of forming covalent bonds with the polymer matrix by methacrylate functional group in addition to physical cross-linking through dispersive interactions. Hence, diverse types of cross-linkings such as physical, covalent and mechanical were possible for PAA-GOBC-0.05 hydrogels simultaneously. Although different kinds of bonding interactions were present in the hydrogel but covalent bonding was dominating allowing the hydrogel for possessing an outstanding tensile strength before breaking.
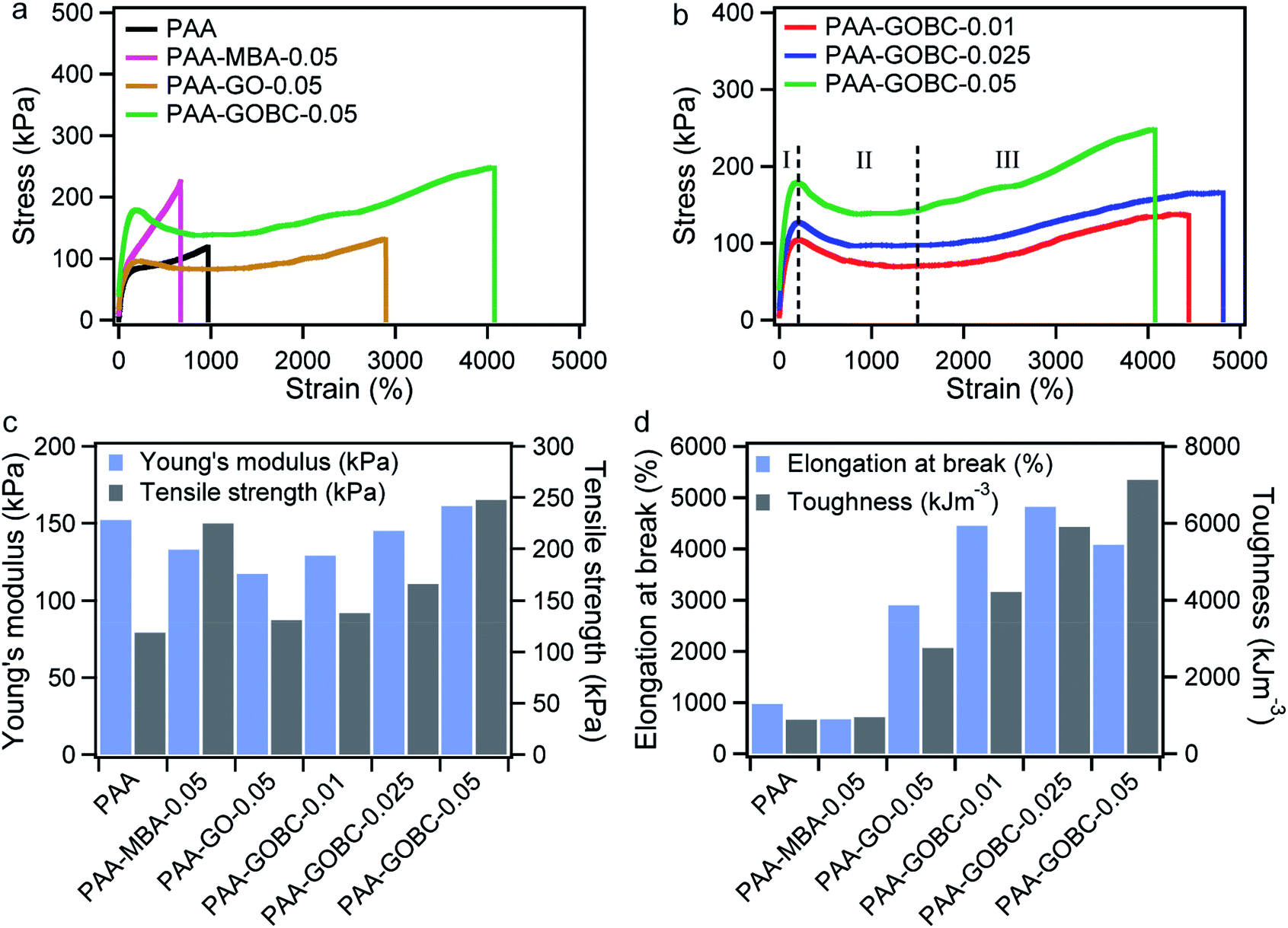 | ||
| Fig. 7 Stress–strain curves of PAA composite hydrogels prepared with different cross-linkers (a) and PAA-GOBC hydrogel with different compositions of GOBC. (b) Comparison of Young's modulus and tensile strength (c), elongation at break and toughness (d) of the hydrogels. | ||
| Samples | Young's modulus (kPa) | Tensile strength (kPa) | Elongation at break (%) | Toughness (kJ m−3) |
|---|---|---|---|---|
| PAA | 152 | 119 | 969 | 881 |
| PAA-MBA-0.05 | 133 | 225 | 670 | 950 |
| PAA-GO-0.05 | 117 | 131 | 2898 | 2748 |
| PAA-GOBC-0.01 | 129 | 137 | 4445 | 4206 |
| PAA-GOBC-0.025 | 145 | 166 | 4817 | 5900 |
| PAA-GOBC-0.05 | 161 | 248 | 4078 | 7124 |
We also observed that the mechanical properties of PAA-GOBC hydrogels were dependent on the concentration of cross-linker. Young's modulus and tensile strength were significantly affected by the increasing concentration of GOBC. The elongation ability increased when the GOBC concentration increased from 0.01 to 0.025%. The enhancement of elongation ability is supportive of the explanation of an increased amount of diversified cross-linking sites in the matrix. However, at a very high concentration (0.05%) decreased elongation of PAA-GOBC-0.05 indicates the negative impact of the GOBC cross-linker over the PAA hydrogel matrix due to the dominance of elastic covalent cross-linking sites.24 On further elaboration of the influence of GOBC cross-linker, the stress–strain response of PAA-GOBC hydrogels during stretching can be divided into three regions (Fig. 7b). The significance of these three regions