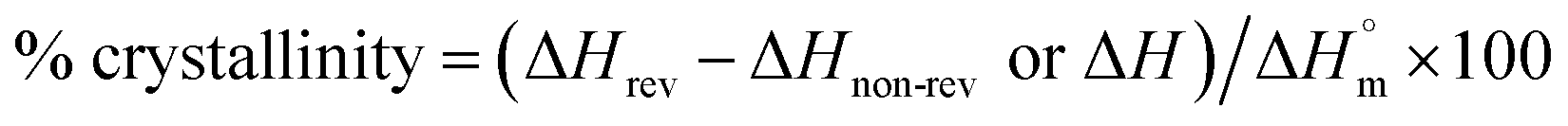DOI:
10.1039/D0RA00098A
(Paper)
RSC Adv., 2020,
10, 10939-10948
Effect of crystallinity on the recovery rate of superhydrophobicity in plasma-nanostructured polymers†
Received
4th January 2020
, Accepted 29th February 2020
First published on 16th March 2020
Abstract
This study explored the optimum conditions to achieve superhydrophobicity in polyethylene terephthalate (PET) in terms of crystallinity and microstructure. Surface superhydrophobicity was achieved by nanostructures induced by oxygen plasma etching and the recovery process of low surface energy through thermal aging of various PETs; semi-crystalline biaxial PET (B-PET) film, amorphous PET (A-PET) film, and semi-crystalline PET (F-PET) fabric. Under the anisotropic plasma etching, the nanostructures on the B-PET film were the longest, followed by the F-PET fabric, which developed a hierarchical micro/nanostructure, then the A-PET film. During thermal aging at 80 °C near Tg, the plasma-treated A-PET film recovered its superhydrophobicity within 3 h, while the plasma-treated B-PET film did not exhibit superhydrophobicity. At 130 °C, higher than Tg, the plasma-treated B-PET film recovered its superhydrophobicity within 1 h, but the plasma-treated A-PET film became opaque as its nanostructures deformed, decreasing its superhydrophobicity. The plasma-treated F-PET fabric exhibited faster recovery and greater superhydrophobicity than the plasma-treated B-PET film, due to its hierarchical micro/nanostructure. In addition, hydrophobic recovery during thermal aging was proved with a decrease in surface polar groups, lowering the surface energy using XPS analysis. Therefore, by designing the ratio of crystal to amorphous regions and surface micro/nanostructures, one can rapidly fabricate superhydrophobic PETs without additional surface finishing.
Introduction
A superhydrophobic surface, as observed naturally in the lotus leaf or water strider, is defined as a surface with a static water contact angle (CA) of >150° and a shedding angle (SA) of <10°. This surface has various functional properties, for instance, water and oil resistance, anti-fogging, anti-freezing, and self-cleaning for water and dirt. In particular, superhydrophobic textiles can be used as functional materials to reduce the frequency of necessary washing and, therefore, provide easy care of textiles. Owing to these advantages, many studies have been conducted on the implementation of superhydrophobic surfaces into various materials requiring breathable, anti-staining, and antibacterial functions.
It is known that a superhydrophobic surface can be fabricated from hierarchical nano- and microstructures with a low-surface-energy coating. Therefore, superhydrophobic surfaces with hierarchical structures have been made by attaching nanoparticles such as SiO2,1 TiO2,2 and carbon nanotubes (CNT)3 onto inherently microscale fabrics and coating them with low surface energy materials, or by dispersing nanoparticles in the low surface energy materials and then applying them to fabrics. However, attached nanostructures are easily removed from fabric surfaces during daily use or washing because of their low adhesion with fabric surfaces, and their byproducts have been associated with harmful effects on biological systems in the environment or human body.4 As for low surface energy coating materials, fluoro compounds containing perfluoroalkyl groups have been most commonly utilized since they can create not only water repellency, but also oil resistance.5 However, fluoro compounds generate carcinogens such as perfluorooctanesulfonic acid or perfluorooctanoic acid during decomposition, which are not easily biodegradable and remain in the human body and environment with harmful effects.6–8 Therefore, international non-governmental environmental organizations (Greenpeace) and researchers have actively informed the public of the risks of using fluoro compounds and tried to restrict and regulate their use, reporting that methods to replace fluoro compounds are required. Consequently, coating materials for direct contact with humans need to be developed without fluoro compounds.
Recent studies have reported a superhydrophobic surface, which is expected to be friendly to the human body and environment, produced by controlling surface energy through thermal aging without using any additional chemicals or coating. Oh et al.9,10 suggested fabricating the superhydrophobic surface using a nanostructuring technique based on oxygen plasma etching or alkaline hydrolysis and the reduction of surface energy with a non-finishing coating, thermal aging. They showed excellent self-cleaning properties with static CAs of >170° and SAs of <10°. These studies were merely focused on the effect of temperature on the recovery of hydrophobicity from the plasma-treated or alkaline hydrolyzed hydrophilic PET surface, which is an external factor. Consequently, it was discovered that the recovery rate of hydrophobicity in polymeric materials is affected by their intrinsic crystallinity and molecular structure, as well as by the fabrication process.
Therefore, in this study, we observed the effect of crystallinity of PET materials on the formation of nanostructures and the rate of subsequent hydrophobic recovery, suggesting the optimum condition to exhibit superhydrophobicity on the plain film or fabric made of microfibers. The materials for this study consist of semi-crystalline biaxial PET (B-PET) film, amorphous PET (A-PET) film, and semi-crystalline microstructured PET (F-PET) fabric. The effects of the crystallinity of the specimen were observed on the formation of nanostructures on each surface by oxygen plasma etching. Subsequently, the hydrophilic PET materials were restored to hydrophobicity or further rendered to superhydrophobicity through thermal aging, in which the effect of crystallinity was in detail analyzed at temperatures under, near, and above the glass transition temperature (Tg). The hydrophobic recovery caused by thermal aging was explored by measurement of the water contact angle (CA) and shedding angle (SA). Surface roughness as well as chemical composition were analyzed with respect to the above temperatures using XPS analysis. Consequently, advantageous and efficient conditions for developing practical superhydrophobic materials were proposed for wider applications.
Experimental
Materials
The PET materials for B-PET film (Goodfellow), A-PET film (Goodfellow), and F-PET fabric (Young Poong Filltex, Korea Rep.) were prepared (Table 1). In order to remove foreign matter from the surface, A-PET, which has low chemical stability due to its low crystallinity, was washed with distilled water and all the other specimens were cleaned using the ultrasonic method for 10 min in acetone (Junsei Chemical Co., Japan) and then dried at room temperature. After that, oxygen plasma etching was performed to obtain the desired surface roughness using 99.99% oxygen gas. No chemicals were used during the thermal aging process to obtain a low surface energy.
Table 1 Characteristics of specimens with the same chemical structure but different micro- or macrostructures
| |
Surface energy (mN m−1) |
Static CA (°) |
Tg (°C) |
Tm (°C) |
Crystallinity (%) |
Thickness (mm) |
| B-PET film |
45.4 |
82.3 ± 1.4 |
79.1 |
252.3 |
37.0 |
0.2 ± 0.05 |
| A-PET film |
41.0 |
89.2 ± 1.4 |
75.6 |
246.5 |
0.5 |
0.2 ± 0.05 |
| F-PET fabric |
43 (ref. 13) |
0.0 ± 0.0 |
82.3 |
253.8 |
36.5 |
0.2 ± 0.05 |
Method
Plasma-based nanostructuring. Nanostructuring by plasma-induced selective etching was performed under the following conditions. Plasma enhanced glow discharge was performed in a custom-made vacuum chamber with both film and fabric specimens of 50 mm × 50 mm × 0.2 mm. Oxygen flowed at 20 sccm at a base pressure of 1.0–1.1 mTorr and the working pressure in the chamber was maintained at 20 mTorr. After that, the specimens were etched for 1, 3, 5, 7, 10, 15, and 20 min with a fixed applied voltage of −400 V.
