
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of benzothiazones containing a hexahydropyrrolo[3,4-c]pyrrol moiety as antitubercular agents against MDR-MTB†
Xican Ma‡
ab,
Bing Han‡ab,
Aoyu Wangc,
Lu Yangb,
Menghao Huangd,
Kushan Chowdhury d,
Jian Gu*a,
Kai Zhang*c and
Kai Lvb
d,
Jian Gu*a,
Kai Zhang*c and
Kai Lvb
aCollege of Pharmacy, Southwest Minzu University, Chengdu, 610041, China. E-mail: gujiancd@163.com
bInstitute of Medicinal Biotechnology, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing, 100050, China
cDepartment of Pharmaceutical Chemistry, School of Pharmacy, Hebei Medical University, Shijiazhuang, 050017, PR China. E-mail: zhk810728@163.com
dDepartment of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, IN 46202, USA
First published on 7th April 2020
Abstract
IMB1603, a spiro-benzothiazone compound discovered by our lab, displayed potent anti-MTB activity in vitro and in vivo. In this study, we reported a series of new BTZs containing the hexahydropyrrolo[3,4-c]pyrrol moiety based on the structure of IMB1603. Among them, BTZs 11 and 24 displayed potent anti-MTB (MIC < 0.035 μM) and MDR-MTB (MIC, 0.053–0.102 μM) activity, good solubility (1.82–1.85 μg mL−1), and low cytotoxicity (CC50 > 200 μM), suggesting BTZs 11 and 24 may serve as promising candidates for further study. The molecular docking study of 11 toward DprE was also investigated, and revealed that 11 mimicked the binding pattern of PBTZ169 in the active site of DprE1.
Tuberculosis (TB) is a chronic infectious disease caused mainly by Mycobacterium tuberculosis (MTB).1 The World Health Organization (WHO) estimated that approximately 10 million people were infected and 1.5 million died from TB worldwide in 2018.2 The current therapy for TB infected patients requires a combination of four front-line drugs for 6–9 months and does not favor patient compliance. Even worse, the prevalence of multidrug-resistant (MDR) TB and extensively drug-resistant (XDR) TB has exacerbated the situation.3–5 Although new drugs with novel mechanisms such as bedaquiline, delamanid and pretomanid have been approved in recent years for the treatment of MDR-TB,6,7 some adverse events or warnings have been noted and limited their use in the clinic.8 Therefore, there is still an urgent unmet medical need for safer and more effective agents for the treatment of TB.
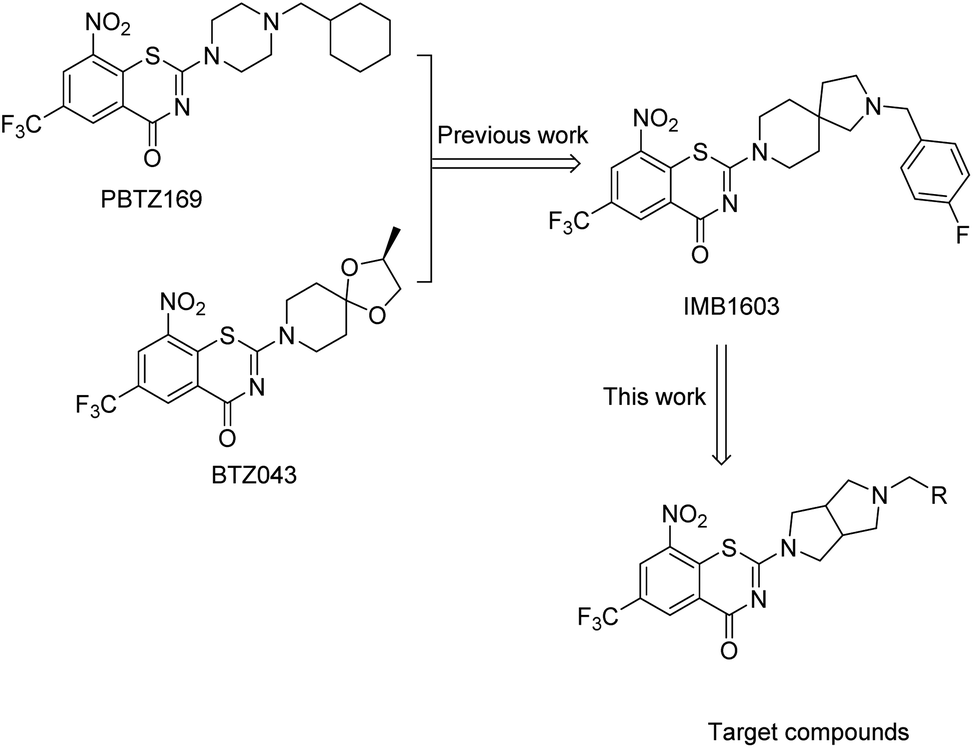
