
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of an anthraquinone-containing polymeric photosensitizer and its application in aerobic photooxidation of thioethers†
Yang Chena,
Jianhua Hu*a and
Aishun Ding *b
*b
aState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, 2005 Songhu Road, Shanghai 200438, P. R. China. E-mail: hujh@fudan.edu.cn; Fax: +86-21-31242888; Tel: +86-21-55665280
bDepartment of Chemistry, Fudan University, 2005 Songhu Road, Shanghai 200438, P. R. China. E-mail: shunzi0522@126.com; Fax: +86-21-31249190; Tel: +86-21-31249190
First published on 13th March 2020
Abstract
Work on the synthesis of a polymeric photosensitizer and its application in the photooxidation of thioethers is reported herein. Firstly, the polymeric photosensitizer was designed and synthesized by the reaction of anthraquinone-2-carbonyl chloride (AQ-2-COCl) with poly(2-hydroxyethyl methacrylate) (PHEMA). Then, the visible light-induced photooxidation of thioethers under aerobic conditions was investigated. The results revealed that the reaction yielded sulfoxides highly chemoselectively in excellent yields with good substrate tolerance. Importantly, AQ-PHEMA could be easily recovered and reused more than 20 times without significant loss of the catalytic activity.
Introduction
Sulfoxides can be used as drugs1 and preservatives2 in daily life. They can also serve as important fragments in organic synthesis3–5 and bioactive molecules.6 Oxidation of thioethers into sulfoxides is the most direct method for the preparation of sulfoxides.7–9 Some other methods for synthesizing sulfoxides were developed in the past few decades, such as organic catalytic oxidation,10 hydrogen peroxide oxidation,11,12 metal complex-catalyzed oxidation,13,14 photooxidation,15,16 etc. However, these methods normally require stoichiometric oxidants, which produce a large amount of environmentally harmful waste. At the same time, another issue of the above methods is controlling the selectivity for sulfoxide and over-oxidation to the sulfone by-product.17 Although some catalytic systems exhibited high chemoselectivity with small molecular catalysts, from a practical application point of view, some of these catalysts could not be easily recovered after one catalytic cycle, or the catalyst was too expensive.10,18 Considering the concept of “green chemistry”, an environmentally friendly, highly catalytically efficient and highly selective oxidation of thioethers into sulfoxides is still urgently needed.Visible light19–32 is an economically green energy. Visible light-induced photocatalysis is a hot research topic33–43 in organic synthesis. For example, Guo’s group reported the cyclopropanation of dibromomalonate derivatives and alkenes. This is the first report on the generation of carbanions under visible light photoredox catalysis.44 Guo’s group also reported the first activation of alkynes using a Lewis acid catalyst together with eosin Y as a small molecular photocatalyst.45 Although small molecule photosensitizers46–48 exhibit good catalytic properties in some reactions, their disadvantages, such as difficulty of separation and reuse as well as high cost, remain unsolved. Therefore, for the purpose of recycling the catalyst, polymeric photosensitizers which were designed and synthesized by linking photosensitive groups onto polymeric chains have drawn much attention from researchers.49–55 Compared with small molecule photosensitizers, polymeric photosensitizers can be easily recycled, which not only reduces the cost, but also simplifies the process. Thus, they have been widely used in the fields of medicine, biology and hydrogels.56–58
But the catalysts could not be easily recovered and reused. Thus, we wondered whether a small molecule photosensitizer could be attached to a polymer, forming a novel polymeric photocatalytic material. This catalytic material could be recycled and reused after catalyzing the photoreaction. With our continuing interest in the synthesis and application of PHEMA,36 AQ-2-COOH59–61 and PHEMA36 were used to synthesize the designed polymeric photosensitizer AQ-PHEMA (Scheme 1) by immobilizing anthraquinone (AQ) onto PHEMA. In the next step, AQ-PHEMA (for synthesis and characterization of AQ-PHEMA, see ESI†) was tested as a photocatalyst in the visible light-induced photooxidation of thioethers under aerobic conditions. Finally, recycling experiments were carefully conducted.