Thermal aging. Thermal aging process was performed on the B-PET and A-PET films and the F-PET fabric specimens in a natural convection oven (ThermoStable ON-32, DAIHAN Scientific Co., Korea) at temperatures below (40 °C), near (80 °C), and above (130 °C) the Tg for 24 h. A schematic illustration of the overall process is shown in Fig. 1.
 |
| | Fig. 1 A schematic illustration of oxygen plasma etching and thermal aging for PET materials. | |
Surface analysis and chemical composition analysis. The changes in the surface structure of the specimens were examined using a field-emission scanning electron microscope (FE-SEM, SIGMA, Carl Zeiss, England). To prevent charging caused by electron accumulation on the surface of the nonconductive specimens, a conductive coating was applied with a thickness of approximately 10 nm by deposition of a Pt layer at 30 mA for 120 s using a sputter coater (EM ACE200, Leica, Austria). Then, the surface and sides of the samples were observed by tilting the SEM stage to 0° and 30° to measure the diameters and distances, and lengths of nanostructures, respectively. The chemical composition and bonding states of the specimens were analyzed with X-ray photoelectron spectroscopy (XPS, AXIS-his, Kratos Inc., USA) by measuring at a depth of 10 nm from the surface at 18 mA and 12 kV before and after chemical etching as well as before and after thermal aging.
Crystallinity of PET. Modulated differential scanning calorimetry (MDSC) was used to analyze the changes in the crystallinity of specimens during both oxygen plasma etching or thermal aging. Compared to general differential scanning calorimetry (DSC), which makes it difficult to detect small changes in crystallinity and the data interpretation are unclear because the heat flow rate is shown as one signal (total heat flow), MDSC has a higher sensitivity, or resolution, to small changes in crystal structure since it can separate kinetic events which occur with temperature changes; this measures the initial crystallinity with a higher accuracy.11,12 For this experiment, the heating rate was set to 4 °C min−1, heating rate amplitude to ±0.424 °C, and period to 40 s. For the specimens that underwent recrystallization, the crystallization enthalpy was determined by subtracting the recrystallization enthalpy value (ΔHnon-rev, exothermic) detected in the non-reversing heat flow from the total crystallization enthalpy value (ΔHrev, endothermic) of the reversing heat flow. However, for the specimens that did not undergo recrystallization, the crystallization enthalpy value from the total heat flow (ΔH) was used. Then, crystallinity (%) was determined by dividing the crystallization enthalpy value by the inherent enthalpy value 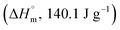 14 of PET and multiplying it by 100, as expressed in the following equation:
14 of PET and multiplying it by 100, as expressed in the following equation:
Wettability measurement. The static CA values were measured using a static CA measuring instrument (Theta Lite Optical Tensiometer, KSV Instruments, Finland).15 The static CAs were measured 10 s after water drops of 3.0 ± 0.5 μl were placed on the sample surface. After five repetitions at different positions on each specimen, an average was calculated. The shedding angle of the samples was evaluated by measuring the dynamic CA using a method proposed by Zimmermann et al.16 The shedding angle can be determined by measuring the minimum angle of tilt at which a 12.5 μl water drop rolls for at least 2 cm after being dropped from 1 cm above the specimen. The shedding angle was obtained by averaging five measurements at different positions on each sample. Superhydrophobicity was determined to be achieved when the static contact angle was over than 150° and the shedding angle was less than 10°.
Results and discussion
Nanostructuring rate with plasma etching time
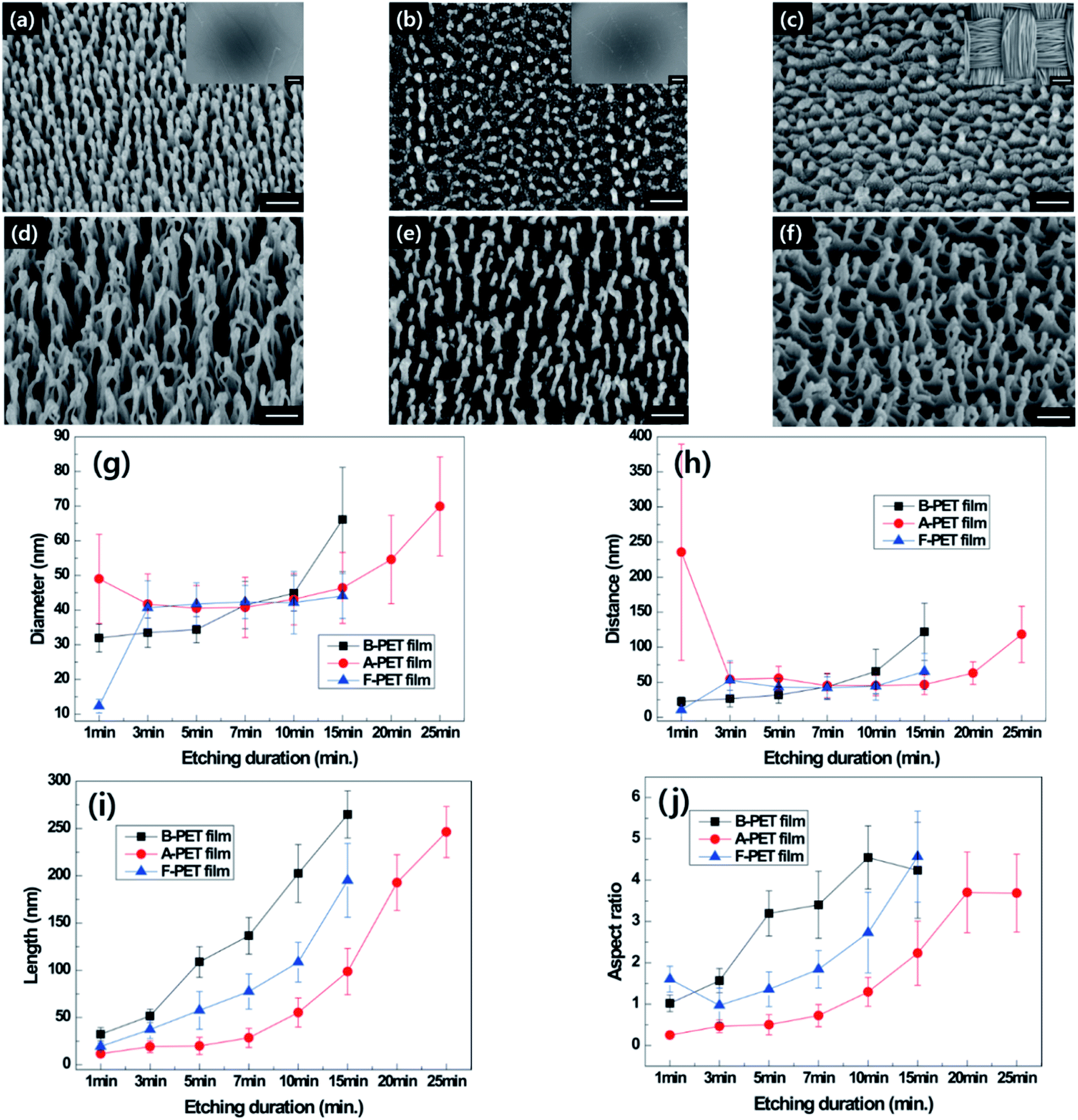
With increasing oxygen plasma etching time, the nanostructures created by the selective plasma etching mechanism17 were formed on the flat surface of every specimen regardless of crystallinity. In general, nanobumps were generated first at the beginning of etching, then the morphology evolved into nanopillars and nanohairs as the etching duration increased (Fig. 2 and ESI Fig. 1–6†). It has been suggested that the Fe or Cr components in the stainless-steel cathode were sputtered and codeposited on the sample surface as the plasma radical ions formed during glow discharge bombarded the cathode plate. Then the metal clusters by diffusion to form a self-etching mask, which prevents the chemical reaction between oxygen plasma radicals and PET at the surface. In contrast, in the specimen areas with no metallic clusters, rapid surface etching occurs, resulting in anisotropic etching. This causes differences in etching speed depending on the position on the specimen surface during oxygen plasma treatment, and the morphology of nanostructures become gradually longer with plasma treatment duration (Fig. 2a–f).18
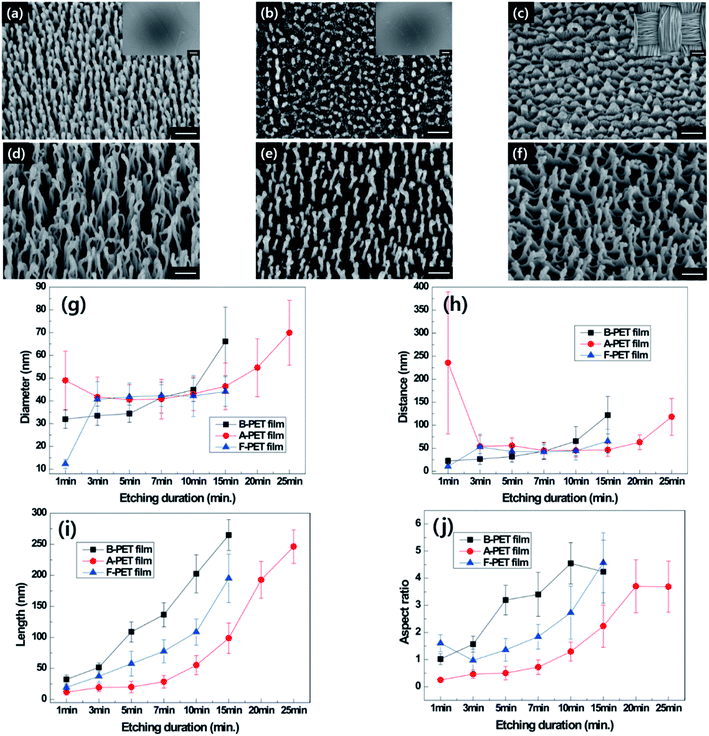 |
| | Fig. 2 Tilted SEM images of B-PET film (a and d), A-PET film (b and e) and F-PET fabric (c and f) for plasma etching durations of 5 and 15 min (×150000, scale bar: 200 nm). Insets are the low magnification images of each sample (×200, scale bar: 100 μm). The diameter (g), distance (h), length (i), and aspect ratio (j) of each PET material measured with oxygen plasma etching duration. | |