Benzothiazinones (BTZs), a novel class of TB agents targeting decaprenylphosphoryl-β-D-ribose 2′-epimerase (DprE1),9,10 exhibited exceptional inhibitory activity against MTB and MDR-MTB strains.11–13 PBTZ169 (macozinone) and BTZ043, the most advanced BTZ candidates, is currently in phase II14 and phase I clinical trial15 for the treatment of both drug-susceptible TB and MDR-TB.16 BTZ scaffold has become a research hot spot throughout the world for the scientists and researchers working in the field of anti-TB.17–21
According to the binding pharmacophore revealed by the crystal structures of BTZ043/PBTZ169 complexed with DprE1, the BTZ core interacted with the active site cavity, whereas the cyclohexane or spirocyclic moiety was located at the protein surface (Fig. 1).13,16 More precisely, the CF3 group was well placed in a small pocket and interacted with Asn392; the nitro group of PBTZ169 or BTZ043 was converted to nitroso which covalently binds to Cys387 or Cys394 residue of DprE1 enzyme, leading to irreversible inactivation of the enzyme.13,16 It appeared that the BTZ core was crucial for the activity, but the spirocyclic or cyclohexane at the C-2 position might be open for structure modification.
 | ||
| Fig. 1 Design of new BTZs. | ||
Based on the binding characteristics and reported structure–activity relationship (SAR), our group focused on the discovery of alternative moieties at the C-2 position of BTZ core.22–25 Recently, we identified IMB1603 with a spiro-heterocyclic segment as a potent anti-TB lead by combining the structure feature of PBTZ169 and BTZ043.23 In this study, replacement of the spiro-heterocyclic group of IMB1603 with hexahydropyrrolo[3,4-c]pyrrol gave a new series of BTZs (Fig. 1). The anti-TB activity, solubility, and toxicity of these new BTZs were evaluated, aiming to identify alternative groups at position 2 of BTZs and find optimized potent anti-TB drug candidates with improved drug-like properties through SAR study.
The synthesis of target compounds 1–43 was shown in Scheme 1. Reductive amination of hexahydropyrrolo[3,4-c]pyrrol A in the presence of aryl aldehydes or acetophenones gave compound B. Thereafter, deprotection of the Boc group in trifluoroacetic (TFA) gave the intermediate C. Coupling of BTZ core D with A or C in the presence of Et3N (TEA) afforded the target compounds 1–35 and 37–43. Removing the Boc group of 35 in TFA gave the target compound 36.
