
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-free and room temperature microwave-assisted direct C7 allylation of indolines via sequential C–H and C–C activation†
Qiuling Wang,
Linlin Shi,
Shuang Liu,
Changlei Zhi,
Lian-Rong Fu,
Xinju Zhu *,
Xin-Qi Hao and
Mao-Ping Song*
*,
Xin-Qi Hao and
Mao-Ping Song*
College of Chemistry, Zhengzhou University, No. 100 of Science Road, Zhengzhou, Henan 450001, P. R. China. E-mail: zhuxinju@zzu.edu.cn; mpsong@zzu.edu.cn
First published on 17th March 2020
Abstract
A Ru or Rh-catalyzed efficient and atom-economic C7 allylation of indolines with vinylcyclopropanes was developed via sequential C–H and C–C activation. A wide range of substrates were well tolerated to afford the corresponding allylated indolines in high yields and E/Z selectivities under microwave irradiation. The obtained allylated indolines could further undergo transformations to afford various value-added chemicals. Importantly, this reaction proceeded at room temperature under solvent-free conditions.
The development of sustainable methodologies is attractive for access to complex molecular architectures in organic chemistry.1 In recent years, various non-conventional techniques, such as microwave irradiation, sonochemistry, mechanical grinding and photochemistry, have achieved remarkable success.2 In particular, microwaves have shown unique advantages with regards to reaction times, energy efficiency, temperature, and reaction media.3 On the other hand, transition-metal-catalyzed activation of C–H4 and C–C5 bonds has been considered as an ideal method for the formation of C–C and C–X bonds. Nevertheless, transition-metal-catalyzed C–H or C–C bond activation under the above non-conventional techniques remains to be explored. It is thus highly imperative to develop a practical strategy in combination of C–H or C–C activation and microwave irradiation.6
Recently, there have significant advances in C–H activation technology by merging C–H functionalization with challenging C–C cleavage strategies.7 Since the pioneering work by Bergman and co-workers8 on the sequential C–H and C–C bond activation, many research groups, including Dong,9 Ackermann,10 Li,11 Cramer,12 and others13 have contributed to C–H/C–C activation. In this content, certain small strained rings are often utilized as an effective synthons to undergo ring-opening reactions driven by strain-release energy.14 Very recently, VCPs (vinylcyclopanes) have been reported as allyl reagents to access various (hetero)aromatic derivatives through sequential C–H and C–C activation (Scheme 1a–d).15
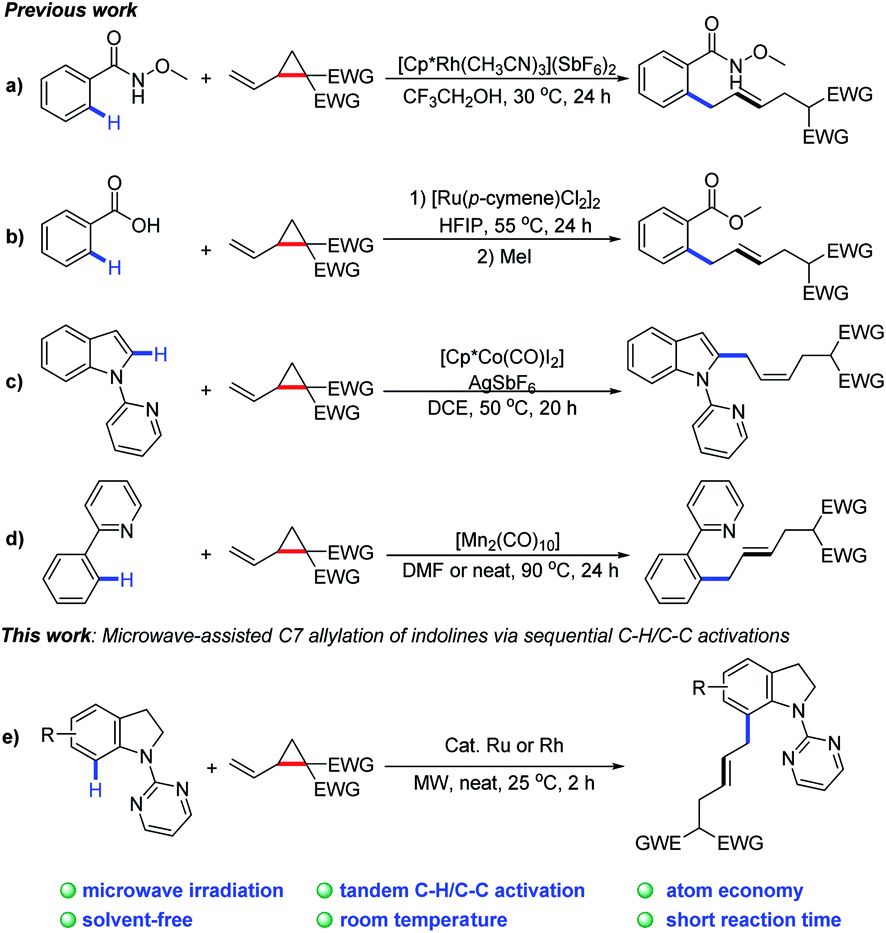 | ||
| Scheme 1 Sequential C–H/C–C activations using VCPs. | ||
As a continuation of our interest in chelation-directed reactions and novel methods for C–H functionalization,16 we herein report a Ru or Rh-catalyzed C-7 allylation of indolines under microwave irradiation using VCPs as the allylating agents (Scheme 1e). This transformation possesses great synthetic potential from the viewpoint of green and sustainable chemistry. Notable features of our protocol include (1) C–H/C–C activation with VCPs by microwave irradiation, (2) broad substrate scope with good regio- and E/Z selectivities, (3) high atom economy, and (4) high efficiency (2 h) at room temperature under solvent-free conditions.
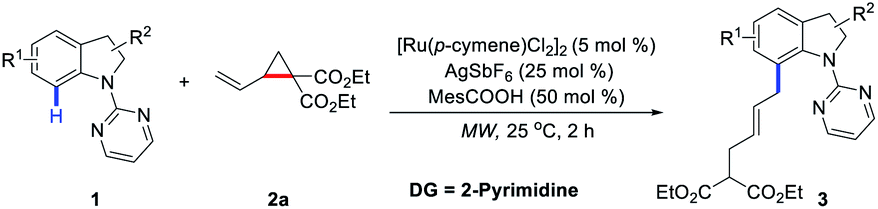
We initiated our investigation by choosing indoline 1a and VCP 2a as model substrates under microwave irradiation conditions (Table 1). To our delight, the allylated product 3aa was isolated in 40% yield in the presence of [Ru(p-cymene)Cl2]2 (5 mol%), AgSbF6 (25 mol%), and AdCOOH (30 mol%) at 25 °C for 1 h (Table 1, entry 1). Other ruthenium salts, such as [Cp*RuCl2]2 and RhCl3·3H2O, were also investigated, which showed no catalytic reactivity (Table 1, entries 2 and 3). Subsequently, the effects of various additives, including MesCOOH, AcOH, NaOAc, PivONa·H2O, and DABCO, were screened, which indicates that MesCOOH was the best choice to afford 3aa in 47% yield (Table 1, entry 4). Increasing the loading of MesCOOH from 30 mol% to 50 mol% resulted in an increased yield (57%) of 3aa (Table 1, entry 9). To improve reaction efficiency, the temperature range was evaluated (Table 1, entries 10–12). It was found that product 3aa was obtained in 83% yield at 50 °C (Table 1, entry 11) and a comparable result could still be achieved at 25 °C (Table 1, entry 12). Finally, the reactivity could be further increased when the reaction was performed at 25 °C for 2 h to deliver the desired product 3aa in 87% yield with excellent E/Z selectivity (Table 1, entry 13). To demonstrate the unique reactivity of Ru catalyst in this protocol, [Cp*Rh(CH3CN)3](SbF6)2 was also tested to give a slightly lower yield with decreased E/Z selectivity at higher temperature (Table 1, entry 14).
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additive (mol%) | T (°C) | Yield (%) |
a Reaction conditions: 1a (0.2 mmol), 2a (0.4 mmol), [Ru(p-cymene)Cl2]2 (5 mol%), additive (30 mol%), MW, 1 h, 90 °C.b MesCOOH (50 mol%).c t = 2 h.d [Cp*Rh(CH3CN)3](SbF6)2 (8 mol%).e The E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z ratio was determined by 1H NMR analysis. MW = microwave irradiation. :Z ratio was determined by 1H NMR analysis. MW = microwave irradiation. |
||||
| 1 | [Ru(p-cymene)Cl2]2 | AdCOOH | 90 | 40 |
| 2 | RuCl3·3H2O | AdCOOH | 90 | N.R |
| 3 | [Cp*RuCl2]2 | AdCOOH | 90 | N.R |
| 4 | [Ru(p-cymene)Cl2]2 | MesCOOH | 90 | 47 |
| 5 | [Ru(p-cymene)Cl2]2 | AcOH | 90 | 40 |
| 6 | [Ru(p-cymene)Cl2]2 | NaOAc | 90 | 20 |
| 7 | [Ru(p-cymene)Cl2]2 | PivONa·H2O | 90 | 21 |
| 8 | [Ru(p-cymene)Cl2]2 | DABCO | 90 | Trace |
| 9b | [Ru(p-cymene)Cl2]2 | MesCOOH | 90 | 57 |
| 10b | [Ru(p-cymene)Cl2]2 | MesCOOH | 70 | 68 |
| 11b | [Ru(p-cymene)Cl2]2 | MesCOOH | 50 | 83 |
| 12b | [Ru(p-cymene)Cl2]2 | MesCOOH | 25 | 65 |
| 13b,c | [Ru(p-cymene)Cl2]2 | MesCOOH | 25 | 87 (>20:1)e |
| 14c,d | [Cp*Rh(CH3CN)3](SbF6)2 | AdCOOH | 80 | 78 (10:1)e |
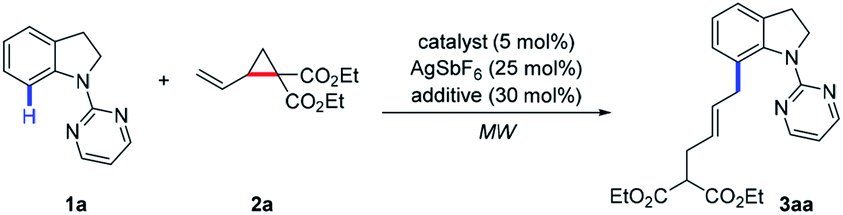
Under the optimized reaction conditions with the ruthenium catalyst, the substrate scope of indolines 1 was investigated (Table 2). C2-alkyl and aryl substituted indolines were initially examined to give products 3ba–fa in 65–96% yields. Indoline bearing methyl group at the C3 position was also tolerated to afford product 3ga in 91% yield. For C4 and C5 substituted indolines, both electron-donating (Me, OMe, and OBn) and electron-withdrawing (F and Cl) groups were well compatible to provide the corresponding allylated products 3ha–la and 3na–ra in 80–96% yields. The presence of bromo and ester functionalities on substrates led to products 3ma (54%), 3sa (73%), and 3ta (60%) in slightly decreased yields. In addition, disubstituted indolines were also tested to deliver products 3ua and 3va in 82% and 89% yields, respectively. Notably, C6-halogen substituted indolines exhibited high activity to afford products 3wa–ya in 78–95% yield. However, C6-methyl substituted substrate turned to be unsuccessful probably due to steric hindrance effect. Moreover, quinoline and carbazole substrates were also employed to afford the allylated products 3za and 3a′a in 71% and 78% yields. Finally, a series of other directing groups such as acetyl, pyridyl, and others were also tried (see in the ESI†). While 3b′a was obtained in 84% yield, others failed to give the coupling products. Overall, indolines with a variety of functionalities ranging from C2 to C6 positions could react with VCP 2a to afford the allylated products in good yields with high E/Z selectivities under the [Ru(p-cymene)Cl2]2 catalytic system. On the other hand, when [RhCp*(CH3CN)3](SbF6)2 was employed as the catalyst instead of Ru analogue, decreased yields accompanied with lower E/Z selectivities was observed in most cases.
|
|---|
| a Reaction conditions [Ru]: 1 (0.2 mmol), 2a (0.4 mmol), [Ru(p-cymene)Cl2]2 (5 mol%), MesCOOH (50 mol%), AgSbF6 (25 mol%), MW, 25 °C, 2 h; [Rh]: 1 (0.2 mmol), 2a (0.4 mmol), [Cp*Rh(CH3CN)3](SbF6)2 (8 mol%), AdCOOH (30 mol%), AgSbF6 (20 mol%), MW, 80 °C, 2 h.b The E:Z ratio was determined by 1H NMR analysis. MW = microwave. |
 |
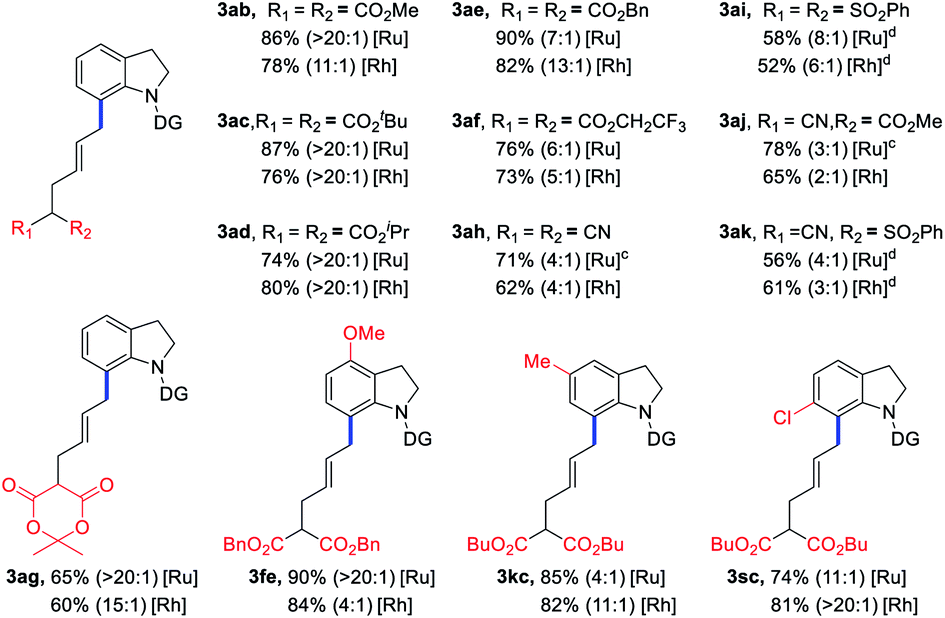
Encouraged by the above results, the scope of VCPs 2 was also explored to further examine the generality of the current C–C/C–H activation strategy with the ruthenium catalyst (Table 3). Various ester-substituted VCPs, including CO2Me, tBuCO2, iPrCO2, and BnCO2 groups, were successfully coupled with indoline 1a to afford products 3ab–ad in 74–90% yields with up to 20:1 E/Z selectivity. For VCPs having CF3CH2CO2 and lactone activating groups, products 3af and 3ag were isolated in decreased yields. When CN and PhSO2 functionalities were introduced into VCP moiety, the corresponding products 3ah–ak were obtained in 56–78% yields with decreased E/Z values. Furthermore, the substituted indolines could also furnish the corresponding products (3fe, 3kc, and 3sc) in 74–90% yields. Finally, Rh catalytic system could give the corresponding products in comparable yields and E/Z ratios.
|
|---|
| a Reaction conditions [Ru]: 1 (0.2 mmol), 2a (0.4 mmol), [Ru(p-cymene)Cl2]2 (5 mol%), MesCOOH (50 mol%), AgSbF6 (25 mol%), MW, 25 °C, 2 h; [Rh]: 1 (0.2 mmol), 2a (0.4 mmol), [Cp*Rh(CH3CN)3](SbF6)2 (8 mol%), AdCOOH (30 mol%), AgSbF6 (20 mol%), MW, 80 °C, 2 h.b The E:Z ratio was determined by 1H NMR analysis.c 40 °C.d [Ru] CH2Cl2 (0.5 mL), 40 °C; [Rh]:MeOH (0.5 mL). MW = microwave. |
 |
To evaluate the practical utility of the current methodology, a gram-scale reaction between 1a (6.0 mmol) and 2a (12.0 mmol) was performed under standard conditions, which delivered the allylated product 3aa in 74% yield (Scheme 2). Meanwhile, the derivatizations of 3aa were also conducted to highlight the synthetic importance of allylated indolines. Firstly, 3aa could undergo decarboxylation in the presence of NaOEt in DMSO to afford compound 4 in 86% yield. Under the condition of KOH in EtOH, the hydrolysis product 5 was obtained in 90% yield. Secondly, in the presence of DDQ in toluene, product 3aa could be oxidized to compound 6 in 38% yield. Thirdly, hydrogenation of 3aa under Pd–C/H2 conditions would afford a reduced product 7, which could further undergo oxidation and decarboxylation transformations to give indole derivatives 8 and 9 in 90% and 82% yields, respectively.
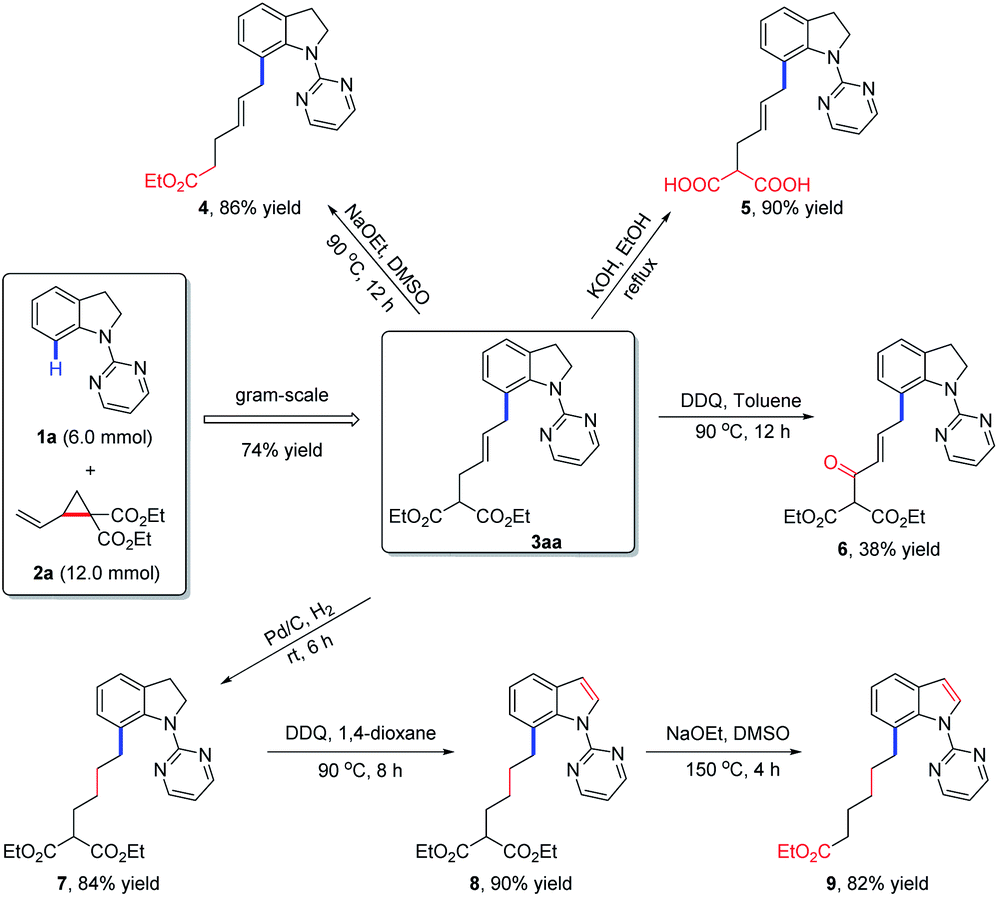 | ||
| Scheme 2 Gram-scale reaction and further derivatization of product 3aa. | ||
To gain insights into the reaction mechanism, a series of control experiments were conducted (Scheme 3). In the H/D exchange experiment, the deuterated [D]-1a was prepared and subjected to the standard conditions. It was found that only negligible amount of deuterium was detected for retrieved 1a. When 2a reacted with [D]-1a for 30 min, a similar result with a distinct D/H exchange was also observed (Scheme 3a). Next, the intermolecular competition experiment between substituted indolines 1o and 1t was conducted. Methoxyl-substituted allylated indoline 3oa was isolated as the major product, indicating that indolines bearing electron-donating groups are more reactive (Scheme 3b). When a radical quencher, such as 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), 2,6-di-tert-butyl-4-methylphenol (BHT), or benzoquinone (BQ), was added, the allylation reaction was suppressed with product 3aa obtained in a decreased yield (Scheme 3c). These results could't indicate that a non-radical mediated reaction pathway. Finally, the KIE values observed in parallel reactions suggest that the C–H bond cleavage is not involved in the limiting step (Scheme 3d).
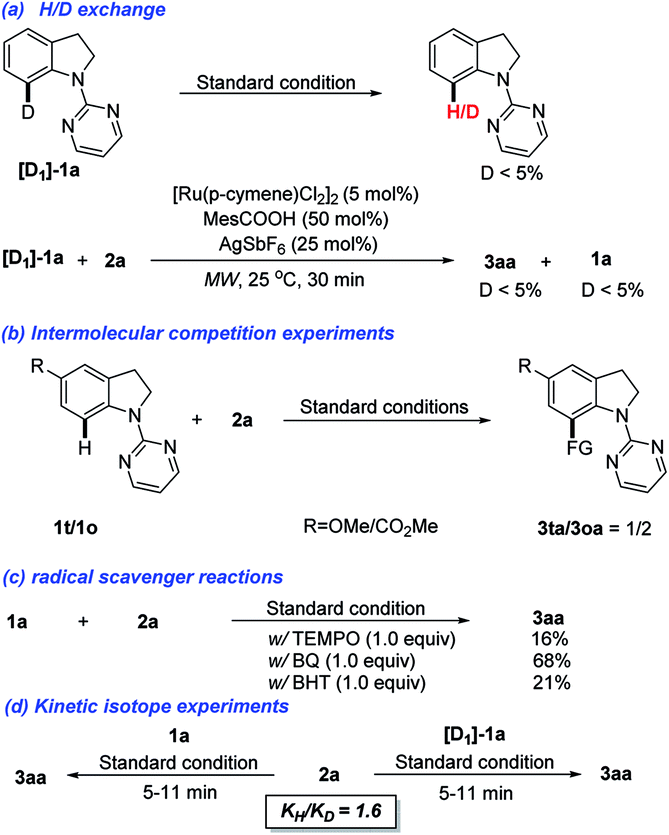 | ||
| Scheme 3 Control experiments. | ||
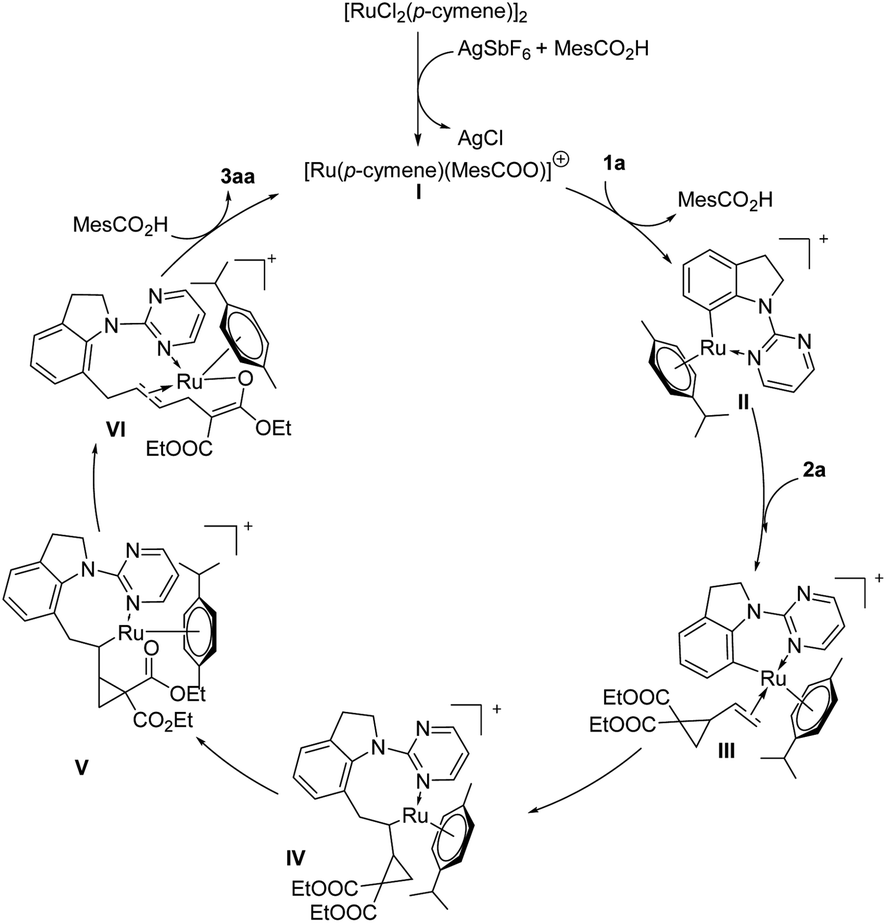
On the basis of above discussion and related reports,17 a plausible catalytic cycle was proposed (Scheme 4). A pyrimidine-directed C–H activation between indoline 1a and an in situ-generated Ru cationic complex I gives a six-membered ruthenacycle II. Coordination of intermediate II with VCP 2a provides intermediate III, which undergoes 1,2-migratory insertion to afford an ruthenium intermediate IV. Then oxygen coordinates to Ru metal center to form intermediate V. After C–C cleavage of VCP 2a, intermediate VI will be generated, followed by protonation to deliver the desired product 3aa and regenerate the active catalyst species I.
 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In summary, we have successfully demonstrated a [Ru(p-cymene)Cl2]2-catalyzed C7 allylation of indolines with VCPs as the allylating reagents via a sequential C–H/C–C process. This protocol is applicable to broad substrate scope with good functional group compatibility and high E/Z selectivity. The practical applications of allylation transformation are highlighted through gram-scale production and multiple derivatizations. Notably, the use of microwave irradiation under room temperature and solvent-free conditions makes this protocol more valuable and operational convenience, which provides a new insight for the formation of indoline derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21672192, 21803059, U1904212, and 21929101) is gratefully appreciated.Notes and references
- (a) B. M. Trost, Acc. Chem. Res., 2002, 35, 695 CrossRef CAS PubMed; (b) P. T. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC; (c) C.-J. Li and P. T. Anastas, Chem. Soc. Rev., 2012, 41, 1413 RSC.
- (a) R. S. Varma, Green Chem., 2014, 16, 2027 RSC; (b) R. Luque and F. L.-Y. Lam, Sustainable Catalysis Energy-Efficient Reactions and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Germany, 2018 CrossRef; (c) W. Zhang and B. W. Cue, Green Techniques for Organic Synthesis and Medicinal Chemistry, John Wiley & Sons, Ltd, New York, 2nd edn, 2018 CrossRef; (d) A. Beillard, X. Bantreil, T.-X. Métro, J. Martinez and F. Lamaty, Chem. Rev., 2019, 119, 7529 CrossRef CAS PubMed.
- (a) A. Daştan, A. Kulkarni and B. Török, Green Chem., 2012, 14, 17 RSC; (b) M. B. Gawande, S. N. Shelke, R. Zboril and R. S. Varma, Acc. Chem. Res., 2014, 47, 1338 CrossRef CAS PubMed; (c) A. K. Rathi, M. B. Gawande, R. Zboril and R. S. Varma, Coord. Chem. Rev., 2015, 291, 68 CrossRef CAS; (d) M. Tsuji, ChemistrySelect, 2017, 2, 805 CrossRef; (e) G. B. Dudley and A. E. Stiegman, Chem. Rec., 2018, 18, 381 CrossRef CAS PubMed.
- (a) L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468 CrossRef CAS PubMed; (b) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (c) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596 CrossRef CAS PubMed; (d) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (e) W. Yu, W. Zhang, Y. Liu, Z. Liu and Y. Zhang, Org. Chem. Front., 2017, 4, 77 RSC; (f) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed; (g) P. Gandeepan, T. Mller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS PubMed; (h) Z. Chen, M.-Y. Rong, J. Nie, X.-F. Zhu, B.-F. Shi and J.-A. Ma, Chem. Soc. Rev., 2019, 48, 4921 RSC.
- (a) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed; (b) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef CAS PubMed; (c) D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977 CrossRef CAS PubMed; (d) F. Song, T. Gou, B.-Q. Wang and Z.-J. Shi, Chem. Soc. Rev., 2018, 47, 7078 RSC.
- S. Laclef, M. Harari, J. Godeau, I. Schmitz-Afonso, L. Bischo, C. Hoarau, V. Levacher, C. Fruit and T. Besson, Org. Lett., 2015, 17, 1700 CrossRef CAS PubMed.
- (a) F. Wang, S. Yu and X. Li, Chem. Soc. Rev., 2016, 45, 6462 RSC; (b) Z. Nairoukh, M. Cormier and I. Marek, Nat. Rev. Chem., 2017, 1, 0035 CrossRef.
- (a) R. A. PerianaRobert and G. Bergman, J. Am. Chem. Soc., 1984, 106, 7272 CrossRef; (b) R. A. PerianaRobert and G. Bergman, J. Am. Chem. Soc., 1986, 108, 7346 CrossRef.
- (a) H. M. Ko and G. Dong, Nat. Chem., 2014, 6, 739 CrossRef CAS PubMed; (b) Y. Xia, G. Lu, P. Liu and G. Dong, Nature, 2016, 539, 546 CrossRef CAS PubMed; (c) Y. Xia, J. Wang and G. Dong, Angew. Chem., Int. Ed., 2017, 56, 2376 CrossRef CAS PubMed.
- (a) A. Masarwa, D. Didier, T. Zabrodski, M. Schinkel, L. Ackermann and I. Marek, Nature, 2014, 505, 199 CrossRef CAS PubMed; (b) Y.-F. Liang, V. Müller, W. Liu, A. Münch, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 9415 CrossRef CAS PubMed; (c) H. Wang, I. Choi, T. Rogge, N. Kaplaneris and L. Ackermann, Nat. Catal., 2018, 1, 993 CrossRef CAS.
- (a) S. Yu and X. Li, Org. Lett., 2014, 16, 1220 CrossRef CAS PubMed; (b) X. Zhou, S. Yu, L. Kong and X. Li, ACS Catal., 2016, 6, 647 CrossRef CAS; (c) L. Kong, S. Yu, G. Tang, H. Wang, X. Zhou and X. Li, Org. Lett., 2016, 18, 3802 CrossRef CAS PubMed.
- (a) T. Seiser, O. A. Roth and N. Cramer, Angew. Chem., Int. Ed., 2009, 48, 6320 CrossRef CAS PubMed; (b) L. Souillart and N. Cramer, Chem. Sci., 2014, 5, 837 RSC; (c) L. Souillart and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 9640 CrossRef CAS PubMed.
- (a) Z. Chai and T. J. Rainey, J. Am. Chem. Soc., 2012, 134, 3615 CrossRef CAS PubMed; (b) H. Zhang, K. Wang, B. Wang, H. Yi, F. Hu, C. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 13234 CrossRef CAS PubMed; (c) A. Masarwa, M. Weber and R. Sarpong, J. Am. Chem. Soc., 2015, 137, 6327 CrossRef CAS PubMed; (d) M. Li and F. Y. Kwong, Angew. Chem., Int. Ed., 2018, 57, 6512 CrossRef CAS PubMed; (e) J.-L. Pan, C. Liu, C. Chen, T.-Q. Liu, M. Wang, Z. Sun and S.-Y. Zhang, Org. Lett., 2019, 21, 2823 CrossRef CAS PubMed; (f) S. Li, P. Shi, R.-H. Liu, X.-H. Hu and T.-P. Loh, Org. Lett., 2019, 21, 1602 CrossRef CAS PubMed.
- (a) B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323 CrossRef CAS PubMed; (b) I. Marek, A. Masarwa, P.-O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414 CrossRef CAS PubMed; (c) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404 CrossRef CAS PubMed; (d) P.-H. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340 CrossRef CAS PubMed.
- (a) J.-Q. Wu, Z.-P. Qiu, S.-S. Zhang, J.-G. Liu, Y.-X. Lao, L.-Q. Gu, Z.-S. Huang, J. Li and H. Wang, Chem. Commun., 2015, 51, 77 RSC; (b) Q. Lu, F. J. R. Klauck and F. Glorius, Chem. Sci., 2017, 8, 3379 RSC; (c) D. Zell, Q. Bu, M. Feldt and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7408 CrossRef CAS PubMed; (d) T. H. Meyer, W. Liu, M. Feldt, A. Wuttke, R. A. Mata and L. Ackermann, Chem.–Eur. J., 2017, 23, 5443 CrossRef CAS PubMed; (e) Z. Hu, X.-Q. Hu, G. Zhang and L. J. Gooßen, Org. Lett., 2019, 21, 6770 CrossRef CAS PubMed.
- (a) X. Zhu, X.-J. Shen, Z.-Y. Tian, S. Lu, L.-L. Tian, W.-B. Liu, B. Song and X.-Q. Hao, J. Org. Chem., 2017, 82, 6022 CrossRef CAS PubMed; (b) Y.-J. Guo, S. Lu, L.-L. Tian, E.-L. Huang, X.-Q. Hao, X. Zhu, T. Shao and M.-P. Song, J. Org. Chem., 2018, 83, 338 CrossRef CAS PubMed; (c) S. Lu, L.-L. Tian, T.-W. Cui, Y.-S. Zhu, X. Zhu, X.-Q. Hao and M.-P. Song, J. Org. Chem., 2018, 83, 13991 CrossRef CAS PubMed; (d) X.-C. Li, C. Du, H. Zhang, J.-L. Niu and M.-P. Song, Org. Lett., 2019, 21, 2863 CrossRef CAS PubMed; (e) L.-L. Tian, S. Lu, Z.-H. Zhang, E.-L. Huang, H.-T. Yan, X. Zhu, X.-Q. Hao and M.-P. Song, J. Org. Chem., 2019, 84, 5213 CrossRef CAS PubMed; (f) Q. Wang, C.-L. Zhi, P. –P. Lu, S. Liu, X. Zhu, X.-Q. Hao and M.-P. Song, Adv. Synth. Catal., 2019, 361, 1253 CrossRef CAS; (g) X.-M. Zhao, E.-L. Huang, Y.-S. Zhu, J. Li, B. Song, X. Zhu and X.-Q. Hao, Org. Biomol. Chem., 2019, 17, 4869 RSC; (h) C. Zhi, Q. Wang, S. Liu, Y. Xue, L. Shi, X. Zhu, X.-Q. Hao and M.-P. Song, J. Org. Chem., 2020, 85, 1022 CrossRef CAS PubMed.
- (a) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (b) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 3635 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02016h |
| This journal is © The Royal Society of Chemistry 2020 |