At the beginning of plasma etching, the specimens did not show differences in the diameters and lengths of nanostructures, but the lengths of the nanostructures varied as oxygen plasma etching time increased (Fig. 2g and i). The nanostructuring rates were compared between specimens through the measurement of nanostructure length (Fig. 2i). The nanostructures on the B-PET film were the longest and its nanostructuring rate was the highest, followed by the F-PET fabric and A-PET film, respectively. This reason for this stems from their difference in crystallinity. The B-PET film or F-PET fabric have a three-phase composition of amorphous, ordered amorphous, and crystalline phases,19 whereas the A-PET film has a single amorphous phase (Table 1). The amorphous regions were etched more uniformly and easily, with a constant and fast etching rate, across the specimens compared with the crystalline regions that have a highly ordered crystal structure and high bond energy between the polymer chains resulting in slower etching.19 Consequently, after plasma etching, the A-PET film had a larger portion of surface uniformly removed by plasma etching compared to the B-PET film and F-PET fabric.20,21 As a result, the A-PET film became thinner, and its nanoscale surface roughness decreased.20 Therefore, it was hypothesized that the anisotropic etching mechanism relies on the crystallinity of the sample as well as on codeposited metal elements on the surface due to the high etching rate of the amorphous region, which surpasses the mask forming rate, making it difficult to form clusters or etching masks there. Therefore, the A-PET film shows a significantly slower surface to obtain nanostructures with an aspect ratio of 3 or higher (Fig. 2b, e and j). However, the B-PET film and F-PET fabric showed well-developed nanostructures with high aspect ratios (Fig. 2a, c, d and f), even after a short period of time, since they have a crystalline region that can act as a deposition site for metal elements. For example, nanostructures with an aspect ratio of 3 or higher were formed within 5 min etching on the B-PET film (Fig. 2j). These results suggest that the nanostructuring rate in oxygen plasma etching is strongly affected by the crystallinity of PET,19 as the crystalline region promotes selective plasma etching.22 It was also found that even though the B-PET film and F-PET fabric have the same chemical compositions and similar thicknesses (0.2 ± 0.05) and crystallinity (37.0% and 36.5%) (Table 1), the B-PET film has a higher nanostructuring rate than the F-PET fabric. This could be because the F-PET fabric has inherent micro-roughness as it is composed of fibers, yarns, and pores (Fig. 2c). Thus, we assume that the etching masks formed by the deposition of metal clusters would not be as regular as those on the B-PET film, resulting in a lower nanostructuring rate. The change in crystallinity for each specimen was measured before and after oxygen plasma etching and ranged from 0.93% to 2.88% for the A-PET film, from 37.0% to 36.8% for the B-PET film, and 36.5% to 36.2% for the F-PET fabric. These results were corresponded with the XRD results as shown in ESI Fig. 7a and b.† Untreated B-PET film showed a strong peak at 2Θ = 26.16° and oxygen plasma etched B-PET had no difference with the peak of untreated B-PET. On the other hand, untreated A-PET film showed 0% crystallinity and the amorphous halo which is a typical pattern of amorphous materials23–27 and the halo area was almost same after oxygen plasma etching. Thus, the crystallinity did not change significantly during the treatment, indicating that oxygen plasma etching has no significant effect on the entire specimen since it only affects the surface region, up to approximately several hundred nanometers deep.28,29
Recovery of surface hydrophobicity of B-PET film and A-PET film
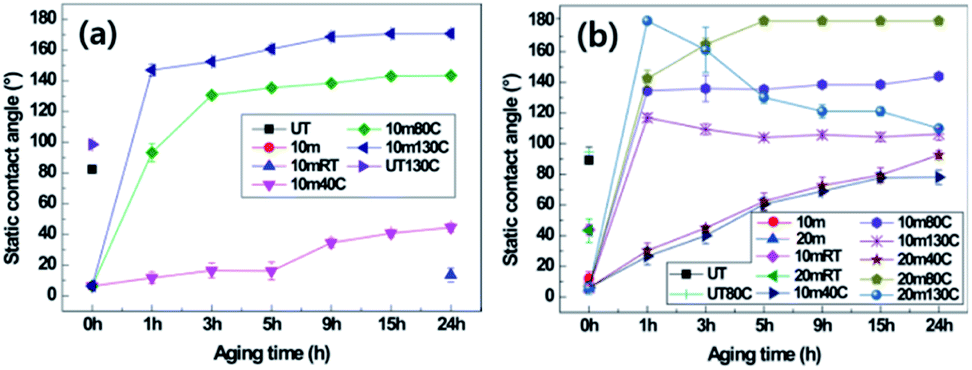
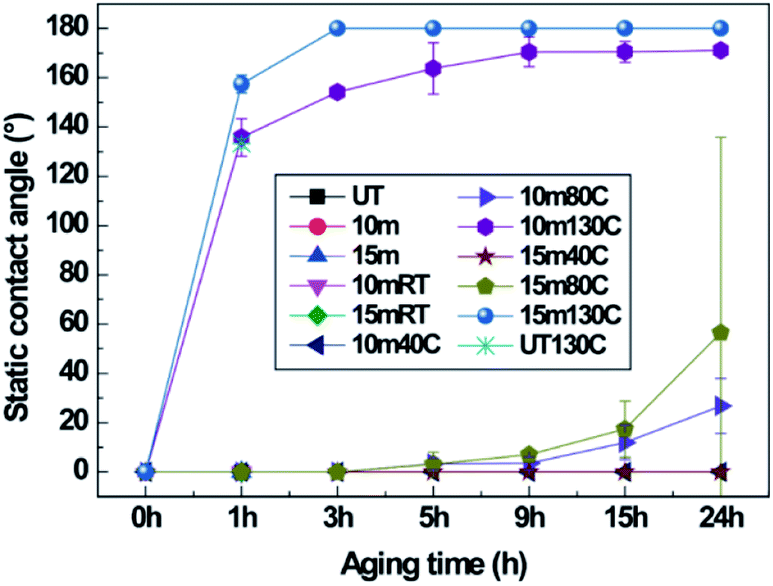
The effects of crystallinity on hydrophobic recovery by thermal aging in nanostructured PET samples were explored. It is known that after surface modification or nanostructuring by oxygen plasma etching, any surface, including on polymeric materials, will be hydrophilic as the surface energy is increased due to free radical or polar groups generated by the oxygen plasma. As the plasma-treated surfaces are exposed to air or thermal condition, the surface energy will be reduced and the water contact angle will increase over time and eventually revert to the original hydrophobic state of the PET material.9 Therefore, hydrophobic recovery was explored on the surfaces after oxygen plasma etching. The static CAs and SAs of two different samples of the 10 min plasma-etched B-PET film and 10 and 20 min plasma-etched A-PET film were measured (Fig. 3, Tables 2 and 3). The PET films with and without nanostructures were treated for 24 h at 40 °C, 80 °C, and 130 °C. Fig. 3a and Table 2 show the static CAs and SAs of the B-PET film that was plasma etched for 10 min and then thermally aged. After thermal aging for 24 h at 40 °C, which is lower than its Tg, the static CA was below 40° and the SA was higher than 45° because the PET films would not change its chemical phase or molecular arrangement, and thus retained hydrophilicity at 40 °C. By contrast, the B-PET film thermally aged at 80 °C, near its Tg, exhibited a sudden increase in hydrophobicity between 1 and 3 h of treatment, with a final static CA of 143.4° ± 1.4° and SA of 18.7° ± 16.1° after 24 h. However, during thermal aging at 130 °C, which is higher than its Tg, it showed a rapid recovery in superhydrophobicity. After thermal aging for 1 h at 130 °C, the B-PET film exhibited a sharp increase in its static CA to 146.9° ± 3.9° and decrease in SA to approximately 4.8° ± 0.8°. After 3 h, both static CA and SA displayed the superhydrophobicity condition and after 24 h changed to 170.0° ± 2.7° and 0.0° ± 0.0°, respectively (Fig. 3a and Table 2). This shows that the higher thermal aging temperature lead fast and higher hydrophobic recovery. In both 10 min and 20 min plasma-stabilize and increased continuously even after thermal aging for 24 h, meaning that hydrophobic recovery was not complete yet. The 10 min plasma-etched B-PET sample thermally aged at 80 °C showed the superhydrophobicity condition and after 24 h changed to 170.0° ± 2.7° and 0.0° ± 0.0°, respectively (Fig. 3a and Table 2). This hydrophobicity in all cases whereas the 20 min plasma-etched A-PET film aged at 80 °C showed high hydrophobicity with a static CA of 180.0° ± 0.0° and a SA of 0.0° ± 0.0° after 5 h, thus exhibiting superhydrophobicity. However, at 130 °C, the static CA of the 10 min plasma-etched A-PET film reached its maximum after 1 h and then gradually decreased over remaining aging duration. Furthermore, the 20 min plasma-etched A-PET film showed its maximum superhydrophobicity after 1 h of thermal aging with a static CA of 164.6° ± 4.1° and a SA of 2.0° ± 0.0°, but with further aging, the static CA drastically decreased to 110.0° ± 1.9° and the SA increased to over 45°.