can be explained from the differences in the contribution of the nanocomposite cross-linking on the structure of PAA in response to the deformation load. During the initial elongation (region I) the stress was mostly tolerated by the covalent cross-linking sites and resulted in elastic deformation. When the gel further deformed, it crossed the elastic barrier by breaking of covalent cross-linking points and demonstrated a necking behavior (region II). In moderate deformation, the PAA chains were able to reassemble over the surface of GOBC through multifunctional physical and chemical bonding, restricting the full release of polymer chains and resulted in elongation without breaking. At very high elongation strain-hardening behavior had been observed (region III). The strain hardening indicates the ease association of PAA polymer chains in closer contact due to the presence of GOBC. The conventional covalent cross-linker (MBA) resulted in fracture at large deformation without demonstrating a significant level of strain-hardening to ensure large elongation without breaking.
We have introduced a new strategy for incorporating the GO in the form of cross-linker in this work to obtain enhanced mechanical properties of the hydrogel. Therefore, a comparison of the mechanical properties of the PAA-GOBC hydrogel with the literature of similar types of hydrogels is necessary. Most of the mechanical parameters, the PAA-GOBC hydrogel had higher magnitudes compared to the hydrogels found in literature as shown in Table 2. For example, the overall toughness of PAA-GOBC hydrogel was five times higher than that of reported for 3D silica-nanoparticle cross-linked hydrogel.29 Other reports showed PAA-GO-Fe3+ hydrogel possessed high tensile strength with very low Young's modulus,53 and PAA-HB hydrogel exhibited a remarkably high Young's modulus with limited elongation.55 In this context, the 2D GOBC cross-linked hydrogel demonstrated a remarkable enhancement in mechanical properties due to its significant and unique ability of chemical and physical cross-linking with PAA polymeric network.