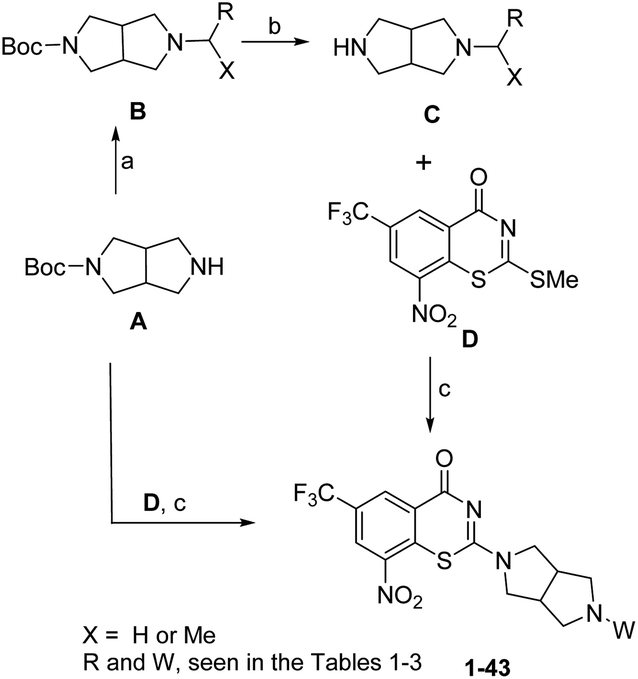 | ||
| Scheme 1 Reagents and conditions: (a): (i) X = H, ArCHO, Na(OAc)3BH, AcOH, CH2Cl2, RT, 3–4 h, 71–85%; OR (ii) X = Me, Ti(OiPr)4, acetophenones, MeOH, NaCNBH3, 40 °C, 5–7 h, 30–55%; (b) TFA, DCM, rt, 3–4 h; (c) (i) Et3N, MeOH, 40 °C, 1–1.5 h, 22–41%, two steps; (ii) TFA, DCM, 70% (Boc deprotection in TFA was only needed for compound 36). | ||
The target compounds 1–43 were initially screened for in vitro activity against MTB H37Rv ATCC 27294 strain using the Microplate Alamar Blue Assay (MABA).26 The minimum inhibitory concentration (MIC) was defined as the lowest concentration effecting a reduction in fluorescence of >90% relative to the mean of replicate bacterium-only controls. The MIC values of the compounds along with isoniazid (INH), rifampicin (RFP) and PBTZ169 were presented in Tables 1–3 for comparison.
|
|||||
|---|---|---|---|---|---|
| Compd | R | MIC (μM) | Compd | R | MIC (μM) |
| 1 | 0.462 | 17 | m-NO2 | 0.414 | |
| 2 | p-F | 0.244 | 18 | m-CF3 | 0.101 |
| 3 | p-Cl | 1.654 | 19 | m-MeO | 3.671 |
| 4 | p-Br | 0.953 | 20 | o-F | 3.595 |
| 5 | p-CN | 0.616 | 21 | p-F, m-F | 0.111 |
| 6 | p-NO2 | 0.451 | 22 | p-Cl, m-Cl | 0.077 |
| 7 | p-CF3 | 0.112 | 23 | p-F, m-Cl | 0.104 |
| 8 | p-MeO | 1.832 | 24 | p-Cl, m-F | <0.035 |
| 9 | p-tBu | 0.218 | 25 | p-F, o-Cl | 0.888 |
| 10 | p-Me | 1.926 | 26 | p-F, o-F | 0.232 |
| 11 | p-CF3O | <0.035 | 27 | p-Cl, o-Cl | 0.566 |
| 12 | H | 0.796 | 28 | p-F, o-Br | 0.783 |
| 13 | m-F | 0.222 | PBTZ169 | <0.035 | |
| 14 | m-Cl | 0.217 | INH | 0.262 | |
| 15 | m-Br | 0.203 | RFP | 0.166 | |
| 16 | m-CN | 0.223 | |||
|
||
|---|---|---|
| Compd | Ar or R′ | MIC (μM) |
| 12 |  |
0.796 |
| 29 | 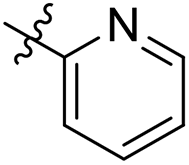 |
1.667 |
| 30 | 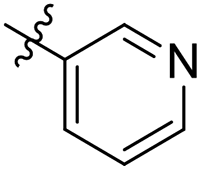 |
1.849 |