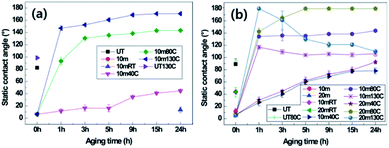 |
| | Fig. 3 Static contact angles of (a) untreated (UT) and 10 min plasma-etched B-PET film, and (b) untreated (UT), 10 min, and 20 min plasma-etched A-PET film depending on thermal aging temperatures at room temperature (RT), 40, 80 and 130 °C with aging duration. | |
Table 2 Shedding angles of 10 min plasma-etched B-PET film depending on thermal aging temperature and treatment duration
| B-PET film |
| Etching duration (min) |
10 |
| |
Thermal aging time (h) |
Temp. (°C) |
| 40 |
80 |
130 |
| Shedding angle (°) |
1 |
>45 |
>45 |
4.8 ± 0.8 |
| 3 |
>45 |
>45 |
1.6 ± 0.8 |
| 5 |
>45 |
>45 |
0.0 ± 0.0 |
| 9 |
>45 |
>45 |
0.0 ± 0.0 |
| 15 |
>45 |
23.5 ± 13.1 |
0.0 ± 0.0 |
| 24 |
>45 |
18.7 ± 16.2 |
0.0 ± 0.0 |
Table 3 Shedding angles of 10 min and 20 min plasma-etched A-PET film depending on thermal aging temperature and treatment duration
| A-PET film |
| Etching duration (min) |
10 |
20 |
| |
Thermal aging time (h) |
Temp. (°C) |
| 40 |
80 |
130 |
40 |
80 |
130 |
| Shedding angle (°) |
1 |
>45 |
>45 |
>45 |
>45 |
23.0 ± 24.1 |
0.0 ± 0.0 |
| 3 |
>45 |
>45 |
>45 |
>45 |
0.3 ± 0.6 |
2.0 ± 0.0 |
| 5 |
>45 |
>45 |
>45 |
>45 |
0.0 ± 0.0 |
>45 |
| 9 |
>45 |
>45 |
>45 |
>45 |
0.0 ± 0.0 |
>45 |
| 15 |
>45 |
>45 |
>45 |
>45 |
0.0 ± 0.0 |
>45 |
| 24 |
>45 |
>45 |
>45 |
>45 |
0.0 ± 0.0 |
>45 |
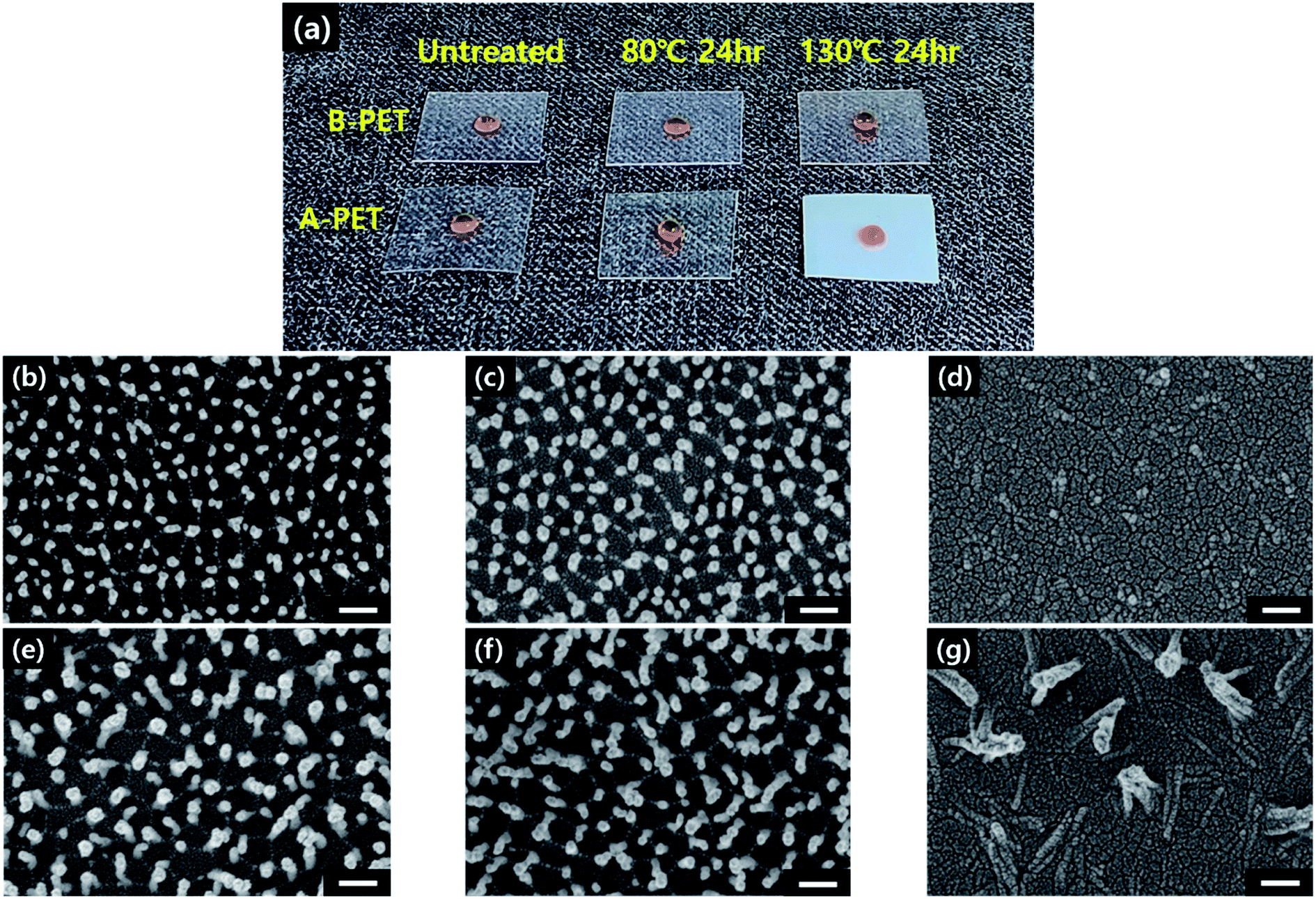
When the surface nanostructures of the A-PET film were examined with respect to aging temperature, it was found that the morphology was mostly unchanged when thermally aged at 80 °C even after 24 h as the Ra and Rq values remained similar at approximately 100 and 77 nm, respectively (ESI Fig. 8c and d†). This confirmed that thermal aging near Tg does not affect the morphology of surface nanostructures. Furthermore, it did not affect the crystallinity or transparency of the A-PET film, resulting in the recovery of superhydrophobicity. On the other hand, the A-PET specimens that underwent thermal aging at 130 °C, above Tg, became opaque and showed decreased hydrophobicity, making the flat-water droplet on the surface (Fig. 4a). This observation revealed that in this condition the nanostructures mostly lay down or disappear as shown in Fig. 4d and g. This is because A-PET film with no crystallinity has low physical and dimensional stability,30 and thus, when it is thermally aged at 130 °C, the Young's modulus decreases until the material becomes rubbery, causing the surface nanostructures morphology to be altered (Fig. 4b–g).31 Additionally, after thermal aging at 130 °C, above the crystallization temperature (Tc, approximately 125.3 °C), crystallinity increased from 0% to 20% as heat-induced spherulitic crystals were generated in the bulk of the A-PET film regardless of oxygen plasma etching.20,32 Therefore, as shown in ESI Fig. 7b† the XRD patterns were changed after thermal aging and crystallinities measured by XRD analysis were changed from 0% to 22.2%, which is similar with the DSC result. It was suggested that for amorphous polymers, thermal aging at a temperature near Tg causes molecular chain rearrangement, moving polar components into the bulk and arranging the non-polar components on the surface which lowers the surface energy9,33 and restores a high surface hydrophobicity. Therefore, for PET materials with low crystallinity, thermal aging at a temperature near or below Tg is more desirable for realizing superhydrophobicity than a temperature above Tg. Hydrophobic recovery behavior for the 10 min plasma-etched B-PET film and the 20 min plasma-etched A-PET film were compared since both have nanostructures with similar geometries of diameters, lengths, and spacings. After oxygen plasma etching and thermal aging at 80 °C for 5 h, the 20 min plasma-etched A-PET film had a static CA of 180.0° ± 0.0° and a shedding angle of 0.0° ± 0.0° (Fig. 3b and Table 3), exhibiting a greater and faster surface hydrophobic recovery than that of the 10 min plasma-etched B-PET film, which showed a static CA of 143.3° ± 1.4° and a shedding angle of 18.7° ± 6.1° (Fig. 3a and Table 2). Furthermore, the time for the A-PET film to reach its maximum static CA was shorter than that for the B-PET film.34 The reason for this is that in the densely packed crystalline region, molecular chain movement is limited as rotational and translational movement of the polymer chains is inhibited,9,33 whereas molecular chain movement is freer in the amorphous region.35 Consistently, during thermal aging at 130 °C A-PET film tended to show greater superhydrophobicity more quickly, within 1 h, than the B-PET film. However, as thermal aging continued, the nanostructures on the A-PET film lay down or melted, causing a decrease in superhydrophobicity as discussed above. On the other hand, the B-PET film could maintain it superhydrophobicity without changing its nanostructures (Fig. 4a). From these results, it can be determined that if crystallinity is high in the PET film, the static CA recovery rate is slow so it takes longer to reach the maximum static CA, but in terms of the robustness under continuous thermal processing and speed of nanostructure formation, a crystal region would be a better condition to exhibit superhydrophobicity.
 |
| | Fig. 4 A photograph of 20 min plasma-etched B-PET and A-PET films before and after thermal aging at 80 and 130 °C for 24 h (a). SEM images of 10 min plasma-etched A-PET film before (b) and after thermal aging at 80 °C (c) and 130 °C (d) for 24 h and 20 min plasma-etched A-PET film before (e) and after thermal aging at 80 °C (f) and 130 °C (g) for 24 h. | |
Recovery of surface hydrophobicity for B-PET film and F-PET fabric
