
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn improved synthesis of telmisartan via the copper-catalyzed cyclization of o-haloarylamidines†
Junchi Zhangab,
Rui Liab,
Fuqiang Zhuc,
Changliang Sun*c and
Jingshan Shen *ab
*ab
aCAS Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences (CAS), 555 Zuchongzhi Road, Shanghai 201203, People's Republic of China. E-mail: shenjingshan@simm.ac.cn
bUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, People's Republic of China
cTopharman Shanghai Co., Ltd, Building 1, No. 388 Jialilue Road, Zhangjiang Hitech Park, Shanghai 201203, People's Republic of China. E-mail: changliang.sun@topharman.cn
First published on 3rd April 2020
Abstract
A concise synthetic route was designed for making telmisartan. The key bis-benzimidazole structure was constructed via the copper-catalyzed cyclization of o-haloarylamidines. By adopting this approach, telmisartan was obtained in a 7-step overall yield of 54% starting from commercially available 3-methyl-4-nitrobenzoic acid, and the use of HNO3/H2SO4 for nitration and polyphosphoric acid (PPA) for cyclization in the reported literatures were avoided.
1. Introduction
Telmisartan 1, a potent and selective angiotensin II type 1 (AT1) receptor antagonist, is one of the top-selling drugs for the treatment of essential hypertension.1,2 This drug is marketed under the brand name of Micardis®, and is characterized by excellent AT1 receptor binding affinity, long half-life, and good tolerability.1–3Take a brief look at the structure of telmisartan, it is assembled from two different benzimidazole subunits and a biphenyl-2-carboxylic acid fragment. The original approach was reported by Ries et al. in 1993 (Scheme 1),4 in which, the central benzimidazole ring was initially constructed stepwise from 4-amino-3-methylbenzoic acid methyl ester 2. Subsequently, after the saponification of 3, the bis-benzimidazole intermediate 5 was formed via the condensation between the free carboxyl group of the central benzimidazole moiety and N-methyl-1,2-benzenediamine 4. Finally, alkylation of 5 with the substituted biphenyl fragment 6 was followed by hydrolysis, and afforded the final product 1 in an eight-step overall yield of 21%.
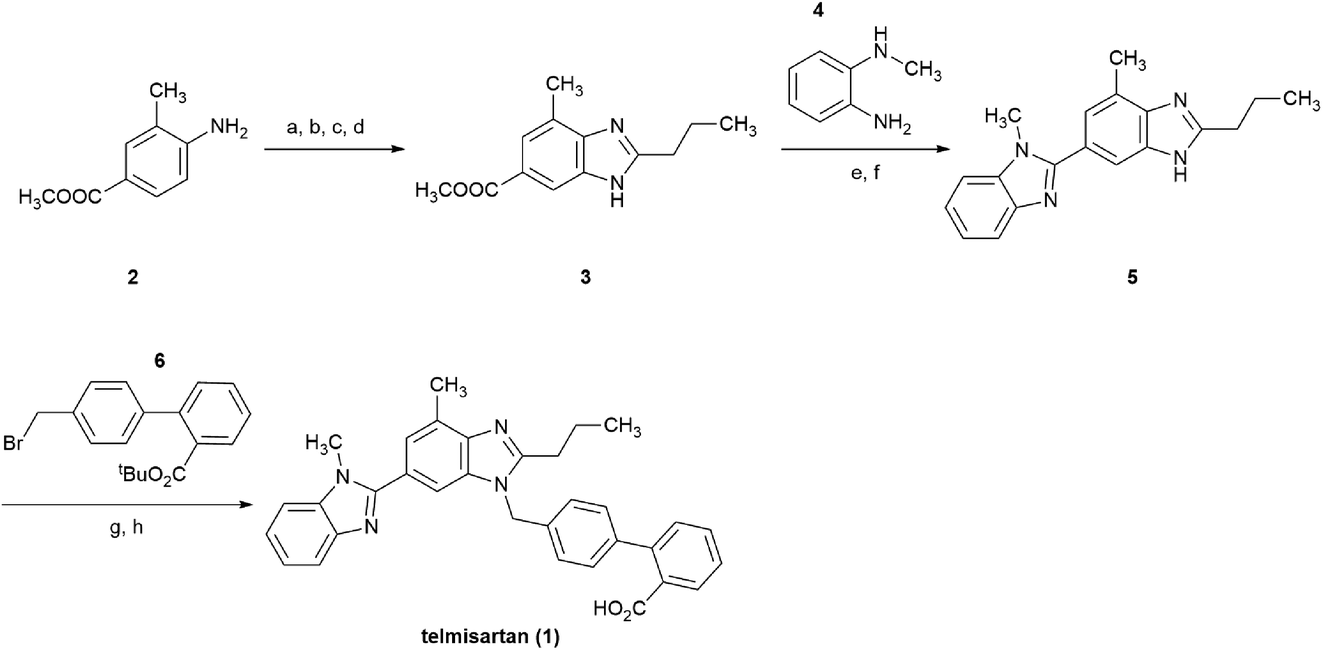 | ||
| Scheme 1 Original approach for the synthesis of telmisartan. Reagents and conditions: (a) n-PrCOCl, C6H5Cl, 100 °C; (b) HNO3/H2SO4, 0 °C; (c) Pd/C, 5 bar H2, MeOH; (d) AcOH, reflux; (e) NaOH, MeOH/H2O, reflux; (f) 4, PPA, 150 °C; (g) 6, t-BuOK, DMF, rt; (h) TFA, DCM, rt. | ||
However, excess amount of nitration reagent HNO3/H2SO4 was utilized among the formation of the central benzimidazole moiety, which might bring concerns to safety and wastewater disposal.5–8 In addition, the crucial intermediate 5 was built using the viscous PPA as the solvent, increased the difficulty of production operation and sewage treatment.9 Moreover, regioisomer impurity was inevitable in the alkylation of 5 with 6, which might cause enhanced difficulty with purification and lose on yield.
Several routes with different synthetic strategies have been reported.2,10–13 Wang et al. adopted a PPA-free method to constructed the methylbenzimidazole ring by employing the cyclocondensation between aromatic aldehyde and o-phenylenediamine.2 In addition, Goossen et al. developed a reductive amination–condensation sequence to build the central benzimidazole moiety,10 completely avoided the regioisomer impurity. However, the problem of the excess use of nitration reagent still remains in the above two routes. Martin et al. reported a convergent approach to the synthesis of telmisartan via a Suzuki cross-coupling, which is direct and efficient.13 However, considering the fact that palladium catalysts were utilized in two steps of the route, the price element might be taken into account.
Transition-metal-catalyzed cross-coupling reactions belong to the frontier areas in modern organic chemistry, among which, the copper-catalyzed Ullmann-type reaction are widely utilized in C–N bond formation due to the high efficiency, low cost, and low toxicity of copper catalysts.14–17 Considerable efforts have been devoted to the copper-catalyzed synthesis of benzimidazole derivatives,18–22 in which the cyclization of o-haloarylamidines has drawn much attention.23–29 Herein, we explored the idea to build the key bis-benzimidazole structure of telmisartan via the copper-catalyzed cyclization of o-haloarylamidines,30,31 which were accessible to be obtained from o-haloarylamines (Scheme 2). This approach was suitable to produce telmisartan with good overall yield while avoiding the shortcomings associated with the reported routes.
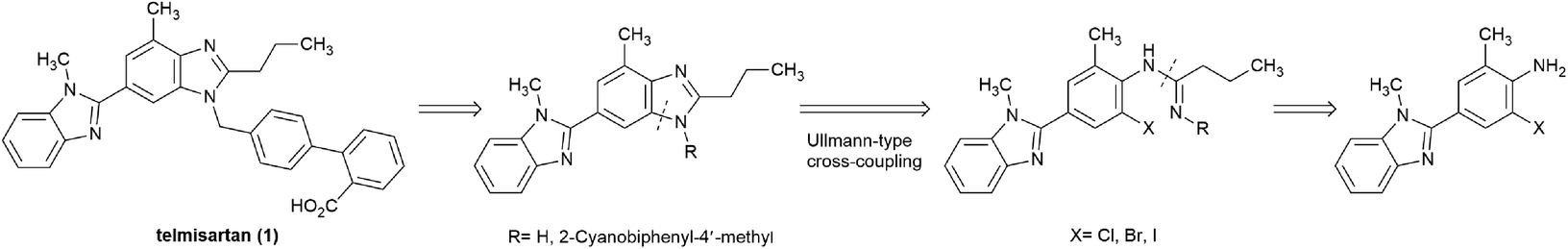 | ||
| Scheme 2 Retrosynthetic analysis of telmisartan. | ||
2. Results and discussion
2.1 Synthesis of o-haloarylamidines 12a–c and 14
Initially, the o-haloarylamines 10a–c were formed in three steps starting from commercially available 3-methyl-4-nitrobenzoic acid 7 (Scheme 3). An amidation–condensation sequence was conducted from compound 7 with 4, afforded 8 in the yield of 85%. Afterwards, reduction of nitro group was easy to occur under Pd/C–H2 condition, and the downstream selective halogenation of 9 was amenable to be carried out by N-halosuccinimides, gave the o-haloarylamines 10a–c with satisfactory regioselectivity and yields, probably due to the higher reactivity on ortho-amino position of 9. Furthermore, the isolated yields of both o-bromoarylamine 10b (86%) and o-iodoarylamine 10c (89%) were higher than o-chloroarylamine 10a (70%).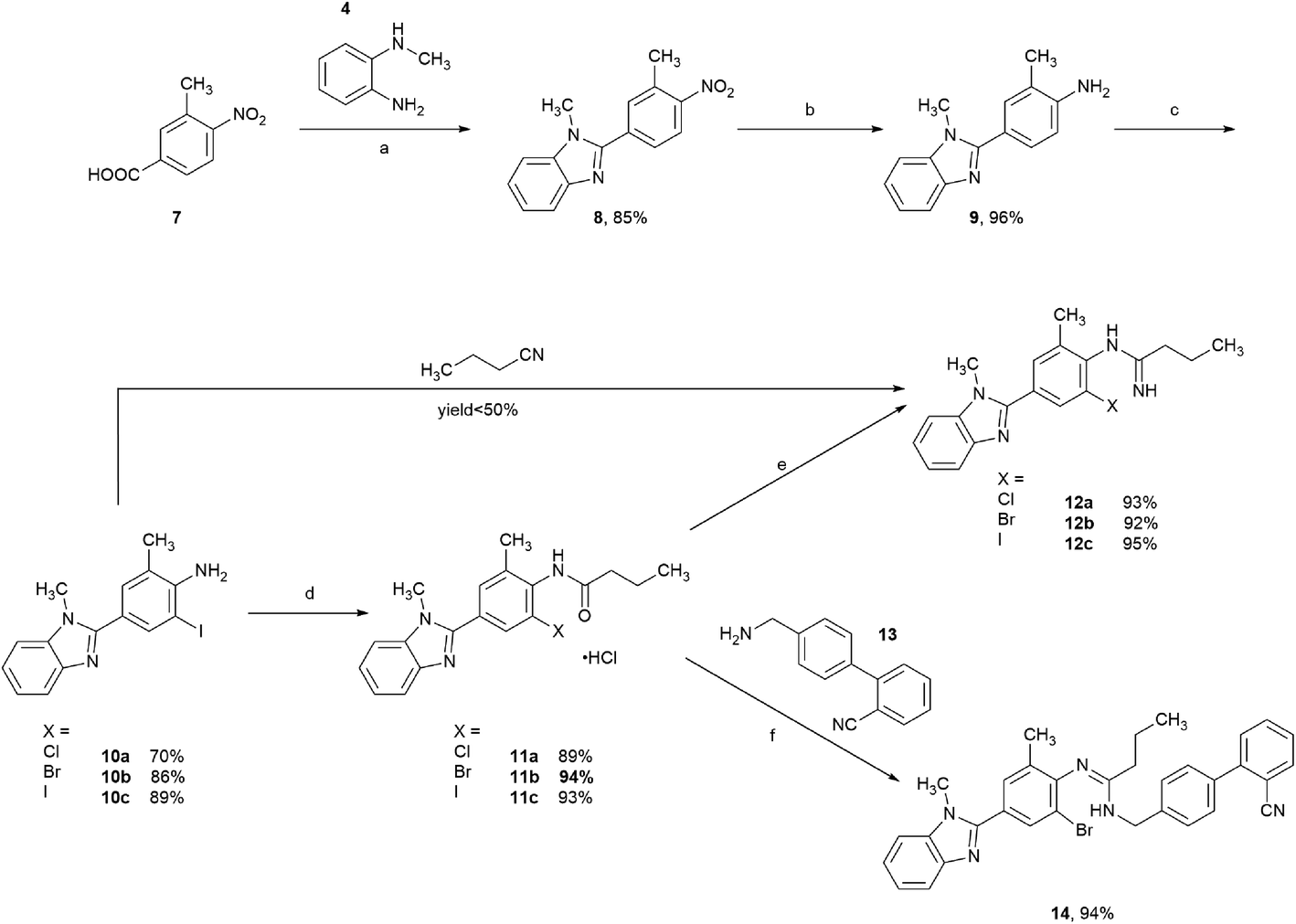 | ||
| Scheme 3 Synthesis of o-haloarylamidines 12a–c and 14. Reagents and conditions: (a) (i) oxalyl chloride, DMF, DCM, 0 °C, rt; (ii) 4, DIPEA, DCM, 0 °C, rt; (iii) TsOH·H2O, toluene, reflux; (b) Pd/C, 5 bar, MeOH, THF, 50 °C; (c) N-halosuccinimide; (d) n-PrCOCl, MeCN, reflux; (e) (i) triphosgene, DMF, MeCN, reflux; (ii) NH3 (7.0 M solution in MeOH); (f) (i) triphosgene, DMF, MeCN, reflux; (ii) 13, Et3N, DCM. | ||
Subsequently, with the o-haloarylamines 10a–c in hand, efforts were focused on the construction of the o-haloarylamidines 12a–c and 14 (Scheme 3). Firstly, 12a–c were directly prepared from 10a–c with n-butyronitrile in the presence of Lewis acids or Brønsted acids,32 however, yields of 12a–c in this method were low (<50%), probably due to the steric hindrance and electronic effect of the ortho-halogen in primary arylamines 10a–c. Hence, we adopted the approach to construct 12a–c and 14 from the arylamides 11a–c, which could be easily prepared from 10a–c through a base-free acylation reaction. The classic method for preparing arylamidines from arylamides usually requires oxalyl chloride/thionyl chloride/phosphorus chloride and so on, however, we attempted herein to introduce triphosgene (also named bis(trichloromethyl)carbonate), regarded broadly as a cleaner alternative, in our synthetic route. Direct reaction of compound 11a–c as free bases with triphosgene resulted in a rapid decomposition of triphosgene, while the monohydrochloride salts of 11a–c facilitated the chlorination step greatly, and the following quenching with NH3 solution in methanol or 4′-(aminomethyl)-[1,1′-biphenyl]-2-carbonitrile 13 (commercially available and is easy to obtain via reported methods33,34), afforded N-monosubstituted o-haloarylamidines 12a–c and N,N′-disubstituted o-haloarylamidine 14 with high yields. Furthermore, based on the construction of the N,N′-disubstituted o-haloarylamidine 14, the biphenyl moiety was introduced prior to the cyclization of the central benzimidazole ring, circumvented the generation of undesirable regioisomer caused by the N-alkylation of the benzimidazole ring in the previously reported method.10
2.2 Optimization of the reaction conditions for the cyclization of o-haloarylamidines
The final cyclization of 12a–c and 14 was conducted via the Ullmann-type cross-coupling reaction. Since the copper-catalyzed cyclization of o-haloarylamidines were the key steps in our research, efforts were devoted to the optimization of reaction conditions for the o-bromoarylamidine 12b (Table 1).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Catalyst | Ligand (0.1 equiv.) | Base | HPLC results in reaction mixtureb (%) | |||
| 5 | 12b | 11b | 10b | ||||||
a The reactions were performed on the scale of 0.5 mmol of 12b under the conditions: 0.1 equiv. of copper catalyst, 3.0 equiv. of base, 12 ml mmol−1 of solvent, heat for 8 h.b Calculated for the reaction mixture from the HPLC area percentage at 220 nm.c Not detected.d The ratio of mixed solvents was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume.e Not added. :1 by volume.e Not added. |
|||||||||
| 1 | DMF | 110 | CuI | DMEDA | Cs2CO3 | 79.4 | 16.9 | 1.2 | 2.5 |
| 2 | DMSO | 110 | CuI | DMEDA | Cs2CO3 | 91.0 | 6.2 | 0.9 | 1.9 |
| 3 | DMSO | 130 | CuI | DMEDA | Cs2CO3 | 98.5 | <0.1 | 0.2 | 1.3 |
| 4 | Toluene | Reflux | CuI | DMEDA | Cs2CO3 | 12.6 | 86.6 | 0.8 | —–c |
| 5 | 1,4-Dioxane | Reflux | CuI | DMEDA | Cs2CO3 | 32.0 | 66.0 | <0.1 | 2.0 |
| 6 | H2O | Reflux | CuI | DMEDA | Cs2CO3 | 4.1 | 94.4 | 1.5 | 0 |
| 7 | 1,4-Dioxane/H2Od | Reflux | CuI | DMEDA | Cs2CO3 | 91.8 | 7.6 | <0.1 | 0.5 |
| 8 | MeCN/H2Od | Reflux | CuI | DMEDA | Cs2CO3 | 63.6 | 34.5 | 1.3 | 0.6 |
| 9 | 2-Me-THF/H2Od | Reflux | CuI | DMEDA | Cs2CO3 | 71.9 | 26.1 | 1.9 | <0.1 |
| 10 | DME/H2Od | Reflux | CuI | DMEDA | Cs2CO3 | 87.9 | 10.8 | 0.4 | 0.9 |
| 11 | DMSO | 130 | —e | —e | Cs2CO3 | 1.4 | 44.0 | 1.2 | 53.4 |
| 12 | DMSO | 130 | CuI | —e | Cs2CO3 | 97.8 | 0.3 | 0.3 | 1.5 |
| 13 | DMSO | 130 | CuBr | —e | Cs2CO3 | 97.6 | 0 | 0.6 | 1.9 |
| 14 | DMSO | 130 | CuCl | —e | Cs2CO3 | 96.7 | 0.5 | 1.0 | 1.8 |
| 15 | DMSO | 130 | Cu2O | —e | Cs2CO3 | 96.4 | 0 | 0.8 | 2.8 |
| 16 | DMSO | 130 | CuBr2 | —e | Cs2CO3 | 82.0 | 10.7 | 1.2 | 6.0 |
| 17 | DMSO | 130 | CuCl2 | —e | Cs2CO3 | 83.1 | 10.5 | 1.2 | 5.2 |
| 18 | DMSO | 130 | CuO | —e | Cs2CO3 | 61.3 | 17.7 | 1.1 | 19.9 |
| 19 | DMSO | 130 | Cu(OAc)2 | —e | Cs2CO3 | 71.4 | 21.7 | 1.3 | 5.6 |
| 20 | DMSO | 130 | CuI | —e | K2CO3 | 83.3 | 12.8 | 1.5 | 2.4 |
| 21 | DMSO | 130 | CuI | —e | KOH | 97.0 | 0 | 0.5 | 2.5 |
As shown in entries 1–10, the impact of solvents was enormous according to the results of HPLC. DMSO exhibited a better result than DMF, dioxane, or toluene, indicating a favored solvent circumstance with higher polarity (Table 1, entries 1–2, entries 4–5), and also a favored condition of higher temperature (Table 1, entries 3). But pure water as solvent worked not effectively due to the low solubility of organic compounds (entry 6). Therefore, biphasic system of water and hydrosoluble solvents instead of DMSO provided a potential solution for the contradiction of polarity and solubility as described above (entries 7–10). The level of 5 reached 91.8% in 1,4-dioxane/H2O (entry 7), and the slightly lower efficiency might be derived from the relative lower boiling point of biphasic solvent systems. Taken all together, DMSO was regarded as the optimal solvent system for further research.
Since several metal-free,35–37 and ligand-free23,26 conditions were reported for the cyclization of o-haloarylamidines, the necessity of copper catalyst and ligand in our route was subsequently investigated (entries 11 and 12). The copper catalyst was proved to be essential, since a significant decline of 5, along with an obvious augment of impurity 10b were observed in the absence of copper (entry 11). Moreover, in a ligand-free condition (entry 12), there was almost no change in the levels of 5 and impurities 10b, 11b, and 12b in comparison with the result in entry 3, suggesting additional ligand could be dispensable in our method, and the coordination effect between copper catalyst and the benzimidazole subunit from substance might play a very similar role in the reaction system.38,39
Afterwards, a series of commonly utilized copper catalysts were investigated in DMSO under a ligand-free condition, using three equivalents of Cs2CO3 as the base (entries 12–19). Copper(I) salts displayed distinct advantages over copper(II) catalysts, and CuI (entry 12) was selected as the optimal choice. In addition, the evaluation of bases was also carried out (entries 12, 20 and 21). Three commonly used bases were employed, and the results showed that both Cs2CO3 and KOH displayed higher yields than K2CO3.
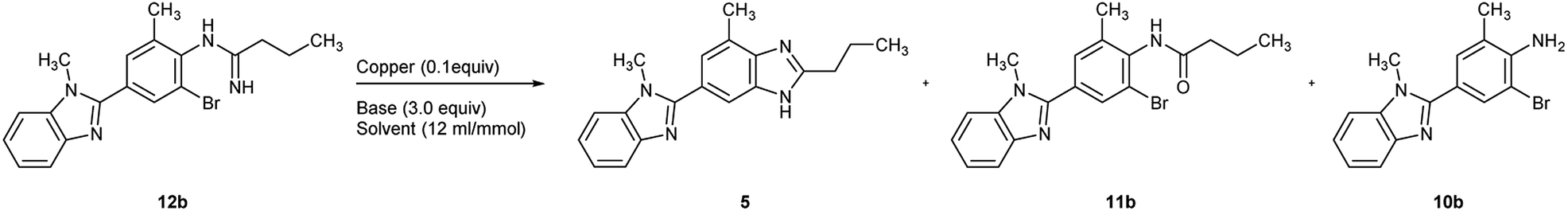
As a consequence, the utilization of CuI (0.1 equiv.) and Cs2CO3 (3.0 equiv.) in DMSO (entry 12) was chosen as the optimal condition for the cyclization of 12b, and the key bis-benzimidazole intermediate 5 was obtained in an isolated yield of 92% in such manner (Scheme 4).
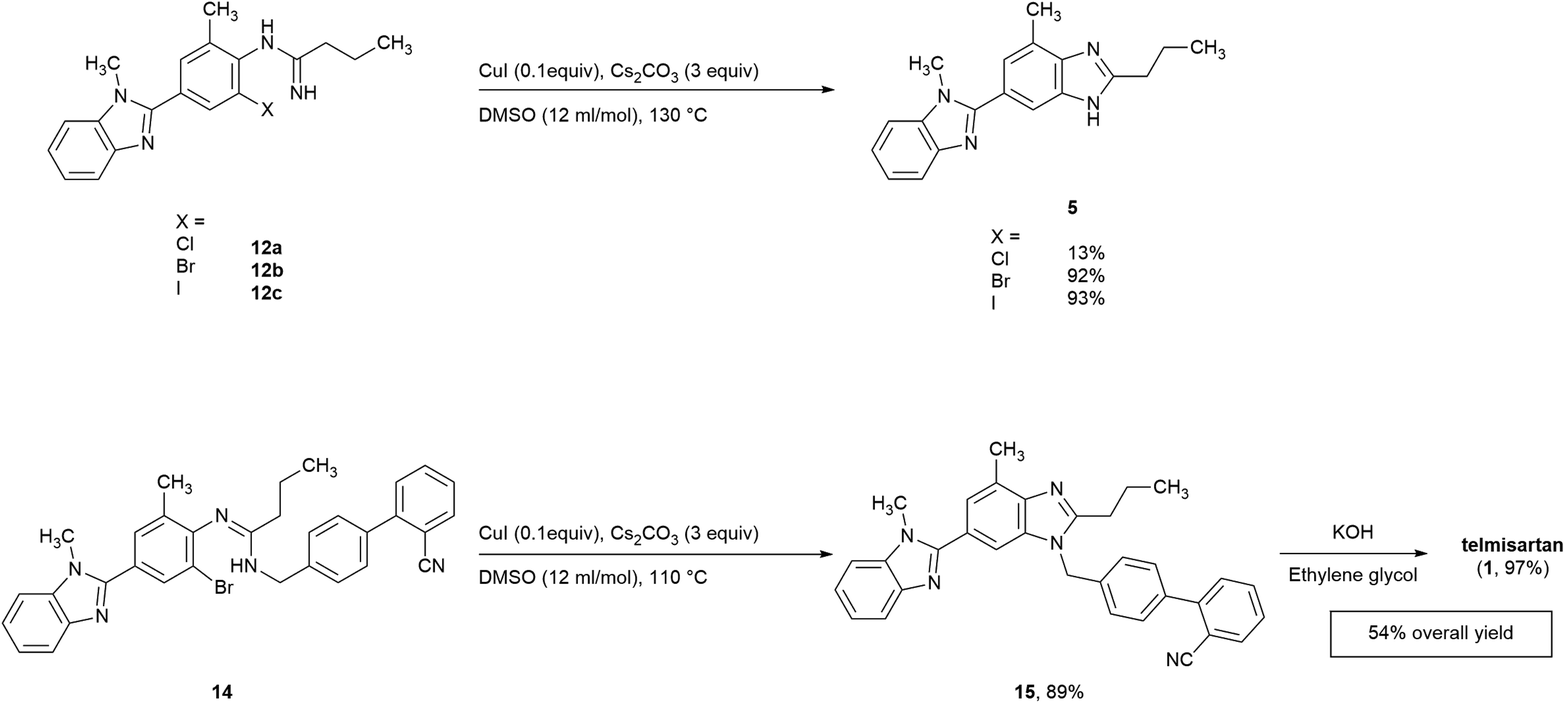 | ||
| Scheme 4 Synthesis of bis-benzimidazole intermediates and telmisartan. | ||
2.3 Synthesis of bis-benzimidazole intermediates and telmisartan
Based on the screening results of 12b, the cyclization of other o-haloarylamidines 12a, 12c, and 14 were subsequently conducted in DMSO under the similar conditions (Scheme 4). The isolated yield of 5 obtained from the iodo precursor 12c (93%) was similar with that from 12b. However, such yield of the cyclization of 12a was merely 13%, which may be caused by the poor leaving group ability of chlorine, while the yield of the impurity 10a could reached the level of 82%, suggesting the competitive relationship between cyclization and hydrolysis reactions in this step.The traditional method for the preparation of compound 15, an industrially widely utilized precursor of telmisartan,40–42 requires the N-alkylation compound of 5 with the 2-cyano-4′-(bromomethyl)biphenyl, resulting in regioisomer impurities. Herein, the cyclization of N,N′-disubstituted o-haloarylamidine 14 was utilized, offering 15 in an isolated yield of 89%. Afterwards, the downstream cyano-group hydrolysis was able to achieve through reported method,41 leading to the formation of the final product with 97% yield after simple purification.
3. Conclusions
In conclusion, an improved synthesis was developed for the preparation of telmisartan, featuring a ligand-free copper-catalyzed cyclization of benzimidazolyl-substituted o-haloarylamidines to form the core bis-benzimidazole fragment, while the use of HNO3/H2SO4 and PPA in the original route was averted. Furthermore, our approach provided a new manner for introducing the biphenyl-4-methyl subunit, which avoided the generation of regioisomer impurities in the traditional N-alkylation method. By adopting this route, telmisartan was obtained in a 7-step overall yield of 54%, while achieving an all-around improvement in safety, waste disposal, and operability. In the long run, such copper-catalyzed cyclization strategy brings a new avenue to the preparation of drugs and their derivatives containing benzimidazole pharmacophores.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by Special Foundation of Chinese Academy of Sciences for Strategic Pilot Technology (Grant No. XDA12050411) and by Science and Technology Commission of Shanghai Municipality (Grant Number: 18431907100).Notes and references
- M. Sharpe, B. Jarvis and K. L. Goa, Drugs, 2001, 61, 1501–1529 CrossRef CAS PubMed.
- P. Wang, G.-j. Zheng, Y.-p. Wang, X.-j. Wang, H.-g. Wei and W.-s. Xiang, Tetrahedron, 2012, 68, 2509–2512 CrossRef CAS.
- A. J. Battershill and L. J. Scott, Drugs, 2006, 66, 51–83 CrossRef CAS PubMed.
- U. J. Ries, G. Mihm, B. Narr, K. M. Hasselbach, H. Wittneben, M. Entzeroth, M. J. C. van, W. Wienen and N. H. Hauel, J. Med. Chem., 1993, 36, 4040–4051 CrossRef CAS PubMed.
- M.-X. Zhang, A. J. DeHope and P. F. Pagoria, Org. Process Res. Dev., 2019, 23, 2527–2531 CrossRef CAS.
- K. Qiao and C. Yokoyama, Chem. Lett., 2004, 33, 808–809 CrossRef CAS.
- K. K. Laali and V. J. Gettwert, J. Org. Chem., 2001, 66, 35–40 CrossRef CAS PubMed.
- L. Lu, J. Xin, C.-S. Woo, T. Cai and H.-I. Lee, Stud. Surf. Sci. Catal., 2006, 159, 353–356 CrossRef CAS.
- J. T. Vicenzi, T. Y. Zhang, R. L. Robey and C. A. Alt, Org. Process Res. Dev., 1999, 3, 56–59 CrossRef CAS.
- L. J. Goossen and T. Knauber, J. Org. Chem., 2008, 73, 8631–8634 CrossRef CAS PubMed.
- A. S. Kumar, S. Ghosh, G. N. Mehta, R. Soundararajan, P. S. R. Sarma and K. Bhima, Synth. Commun., 2009, 39, 4149–4157 CrossRef CAS.
- A. Sanjeev Kumar, S. Ghosh and G. N. Mehta, Beilstein J. Org. Chem., 2010, 6, 25 Search PubMed.
- A. D. Martin, A. R. Siamaki, K. Belecki and B. F. Gupton, J. Org. Chem., 2015, 80, 1915–1919 CrossRef CAS PubMed.
- S. Bhunia, G. G. Pawar, S. V. Kumar, Y. W. Jiang and D. W. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS PubMed.
- S. H. Cho, J. Yoon and S. Chang, J. Am. Chem. Soc., 2011, 133, 5996–6005 CrossRef CAS PubMed.
- K. Sun, S. Q. Mu, Z. H. Liu, R. R. Feng, Y. L. Li, K. Pang and B. Zhang, Org. Biomol. Chem., 2018, 16, 6655–6658 RSC.
- F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed.
- T. Liu and H. Fu, Synthesis, 2012, 44, 2805–2824 CrossRef CAS.
- M. Largeron and K. M. H. Nguyen, Synthesis, 2018, 50, 241–253 CrossRef CAS.
- Y. Y. Qu, L. Pan, Z. Q. Wu and X. G. Zhou, Tetrahedron, 2013, 69, 1717–1719 CrossRef CAS.
- J. H. Li, S. Benard, L. Neuville and J. P. Zhu, Org. Lett., 2012, 14, 5980–5983 CrossRef CAS PubMed.
- D. Yu, Q. You, X. M. Zhang, G. D. Tao and W. Zhang, Appl. Organomet. Chem., 2016, 30, 695–698 CrossRef CAS.
- P. Saha, T. Ramana, N. Purkait, M. A. Ali, R. Paul and T. Punniyamurthy, J. Org. Chem., 2009, 74, 8719–8725 CrossRef CAS PubMed.
- J. S. Peng, M. Ye, C. J. Zong, F. Y. Hu, L. T. Feng, X. Y. Wang, Y. F. Wang and C. X. Chen, J. Org. Chem., 2011, 76, 716–719 CrossRef CAS PubMed.
- J. T. Zhu, H. B. Xie, Z. X. Chen, S. Li and Y. M. Wu, Chem. Commun., 2009, 2338–2340, 10.1039/b900984a.
- B. G. Szczepankiewicz, J. J. Rohde and R. Kurukulasuriya, Org. Lett., 2005, 7, 1833–1835 CrossRef CAS PubMed.
- N. Mishra, A. S. Singh, A. K. Agrahari, S. K. Singh, M. Singh and V. K. Tiwari, ACS Comb. Sci., 2019, 21, 389–399 CrossRef CAS PubMed.
- J. Yu, Y. Xia and M. Lu, Appl. Organomet. Chem., 2014, 28, 764–767 CrossRef CAS.
- K. Liubchak, K. Nazarenko and A. Tolmachev, Tetrahedron, 2012, 68, 2993–3000 CrossRef CAS.
- M. De Greef, B. Peter and R. Stumpf, WO2017017096A1, 2017.
- T. Schaefer, M. Kawamura and H. Nagashima, WO2017056052A1, 2017.
- G. T. Lee, K. Prasad and O. Repic, Tetrahedron Lett., 2002, 43, 3255–3257 CrossRef CAS.
- C. Lamanna, A. Catalano, A. Carocci, A. Di Mola, C. Franchini, V. Tortorella, P. M. L. Vanderheyden, M. S. Sinicropi, K. A. Watson and S. Sciabola, ChemMedChem, 2007, 2, 1298–1310 CrossRef CAS PubMed.
- CN107325092A, 2017.
- H. Baars, A. Beyer, S. V. Kohlhepp and C. Bolm, Org. Lett., 2014, 16, 536–539 CrossRef CAS PubMed.
- C. Chen, C. Chen, B. Li, J. Tao and J. Peng, Molecules, 2012, 17, 12506–12520 CrossRef CAS PubMed.
- S.-K. Xiang, W. Tan, D.-X. Zhang, X.-L. Tian, C. Feng, B.-Q. Wang, K.-Q. Zhao, P. Hu and H. Yang, Org. Biomol. Chem., 2013, 11, 7271–7275 RSC.
- J. C. Geng, L. Qin, C. H. He and G. H. Cui, Transition Met. Chem., 2012, 37, 579–585 CrossRef CAS.
- X. Xu, Z. Xi, W. Chen and D. Wang, J. Coord. Chem., 2007, 60, 2297–2308 CrossRef CAS.
- U. A. Amarnath and U. S. Suryakiran, WO2014027280A1, 2014.
- P. C. Ray, S. Nigam, A. K. Pandey, P. Patil, J. M. Reddy and N. Oruganti, WO2011077444A1, 2011.
- M. Wu, J. Li, W. Chen, G. Tian, F. Zhu, J. Suo and J. Shen, WO2014067237A1, 2014.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00886a |
| This journal is © The Royal Society of Chemistry 2020 |