| 31 | 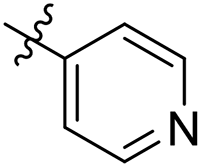 |
1.981 |
| 32 | 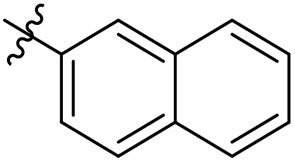 |
0.463 |
| 33 | 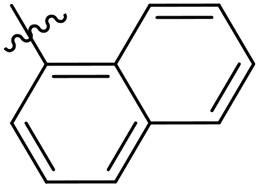 |
0.899 |
| 34 | 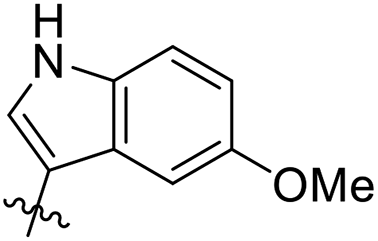 |
27.350 |
| 35 | Boc | 1.720 |
| 36 | H | >40 |
| PBTZ169 | <0.035 | |
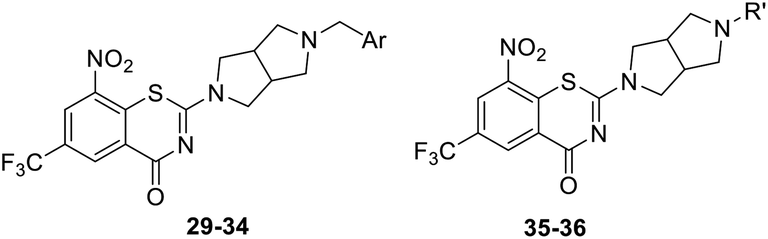
|
||
|---|---|---|
| Compd | R′′ | MIC (μM) |
| 37 | p-CF3 | 0.202 |
| 38 | p-OCF3 | 0.209 |
| 39 | p-F, m-F | 0.110 |
| 40 | p-Cl, m-F | 0.219 |
| 41 | p-F, m-Cl | 0.108 |
| 42 | m-F, m-F | 0.205 |
| 43 | p-F, m-F, m-F | 0.189 |
| PBTZ169 | <0.035 | |
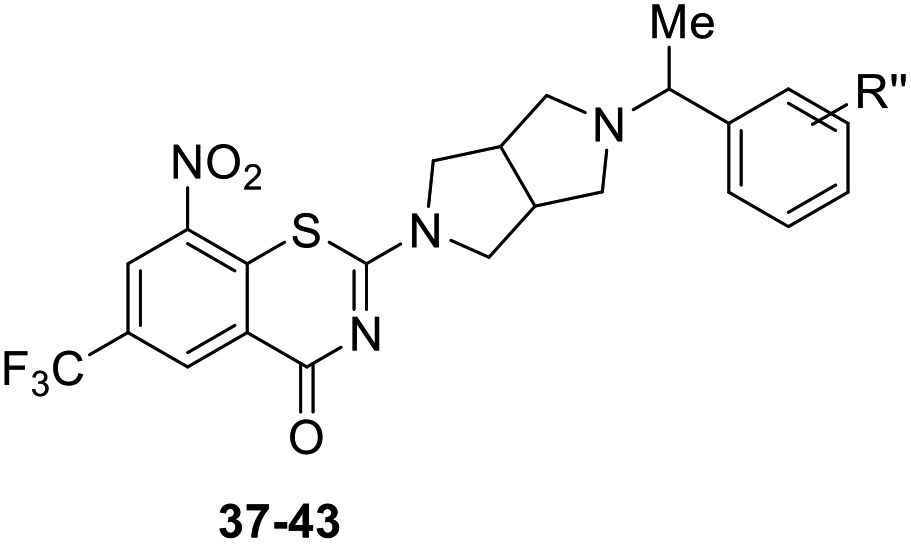
As shown in Table 1, BTZs 1 and 2 with cyclohexyl and p-fluorobenzyl group from PBTZ169 and IMB1603 were initially synthesized and evaluated. The MIC value of BTZ 2 was lower than that of 1, and comparable to that of INH. According to our previous SAR findings of spiro-BTZs, the substituents at the phenyl ring were crucial for anti-TB activity.23 In parallel with the spiro-BTZ series, we synthesized compounds 3–28 with diversity R substituents at the phenyl ring. To our delight, compounds 7, 11, 18 and 21–24 displayed increased potency (MIC < 0.15 μM). Especially, compound 11 with p-CF3O and 24 with p-Cl, m-F were found to exhibit comparable anti-TB activity to PBTZ169 (MIC < 0.035 μM). It is interesting that moving the para substituent to meta position led to an improved anti-TB activity (2–7 vs. 13–19), while transferring to the ortho position seemed to result in a decreased potency (2 vs. 20). Notably, BTZs with double substituents at the para and meta position of the benzyl moiety showed better anti-TB activity (MIC < 0.15 μM) than the corresponding mono-substituted BTZs (21–24 vs. 2–3 and 13–14), whereas introducing double substituents to the para and ortho position of benzyls were not well tolerated (25–28, MIC > 0.2 μM).