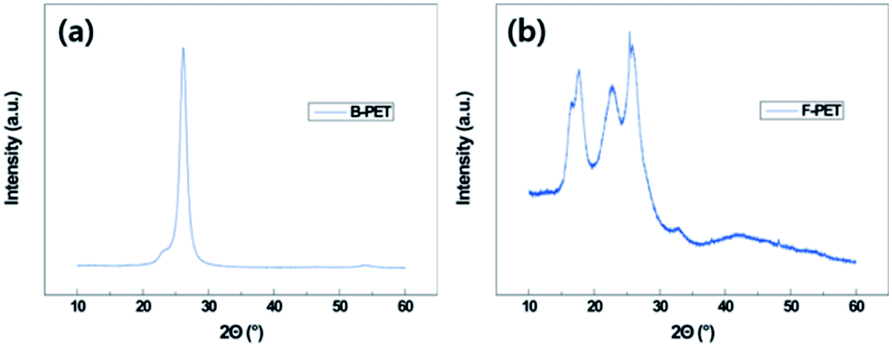
During oxygen plasma etching, a nanoscale roughness was formed on the B-PET film as shown in Fig. 2a, while a hierarchical structure was developed on the F-PET fabric where fibers several tens of micrometers in length have nanohairy structures on its surfaces (Fig. 2c). Therefore, in order to investigate the effect of micro roughness on thermal aging, the 10 min plasma-etched B-PET film and the 15 min plasma-etched F-PET film were compared as shown in Fig. 3a and 5. Specimens were chosen to have similar nanostructure morphology for the diameter, length, and spacing between nanostructures (Fig. 2g–i). Before discussing the hydrophobic recovery between B-PET film and F-PET fabric, the surface wettability change on the F-PET was analysed depending on the plasma etching duration or thermal aging. Fig. 5 shows the surface wettability of the 10 min and 15 min plasma-etched F-PET fabric according to the thermal aging temperature and time. None of the F-PET fabrics showed hydrophobic recovery at 40 °C. After thermal aging at 80 °C for 24 h, the static CA increased noticeably, but did not reach equilibrium. When thermal aged at 130 °C, however, superhydrophobicity was achieved. After treated at 130 °C for 1 h, the 15 min plasma-etched F-PET fabric recovered a static CA of 157.5° ± 3.7° and a SA of 0.6° ± 0.5° and after 3 h, it showed extreme superhydrophobicity with a static CA of 180.0° ± 0.0° and SA of 0.0° ± 0.0° (Fig. 5 and Table 4). In order to illustrate the effect of micro-etched F-PET fabric. After thermal aging performed at 80 °C for 24 h, the 10 min plasma-etched B-PET film showed a static CA of 143.4° ± 1.4° and a SA of 18.7° ± 16.2°, exhibiting higher surface hydrophobic recovery than that of the 15 min plasma-etched F-PET fabric which showed a static CA of 56.4° ± 67.5° and a SA of >45°. Considering that the 10 min plasma-etched B-PET film and the 15 min plasma-etched F-PET fabric showed similar levels of crystallinities and nano-roughness before and after thermal aging (ESI Fig. 7a, c, 8a and b†), this difference in hydrophobic recovery must be due to the difference in polymer orientation caused by different production processes. In other words, this study used B-PET film produced not by simultaneous biaxial stretching35 but sequential biaxial stretching. According to the X-ray diffraction (XRD) graph (Fig. 6), B-PET film showed a narrow peak at 2Θ = 26.16°, indicating that the crystal structure is formed only in (100) planes36 and is highly oriented. Contrarily, for F-PET fabric, which is produced by spinning and drawing, the molecular chains are arranged in the axial direction of fibers, and the peaks were detected at 2Θ = 17.65°, 22.60° and 25.42°. Thus, the crystals exist in the (010), (−101), and (100) planes.37,38 Moreover, it was found that the orientation was weaker than in the B-PET film when the peak widths were compared. Therefore, the movement of molecular chains in the B-PET film, which are highly oriented in one plane is easier than in the F-PET fabric which is less oriented in multiple planes.39 Consequently, the B-PET fabric shows a higher surface hydrophobicity recovery after thermal aging at 40 °C and 80 °C for 24 h than the F-PET fabric. After thermal aging at 130 °C for 24 h, the 10 min plasma-etched B-PET film had a static CA of 170.0° ± 2.7° and a SA of 0.0° ± 0.0° and the 15 min plasma-etched F-PET fabric had a static CA of 180.0° ± 0.0° and a SA of 0.0° ± 0.0°. It was shown that both had greater hydrophobicity recovery at this temperature than at temperatures below and near Tg (Fig. 3a, 5 and Tables 2 and 4), which is the temperature at which hard polymers begin to become soft like rubber40,41 and molecular chain rearrangement occurs. Moreover, when the micro-roughness effect of F-PET fabric was added, higher superhydrophobicity was manifested. As shown in Table 1, the untreated F-PET fabric showed a static contact angle of approximately 0° due to the inherent micro-roughness of fabric, but only after thermal aging at 130 °C for 24 h, its static contact angle increased to 133.4° ± 1.9° due to the surface molecular chain rearrangement and inherent micro-roughness of the fabric. However, after thermal aging at 130 °C, the original static CA of the B-PET film, which was approximately 82.3° ± 1.4° increased to 98.5° ± 1.8° only by surface molecular chain rearrangement. Additionally, after thermal aging at 130 °C, the SA of B-PET film was 0.0° ± 0.0° from 5 h of thermal aging, whereas that of the F-PET fabric became 0.0° ± 0.0° after 3 h of thermal aging. Thus, even though they have the same chemical structure and similar nanostructure diameters, lengths, and distances of separation, as well as crystallinity, the recovery of superhydrophobicity of the F-PET fabric was faster than that of the B-PET film. This originates from the fact that the F-PET fabric has a small contact area and adhesion force with water drops due to its hierarchical structure.42,43 On this note, Su et al.44 mentioned that nano-roughness in hierarchical structures gives a force that can endure high pressures and helps water drops maintain the Cassie–Baxter model because micro-roughness drastically decreases the adhesion area with water droplets. Thus, micro-roughness as well as nano-roughness are favorable for satisfying the superhydrophobic surface condition. The above results imply that micro-roughness enables superhydrophobicity to be achieved with shorter oxygen plasma etching and thermal aging times, which will make it more efficient in reaching superhydrophobicity compared to flat specimens.