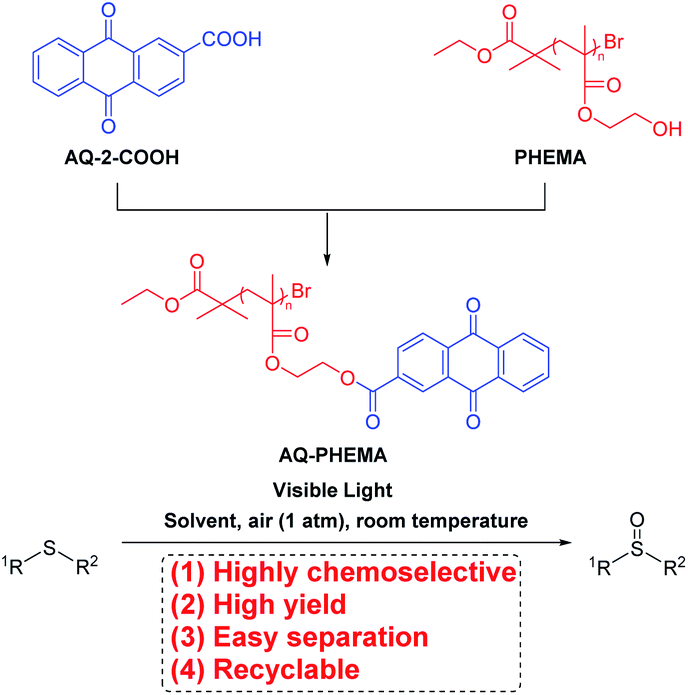 | ||
| Scheme 1 The synthetic procedure of AQ-PHEMA and its application in the photooxidation of thioethers. | ||
Results and discussion
Optimization and scope investigation
In the beginning, methyl phenyl thioether (1a) was chosen as the model substrate for the investigation of AQ-PHEMA-catalyzed photooxidation. The initial attempt was carried out in cyclohexane under air atmosphere at room temperature. After 72 hours, the desired product methyl phenyl sulfoxide (2a) was formed in 21% NMR yield, with 29% of 1a remaining unreacted and 2% of the over-oxidized by-product methyl phenyl sulfone (3a) being observed (entry 1, Table 1). Encouraged by this result, a survey of solvents was carried out (entries 2–10, Table 1). When toluene or THF was tested, the NMR yield of 2a increased. But 3a was also produced in a higher yield (entries 2 and 3, Table 1). Then, ethyl acetate, dichloromethane, acetone, and ether were tested. Although a small amount of 3a was formed, the NMR yield of 2a was remarkably increased (entries 4–7, Table 1). When CH3CN or CH3NO2 was chosen as the solvent, 2a was formed in an excellent yield with less than 5% 3a generated (entries 8 and 9, Table 1). Fortunately, the reaction in CH3OH gave a 99% NMR yield of 2a with only 1% 3a detected (entry 10, Table 1). Thus, CH3OH was chosen as the optimal solvent for this reaction. Notably, AQ-PHEMA is insoluble in all the above tested solvents and can only be dispersed in the solvent, so it can be easily recovered by a simple filtration. Subsequently, modifications of the catalytic amount of AQ-PHEMA were conducted (entries 10–13, Table 1). The results showed that the catalytic amount of AQ-PHEMA could be reduced to 1 mol% (entry 12, Table 1). Next, control experiments were carried out and the results demonstrated that both the catalyst and light were necessary for this transformation (entries 14 and 15, Table 1). Furthermore, taking into account the thermal effect of the purple LED light, the reaction was carried out at 50 °C without light. The results confirmed that no reaction occurred at all (entry 16, Table 1). Next, the oxidation using AQ as catalyst was investigated. The reaction was completed in 9 hours with high chemoselectivity and excellent yield (entry 17, Table 1). However, AQ was difficult to recover due to its solubility in CH3OH. Finally, to test whether the bromine atom derived from the initiator in the synthesis of PHEMA has some effect on the reaction, the oxidation using PHEMA as catalyst was investigated. No reaction took place (entry 18, Table 1). The above two results clearly demonstrated that the bromine atom did not play an important role in this photocatalytic reaction and AQ was the key catalyst in this transformation. Thus, Condition A (1 mol% of AQ-PHEMA, CH3OH, purple LED, air (1 atm), and room temperature) was considered as the optimized conditions for further studies.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (mol%) | Time (h) | NMR yieldb (%) | ||
| 1a | 2a | 3a | ||||
| a The reaction was carried out using 1a (1 mmol) and AQ-PHEMA in solvents (5 mL) irradiated by a purple LED under air atmosphere at rt. (Based on AQ anchored on PHEMA, the mass of 5 mol% AQ-PHEMA is 17 mg; the mass of 3 mol% AQ-PHEMA is 10 mg; the mass of 1 mol% AQ-PHEMA is 3 mg; and the mass of 0.5 mol% AQ-PHEMA is 2 mg.)b Yield determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 (1 mmol) as internal standard.c Isolated yield of 2a.d The reaction was carried out without light.e The reaction was carried out at 50 °C.f The reaction was carried out using 1 mol% AQ as catalyst.g The reaction was carried out in the presence of 4 mg of PHEMA. | ||||||
| 1 | Cyclohexane | 5 | 72 | 29 | 21 | 2 |
| 2 | Toluene | 5 | 57 | 0 | 30 | 16 |
| 3 | THF | 5 | 48 | 0 | 39 | 54 |
| 4 | Ethyl acetate | 5 | 49 | 0 | 50 | 19 |
| 5 | DCM | 5 | 72 | 0 | 51 | 3 |
| 6 | Acetone | 5 | 39 | 0 | 66 | 7 |
| 7 | Et2O | 5 | 72 | 0 | 74 | 1 |
| 8 | CH3CN | 5 | 36 | 0 | 95 | 5 |
| 9 | CH3NO2 | 5 | 28 | 0 | 97 | 3 |
| 10 | CH3OH | 5 | 16 | 0 | 99 | 1 |
| 11 | CH3OH | 3 | 18 | 0 | 99 | 1 |
| 12 | CH3OH | 1 | 24 | 0 | 99 (96)c | 1 |
| 13 | CH3OH | 0.5 | 24 | 48 | 44 | 6 |
| 14 | CH3OH | — | 24 | 99 | 0 | 0 |
| 15d | CH3OH | 1 | 24 | 99 | 0 | 0 |
| 16d,e | CH3OH | 1 | 24 | 99 | 0 | 0 |
| 17f | CH3OH | 1 | 9 | 0 | 99 | 1 |
| 18g | CH3OH | — | 16 | 99 | 0 | 0 |
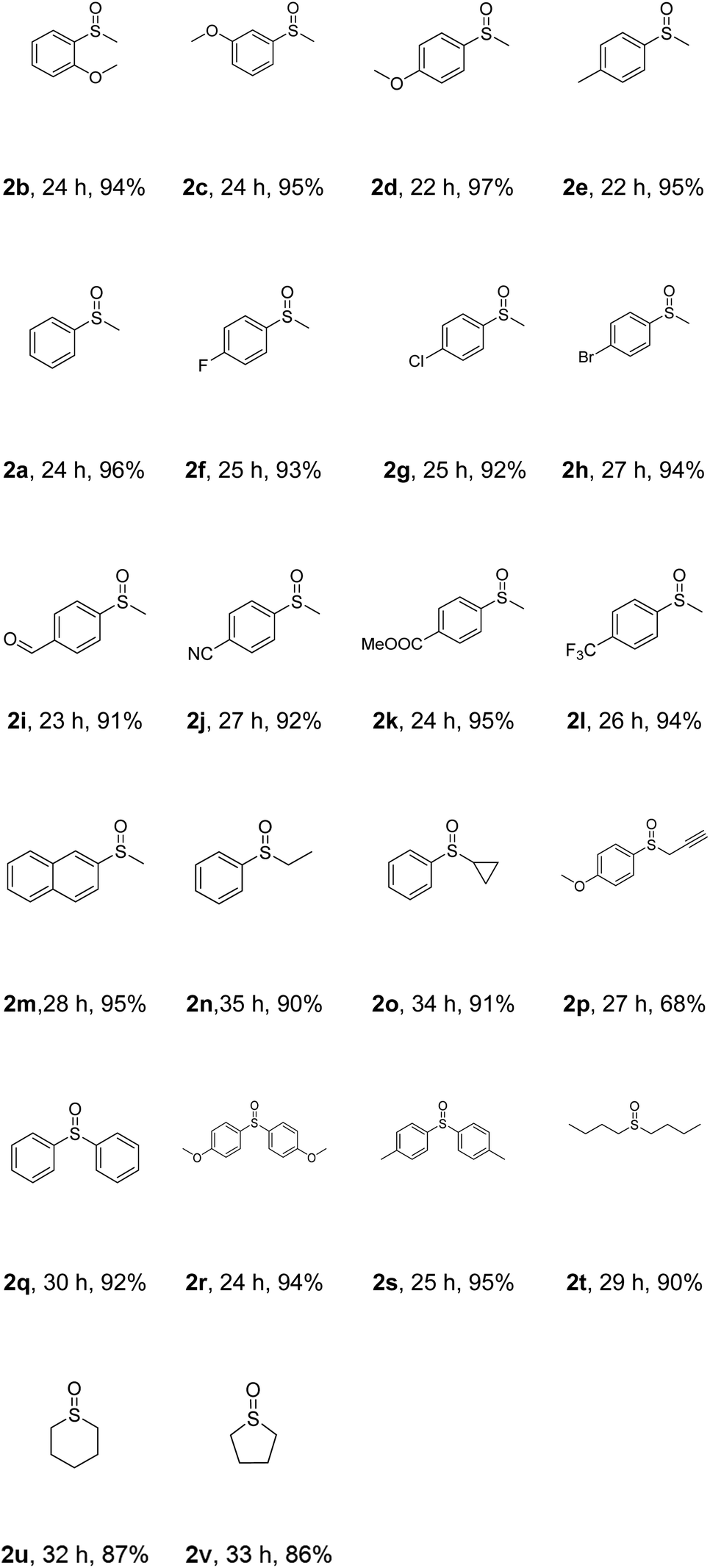
With the optimized reaction conditions in hand, the scope of this photoreaction was explored (Table 2). Firstly, the electronic effect of the aryl group in methyl aryl thioether was examined. With strong electron donating groups, like methoxy, attached to the aryl ring, excellent yields were obtained for the ortho-, meta- and para-methoxyphenyl thioethers (2b–d). For weak electron donating groups, like methyl, the corresponding product (2e) could be generated in an excellent yield. Good reactivity was also observed with halogen-attached substrates (2f–h). Then, we tested substrates with strong electron withdrawing groups. The results showed that substrates with aldehyde (2i), nitrile (2j), methoxycarbonyl (2k) or trifluoromethyl (2l) worked very well. Notably, the naphthyl group was also tolerated under Condition A (2m). Next, the effect of different alkyl groups was examined. Reactants with ethyl, cyclopropyl and propynyl groups yielded the desired products (2n–p) in 90%, 91% and 68% isolated yield, respectively. The reactivity of diaryl thioethers was also investigated. Excellent chemoselectivity as well as excellent yields were observed (2q–s). Finally, reactions with dialiphatic thioethers were studied. Di-n-butyl thioether gave an excellent yield of 2t, while tetrahydro-2H-thiopyran and tetrahydrothiophene led to slightly lower yields of 2u and 2v, respectively. The above results indicated that this reaction showed very good substrate scope and functional group tolerance.
|
|---|
| a All reactions were carried out using 1 (1 mmol) and AQ-PHEMA (1 mol%) in CH3OH (5 mL) irradiated by a purple LED light at rt under air atmosphere. The isolated yield is reported. |
 |
Recycling experiments
To investigate the recyclability of AQ-PHEMA in this transformation, 4-methoxyphenyl methyl thioether (1d) was selected as the model substrate for the AQ-PHEMA recycling experiments under Condition A. 1d could be completely consumed after 24 hours and the NMR yield of the product 4-methoxyphenyl methyl sulfoxide (2d) was 99% in the first cycle of the photocatalytic reaction. After the first cycle, the catalyst AQ-PHEMA could be easily separated and recovered by simple filtration and directly used for the second cycle. The following reaction cycles were performed using the same procedure (for the specific plan for the recycling experiments, see ESI†). The results showed that AQ-PHEMA could be reused more than 20 times without significant loss of the catalytic activity (Fig. 1).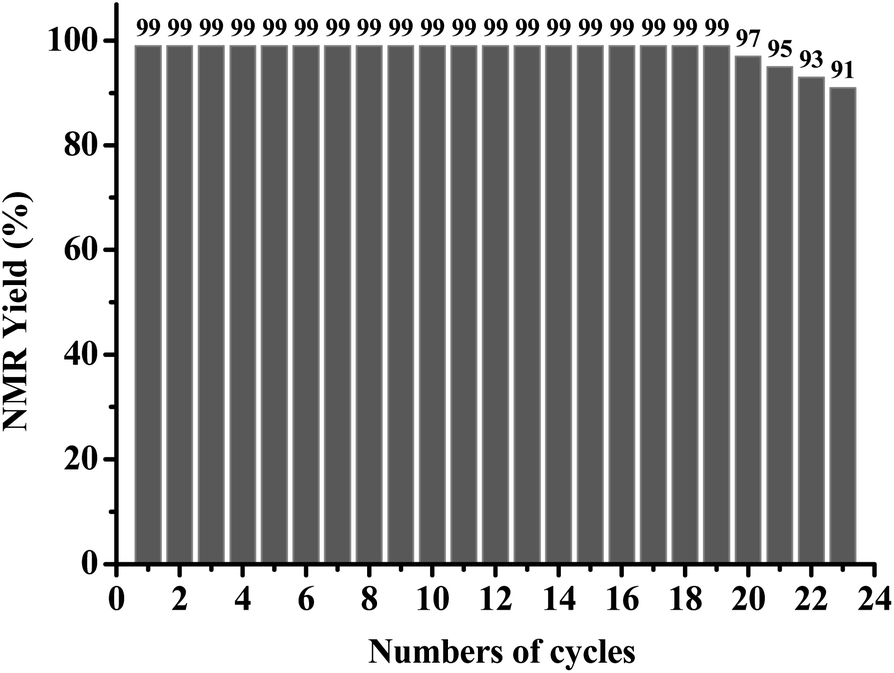 | ||
| Fig. 1 Recycling experiments for the photocatalytic reaction of 1d. | ||
At the end of the recycling experiments, the recovered AQ-PHEMA (after 23 cycles) was characterized by 1H NMR and GPC. As shown in Fig. 2, no significant changes were observed in the 1H NMR spectrum compared to the original AQ-PHEMA. The molecular weight Mn,GPC and molecular distribution Mw/Mn of the recovered AQ-PHEMA (after 23 cycles) were very close to those of the original AQ-PHEMA (Fig. 3). These results clearly indicated that the structure of AQ-PHEMA did not change during the whole catalytic procedure.
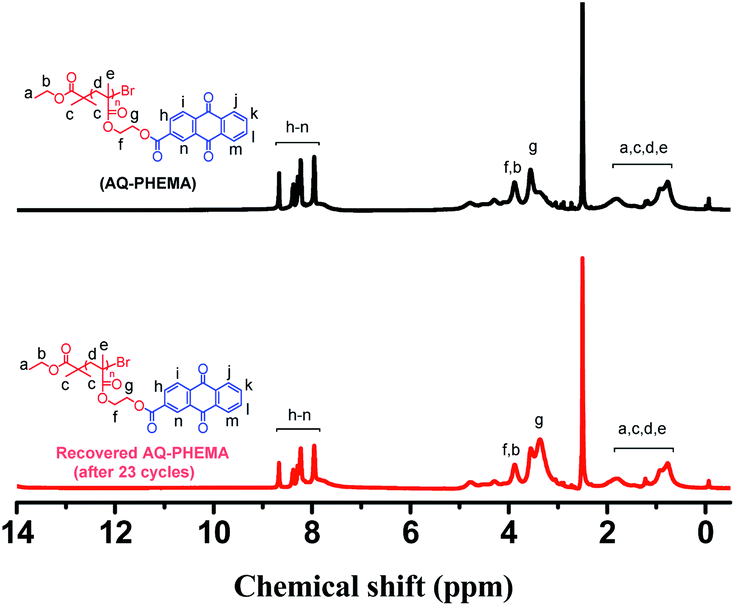 | ||
| Fig. 2 1H NMR spectra for AQ-PHEMA and the recovered AQ-PHEMA (after 23 cycles) in d6-DMSO. | ||
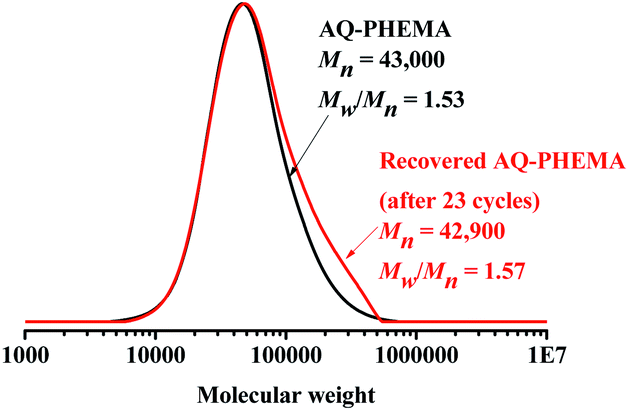 | ||
| Fig. 3 The GPC traces of AQ-PHEMA and the recovered AQ-PHEMA (after 23 cycles). | ||
Proposed mechanism
Based on the Guo group’s outstanding work,9 literature precedents59–64 and the cyclic voltammogram of AQ-PHEMA under irradiation (see Fig. S2†), the following possible reaction mechanism was proposed as shown in Fig. 4. AQ-PHEMA was excited under visible light irradiation and subsequently sensitized oxygen to singlet oxygen which was more oxidative than normal triplet oxygen. Singlet oxygen could take an electron from the lone pair of electrons in sulfide 1 to form a sulfide radical cation 3 and a superoxide radical anion, and then 3 further interacted with the superoxide radical anion to obtain intermediate 4. 4 reacted with another molecule of thioether 1 to form the final sulfoxide product 2.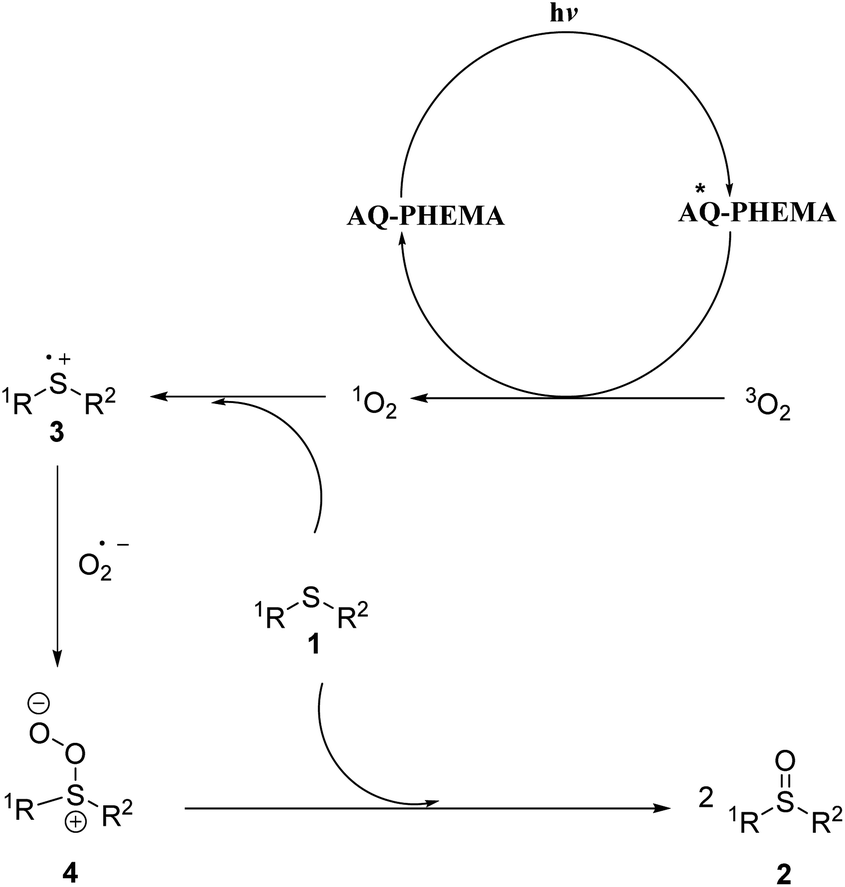 | ||
| Fig. 4 A possible reaction mechanism. | ||
Conclusions
In conclusion, we successfully synthesized the polymeric photosensitizer AQ-PHEMA by immobilizing the AQ functional group onto the precursor PHEMA. AQ-PHEMA could be used as a visible light sensitizer to catalyze the oxidation of thioethers into sulfoxides under aerobic conditions. The reaction exhibited high efficiency and chemoselectivity with extensive functional group tolerance. A possible mechanism was proposed. The AQ-PHEMA catalyst could be easily recovered and reused more than 20 times. The excellent photocatalytic properties, easy separation, and recyclability revealed the great potential of this catalyst in industrial applications and environment protection.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We greatly acknowledge the financial support from Shanghai Scientific and Technological Innovation Project (18YF1428800).References
- Q. L. Zeng, S. Gao and A. K. Chelashaw, Mini-Rev. Org. Chem., 2013, 10, 198–206 CrossRef CAS.
- J. J. Tarrand, P. R. LaSala, X. Y. Han, K. V. Rolston and D. P. Kontoyiannis, J. Clin. Microbiol., 2012, 50, 1552–1557 CrossRef CAS PubMed.
- M. C. Carreno, G. Hernandez-Torres, M. Ribagorda and A. Urbano, Chem. Commun., 2009, 6129–6144, 10.1039/b908043k.
- J. Magolan, E. Jones-Mensah and M. Karki, Synthesis, 2016, 48, 1421–1436 CrossRef.
- X.-F. Wu and K. Natte, Adv. Synth. Catal., 2016, 358, 336–352 CrossRef CAS.
- J. Legros, J. R. Dehli and C. Bolm, Adv. Synth. Catal., 2005, 347, 19–31 CrossRef CAS.
- K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetrahedron, 2005, 61, 8315–8327 CrossRef CAS.
- P. Kowalski, K. Mitka, K. Ossowska and Z. Kolarska, Tetrahedron, 2005, 61, 1933–1953 CrossRef CAS.
- C. Ye, Y. Zhang, A. Ding, Y. Hu and H. Guo, Sci. Rep., 2018, 8, 2205 CrossRef PubMed.
- S. Murahashi, D. Zhang, H. Iida, T. Miyawaki, M. Uenaka, K. Murano and K. Meguro, Chem. Commun., 2014, 50, 10295–10298 RSC.
- S. Liao, I. Coric, Q. Wang and B. List, J. Am. Chem. Soc., 2012, 134, 10765–10768 CrossRef CAS PubMed.
- B. J. Marsh and D. R. Carbery, Tetrahedron Lett., 2010, 51, 2362–2365 CrossRef CAS.
- L. Zhao, H. Zhang and Y. Wang, J. Org. Chem., 2016, 81, 129–136 CrossRef CAS PubMed.
- S. R. Gogoi, J. J. Boruah, G. Sengupta, G. Saikia, K. Ahmed, K. K. Bania and N. S. Islam, Catal. Sci. Technol., 2015, 5, 595–610 RSC.
- D. Chao and M. Zhao, ChemSusChem, 2017, 10, 3358–3362 CrossRef CAS PubMed.
- T. Neveselý, E. Svobodová, J. Chudoba, M. Sikorski and R. Cibulka, Adv. Synth. Catal., 2016, 358, 1654–1663 CrossRef.
- B. Sreedhar, P. Radhika, B. Neelima, N. Hebalkar and A. K. Mishra, Catal. Commun., 2008, 10, 39–44 CrossRef CAS.
- S. Liao and B. List, Adv. Synth. Catal., 2012, 354, 2363–2367 CrossRef CAS.
- T. Bach, Angew. Chem., Int. Ed., 2015, 54, 11294–11295 CrossRef CAS PubMed.
- J. W. Beatty and C. R. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed.
- X. Deng, Z. Li and H. Garcia, Chem.–Eur. J., 2017, 23, 11189–11209 CrossRef CAS PubMed.
- A. Ding, Y. Zhang, Y. Chen, R. Rios, J. Hu and H. Guo, Tetrahedron Lett., 2019, 60, 660–663 CrossRef CAS.
- R. Jin, Y. Chen, W. Liu, D. Xu, Y. Li, A. Ding and H. Guo, Chem. Commun., 2016, 52, 9909–9912 RSC.
- C. B. Larsen and O. S. Wenger, Chem.–Eur. J., 2018, 24, 2039–2058 CrossRef CAS PubMed.
- Y. Liu, Y. Song, Y. You, X. Fu, J. Wen and X. Zheng, J. Saudi Chem. Soc., 2018, 22, 439–448 CrossRef CAS.
- S. Poplata, A. Troster, Y. Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- E. R. Shilpa and V. Gayathri, J. Saudi Chem. Soc., 2018, 22, 678–691 CrossRef CAS.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193–5203 RSC.
- Y. Zhang, C. Ye, S. Li, A. Ding, G. Gu and H. Guo, RSC Adv., 2017, 7, 13240–13243 RSC.
- R. Alonso and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 4368–4371 CrossRef CAS PubMed.
- A. M. Asiri, M. S. Al-Amoudi, S. A. Bazaid, A. A. Adam, K. A. Alamry and S. Anandan, J. Saudi Chem. Soc., 2014, 18, 155–163 CrossRef.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- A. Ding, Y. Chen, G. Wang, Y. Zhang, J. Hu and H. Guo, Polymer, 2019, 174, 101–108 CrossRef CAS.
- C. Feng and X. Huang, Acc. Chem. Res., 2018, 51, 2314–2323 CrossRef CAS PubMed.
- A. B. Lavand and Y. S. Malghe, J. Saudi Chem. Soc., 2015, 19, 471–478 CrossRef.
- J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 102–113 RSC.
- M. H. Shaw, J. Twilton and D. W. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- G. Yilmaz, B. Aydogan, G. Temel, N. Arsu, N. Moszner and Y. Yagci, Macromolecules, 2010, 43, 4520–4526 CrossRef CAS.
- G. Liao, J. Fang, Q. Li, S. Li, Z. Xu and B. Fang, Nanoscale, 2019, 11, 7062–7096 RSC.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- Y. Zhang, R. Qian, X. Zheng, Y. Zeng, J. Sun, Y. Chen, A. Ding and H. Guo, Chem. Commun., 2015, 51, 54–57 RSC.
- R. Jin, Y. Chen, W. Liu, D. Xu, Y. Li, A. Ding and H. Guo, Chem. Commun., 2016, 52, 9909–9912 RSC.
- Y. J. Wang, G. Chang, Q. Chen, G. J. Yang, S. Q. Fan and B. Fang, Chem. Commun., 2015, 51, 685–688 RSC.
- S.-C. Yang, G. Chang, G.-J. Yang, Y.-J. Wang and B. Fang, Catal. Sci. Technol., 2015, 5, 228–233 RSC.
- D. Xu, Q. Chu, Z. Wu, Q. Chen, S.-Q. Fan, G.-J. Yang and B. Fang, J. Catal., 2015, 325, 118–127 CrossRef CAS.
- C. Cepraga, S. Marotte, E. Ben Daoud, A. Favier, P. H. Lanoe, C. Monnereau, P. Baldeck, C. Andraud, J. Marvel, M. T. Charreyre and Y. Leverrier, Biomacromolecules, 2017, 18, 4022–4033 CrossRef CAS PubMed.
- B. Xu, C. Feng, J. Hu, P. Shi, G. Gu, L. Wang and X. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 6685–6692 CrossRef CAS PubMed.
- B. Xu, C. Feng and X. Huang, Nat. Commun., 2017, 8, 333 CrossRef PubMed.
- Y. Zhao, Z. Lu, X. Dai, X. Wei, Y. Yu, X. Chen, X. Zhang and C. Li, Bioconjugate Chem., 2018, 29, 3222–3230 CrossRef CAS PubMed.
- W. Zhong, J. Huang, S. Liang, J. Liu, Y. Li, G. Cai, Y. Jiang and J. Liu, ACS Energy Lett., 2020, 5, 31–38 CrossRef CAS.
- W. Zhong, S. Shen, M. He, D. Wang, Z. Wang, Z. Lin, W. Tu and J. Yu, Appl. Catal., B, 2019, 258, 117967 CrossRef CAS.
- W. Zhong, S. Shen, S. Feng, Z. Lin, Z. Wang and B. Fang, CrystEngComm, 2018, 20, 7851–7856 RSC.
- Y. Liu, X. Huang, K. Han, Y. Dai, X. Zhang and Y. Zhao, ACS Sustainable Chem. Eng., 2019, 7, 4004–4011 CrossRef CAS.
- Z. Lu, X. Zhang, Y. Zhao, Y. Xue, T. Zhai, Z. Wu and C. Li, Polym. Chem., 2015, 6, 302–310 RSC.
- E. Yaghini, R. Dondi, K. M. Tewari, M. Loizidou, I. M. Eggleston and A. J. MacRobert, Sci. Rep., 2017, 7, 6059 CrossRef PubMed.
- L. Cui, Y. Matusaki, N. Tada, T. Miura, B. Uno and A. Itoh, Adv. Synth. Catal., 2013, 355, 2203–2207 CrossRef CAS.
- M. Taguchi, Y. Nagasawa, E. Yamaguchi, N. Tada, T. Miura and A. Itoh, Tetrahedron Lett., 2016, 57, 230–232 CrossRef CAS.
- Q. Zhou, S. Xu and R. Zhang, Tetrahedron Lett., 2019, 60, 734–738 CrossRef CAS.
- C. C. B. Enrico and L. Osvaldo, Tetrahedron, 1997, 53, 4469–4478 CrossRef.
- J. Dad’ová, E. Svobodová, M. Sikorski, B. König and R. Cibulka, ChemCatChem, 2012, 4, 620–623 CrossRef.
- W. Li, Z. Xie and X. Jing, Catal. Commun., 2011, 16, 94–97 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, and characterization data for all compounds. See DOI: 10.1039/d0ra00880j |
| This journal is © The Royal Society of Chemistry 2020 |