As a continuing SAR study, BTZs 29–34 with other aromatic cyclic groups as replacement of the benzyl moiety were explored. Compared to BTZ 12, the pyridyl analogues 29–31 and β-naphthalene 33 showed decreased potency, whereas the α-naphthalene 32 displayed a slightly increased anti-TB activity. The indole moiety was also not favored, leading to a dramatically decreased anti-TB activity (34, MIC = 27.35 μM). In addition, BTZ 35 with Boc-group was synthesized and proved to display moderate anti-TB potency (MIC < 2 μM). Removal of the R′ group, as in compound 36, led to a total loss of anti-TB potency (MIC > 40 μM), indicating the existence of N–H bond might not be tolerated at the C2-position of the BTZ core.
Finally, as shown in Table 3, BTZs with a methyl group at the linker were investigated in this set. The R′′ group of 37–41 were selected from the BTZs with potent anti-TB activity in Table 1 (MIC < 0.15 μM). Compared to the corresponding BTZs in Table 1, the introduction of a methyl group resulted in a decreased anti-TB potency (37–41 vs. 7, 11, 21, 23–24). In addition, BTZs 42 with double m-F and 43 with p-F, double m-F, were designed and synthesized according above SAR findings. However, the anti-TB potency of BTZs 42–43 was also not as good as we expected, both of them displayed lower anti-TB activity than 39.
On the bases of above studies, BTZs 7, 11, 18, 21–24, 39, and 41 with potent anti-TB activity (MIC < 0.15 μM) were evaluated against two clinical isolated MDR-MTB (16833 and 16995) strains resistant to both INH and RFP.§ As shown in Table 4, although the MIC values of these new BTZs were all lower than that of PBTZ169 (MIC < 0.035 μM), most of them displayed considerable anti-MDR-TB activity (MIC < 0.15 μM).
| Compd | MIC (μM) | CC50b (μM) | Water solubilityc (mg mL−1) | |
|---|---|---|---|---|
| MDR-MTB1a | MDR-MTB2a | |||
| a MDR-MTB1 (MDR-MTB 16833) and MDR-MTB2 (MDR-MTB 16995) were obtained from the State Laboratory of Tuberculosis Reference of China.b The 50% cytotoxic concentration.c The water solubility was tested in 0.01 M HCl solution (approximate pH 2.0).d This data was from ref. 24. | ||||
| 7 | 0.080 | 0.053 | 166.37 | 2.30 |
| 11 | 0.085 | 0.053 | >200 | 1.85 |
| 18 | 0.209 | 0.112 | 55.77 | 2.29 |
| 21 | 0.119 | 0.058 | 38.63 | 3.50 |
| 22 | 0.110 | 0.056 | 27.31 | 1.94 |
| 23 | 0.115 | 0.058 | 40.79 | 2.01 |
| 24 | 0.102 | 0.073 | >200 | 1.82 |
| 39 | 0.121 | 0.087 | 54.98 | 2.97 |
| 41 | 0.188 | 0.097 | 50.68 | 2.55 |
| PBTZ169 | <0.035 | <0.035 | >200 | 0.90d |
| INH | >40 | >40 | ND | NT |
| RFP | >40 | >40 | ND | NT |
These compounds were then tested for mammalian cell cytotoxicity using Vero cells¶ measured as a concentration inhibiting 50% growth (CC50) as compared to a no-treatment control and the results were reported in Table 4. Most of the tested targets displayed higher cytotoxicity than that of PBTZ169. However, to our delight, compounds 11 and 24 showed low cytotoxicity (CC50 > 200 μM). Subsequently, considering acidic gastrointestinal environments, these new BTZs were evaluated for their water solubility at pH 2 (0.01 M HCl solution) by using an HPLC-UV method.23 All of them displayed good solubility (1.82–3.50 μg mL−1), and were more water-soluble than PBTZ169 (0.90 μg mL−1).
It was reported that BTZs bound to the DprE1 dimer–dimer interface,13,16 indicating that the new BTZs might also bind the same site. Thus, we predicted the binding mode of compound 11 in the DprE1 dimer–dimer interface (PDB code: 4NCR) through molecular docking using CDDOCK module of Discovery Studio 3.5. Since the intrinsic ligand PBTZ169 was crystallized as a covalent adduct with DprE1, it was necessary to optimize the docking protocol of 11 into DprE1. Briefly, the benzothiazinone ligand (semimercaptal) present in the crystal structure was deleted and no hydrogen was added to the formed thiolate of Cys 387. The reduced hydroxylamine form of 11 was docked in the same binding pocket forming the semimercaptal adduct as reported before.17 The docking study revealed that 11 mimicked the binding pattern of PBTZ169 in the active site of DprE1 (Fig. 2B). The (trifluoromethoxy)benzyl moiety of 11 was placed outside the pocket and showed high flexibility as the cyclohexylmethyl moiety of PBTZ169, whereas the BTZ core occupied the inner part of the cavity (Fig. 2A).
 | ||
| Fig. 2 Overlay of the docking hydroxylamine intermediate of 11 (carbons are green) on the crystallized semimercaptal adduct (carbons are off white). (A) The overall view of the binding pattern; (B) close-up view of the DprE1 active site. | ||
Conclusions
In summary, a series of new BTZs containing hexahydropyrrolo[3,4-c]pyrrol moiety were designed and synthesized based on the spiro-BTZ IMB1603 discovered in our lab. Many of them exhibited potent in vitro anti-TB activity. Especially, compounds 11 and 24 were found to display excellent anti-MTB activity against the drug-sensitive MTB strain H37Rv (MIC < 0.035 μM), and also potent anti-MDR-MTB activity against the two drug-resistant clinical isolates (MIC, 0.053–0.102 μM). In addition, BTZs 11 and 24 showed low cytotoxicity (CC50 > 200 μM), and exhibited better water solubility than PBTZ169, suggesting both of them may serve as new and promising candidates for further antitubercular drug discovery. The molecular docking results suggested that 11 mimicked the binding pattern of PBTZ169 in the active site of DprE1. Studies to determine the PK profiles and in vivo efficacy of 11 and 24 are currently under way.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Mega-project for Innovative Drugs (2018ZX09721001-004-007; 2018ZX09711001-007-002), National Natural Science Foundation of China (81872753).References
- C. L. Daley, Thorac. Surg. Clin., 2019, 29, 19–25 CrossRef PubMed.
- World Health Organization, Global Tuberculosis Report, 2019, www.who.int/tb/publications/global_report/en/ Search PubMed.
- M. AlMatar, H. AlMandeal, I. Var, B. Kayar and F. Koksal, Biomed. Pharmacother., 2017, 91, 546–558 CrossRef CAS PubMed.
- J. Herrmann, J. Rybniker and R. Muller, Curr. Opin. Biotechnol., 2017, 48, 94–101 CrossRef CAS PubMed.
- S. Tiberi, N. du Plessis, G. Walzl, M. J. Vjecha, M. Rao, F. Ntoumi, S. Mfinanga, N. Kapata, P. Mwaba, T. D. McHugh, G. Ippolito, G. B. Migliori, M. J. Maeurer and A. Zumla, Lancet Infect. Dis., 2018, 18, e183–e198 CrossRef PubMed.
- D. T. Hoagland, J. Liu, R. B. Lee and R. E. Lee, Adv. Drug Delivery Rev., 2016, 102, 55–72 CrossRef CAS PubMed.
- N. J. Ryan and J. H. Lo, Drugs, 2014, 74, 1041–1045 CrossRef CAS PubMed.
- Side Effects of bedaquiline and pretomanid, https://www.drugs.com/sfx/pretomanid-side-effects.html, https://www.drugs.com/sfx/bedaquiline-side-effects.html.
- M. Brecik, I. Centarova, R. Mukherjee, G. S. Kolly, S. Huszar, A. Bobovska, E. Kilacskova, V. Mokosova, Z. Svetlikova, M. Sarkan, J. Neres, J. Kordulakova, S. T. Cole and K. Mikusova, ACS Chem. Biol., 2015, 10, 1631–1636 CrossRef CAS PubMed.
- R. V. Chikhale, M. A. Barmade, P. R. Murumkar and M. R. Yadav, J. Med. Chem., 2018, 61, 8563–8593 CrossRef CAS PubMed.
- V. Makarov, G. Manina, K. Mikusova, U. Mollmann, O. Ryabova, B. Saint-Joanis, N. Dhar, M. R. Pasca, S. Buroni, A. P. Lucarelli, A. Milano, E. De Rossi, M. Belanova, A. Bobovska, P. Dianiskova, J. Kordulakova, C. Sala, E. Fullam, P. Schneider, J. D. McKinney, P. Brodin, T. Christophe, S. Waddell, P. Butcher, J. Albrethsen, I. Rosenkrands, R. Brosch, V. Nandi, S. Bharath, S. Gaonkar, R. K. Shandil, V. Balasubramanian, T. Balganesh, S. Tyagi, J. Grosset, G. Riccardi and S. T. Cole, Science, 2009, 324, 801–804 CrossRef CAS PubMed.
- V. Makarov, S. T. Cole and K. Johnsson, J. Am. Chem. Soc., 2010, 132, 13663–13665 CrossRef PubMed.
- J. Neres, F. Pojer, E. Molteni, L. R. Chiarelli, N. Dhar, S. Boy-Rottger, S. Buroni, E. Fullam, G. Degiacomi, A. P. Lucarelli, R. J. Read, G. Zanoni, D. E. Edmondson, E. De Rossi, M. R. Pasca, J. D. McKinney, P. J. Dyson, G. Riccardi, A. Mattevi, S. T. Cole and C. Binda, Sci. Transl. Med., 2012, 4, 150ra121 Search PubMed.
- Phase 2a study of PBTZ169, https://clinicaltrials.gov/ct2/show/NCT03334734.
- Phase 1 study of BTZ043, https://clinicaltrials.gov/ct2/show/NCT04044001.
- V. Makarov, B. Lechartier, M. Zhang, J. Neres, A. M. van der Sar, S. A. Raadsen, R. C. Hartkoorn, O. B. Ryabova, A. Vocat, L. A. Decosterd, N. Widmer, T. Buclin, W. Bitter, K. Andries, F. Pojer, P. J. Dyson and S. T. Cole, EMBO Mol. Med., 2014, 6, 372–383 CrossRef CAS PubMed.
- R. Tiwari, P. A. Miller, L. R. Chiarelli, G. Mori, M. Sarkan, I. Centarova, S. H. Cho, K. Mikusova, S. G. Franzblau, A. G. Oliver and M. J. Miller, ACS Med. Chem. Lett., 2016, 7, 266–270 CrossRef CAS PubMed.
- L. Xiong, C. Gao, Y. J. Shi, X. Tao, J. Rong, K. L. Liu, C. T. Peng, N. Y. Wang, Q. Lei, Y. W. Zhang, L. T. Yu and Y. Q. Wei, RSC Adv., 2018, 8, 11163–11176 RSC.
- P. Li, B. Wang, X. Zhang, S. M. Batt, G. S. Besra, T. Zhang, C. Ma, D. Zhang, Z. Lin, G. Li, H. Huang and Y. Lu, Eur. J. Med. Chem., 2018, 160, 157–170 CrossRef CAS PubMed.
- R. Tiwari, P. A. Miller, S. Cho, S. G. Franzblau and M. J. Miller, ACS Med. Chem. Lett., 2015, 6, 128–133 CrossRef CAS PubMed.
- T. Karoli, B. Becker, J. Zuegg, U. Mollmann, S. Ramu, J. X. Huang and M. A. Cooper, J. Med. Chem., 2012, 55, 7940–7944 CrossRef CAS PubMed.
- R. Zhang, K. Lv, B. Wang, L. Li, B. Wang, M. Liu, H. Guo, A. Wang and Y. Lu, RSC Adv., 2017, 7, 1480–1483 RSC.
- K. Lv, X. You, B. Wang, Z. Wei, Y. Chai, B. Wang, A. Wang, G. Huang, M. Liu and Y. Lu, ACS Med. Chem. Lett., 2017, 8, 636–641 CrossRef CAS PubMed.
- K. Lv, Z. Tao, Q. Liu, L. Yang, B. Wang, S. Wu, A. Wang, M. Huang, M. Liu and Y. Lu, Eur. J. Med. Chem., 2018, 151, 1–8 CrossRef CAS PubMed.
- A. Wang, K. Lv, Z. Tao, J. Gu, L. Fu, M. Liu, B. Wan, S. G. Franzblau, C. Ma, X. Ma, B. Han, A. Wang, S. Xu and Y. Lu, Eur. J. Med. Chem., 2019, 181, 111595 CrossRef PubMed.
- C. Trefzer, M. Rengifo-Gonzalez, M. J. Hinner, P. Schneider, V. Y. Lu, M. Zheng, B. Wang, L. Fu, W. Zhao, P. Li, J. Xu, H. Zhu, H. Jin, D. Yin, H. Huang, A. M. Upton and Z. Ma, Antimicrob. Agents Chemother., 2011, 55, 5185–5193 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00750a |
| ‡ These authors contributed equally to this work. |
| § The M. tuberculosis strains used in these studies included the laboratory strain H37Rv (ATCC 27294; American Type Culture Collection, Rockville, MD) and drug-resistant clinical isolates. All isolates were obtained from the State Laboratory of Tuberculosis Reference of China. |
| ¶ African green monkey kidney (Vero) cells were purchased from the American Type Culture Collection (ATCC), and were cultured in Minimum Essential Medium (MEM) supplemented with 10% fetal bovine serum (FBS) (GBICO) and antibiotics (100 U per mL penicillin and 100 mg per mL streptomycin) at 37 °C in a 5% CO2 incubator. |
| This journal is © The Royal Society of Chemistry 2020 |