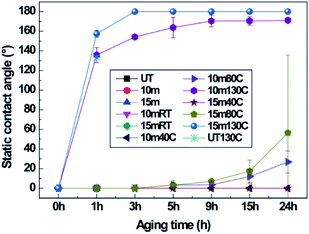 |
| | Fig. 5 Static CAs of untreated, 10 min, and 15 min plasma-etched PET fabric (F-PET) depending on thermal aging temperature and duration. | |
Table 4 Shedding angles of 10 min and 15 min plasma-etched PET fabric (F-PET) depending on thermal aging temperature and duration
| F-PET fabric |
| Etching duration (min) |
10 |
15 |
| |
Thermal aging time (h) |
Temp. (°C) |
| 40 |
80 |
130 |
40 |
80 |
130 |
| Shedding angle (°) |
1 |
>45 |
>45 |
6.0 ± 1.9 |
>45 |
>45 |
0.6 ± 0.5 |
| 3 |
>45 |
>45 |
3.4 ± 0.9 |
>45 |
>45 |
0.0 ± 0.0 |
| 5 |
>45 |
>45 |
3.3 ± 0.6 |
>45 |
>45 |
0.0 ± 0.0 |
| 9 |
>45 |
>45 |
3.3 ± 1.2 |
>45 |
>45 |
0.0 ± 0.0 |
| 15 |
>45 |
>45 |
3.0 ± 1.0 |
>45 |
>45 |
0.0 ± 0.0 |
| 24 |
>45 |
>45 |
1.5 ± 1.0 |
>45 |
>45 |
0.0 ± 0.0 |
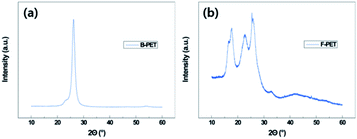 |
| | Fig. 6 XRD patterns of untreated biaxial PET (B-PET) film (a) and untreated PET fabric (F-PET) (b). | |
Changes in chemical composition before and after oxygen plasma etching and thermal aging
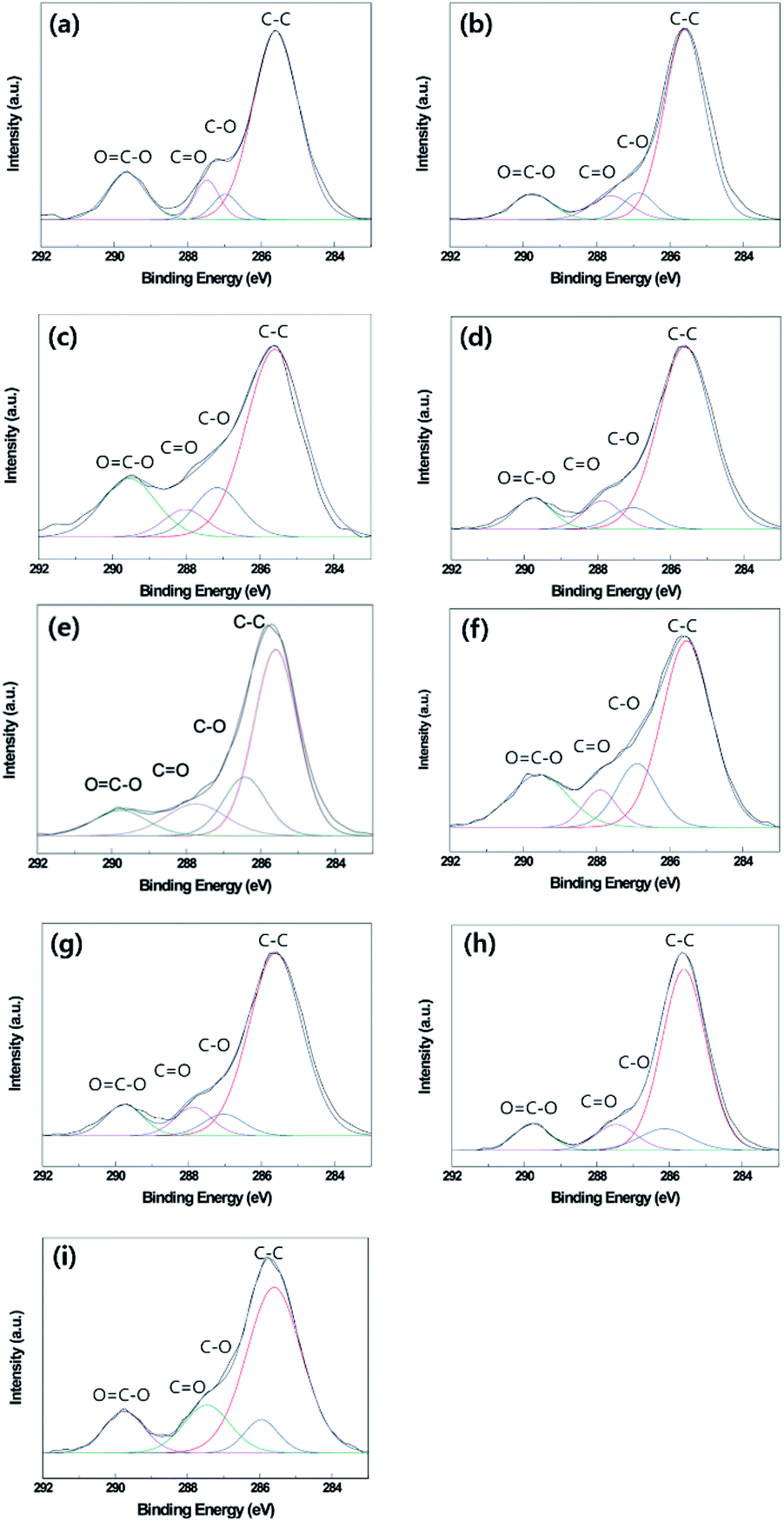
Fig. 7 and Table 5 show the surface chemical compositions as determined by XPS in optimum oxygen plasma etching or thermal aging conditions of the B-PET film, A-PET film, and F-PET fabric. Similar peaks were detected on the surfaces of the untreated B-PET film, A-PET film, and F-PET fabric at 285.60 eV, 286.91 eV, 287.41 eV, and 289.58 eV, as seen in Fig. 7a–c, which indicate C–C, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and OC–O bonds, respectively.45 Based on the bond ratio, the C–C bond from the original molecular structure of the PET shows the highest contribution across all the specimens (Table 5a–c). However, after oxygen plasma etching at the optimum condition for each specimen, the number of C–C bonds decreased because of cutting of the surface molecular chain,46 while the number of C–O, CO, or OC–O bonds increased due to the introduced polar components (Fig. 7d–f and Table 5d–f).9 This suggests that all the specimens become hydrophilic because of the introduced polar groups after oxygen plasma etching. When thermal aging was performed for 24 h at the optimum temperatures of each specimen, the number of C–C bonds increased but the number of C–O, CO, or OC–O bonds decreased (Fig. 7g–i and Table 5g–i). It is suggested that the polymer chains were rotated to lower the surface energy during thermal aging. As a result, the polar and non-polar components moved towards the bulk and surface, respectively.9,10 This indicates that the increase in hydrophobicity from thermal aging after oxygen plasma etching is due to surface molecular chain rearrangement.33,47
O, and OC–O bonds, respectively.45 Based on the bond ratio, the C–C bond from the original molecular structure of the PET shows the highest contribution across all the specimens (Table 5a–c). However, after oxygen plasma etching at the optimum condition for each specimen, the number of C–C bonds decreased because of cutting of the surface molecular chain,46 while the number of C–O, CO, or OC–O bonds increased due to the introduced polar components (Fig. 7d–f and Table 5d–f).9 This suggests that all the specimens become hydrophilic because of the introduced polar groups after oxygen plasma etching. When thermal aging was performed for 24 h at the optimum temperatures of each specimen, the number of C–C bonds increased but the number of C–O, CO, or OC–O bonds decreased (Fig. 7g–i and Table 5g–i). It is suggested that the polymer chains were rotated to lower the surface energy during thermal aging. As a result, the polar and non-polar components moved towards the bulk and surface, respectively.9,10 This indicates that the increase in hydrophobicity from thermal aging after oxygen plasma etching is due to surface molecular chain rearrangement.33,47
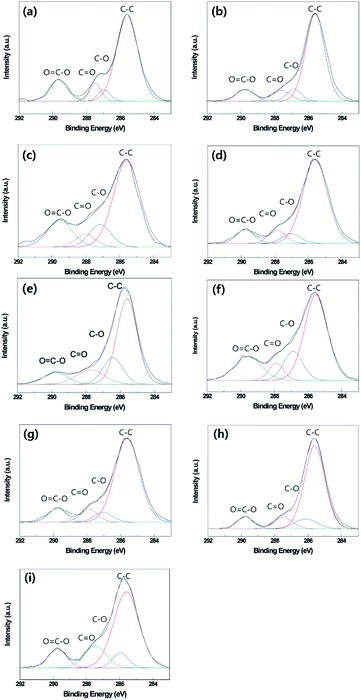 |
| | Fig. 7 XPS survey spectra of untreated B-PET film (a), A-PET film (b), and F-PET fabric (c). 10 min plasma-etched B-PET film (d), 20 min plasma-etched A-PET film (e), 15 min plasma-etched F-PET fabric (f), 10 min plasma-etched and thermally aged at 130 °C for 24 h B-PET film (g), 20 min plasma-etched and thermally aged at 80 °C for 24 h A-PET film (h), 15 min plasma-etched and thermally aged at 130 °C for 24 h F-PET fabric (i). | |
Table 5 Bonding contribution of untreated B-PET film (a), untreated A-PET film (A-PET) (b), untreated F-PET fabric (c), 10 min plasma-etched B-PET film (d), 20 min plasma-etched A-PET film (e), 15 min plasma-etched F-PET fabric (f), 10 min plasma-etched and thermally aged at 130 °C for 24 h B-PET film (g), 20 min plasma-etched and thermal aged at 80 °C for 24 h A-PET film (h), 15 min plasma-etched and thermally aged at 130 °C for 24 h F-PET fabric (i)
| Bonding contribution (at%) |
| Binding energy (eV) |
285.60 |
286.91 |
287.41 |
289.58 |
| Specimens |
C–C |
C–O |
CO |
OC–O |
| (a) Untreated B-PET film |
71.67 |
5.38 |
8.02 |
14.93 |
| (b) Untreated A-PET film |
73.36 |
7.88 |
9.07 |
9.69 |
| (c) Untreated F-PET fabric |
61.75 |
13.28 |
6.74 |
18.23 |
| (d) B-PET film 10 m |
54.01 |
9.52 |
21.20 |
15.27 |
| (e) A-PET film 20 m |
58.61 |
17.92 |
13.71 |
9.76 |
| (f) F-PET fabric 15 m |
57.45 |
16.08 |
7.58 |
18.89 |
| (g) B-PET film 10 m 130 °C |
75.63 |
6.89 |
8.32 |
9.16 |
| (h) A-PET film 20 m 80 °C |
72.77 |
10.63 |
9.44 |
7.16 |
| (i) F-PET fabric 15 m 130 °C |
76.09 |
7.73 |
6.16 |
10.39 |
Conclusions
The rapid achievement of superhydrophobicity was explored in three different PET materials through plasma-induced nanostructuring and subsequent thermal aging, which are the main processes for affecting the hydrophobic recovery and environment/human friendly methods. Various oxygen plasma etching durations and thermal aging conditions were applied to each specimen, and the degree of hydrophobic recovery was compared by measuring the static CAs and SAs. Observation of nanostructure formation by oxygen plasma etching revealed that the B-PET film, similar to the F-PET fabric, formed nanostructures more rapidly than the A-PET film. This suggests that the nanostructuring rate in oxygen plasma etching is strongly affected by the crystallinity of PET materials as crystalline regions promote selective plasma etching. After oxygen plasma etching, the A-PET film effectively recovered its superhydrophobicity when thermally aged near its Tg (80 °C). However, B-PET film and F-PET fabric having a higher degree of crystallinity were required a higher temperature (130 °C) than the Tg to achieve superhydrophobicity, since the crystal regions hinder chain mobility. In the case of the A-PET film, when thermally aged at 130 °C, superhydrophobicity was reached within 1 h, but after 1 h, the nanostructures on the A-PET film deformed and decreasing superhydrophobicity due to less thermal stability for nanostructures. Therefore, with respect to superhydrophobic robustness, some degree of crystallinity could be necessary, because the PET material with some degree of crystallinity forms rapidly and has sturdy nanostructures. Compared to the B-PET film, F-PET fabric showed a faster and greater superhydrophobicity because of its hierarchical structure of micro- and nano-roughness. The intensive XPS examination of the surface characteristics of PETs before and after oxygen plasma etching or thermal aging showed that, regardless of samples, hydrophobic recovery is also led by a decrease in the surface energy as the polar components introduced by oxygen plasma etching were rearranged into the bulk. From these results, when the PET materials, and possibly other polymers, have moderately high crystallinity, nano-roughness can be achieved rapidly with a high aspect ratio by oxygen plasma etching and superhydrophobic recovery can be robustly achieved with subsequent thermal aging and no additional chemical coating. From these results, we assume that these findings could be widely applicable for functional clothing textiles and biomedical goods, as well as many other products made of polymeric materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by MSIP (No. 2016M3A7B4910940 and 2018R1A2B6003526) and a grant (KCG-01-2017-02) by the Korea Coast Guard of the Korean government.
References
- J. Li, H. Wan, Y. Ye, H. Zhou and J. Chen, Appl. Surf. Sci., 2012, 261, 470–472 CrossRef CAS.
- X. Feng, J. Zhai and L. Jiang, Angew. Chem., Int. Ed., 2005, 44, 5115–5118 CrossRef CAS PubMed.
- M. H. Shim, J. Kim and C. H. Park, Text. Res. J., 2014, 84, 1268–1278 CrossRef.
- A. Elsaesser and C. V. Howard, Adv. Drug Delivery Rev., 2012, 64, 129–137 CrossRef CAS PubMed.
- F. Geyer, C. Schönecker, H. Butt and D. Vollmer, Adv. Mater., 2017, 29, 1603524 CrossRef PubMed.