| Hydrogel | Young's modulus (kPa) | Tensile strength (kPa) | Elongation at break (%) | Toughness (kJ m−3) | Reference |
|---|---|---|---|---|---|
| a HB = hyperbranched bis-MPA polyester-64-hydroxyl. | |||||
| PAA-cellulose nanocrystals | 27 | 142 | 1107 | — | 56 |
| PAA-silica nanoparticles | 30 | 157 | 987 | 1385 | 29 |
| PAA-GO-Fe3+ | 32 | 272 | 2185 | 3400 | 14 |
| PAA-HBa | 247 | 188 | 513 | — | 55 |
| PAA-MBA | 133 | 225 | 670 | 950 | This work |
| PAA-GOBC | 161 | 248 | 4078 | 7124 | This work |
3.4 Swelling behavior of PAA composite hydrogels
The influence of GOBC and MBA contents on the swelling behavior of PAA hydrogels was compared. The swelling kinetics of the PAA with different cross-linkers hydrogels has been shown in Fig. 8. Generally, the cross-linking density of the hydrogel network plays an important role in the swelling ratio.57,58 When the PAA hydrogel with an MBA cross-linker was immersed in deionized water, it exhibited a significant level of swelling over time due to the absorption of a large amount of water and it reached an equilibrium value after a long time. The PAA hydrogels with GOBC cross-linkers also followed similar kinetics. Additionally, with the increase of GOBC cross-linker content, from 0.01% to 0.05% in PAA hydrogels, the equilibrium swelling capacity was found to be decreased.24 It exhibited that 0.05% GOBC was enough to restrict the disintegration of hydrogels, however, demonstrated good swelling capacity greater than MBA cross-linked hydrogel. The difference in equilibrium swelling capacity of GOBC and MBA cross-linked hydrogels had been attributed to the difference in the cross-linker binding interaction. The cross-linking ability of GOBC was not enough to restrict the swelling of PAA hydrogel to the same extent as for MBA. Large numbers of diverse cross-linking sites were present in PAA-GOBC hydrogel in addition to a limited number of strong covalent cross-linking sites. The wide distribution of cross-linking of PAA-GOBC hydrogel facilitated large elongation without breaking while restricting the swelling of hydrogel. Nevertheless, the increase in GOBC cross-linker amount was supportive of restricting the swelling capacity of PAA hydrogel by increasing the number of cross-linker sites per unit volume in the matrix.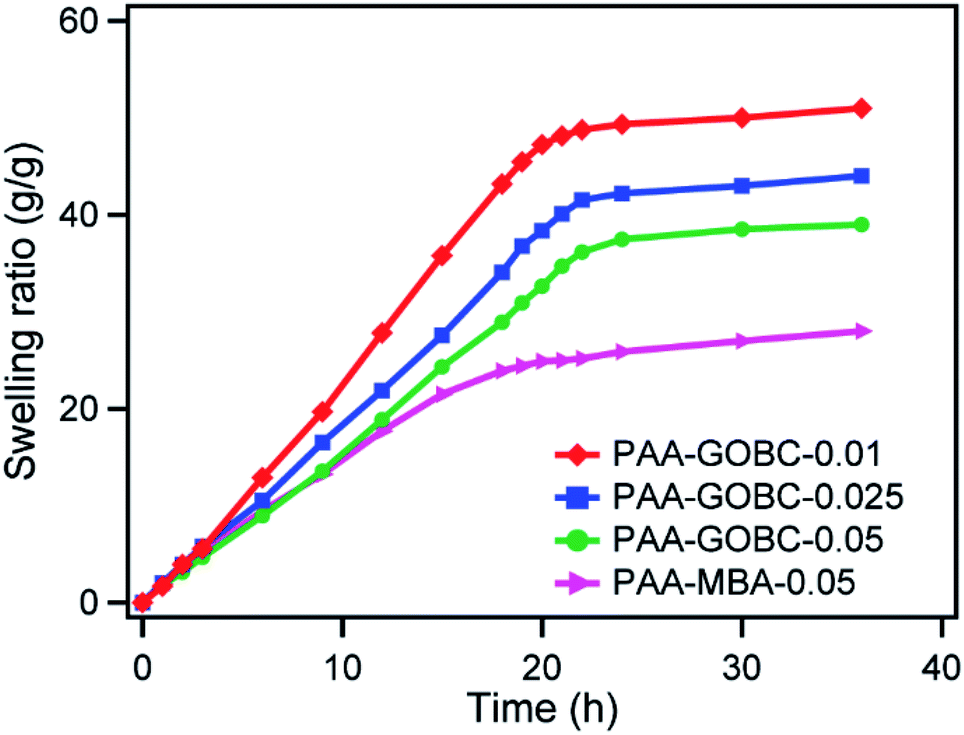 | ||
| Fig. 8 Swelling ratio with respect to time of PAA composite hydrogels. | ||
4. Conclusion
In summary, we developed a new strategy for the modification of GO with vinyl groups, which are capable of forming covalent cross-linking with acrylic acid and similar monomers. The as-synthesized GOBC cross-linker exhibited excellent ability to form both covalent and non-covalent bonds with polymer chains. Incorporation of GOBC into PAA hydrogel instigated very high extensibility along with large toughness into the composite hydrogels. The improvement of the mechanical performance of PAA-GOBC with the increased addition of GOBC was associated with the viscoelastic contribution of the cross-linker. The remarkable improvement in the mechanical performance of the hydrogels is attributed to the unique combination of physical and chemical cross-linking sites present in GOBC. This kind of multiple cross-linking thus accounted for the outstanding tensile extensibility, which is much desired for artificial muscle type materials.59 Moreover, the interactions of the cross-linker and polymer chains offer a homogeneous stress distribution in the matrix of hydrogel to restrict crack propagation. The bifunctional cross-linking bonding of GOBC improved the mechanical properties of hydrogel keeping scope for moderate swelling of hydrogel for water uptake. The as-synthesized GOBC has the ability to be a general platform for cross-linking many other vinyl or acrylic polymers such as polyvinyl alcohol, polyacrylamide, etc. and thus for obtaining high mechanical performance. This strategy, therefore, will pave the way for exploring the advanced applications of hydrogels in biomedical and materials engineering.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support from BUET and the Ministry of Science and Technology, People's Republic of Bangladesh. We are also thankful to the World Academy of Sciences (TWAS) for providing the funds for purchasing the UTM.References
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed.
- J. J. Schmidt, J. Rowley and H. J. Kong, J. Biomed. Mater. Res., Part A, 2008, 87, 1113–1122 CrossRef PubMed.
- A. K. Agarwal, L. Dong, D. J. Beebe and H. Jiang, Lab Chip, 2007, 7, 310–315 RSC.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- I. Tokarev and S. Minko, Adv. Mater., 2010, 22, 3446–3462 CrossRef CAS PubMed.
- C. Huang, H. Bai, C. Li and G. Shi, Chem. Commun., 2011, 47, 4962–4964 RSC.
- N. Sahiner, Prog. Polym. Sci., 2013, 38, 1329–1356 CrossRef CAS.
- Q. Guo, Z. Shi, H. Xu, X. Ma, J. Yin and M. Tian, Macromol. Chem. Phys., 2017, 218, 1600549 CrossRef.
- S. Wada, N. Kitamura, T. Nonoyama, R. Kiyama, T. Kurokawa, J. P. Gong and K. Yasuda, Acta Biomater., 2016, 44, 125–134 CrossRef CAS PubMed.
- J. Maitra and V. K. Shukla, Am. J. Polym. Sci., 2014, 4, 25–31 CAS.
- D. Zhao, J. Huang, Y. Zhong, K. Li, L. Zhang and J. Cai, Adv. Funct. Mater., 2016, 26, 6279–6287 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- J. Yang, X.-P. Wang and X.-M. Xie, Soft Matter, 2012, 8, 1058–1063 RSC.
- J. Yang, F.-K. Shi, C. Gong and X.-M. Xie, J. Colloid Interface Sci., 2012, 381, 107–115 CrossRef CAS PubMed.
- Z. Liu, Z. Yang and Y. Luo, Polym. Compos., 2012, 33, 665–674 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS PubMed.
- H. Chen, M. B. Müller, K. J. Gilmore, G. G. Wallace and D. Li, Adv. Mater., 2008, 20, 3557–3561 CrossRef CAS.
- B. Adhikari and A. Banerjee, Soft Matter, 2011, 7, 9259–9266 RSC.
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS PubMed.
- Y. Pan, H. Bao, N. G. Sahoo, T. Wu and L. Li, Adv. Funct. Mater., 2011, 21, 2754–2763 CrossRef CAS.
- L. Zhang, Z. Wang, C. Xu, Y. Li, J. Gao, W. Wang and Y. Liu, J. Mater. Chem., 2011, 21, 10399–10406 RSC.
- Y. Huang, M. Zhang and W. Ruan, J. Mater. Chem. A, 2014, 2, 10508–10515 RSC.
- J. Shen, B. Yan, T. Li, Y. Long, N. Li and M. Ye, Soft Matter, 2012, 8, 1831–1836 RSC.
- H. P. Cong, P. Wang and S. H. Yu, Small, 2014, 10, 448–453 CrossRef CAS PubMed.
- H. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- H.-P. Cong, P. Wang and S.-H. Yu, Chem. Mater., 2013, 25, 3357–3362 CrossRef CAS.
- R. Liu, S. Liang, X.-Z. Tang, D. Yan, X. Li and Z.-Z. Yu, J. Mater. Chem., 2012, 22, 14160–14167 RSC.
- M. I. Sujan, S. D. Sarkar, S. Sultana, L. Bushra, R. Tareq, C. K. Roy and M. S. Azam, RSC Adv., 2020, 10, 6213–6222 RSC.
- H. Tetsuka, R. Asahi, A. Nagoya, K. Okamoto, I. Tajima, R. Ohta and A. Okamoto, Adv. Mater., 2012, 24, 5333–5338 CrossRef CAS PubMed.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- M. R. Islam, M. Ferdous, M. I. Sujan, X. Mao, H. Zeng and M. S. Azam, J. Colloid Interface Sci., 2020, 562, 52–62 CrossRef CAS PubMed.
- K.-W. Park, J. Mater. Chem. A, 2014, 2, 4292–4298 RSC.
- Q. Xu, M. Zeng, Z. Feng, D. Yin, Y. Huang, Y. Chen, C. Yan, R. Li and Y. Gu, RSC Adv., 2016, 6, 31484–31496 RSC.
- H. Weng, F. Liao, M. Wang, M. Lin and X. Ge, RSC Adv., 2016, 6, 59684–59691 RSC.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- J. Chen, B. Yao, C. Li and G. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS.
- Z. Zhu, L. Ma, M. Su, D. Liu and Z. Wang, J. Mater. Chem. B, 2013, 1, 1432–1438 RSC.
- H. Zhang, R. Huang, H. Cang, Z. Cai and B. Sun, J. Mater. Chem. B, 2014, 2, 1742–1750 RSC.
- T.-F. Yeh, S.-J. Chen, C.-S. Yeh and H. Teng, J. Phys. Chem. C, 2013, 117, 6516–6524 CrossRef CAS.
- W. Zhang, J. Ma, D. Gao, Y. Zhou, C. Li, J. Zha and J. Zhang, Prog. Org. Coat., 2016, 94, 9–17 CrossRef CAS.
- A. Navaee and A. Salimi, RSC Adv., 2015, 5, 59874–59880 RSC.
- H. Yu, B. Zhang, C. Bulin, R. Li and R. Xing, Sci. Rep., 2016, 6, 36143 CrossRef CAS PubMed.
- F. Ma, J. Nian, C. Bi, M. Yang, C. Zhang, L. Liu, H. Dong, M. Zhu and B. Dong, J. Solid State Chem., 2019, 277, 9–16 CrossRef CAS.
- C. Wang, X. Wang, T. Lu, F. Liu, B. Guo, N. Wen, Y. Du, H. Lin, J. Tang and L. Zhang, RSC Adv., 2016, 6, 22461–22468 RSC.
- A. A. Alhwaige, S. M. Alhassan, M. S. Katsiotis, H. Ishida and S. Qutubuddin, RSC Adv., 2015, 5, 92719–92731 RSC.
- A. Kaniyoor, T. T. Baby, T. Arockiadoss, N. Rajalakshmi and S. Ramaprabhu, J. Phys. Chem. C, 2011, 115, 17660–17669 CrossRef CAS.
- F. Baldovino, A. Quitain, N. P. Dugos, S. A. Roces, M. Koinuma, M. Yuasa and T. Kida, RSC Adv., 2016, 6, 113924–113932 RSC.
- S. Yu, J. Liu, W. Zhu, Z.-T. Hu, T.-T. Lim and X. Yan, Sci. Rep., 2015, 5, 16369 CrossRef CAS PubMed.
- S. Chakraborty, S. Saha, V. Dhanak, K. Biswas, M. Barbezat, G. P. Terrasi and A. K. Chakraborty, RSC Adv., 2016, 6, 67916–67924 RSC.
- S. Li and S. Yan, RSC Adv., 2016, 6, 33426–33432 RSC.
- Z.-Q. Zhu, H.-X. Sun, X.-J. Qin, L. Jiang, C.-J. Pei, L. Wang, Y.-Q. Zeng, S.-H. Wen, P.-Q. La and A. Li, J. Mater. Chem., 2012, 22, 4811–4817 RSC.
- M. Zhong, Y.-T. Liu and X.-M. Xie, J. Mater. Chem. B, 2015, 3, 4001–4008 RSC.
- J.-F. Dai, G.-J. Wang, L. Ma and C.-K. Wu, Rev. Adv. Mater. Sci., 2015, 40, 60–71 CAS.
- Y. Yu, L. C. X. De Andrade, L. Fang, J. Ma, W. Zhang and Y. Tang, J. Mater. Sci., 2015, 50, 3457–3466 CrossRef CAS.
- J. Yang, C.-R. Han, J.-F. Duan, M.-G. Ma, X.-M. Zhang, F. Xu, R.-C. Sun and X.-M. Xie, J. Mater. Chem., 2012, 22, 22467–22480 RSC.
- B. H. Cipriano, S. J. Banik, R. Sharma, D. Rumore, W. Hwang, R. M. Briber and S. R. Raghavan, Macromolecules, 2014, 47, 4445–4452 CrossRef CAS.
- J. Zhang, M. W. Sun, L. Zhang and X. M. Xie, J. Appl. Polym. Sci., 2003, 90, 1851–1856 CrossRef CAS.
- X. Bourges, P. Weiss, A. Coudreuse, G. Daculsi and G. Legeay, Biopolymers, 2002, 63, 232–238 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |