
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlumina-promoted oxodefluorination†
Akhmetov Vladimir *a,
Feofanov Mikhaila and
Konstantin Amsharovab
*a,
Feofanov Mikhaila and
Konstantin Amsharovab
aFriedrich-Alexander University Erlangen-Nuernberg, Department of Chemistry and Pharmacy, Organic Chemistry II, Nikolaus-Fiebiger Str. 10, 91058 Erlangen, Germany. E-mail: vladimir.akhmetov@fau.de
bInstitute of Chemistry, Organic Chemistry, Martin-Luther-University Halle-Wittenberg, Kurt-Mothes-Strasse 2, D-06120 Halle, Germany
First published on 17th March 2020
Abstract
A simple protocol for the clean preparation of heterocyclic compounds containing dibenzofuran's core via oxodefluorination of fluoroarenes on activated γ-Al2O3 is reported. Alumina can be considered as a reliable oxygen source enabling one-pot substitution of fluorine atoms and yielding benzoannulated furan derivatives. The corresponding C–F bond activation is selective towards less stable C–Br/C–I and occurs under metal- and solvent-free conditions.
Dibenzo[b,d]furan is an important moiety as its derivatives constitute a long list of natural products.1,2 Moreover, multiple synthetic compounds bearing such a fragment also show biological activity3–5 e.g. mimic fragments of complex biological structures.6,7 Due to its rigidity, dibenzofuran is oftentimes used as a bridge fixing fragment of the molecule at a desired distance giving rise to various chelating ligands,8–13 cryptands,14 and other supramolecular hosts.15–19 Furthermore, the stable dibenzofuran core enables different patterns of substitution. These features, in combination with appropriate electronic properties, make dibenzofuran and its derivatives potentially valuable components of phosphorescent OLEDs20–22 thermally activated delayed fluorescence emitters,23–25 perovskite solar cells26 etc. Thus, functionalized dibenzofurans appear to be important building blocks.
Despite numerous synthetic approaches to this class of compounds, the construction of functionalized dibenzofurans is far from being trivial since all existing methods are generally reduced to either C–O or C–C coupling (Fig. 1). This is mainly connected to the fact that oxygen has to be present in the precursor's structure because there is no reliable oxygen source that would enable double C–O bond incorporation. The majority of the available methods are based on Pd-catalysis,27–34 while others exploit strong bases/acids and toxic solvent.35–38 In addition, these methods rarely tolerate C–Br and C–I functionalities, which could have been used for further modifications.
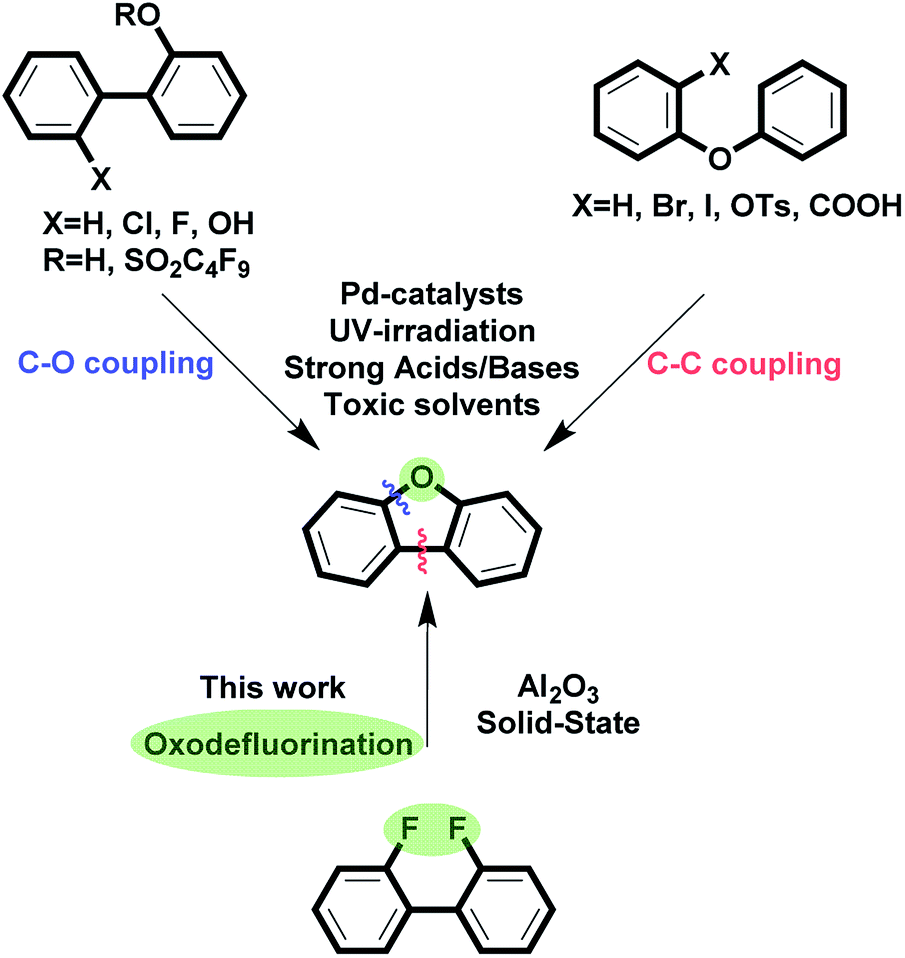 | ||
| Fig. 1 Synthesis of dibenzofuran. Existing approaches vs. oxodefluorination. | ||
Herein, we report a transformation of fluorinated biphenyls into dibenzofuran's derivatives. The reaction occurs on the surface of activated γ-Al2O3 and does not require other reagents. The scope and limitations of the reactions are considered: the technique is compatible with C–Br and C–I functionalities, enables double oxodefluorination and incorporation of hexagons, thus yielding ladder-type π-conjugated heteroacenes and xanthene's derivatives, respectively.
As a part of our ongoing research on alumina mediated C–F activation,39,40 we have observed an interesting competition between C(aryl)–C(aryl) coupling and formation of oxygen-containing side products.41 In certain cases (i.e. when two fluorine atoms are dislocated in bay-region) the side products turned out to contain dibenzofuran's moiety.42 Some additional data on this competition are briefly discussed in ESI.†
To investigate the formation of dibenzofuran's core in more details, we have excluded one of the competitors i.e. the possibility for intramolecular C(aryl)–C(aryl) coupling. The simplest model meeting the requirements is 2,2′-difluorobiphenyl (1a), which upon exposure to activated alumina transforms into dibenzofuran 1 in 65% yield. As a matter of fact, the desired 1 is the only product that can be extracted from the reactionary mixture with aprotic solvent such as toluene (Fig. 2). Thus, flash chromatography is not required, since reactionary alumina already plays the role of a plug enabling effective separation from intermediate and side products, which can be extracted with methanol afterwards (see ESI†). Notably, eliminated fluorine atoms are most likely to be bound to alumina in the form of extremely strong and naturally occurring Al–F bond yielding partially fluorinated alumina, which is of interest in multiple fields including catalysis.43,44
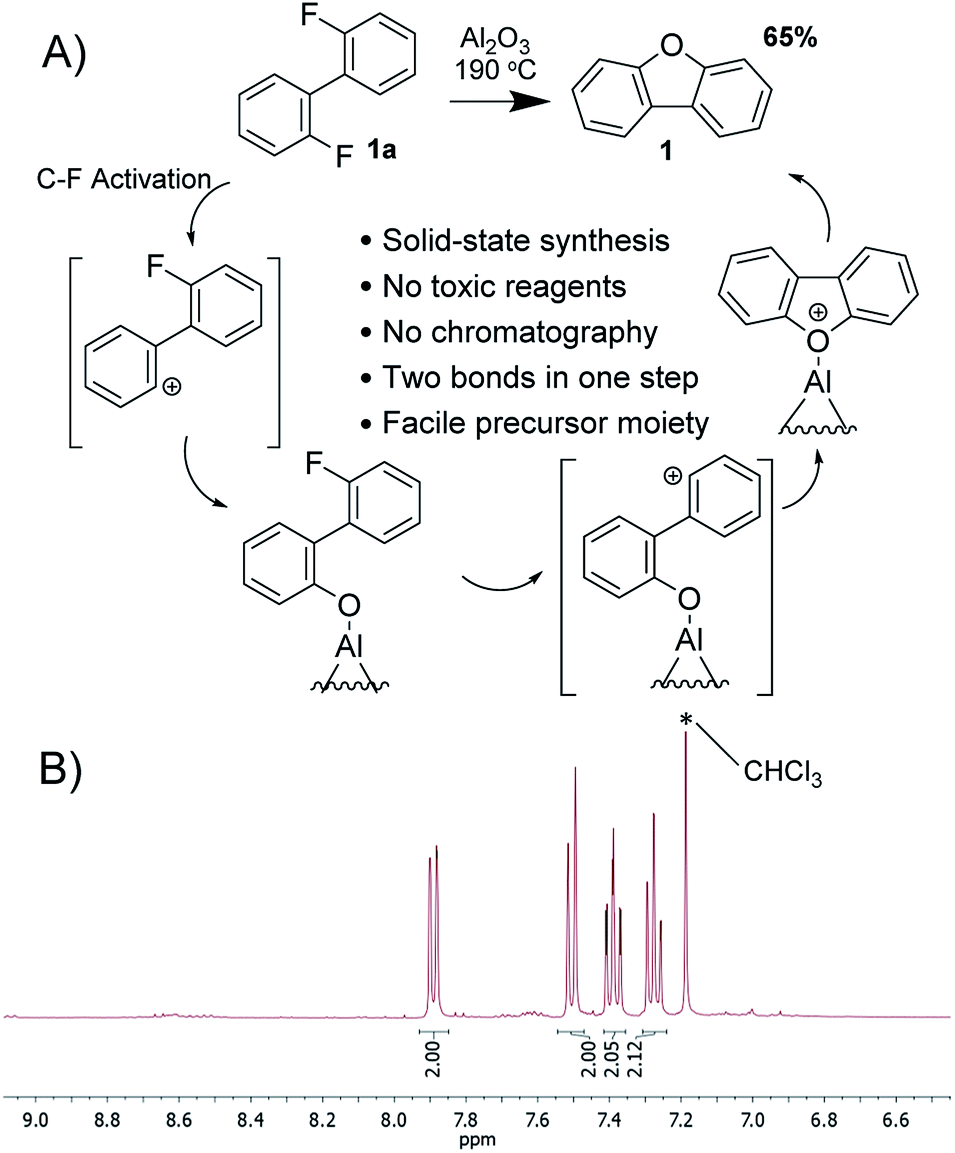 | ||
| Fig. 2 (A) Synthesis of dibenzofuran and suggested mechanism of the reaction. (B) 1H NMR of the reactionary mixture as obtained after extraction with toluene. | ||
As any other heterogeneous process, this reaction is quite likely to have a complicated manner. However, some pieces of evidence on C(aryl)–F polarization41,45,46 allow us to suggest the key steps of the transformation. Alumina induces a partial positive charge on carbon generating incipient phenyl cation species,47 which are to interact with proximal moieties. In the case of 2,2′-difluorobiphenyl, no aryl functionalities are available for intramolecular C(aryl)–C(aryl) coupling; therefore oxygen on alumina surface serves as an alternative nucleophile interacting with cation-like species. Such hydrolysis of C–F bond has already been observed as a side process.41 Unlike the reported transformation, the current precursor contains two fluorine atoms, whereas the polarization of the second C(aryl)–F bond induces the second C–O coupling, this time intramolecular, and subsequent covalent detachment of the product. Some other possible mechanisms occurring via aryne-particles or dehydration were excluded (see ESI†). Noteworthy, the outcome of the reaction resembles double SNAr, which can be generally achieved in activated arenes i.e. containing multiple electron-withdrawing groups. Such restriction does not apply to the alumina-promoted oxodefluorination due to its cationic nature.
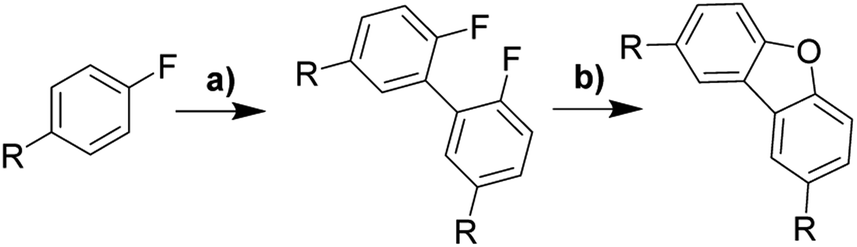
An interesting aspect of C(aryl)–F bond is that it can be used as both directing and functional groups, although it is seldom considered as the latter. In accordance with Table 1, para-substituted fluorobenzenes undergo directed ortho-metalation enabling simple one-pot synthesis of functionalized biphenyls bearing two fluorine atoms in 2,2′-positions. This gives a facile approach to several precursors, which were used to investigate the scope and limitation of the alumina-promoted oxodefluorination. After heating with activated alumina at 190 °C under vacuum, the reactionary mixture was extracted with toluene. The obtained extract containing the target molecule did not require flash chromatography unless stated otherwise.
|
|---|
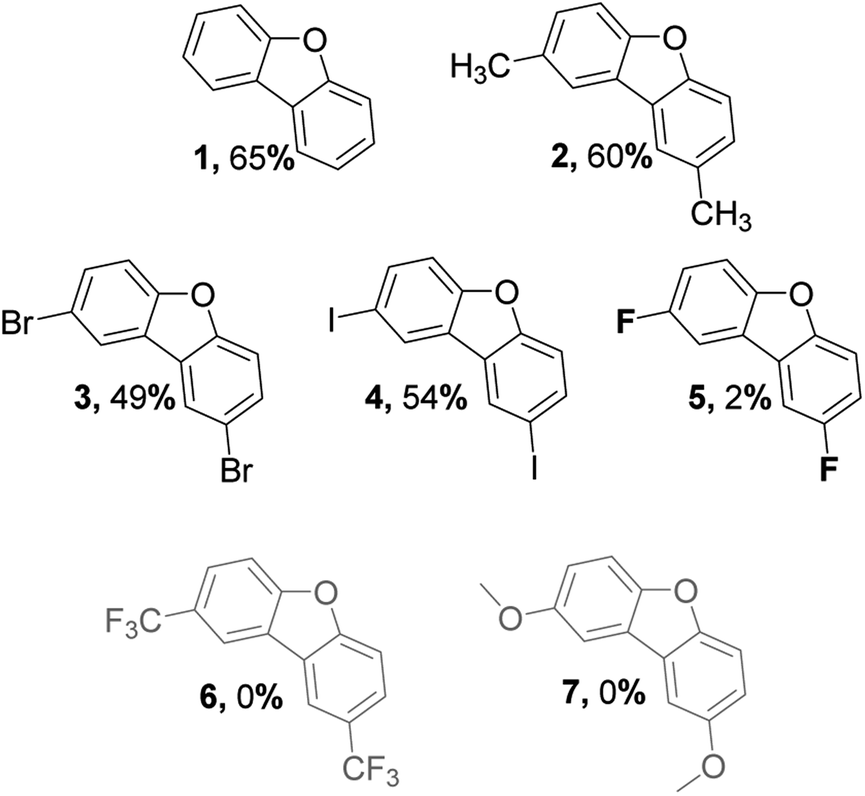 |
While tolerance to methyl group comes as no surprise, selective C–F activation in presence of C–Br and more interestingly C–I is a quite notable feature since fluorine forms stronger bonds with carbon in comparison to other halogens. At the same time, the reaction has a rather poor selectivity to C(aryl)–F bonds since only traces of 5 were extracted, whereas the rest of the precursor has remained on the surface in forms of hydrolyzed side products. A similar, although anticipated, outcome was observed in the case of 6, where nothing could be extracted with toluene due to the rapid hydrolysis of CF3 groups.48 As of methoxy groups, the corresponding toluene extract revealed no desired product 7. Presumably, it is connected to the fact that OCH3 occupies reactive sites of alumina more rapidly than C–F polarization takes place.
This observation is especially interesting in light of the following evidence. Oligophenylenes 8a, 9a, and 10a synthesized in one step via Suzuki coupling undergo double annulation yielding respective ladder-type π-conjugated compounds 8–10, which are promising candidates as organic semiconductors (Scheme 1).37 Here, the formation of the first dibenzofuran's moiety, apparently, does not prevent the second region from annulation.
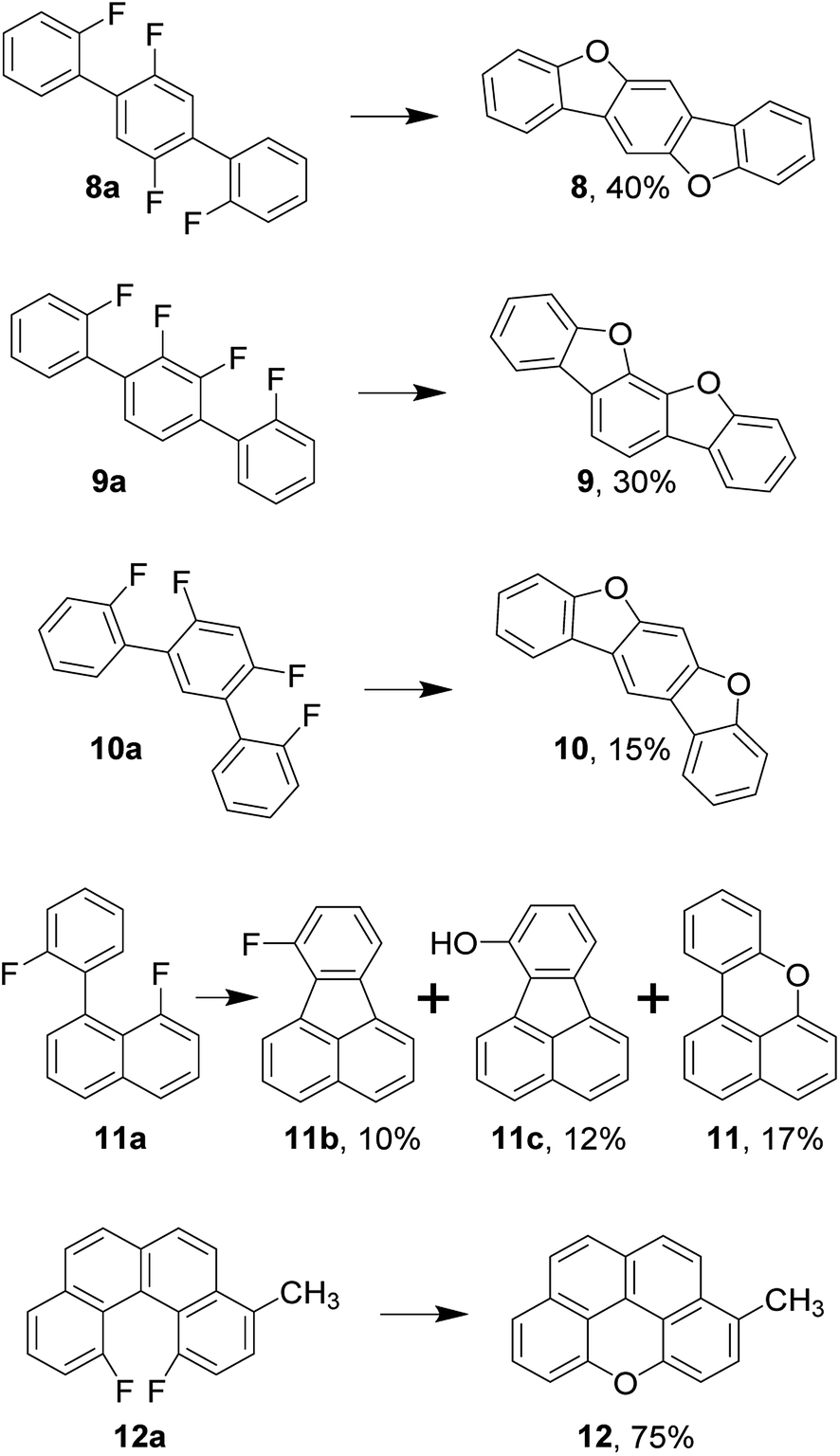 | ||
| Scheme 1 Synthesis of ladder-type π-conjugated compounds, benzo[kl]xanthene and incorporation of oxygen into cove region. | ||
To investigate whether the approach is applicable to the synthesis of xanthene's derivatives such as benzo[kl]xanthene (11), we have designed the corresponding precursor 11a having a choice between C–F and C–C couplings. Due to this competition, several products were extracted with toluene in this particular case. Therefore, toluene and methanol extracts were combined and separated by means of flash chromatography to reveal three major products 11b, 11c, and 11. To preclude the variability, we have synthesized rigid precursor 12a with two fluorine atoms in cove region. Thus, 12a transforms into 12 in considerable 75% yield enabling the synthesis of elusive naphtho[2,1,8,7-klmn]xanthene's core.49
Conclusions
In summary, we have occasionally come across an attractive method enabling the incorporation of oxygen in place of two C(aryl)–F bonds, where alumina serves as a reliable oxygen source. The simplicity of the required fluorinated moiety makes precursors, in general, readily available via facile one-step synthesis. The method allows the synthesis of dibenzofuran's, xanthene's, and ladder-type π-conjugated derivatives under metal- and solvent-free conditions. In principle, alumina-promoted oxodefluorination tolerates such useful functionalities as C–Br and C–I. The synthetic procedure is straightforward and requires activated γ-Al2O3 as a reagent and simple extraction to yield high purity target products without flash chromatography and respective solvent and silica wastes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG) – Projektnummer 182849149 – SFB 953, AM407.References
- C.-H. Lin, H.-S. Chang, C.-H. Liao, T.-H. Ou, I.-S. Chen and I.-L. Tsai, J. Nat. Prod., 2010, 73, 1628–1631 CrossRef CAS PubMed.
- M. Millot, A. Dieu and S. Tomasi, Nat. Prod. Rep., 2016, 33, 801–811 RSC.
- L. Yurttaş, U. Abu Mohsen, Y. Ozkan, S. Cobanoglu, S. Levent and Z. A. Kaplancikli, J. Enzyme Inhib. Med. Chem., 2016, 31, 1177–1183 CrossRef PubMed.
- Y. Ma, H.-Y. Wei, Y.-Z. Zhang, W.-Y. Jin, H.-L. Li, H. Zhou, X.-C. Cheng and R.-L. Wang, Oncotarget, 2017, 8(24), 38466–38481 Search PubMed.
- N. Goyal, J. P. Donahue, C. Do, T. Perry, K. Bongay-Williams and M. Foroozesh, IUCrData, 2018, 3, x181306 CrossRef CAS PubMed.
- F. Jean, E. Buisine, O. Melnyk, H. Drobecq, B. Odaert, M. Hugues, G. Lippens and A. Tartar, J. Am. Chem. Soc., 1998, 120, 6076–6083 CrossRef CAS.
- H. M. Petrassi, S. M. Johnson, H. E. Purkey, K. P. Chiang, T. Walkup, X. Jiang, E. T. Powers and J. W. Kelly, J. Am. Chem. Soc., 2005, 127, 6662–6671 CrossRef CAS PubMed.
- P. L. Caradoc-Davies and L. R. Hanton, Dalton Trans., 2003, 1754–1758 RSC.
- M. L. Hlavinka and J. R. Hagadorn, Chem. Commun., 2003, 2686 RSC.
- E. Deschamps, B. Deschamps, J. Laure Dormieux, L. Ricard, N. Mézailles and P. Le Floch, Dalton Trans., 2006, 594–602 RSC.
- S. He, F. Wang, W.-L. Tong, S.-M. Yiu and M. C. W. Chan, Chem. Commun., 2016, 52, 1017–1020 RSC.
- S. T. Li, B. Braun-Cula, S. Hoof and C. Limberg, Dalton Trans., 2018, 47, 544–560 RSC.
- K. Itoh and M. P. Sibi, Org. Biomol. Chem., 2018, 16, 5551–5565 RSC.
- C. Bazzicalupi, A. Bencini, S. Ciattini, F. Denat, P. Désogère, C. Goze, I. Matera and B. Valtancoli, Dalton Trans., 2010, 39, 11643 RSC.
- M. Asakawa, P. R. Ashton, C. L. Brown, M. C. T. Fyfe, S. Menzer, D. Pasini, C. Scheuer, N. Spencer, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem.–Eur. J., 1997, 3, 1136–1150 CrossRef CAS.
- B. J. Pistorio, C. J. Chang and D. G. Nocera, J. Am. Chem. Soc., 2002, 124, 7884–7885 CrossRef CAS PubMed.
- K. M. Kadish, L. Frémond, Z. Ou, J. Shao, C. Shi, F. C. Anson, F. Burdet, C. P. Gros, J.-M. Barbe and R. Guilard, J. Am. Chem. Soc., 2005, 127, 5625–5631 CrossRef CAS PubMed.
- L. E. Garner, H. Zhu, M. L. Hlavinka, J. R. Hagadorn and E. Y.-X. Chen, J. Am. Chem. Soc., 2006, 128, 14822–14823 CrossRef CAS PubMed.
- J. Rosenthal, T. D. Luckett, J. M. Hodgkiss and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 6546–6547 CrossRef CAS PubMed.
- F. May, M. Al-Helwi, B. Baumeier, W. Kowalsky, E. Fuchs, C. Lennartz and D. Andrienko, J. Am. Chem. Soc., 2012, 134, 13818–13822 CrossRef CAS PubMed.
- J. H. Yun, Y. J. Kang, S. H. Han and J. Y. Lee, J. Mater. Chem. C, 2018, 6, 320–325 RSC.
- J. H. Yun, Y. J. Kang, S. H. Han and J. Y. Lee, Dyes Pigm., 2019, 162, 1–7 CrossRef CAS.
- J. G. Yu, S. H. Han, H. L. Lee, W. P. Hong and J. Y. Lee, J. Mater. Chem. C, 2019, 7, 2919–2926 RSC.
- M. Jung, K. H. Lee, W. P. Hong and J. Y. Lee, J. Mater. Chem. C, 2019, 7, 7760–7767 RSC.
- W.-W. Tao, K. Wang, J.-X. Chen, Y.-Z. Shi, W. Liu, C.-J. Zheng, Y.-Q. Li, J. Yu, X.-M. Ou and X.-H. Zhang, J. Mater. Chem. C, 2019, 7, 4475–4483 RSC.
- Y. Shi, K. Hou, Y. Wang, K. Wang, H. Ren, M. Pang, F. Chen and S. Zhang, J. Mater. Chem. A, 2016, 4, 5415–5422 RSC.
- M. Parisien, D. Valette and K. Fagnou, J. Org. Chem., 2005, 70, 7578–7584 CrossRef CAS PubMed.
- K. Kawaguchi, K. Nakano and K. Nozaki, J. Org. Chem., 2007, 72, 5119–5128 CrossRef CAS PubMed.
- B. Liégault, D. Lee, M. P. Huestis, D. R. Stuart and K. Fagnou, J. Org. Chem., 2008, 73, 5022–5028 CrossRef PubMed.
- C. Wang, I. Piel and F. Glorius, J. Am. Chem. Soc., 2009, 131, 4194–4195 CrossRef CAS PubMed.
- B. Xiao, T.-J. Gong, Z.-J. Liu, J.-H. Liu, D.-F. Luo, J. Xu and L. Liu, J. Am. Chem. Soc., 2011, 133, 9250–9253 CrossRef CAS PubMed.
- C. S. Nervig, P. J. Waller and D. Kalyani, Org. Lett., 2012, 14, 4838–4841 CrossRef CAS PubMed.
- T. Okazaki, M. Nakagawa, T. Futemma and T. Kitagawa, J. Phys. Org. Chem., 2016, 29, 107–111 CrossRef CAS.
- H. Kaida, T. Satoh, Y. Nishii, K. Hirano and M. Miura, Chem. Lett., 2016, 45, 1069–1071 CrossRef CAS.
- T. Yamato, C. Hideshima, G. K. S. Prakash and G. A. Olah, J. Org. Chem., 1991, 56, 3192–3194 CrossRef CAS.
- K. Nakanishi, D. Fukatsu, K. Takaishi, T. Tsuji, K. Uenaka, K. Kuramochi, T. Kawabata and K. Tsubaki, J. Am. Chem. Soc., 2014, 136, 7101–7109 CrossRef CAS PubMed.
- M. A. Truong and K. Nakano, Beilstein J. Org. Chem., 2016, 12, 805–812 CrossRef PubMed.
- P. Camargo Solórzano, F. Brigante, A. B. Pierini and L. B. Jimenez, J. Org. Chem., 2018, 83, 7867–7877 CrossRef PubMed.
- O. Papaianina, V. A. Akhmetov, A. A. Goryunkov, F. Hampel, F. W. Heinemann and K. Y. Amsharov, Angew. Chem., Int. Ed., 2017, 56, 4834–4838 CrossRef CAS PubMed.
- V. Akhmetov and K. Amsharov, Phys. Status Solidi, 2019, 256, 1900254 CrossRef CAS.
- V. Akhmetov, M. Feofanov, O. Papaianina, S. Troyanov and K. Amsharov, Chem.–Eur. J., 2019, 25, 11609–11613 CrossRef CAS PubMed.
- V. Akhmetov, M. Feofanov, S. Troyanov and K. Amsharov, Chem.–Eur. J., 2019, 25, 7607–7612 CrossRef CAS PubMed.
- T. Skapin, J. Mater. Chem., 1995, 5, 1215–1222 RSC.
- R. Padhye, A. J. A. Aquino, D. Tunega and M. L. Pantoya, ACS Appl. Mater. Interfaces, 2017, 9, 24290–24297 CrossRef CAS PubMed.
- O. Allemann, K. K. Baldridge and J. S. Siegel, Org. Chem. Front., 2015, 2, 1018–1021 RSC.
- M. Lu, O. Allemann, J. Xu, A. Linden, K. K. Baldridge and J. S. Siegel, Org. Chem. Front., 2019, 6, 2640–2646 RSC.
- D. Sharapa, A.-K. Steiner and K. Amsharov, Phys. Status Solidi, 2018, 1800189, 1800189 CrossRef.
- O. Papaianina and K. Y. Amsharov, Chem. Commun., 2016, 52, 1505–1508 RSC.
- P. M. Donovan and L. T. Scott, J. Am. Chem. Soc., 2004, 126, 3108–3112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01369b |
| This journal is © The Royal Society of Chemistry 2020 |