- U. N. Joensen, R. Bossi, H. Leffers, A. A. Jensen, N. E. Skakkebæk and N. Jørgensen, Environ. Health Perspect., 2009, 117, 923–927 CrossRef CAS PubMed.
- B. D. Key, R. D. Howell and C. S. Criddle, Environ. Sci. Technol., 1997, 31, 2445–2454 CrossRef CAS.
- A. B. Lindstrom, M. J. Strynar and E. L. Libelo, Environ. Sci. Technol., 2011, 45, 7954–7961 CrossRef CAS PubMed.
- J.-H. Oh, T.-J. Ko, M.-W. Moon and C. H. Park, RSC Adv., 2017, 7, 25597–25604 RSC.
- J. Oh and C. H. Park, Macromol. Mater. Eng., 2018, 303, 1700673 CrossRef.
- L. C. Thomas, Modulated DSC® Paper #1, Modulated DSC® Basics; An Overview and Summary of Advantages and Disadvantages Relative to Traditional DSC, TA Instruments, 2005, pp. 1–8 Search PubMed.
- B. Molnar and F. Ronkay, Investigation of Morphology of Recycled PET by Modulated DSC, Trans Tech Publ, 2017, vol. 885, pp. 263–268 Search PubMed.
- W. Wu, R. F. Giese Jr and C. J. van Oss, Langmuir, 1995, 11, 379–382 CrossRef CAS.
- W. Sichina, DSC as Problem Solving Tool: Measurement of Percent Crystallinity of Thermoplastics, Perkin Elmer Instruments, PETTech-40, 2000, pp. 1–4 Search PubMed.
- C. M. Weikart and H. K. Yasuda, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3028–3042 CrossRef CAS.
- J. Zimmermann, S. Seeger and F. A. Reifler, Text. Res. J., 2009, 79, 1565–1570 CrossRef CAS.
- T. Ko, K. H. Oh and M. Moon, Adv. Mater. Interfaces, 2015, 2, 1400431 CrossRef.
- T.-J. Ko, W. Jo, H. J. Lee, K. H. Oh and M.-W. Moon, Thin Solid Films, 2015, 590, 324–329 CrossRef CAS.
- S. R. Matthews, Y. J. Hwang, M. G. McCord and M. A. Bourham, J. Appl. Polym. Sci., 2004, 94, 2383–2389 CrossRef CAS.
- I. Junkar, U. Cvelbar, A. Vesel, N. Hauptman and M. Mozetič, Plasma Processes Polym., 2009, 6, 667–675 CrossRef CAS.
- T. Jacobs, N. De Geyter, R. Morent, S. Van Vlierberghe, P. Dubruel and C. Leys, Surf. Coat. Technol., 2011, 205, S511–S515 CrossRef CAS.
- L. Zhu, W. Teng, H. Xu, Y. Liu, Q. Jiang, C. Wang and Y. Qiu, Surf. Coat. Technol., 2008, 202, 1966–1974 CrossRef CAS.
- P. G. Karagiannidis, A. C. Stergiou and G. P. Karayannidis, Eur. Polym. J., 2008, 44, 1475–1486 CrossRef CAS.
- I. N. Strain, Q. Wu, A. M. Pourrahimi, M. S. Hedenqvist, R. T. Olsson and R. L. Andersson, J. Mater. Chem. A, 2015, 3, 1632–1640 RSC.
- C. A. Massa, S. Capaccioli, A. Manariti and M. Bertoldo, Eur. Polym. J., 2014, 60, 286–296 CrossRef CAS.
- V. Causin, Anal. Methods, 2010, 2, 792–804 RSC.
- F. Hamonic, V. Miri, A. Saiter and E. Dargent, Eur. Polym. J., 2014, 58, 233–244 CrossRef CAS.
- J.-H. Oh, T.-J. Ko, M.-W. Moon and C. H. Park, RSC Adv., 2014, 4, 38966–38973 RSC.
- S. Kwon, T.-J. Ko, E. Yu, J. Kim, M.-W. Moon and C. H. Park, RSC Adv., 2014, 4, 45442–45448 RSC.
- Y. Kong and J. Hay, Polymer, 2002, 43, 3873–3878 CrossRef CAS.
- C. Buckley and D. Jones, Polymer, 1995, 36, 3301–3312 CrossRef CAS.
- B. Haworth, Z. Dong and P. Davidson, Polym. Int., 1993, 32, 325–335 CrossRef CAS.
- M. Mortazavi and M. Nosonovsky, Appl. Surf. Sci., 2012, 258, 6876–6883 CrossRef CAS.
- C. Canal, R. Molina, E. Bertran and P. Erra, J. Adhes. Sci. Technol., 2004, 18, 1077–1089 CrossRef CAS.
- Y. I. Yun, K. S. Kim, S.-J. Uhm, B. B. Khatua, K. Cho, J. K. Kim and C. E. Park, J. Adhes. Sci. Technol., 2004, 18, 1279–1291 CrossRef CAS.
- H. Chang, J. Schultz and R. Gohil, J. Macromol. Sci., Part B: Phys., 1993, 32, 99–123 CrossRef.
- P. Chandran and S. Jabarin, Adv. Polym. Technol., 1993, 12, 153–165 CrossRef CAS.
- S. G. Prasad, A. De and U. De, Int. J. Spectrosc., 2011, 2011, 1–7 CrossRef.
- M. D. Shelby, A. J. Hill, M. I. Burgar and G. L. Wilkes, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 32–46 CrossRef CAS.
- Y. Lu, L. Xue and F. Li, Appl. Surf. Sci., 2011, 257, 3135–3139 CrossRef CAS.
- V. Babaahmadi, M. Montazer and W. Gao, Carbon, 2017, 118, 443–451 CrossRef CAS.
- K. Koch, B. Bhushan, Y. C. Jung and W. Barthlott, Soft Matter, 2009, 5, 1386–1393 RSC.
- A. Richard, Faraday Discuss., 1994, 98, 219–230 RSC.
- Y. Su, B. Ji, K. Zhang, H. Gao, Y. Huang and K. Hwang, Langmuir, 2010, 26, 4984–4989 CrossRef CAS PubMed.
- A. Lafuma and D. Quéré, Nat. Mater., 2003, 2, 457–460 CrossRef CAS PubMed.
- Z. Xing, Z. Ju, Y. Zhao, J. Wan, Y. Zhu, Y. Qiang and Y. Qian, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- Y. P. Li and M. K. Lei, J. Mater. Sci. Technol., 2014, 30, 965–972 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00098a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c and
Chung Hee Park
*c and
Chung Hee Park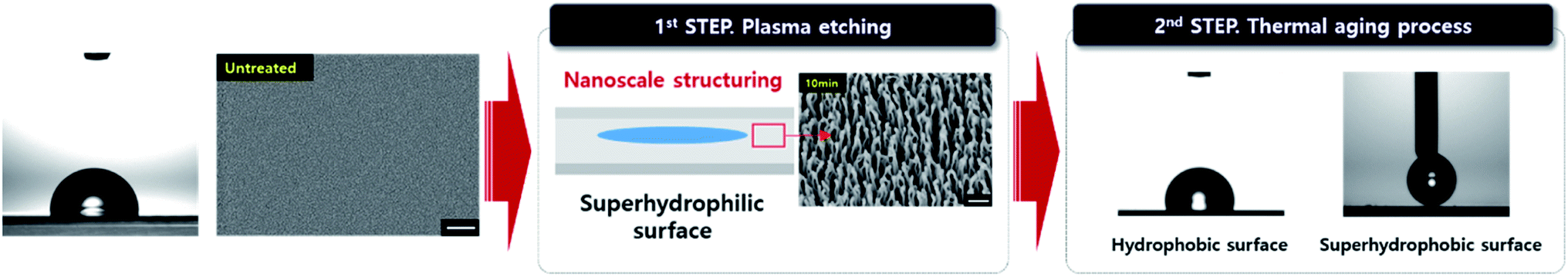
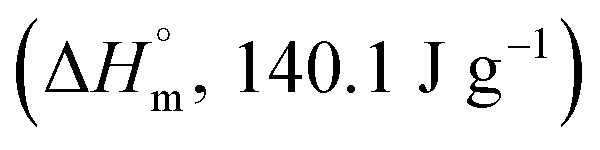 14 of PET and multiplying it by 100, as expressed in the following equation:
14 of PET and multiplying it by 100, as expressed in the following equation: