
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficiency and stability enhancement of perovskite solar cells using reduced graphene oxide derived from earth-abundant natural graphite†
Selengesuren Suragtkhuua,
Odonchimeg Tserendavaga,
Ulziibayar Vandandoobc,
Abdulaziz S. R. Bati d,
Munkhjargal Bat-Erdened,
Joseph G. Shapter*d,
Munkhbayar Batmunkh*de and
Sarangerel Davaasambuu*a
d,
Munkhjargal Bat-Erdened,
Joseph G. Shapter*d,
Munkhbayar Batmunkh*de and
Sarangerel Davaasambuu*a
aDepartment of Chemistry, School of Arts and Sciences, National University of Mongolia, Ulaanbaatar 14200, Mongolia. E-mail: sarangerel@num.edu.mn
bSchool of Applied Sciences, Mongolian University of Science and Technology, Ulaanbaatar 14191, Mongolia
cInstitute of Mathematics and Digital Technology, Mongolian Academy of Sciences, Ulaanbaatar, Mongolia
dAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, Brisbane, Queensland 4072, Australia. E-mail: j.shapter@uq.edu.au; m.batmunkh@uq.edu.au
eCentre for Clean Environment and Energy, Griffith University, Gold Coast, Queensland 4222, Australia
First published on 3rd March 2020
Abstract
Graphene – two-dimensional (2D) sheets of carbon atoms linked in a honeycomb pattern – has unique properties that exhibit great promise for various applications including solar cells. Herein we prepared two-dimensional (2D) reduced graphene oxide (rGO) nanosheets from naturally abundant graphite flakes (obtained from Tuv aimag in Mongolia) using solution processed chemical oxidation and thermal reduction methods. As a proof of concept, we used our rGO as a hole transporting material (HTM) in perovskite solar cells (PSCs). Promisingly, the use of rGO in the hole transporting layer (HTL) not only enhanced the photovoltaic efficiency of PSCs, but also improved the device stability. In particular, the best performing PSC employing rGO nanosheets exhibited a power conversion efficiency (PCE) of up to 18.13%, while the control device without rGO delivered a maximum efficiency of 17.26%. The present work demonstrates the possibilities for solving PSC issues (stability) using nanomaterials derived from naturally abundant graphite sources.
Introduction
The conversion of sunlight into electricity using photovoltaic (PV) cells (known as solar cells) is now a mainstream renewable energy source. At present, the PV market is mainly dominated by crystalline silicon solar cells. This class of solar cells is a mature technology and can efficiently convert the sun's energy into electrical power, but they still suffer from high manufacturing, installation, and material costs.1 Therefore, the search for novel solar technologies that can deliver high power conversion efficiency at low-cost has been the subject of intense investigation over the past several decades.2–6 Dye-sensitized solar cells (DSSCs), organic solar cells (OSCs) and perovskite solar cells (PSCs) have received a great deal of attention from the PV community.7–13As an emerging PV, PSCs have attracted tremendous interest from the PV community due to the potential they offer in terms of low manufacturing cost and high performance.14,15 Since the discovery in 2009,16 PSCs have been sitting in the spotlight as a promising clean and renewable energy technology and have become one of the most popular topics in scientific research. The rapidly growing popularity of PSCs is due to the unprecedented progress in the power conversion efficiency (PCE) that has been made within only ten years.17 A certified PCE of 25.2% has been demonstrated for single-junction architecture PSC.18 Moreover, PSCs can be fabricated with high flexibility, tunable colour and lightweight design, all of which are attractive features in the PV industry.
PSCs can be classified into two main categories (n–i–p and p–i–n) based on their device architecture.19,20 Of particular interest is the n–i–p structured devices owing to the high performance. A typical n–i–p PSC is made of a transparent conducting oxide (TCO) (indium-doped and/or fluorine-doped tin oxide (ITO or FTO)) electrode, a n-type semiconducting oxide layer (TiO2 or SnO2), a perovskite layer, a p-type hole transporting layer (HTL) and a metal contact (Au or Ag).21,22 In such a device structure, the hole transporting material (HTM) plays an important role in selectively extracting photogenerated holes from the perovskite and transporting them to the conductive metal electrode.23 A p-type organic hole conductor, 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (Spiro-OMeTAD) is the most widely used HTM for n–i–p PSCs.24 However, due to the high cost of Spiro-OMeTAD, a wide range of organic and inorganic HTMs have been explored as alternatives candidates such as poly(3-hexylthiophene) P3HT, poly(3,4-ethylenedioxythiophene)polystyrene sulfonate (PEDOT:PSS), poly(triarylamine) (PTAA), modified fluorene–dithiophene (FDT), copper(I) thiocyanate (CuSCN), nickel(II) oxide (NiO) and others.25 Despite the great advances in the development of novel HTMs, Spiro-OMeTAD still remains the material of choice for PSCs owing to its ability to deliver high device efficiency.
It is well known that pristine Spiro-OMeTAD suffers significantly from low intrinsic charge carrier mobility,26 which is usually addressed by increasing the carrier density via oxidative doping using lithium bis-(trifluoromethane)sulfonimide (Li-TFSI) and 4-tert-butylpyridine (4-tbp).27 While 4-tbp is a corrosive chemical, the hygroscopic nature of the Li-TFSI dopant accelerates the moisture-induced degradation of PSCs.28 One promising strategy to improve the stability of PSCs, while maintaining high efficiency, is to incorporate functional materials into the HTM.27,28 For instance, polymer-functionalized carbon nanotubes (CNTs),28 bamboo-structured CNTs,29 MoS2 (ref. 30) and others have been used to improve the efficiency and stability of the PSCs.
Graphene exhibits remarkable properties such as excellent conductivity, superior strength to other material, good flexibility and high transparency, all of which holds specific promise for many applications including PSCs.31 Excellent progress has been made in the use of graphene derivatives for both n–i–p and p–i–n PSCs.32–39 Reduced graphene oxide (rGO) was used as a sole HTM to replace the traditional Spiro-OMeTAD in PSCs by Palma et al.40 who demonstrated excellent PSC stability using rGO. Although PSCs with rGO based HTM showed excellent stability, these PSCs showed very low PCEs (3–6%). Luo et al. combined rGO (reduced by FeI2) with dopant-free Spiro-OMeTAD and achieved 10.6%, while showing good stability.41 Cho et al.42 incorporated rGO in the doped Spiro-OMeTAD and obtained a PCE of 18.76%, while the control device without rGO displayed a similar cell efficiency (18.89%). However, no stability test was conducted in this work.42 Indeed, there has been no effort exploring the stability of high efficiency PSCs fabricated with rGO integrated Spiro-OMeTAD. Moreover, all of these reports used chemical reductions to prepare their rGO samples. However, it is well documented that the chemically reduced GO suffers from low electrical conductivity.43 In addition, the commercial graphite available in the market is generally used as a starting material for the rGO production. However, exploring new graphite sources that are abundantly available for use and could provide promising properties would be of great value.
In this work, we used naturally abundant graphite obtained from a Mongolian mine to prepare rGO using chemical oxidation method, followed by thermal reduction. The as-prepared rGO has been used as a HTM for PSCs. The best-performing device with rGO incorporated Spiro-OMeTAD based HTL delivered a PCE of 18.13% and showed improved stability as compared to the control PSCs. In particular, the PSC fabricated with rGO incorporated Spiro-OMeTAD based HTL preserved 75% of the initial device performance after storing 500 h in ambient condition (40–60% humidity), while the reference device retained only 54% of its initial performance.
Experimental section
Preparation of GO and rGO
The raw graphite sample was obtained from a mine located in Tuv aimag (Bayan soum), Mongolia. Specifically, the mine name: Zulegt, longitude: 47 04 00, latitude: 107 39 05, above sea level: 1400–1460 m. Firstly, the raw graphite was chemically oxidized to produce graphite oxide according to an improved Hummers' method with a slight modification.44 Briefly, a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of sulfuric acid (98% H2SO4; XiLong Chemical Factory Co. Ltd.) and phosphoric acid (85%, H3PO4; UnionLab Chemical Factory Co. Ltd.) (360:40 mL) was added to a mixture of raw graphite (3 g) and potassium permanganate (99.5%, KMnO4) (18 g). The oxidation process was accomplished by stirring the mixture at 330 rpm at 50 °C for 12 h. Then, the reaction mixture was cooled down to room temperature and poured onto ice (300 mL) with hydrogen peroxide (30%, H2O2; XiLong Chemical Factory Co. Ltd.) (3 mL). The mixture was then washed twice with distilled (DI) water, then twice with hydrochloric acid (30%, HCl; XiLong Chemical Factory Co. Ltd.) and finally twice with ethyl alcohol. After centrifugation, a white, insoluble, solid residue was observed at the bottom of the centrifuge tube and was removed from the sample during the washing process. Then the final GO solution was centrifuged at 4500 rpm for 90 min and the supernatant decanted away. The formed light brown GO was freeze-dried (LGJ-12) at −46 °C for 12 h. Further, the obtained GO was thermally reduced to rGO in a tube furnace (OTF-1200X-II-UL, quartz tube) at 900 °C for 3 h under an Ar gas flow.
:1 (v/v) mixture of sulfuric acid (98% H2SO4; XiLong Chemical Factory Co. Ltd.) and phosphoric acid (85%, H3PO4; UnionLab Chemical Factory Co. Ltd.) (360:40 mL) was added to a mixture of raw graphite (3 g) and potassium permanganate (99.5%, KMnO4) (18 g). The oxidation process was accomplished by stirring the mixture at 330 rpm at 50 °C for 12 h. Then, the reaction mixture was cooled down to room temperature and poured onto ice (300 mL) with hydrogen peroxide (30%, H2O2; XiLong Chemical Factory Co. Ltd.) (3 mL). The mixture was then washed twice with distilled (DI) water, then twice with hydrochloric acid (30%, HCl; XiLong Chemical Factory Co. Ltd.) and finally twice with ethyl alcohol. After centrifugation, a white, insoluble, solid residue was observed at the bottom of the centrifuge tube and was removed from the sample during the washing process. Then the final GO solution was centrifuged at 4500 rpm for 90 min and the supernatant decanted away. The formed light brown GO was freeze-dried (LGJ-12) at −46 °C for 12 h. Further, the obtained GO was thermally reduced to rGO in a tube furnace (OTF-1200X-II-UL, quartz tube) at 900 °C for 3 h under an Ar gas flow.
Device fabrication
ITO (Xin Yan Technology Ltd) coated glass substrates were cleaned with a detergent, distilled water, acetone and isopropanol for 10 min each using ultrasonication, followed by UV-ozone cleaning for 20 min. Then, the substrate was spin coated with a thin layer of SnO2 nanoparticles (2.67 wt%; Alfa Aesar) at 4000 rpm for 30 s, and annealed in ambient air at 175 °C for 30 min. Meanwhile, the mixed perovskite precursor solution was prepared according following the approach of Saliba et al.45 In brief, FAI (1 M) (Greatcell Energy), PbI2 (1.1 M) (TCI), PbBr2 (0.2 M) (TCI) and MABr (0.2 M) (Greatcell Energy) were mixed in anhydrous DMF:DMSO 4:1 (v/v) (Sigma). Then, CsI (Sigma), pre-dissolved as a 1.5 M stock solution in DMSO, was added to the perovskite precursor. The perovskite solution was filtered before being spin coated onto the SnO2 coated ITO substrates. Before the deposition of perovskite, SnO2 coated electrodes were UV-ozone treated for 10 min (this is a very important step for successful perovskite deposition). The spin-coating recipe includes two steps, first 1000 rpm for 10 s with a ramp of 250 rpm s−1, then 5000 rpm for 25 s with a ramp of 2000 rpm s−1. 10 s before the end of the spin-coating program, anhydrous chlorobenzene (Sigma) (100 μL) was dropped on the center of spinning substrate. The films were then heated at 100 °C for 45 min in the glovebox.
Spiro-OMeTAD based HTM solution was prepared by dissolving 58 mg Spiro-OMeTAD (Merck), 23 μL 4-tert-butylpyridine (tBP) (Sigma), 14 μL of a stock solution of 520 mg mL−1 lithium bis(trifluoromethylsulphonyl)imide (Li-TFSI) (Sigma) in acetonitrile (Sigma) and 16.5 μL of a stock solution of 100 mg mL−1 FK102 Co(III) PF6 salt (Greatcell Energy) in acetonitrile, in 800 μL chlorobenzene. The rGO solution was prepared by dispersing 10 mg rGO in 5 mL chlorobenzene using ultrasonication for 45 min. The concentration of rGO added to the Spiro-OMeTAD solution was 0.145 mg mL−1. For the fabrication of rGO-only HTM based devices, rGO dispersion with a concentration of 1 mg mL−1 was prepared and used. All HTMs (Spiro-OMeTAD, rGO and rGO + Spiro-OMeTAD) were deposited on the perovskite layer by spin coating at 4000 rpm for 20 s. After the HTM deposition, the films were stored overnight in a dry air desiccator. Finally, 80 nm gold was thermally evaporated at a rate of 0.4 Å s−1.
Characterization
Raman spectra were acquired using a WITec alpha300 RA + S Raman microscope using an excitation laser wavelength of 532 nm with a 20× objective. The grating used was 600 grooves mm−1. Atomic force microscopy (AFM) was performed in air using Asylum Research Cypher S with Asylum Research software, operating in standard tapping mode configuration using AIR cantilever holder. The AFM probe used was high accuracy noncontact composite probe with silicon body, polysilicon lever and silicon high resolution tip (tip curvature radius: <10 nm) from TipsNano. X-ray diffraction (XRD) patterns were obtained using an Enraf Nonius Delft 583 X-ray diffractometer with CuKα source with a step size of 0.02° (40 kV, 30 mA). X-ray photoelectron spectroscopy (XPS) data were acquired using a Kratos Axis ULTRA X-ray Photoelectron Spectrometer incorporating a 165 mm hemispherical electron energy analyzer. The incident radiation was monochromatic Al Kα X-rays (1486.6 eV) at 225 W (15 kV, 15 mA). Base pressure in the analysis chamber was 1.0 × 10−9 torr and during sample analysis 1.0 × 10−8 torr.The current density–voltage (J–V) characteristics of the devices were measured in a N2-filled glovebox using a Keithley 2400 source measure unit (SMU) and with simulated air mass index 1.5 global light with an intensity of 100 mW cm−2. The system was calibrated with a National Renewable Energy Laboratory (NREL) certified, spectral mismatch-corrected standard silicon photodiode. The J–V measurements were carried out in reverse scan direction from +1.2 V to −0.1 V. The active area of the devices was 0.2 cm2.
Results and discussion
A raw sample, which was obtained from Mongolian mining field, was first characterized using XRD. Fig. 1 shows the XRD pattern of the raw sample. As shown in Fig. 1, a sharp peak at 2θ of around 26.6° reveals that the sample contains graphite and other minerals. According to the XRD, the interlayer spacing of the (002) plane (3.35 Å) was observed and is consistent with reported d-spacing of graphite in the literature.46 In addition to the (002) plane of graphite, the peak at around 26.7° was observed and can be assigned to the (101) plane (3.34 Å) for quartz (SiO2). Moreover, our XRD confirmed that noticeable peaks at 20.9°, 23.5°, 25.7° and 27.6° are due to the SiO2 and other minerals such as albite, orthoclase and biotite.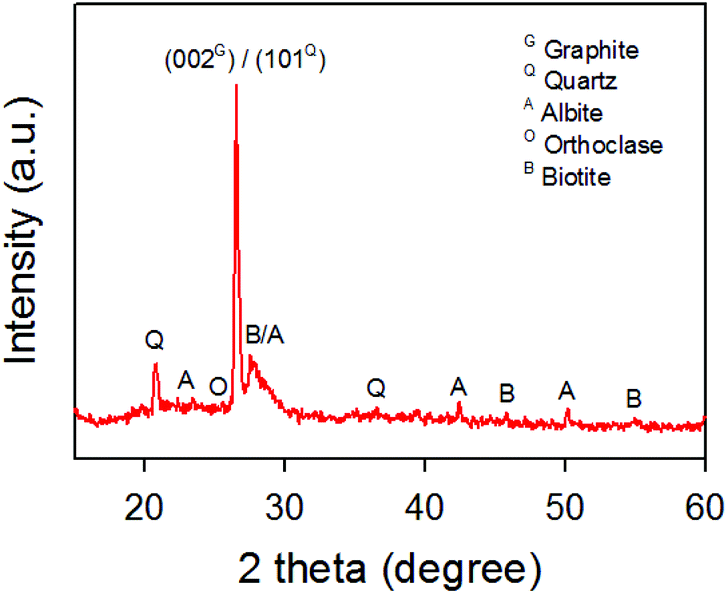 | ||
| Fig. 1 XRD pattern of raw graphite samples obtained from a Mongolian mine. | ||
The as-received raw sample was chemically oxidized according to an improved Hummers' method using concentrated H2SO4, H3PO4, KMnO4 and H2O2.44 During the oxidation and washing processes, a white solid residue was observed and separated from the graphite oxide by repeatedly centrifuging and washing. The white residue was then characterized using XRD (Fig. S1†), which confirmed the removal of the minerals from the graphite oxide. The as-prepared graphite oxide was further exfoliated using ultrasonication to obtain GO nanosheets which were then thermally reduced to rGO at 900 °C for 3 h under an Ar atmosphere.
In order to confirm the successful separation of our GO and white residue, XPS was used to characterize the raw graphite, GO and rGO. XPS is a powerful tool to explore chemical compositions and atomic bonding. Fig. 2a depicts the XPS survey spectra of the three samples. It can be clearly seen that the raw graphite sample showed strong carbon (C) and oxygen (O) peaks in addition to weak peaks for aluminum (Al), silicon (Si), iron (Fe) and sodium (Na). The intensity of the O peak was higher than that of the C peak due to the content of SiO2 and other minerals in the raw sample.
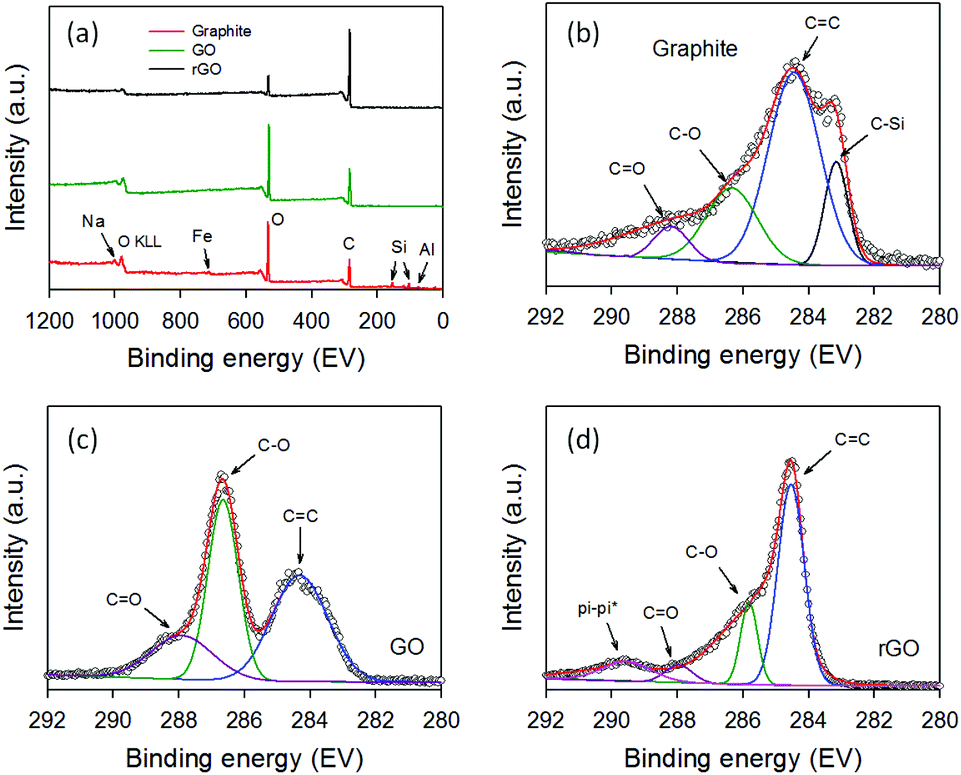 | ||
| Fig. 2 (a) XPS survey spectra of raw graphite, GO and rGO. High resolution XPS C 1s spectra of (b) graphite, (c) GO and (d) rGO. | ||
Importantly, none of these peaks associated with the minerals was found in the XPS survey scan of GO and rGO, confirming that the minerals were successfully separated from the graphite oxide. As shown in Fig. 2a, the O peak intensity decreased dramatically after thermal reduction of GO at 900 °C for 3 h, as expected. Further, Fig. 2b–d display the high-resolution XPS C 1s spectra of raw graphite, GO and rGO. All three samples showed the same peak at ∼284.4 eV which can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C binding peak, while exhibiting C–O and CO binding peaks at ∼286.7 eV and ∼288.1 eV, respectively.47 However, an additional peak at ∼283.2 eV, which can be assigned to C–Si bonding, was observed in the raw graphite containing the minerals (Fig. 2b).48 The disappearance of C–Si peak in the GO and rGO samples further confirms the successful separation of the minerals from the graphite oxide. As shown in Fig. 2c, the intensities of the C–O and CO bonding in the GO sample were very strong, suggesting successful oxidation of graphite. These strong peaks (C–O and CO) decreased significantly upon thermal reduction of GO (Fig. 2d).
C binding peak, while exhibiting C–O and CO binding peaks at ∼286.7 eV and ∼288.1 eV, respectively.47 However, an additional peak at ∼283.2 eV, which can be assigned to C–Si bonding, was observed in the raw graphite containing the minerals (Fig. 2b).48 The disappearance of C–Si peak in the GO and rGO samples further confirms the successful separation of the minerals from the graphite oxide. As shown in Fig. 2c, the intensities of the C–O and CO bonding in the GO sample were very strong, suggesting successful oxidation of graphite. These strong peaks (C–O and CO) decreased significantly upon thermal reduction of GO (Fig. 2d).
Fig. 3a illustrates the Raman spectra of our GO and rGO. Both samples exhibit two characteristic peaks located at ∼1352 cm−1 and ∼1606 cm−1 which are ascribed to the disorder peak (D band) arises from the defects in the sp2 carbon and graphitic peak (G band) arises from the in-plane vibrational E2g mode of the sp2 carbon, respectively.49–51 It is well established that the intensity ratio of D to G band (ID/IG) is used to determine the level of defects.35 The ID/IG for the GO and rGO were 0.96 and 1.04, respectively, which are very consistent with the values reported in the literature.52 A slight increase in the ID/IG observed upon reduction of GO is well accepted due to the defects introduced to the sp2 carbon during the reduction.52
 | ||
| Fig. 3 (a) Raman spectra of GO and rGO samples. (b) AFM image and (c) the corresponding height profile of rGO nanosheet. | ||
The AFM image of GO is shown in Fig. S2.† The thickness and lateral size of our GO was measured to be ∼4.4 nm and ∼400 nm, respectively. No significant difference in the thickness and flake size was observed after reduction of GO. Fig. 3b illustrates the AFM image of a rGO nanosheet and the corresponding height measurement is depicted in Fig. 3c. The lateral dimension of the rGO was around 400 nm, while its thickness was ∼5.6 nm. Moreover, the as-prepared rGO nanosheets were highly dispersible in organic solvents such as dimethylformamide (DMF) and chlorobenzene (Fig. S3†), making it applicable for a wide range of solution processable applications. Clearly, all of these characterization results reveal that both GO and rGO can successfully be obtained from naturally available mining source in Mongolia. Therefore, further functionalization and doping techniques should be explored in order to precisely tune the chemical and physical properties of graphene derivatives obtained from this graphite source.
As a proof of concept, we used our rGO as a HTM for n–i–p structured PSCs. In this work, we used cesium-doped triple cation mixed perovskite as an active light harvesting material which was deposited on SnO2 electron transporting layer (see Fig. S4†). Firstly, only rGO was directly used to substitute the traditional Spiro-OMeTAD. As such, rGO dispersion in chlorobenzene (1 mg mL−1) was prepared and spin coated at 4000 rpm for 20 s directly on perovskite layer. We repeated the spin coatings several times to optimize the device fabrication (Fig. S5†). The photocurrent density–voltage (J–V) characteristic of the best-performing PSC fabricated using rGO-only as a HTM (2 times spin coating) is displayed in Fig. S6.† This rGO HTM based device showed a short-circuit current (Jsc) of 16.20 mA cm−2, open-circuit voltage (Voc) of 0.72 V and fill factor (FF) of 0.43, yielding a PCE of 4.98%. A scanning electron microscopy (SEM) image (top-view) of the spin coated rGO suggests that the perovskite surface cannot be covered fully using rGO (Fig. S6† inset). This results in a direct contact between perovskite and Au electrode, which explains the poor efficiency of PSC fabricated using rGO-only as a HTM. Despite this, this efficiency (4.98%) was significantly higher than that (2.09%) of the device fabricated without an HTM, demonstrating the ability of rGO acting as a HTM.
Further, we used our rGO as an additive to the traditional Spiro-OMeTAD in the HTL for PSCs. Cross section SEM image of the PSC device fabricated with rGO + Spiro-OMeTAD is depicted in Fig. 4a. Fig. 4b also illustrates the layered structure of the device and the corresponding energy level diagram is shown in Fig. S7.† For comparison, we also fabricated reference PSCs with a device architecture of ITO/SnO2/perovskite/Spiro-OMeTAD/Au. The PV characteristics of the fabricated devices were studied under an air mass (AM) 1.5 illumination at 100 mW cm−2.
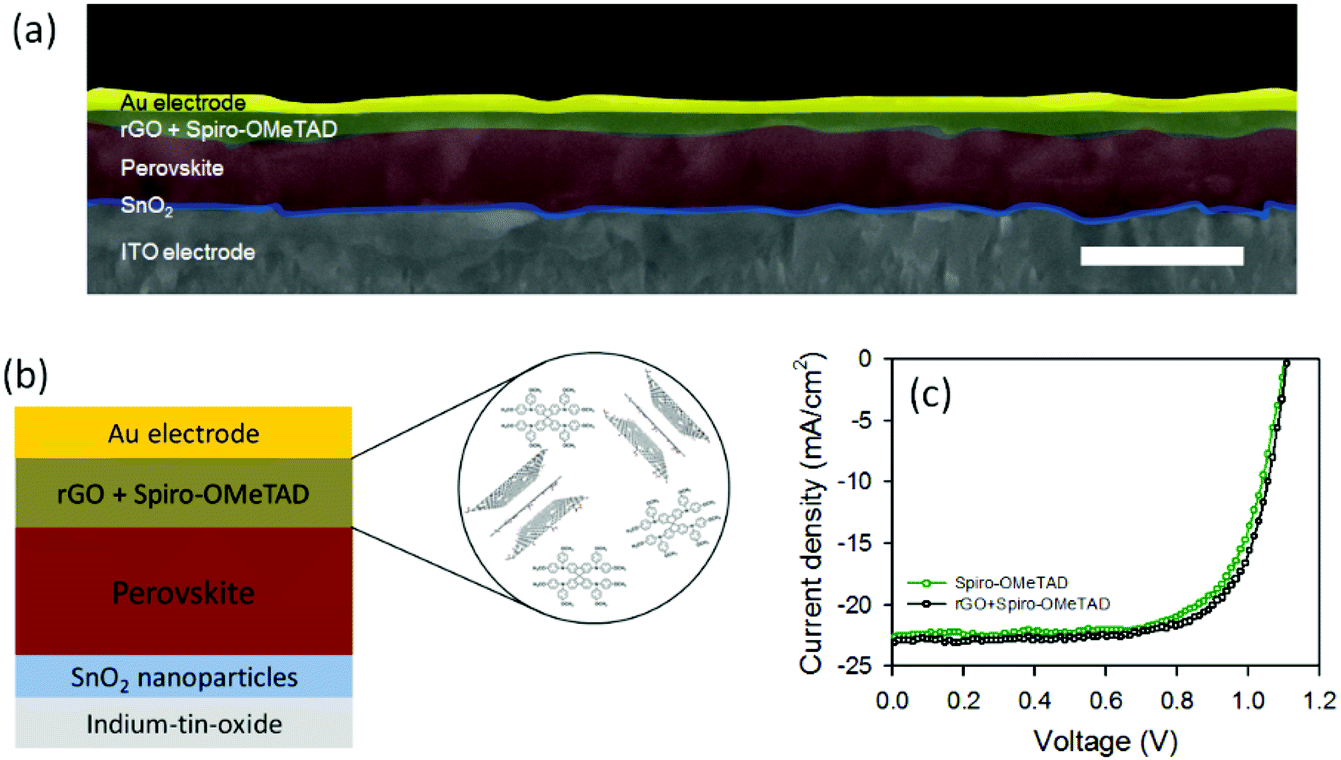 | ||
| Fig. 4 (a) Cross sectional SEM image of the PSC fabricated with rGO + Spiro-OMeTAD HTM. Scale bar: 1 μm. (b) PSC architecture of the fabricated devices using rGO as an additive to the Spiro-OMeTAD. (c) J–V curves of the best-performing PSCs fabricated with and without rGO in the HTL. | ||
Fig. 4c depicts the J–V characteristics of the best-performing devices with and without rGO in the Spiro-OMeTAD HTL and the corresponding PV parameters have been summarized in Table 1. The observed Jsc, Voc, and FF values for the best reference device based on Spiro-OMeTAD were 22.56 mA cm−2, 1.10 V, and 0.70, respectively, yielding a PCE of 17.26%. The average efficiency of the reference cells was measured to be 16.84 ± 0.37%. The highest efficiency, in this study, was achieved using our rGO incorporated Spiro-OMeTAD based PSC. Particularly, the device exhibited a Jsc of 23.05 mA cm−2, Voc of 1.11 V, FF of 0.71 and a PCE of 18.13%. Moreover the measured average efficiency of the rGO + Spiro-OMeTAD based PSCs was 17.82%, which was higher than that of the reference devices, suggesting that the addition of rGO can slightly increase the PV efficiency. This slight enhancement in the efficiency may be due to the higher electrical conductivity and suitable energy band alignment of our rGO in the device.31,41 It should be noted that Cho et al.42 introduced rGO prepared from commercial graphite powder (Alfa Aesar, ∼200 mesh) in the doped Spiro-OMeTAD. However, the efficiency of the rGO incorporated device was slightly lower than that of the control device without rGO. This indicates the effectiveness of our rGO as a HTM for PSCs.
| Device | Jsc (mA cm−2) | Voc (V) | FF | PCE (%) |
|---|---|---|---|---|
| Spiro-OMeTAD (reference) | 22.56; 21.37 ± 0.65 | 1.10; 1.11 ± 0.01 | 0.70; 0.71 ± 0.01 | 17.26; 16.84 ± 0.37 |
| rGO + Spiro-OMeTAD | 23.05; 22.54 ± 0.53 | 1.11; 1.12 ± 0.01 | 0.71; 0.71 ± 0.01 | 18.13; 17.82 ± 0.29 |
Although the observed enhancement in the efficiency was not significant, the main aim of this work was to explore the effect of rGO produced from natural sources on the PSC stability. We, therefore, tested the PV parameters of three different PSCs, namely rGO + Spiro-OMeTAD, reference (Spiro-OMeTAD only) and rGO-only, after storing devices in ambient conditions (in the dark) for 21 days (500 h). The stability of these devices after 500 h is plotted in Fig. 5 and has been summarized in Table S1.† It can be observed that the efficiency of rGO-only based cell dropped from 4.98% to 0.41%, losing more than 90% of its initial efficiency. This is due to the direct exposure of perovskite to the air caused by the incomplete coverage of rGO on the perovskite surface. On the other hand, the reference device retained 54% of the initial cell performance (PCE dropped from 17.26% to 9.31%) after 500 h storage in ambient conditions. Interestingly, our PSC employing rGO + Spiro-OMeTAD based HTM preserved 75% of its initial efficiency (PCE dropped from 18.13% to 13.68%), showing significant improvement in the device stability. We attribute this improved stability to the hydrophobic nature of the rGO which protects perovskite from the moisture. This result suggests that the use of rGO as an additive to high performance HTMs such as Spiro-OMeTAD improves the device stability significantly, while maintaining high cell efficiency.
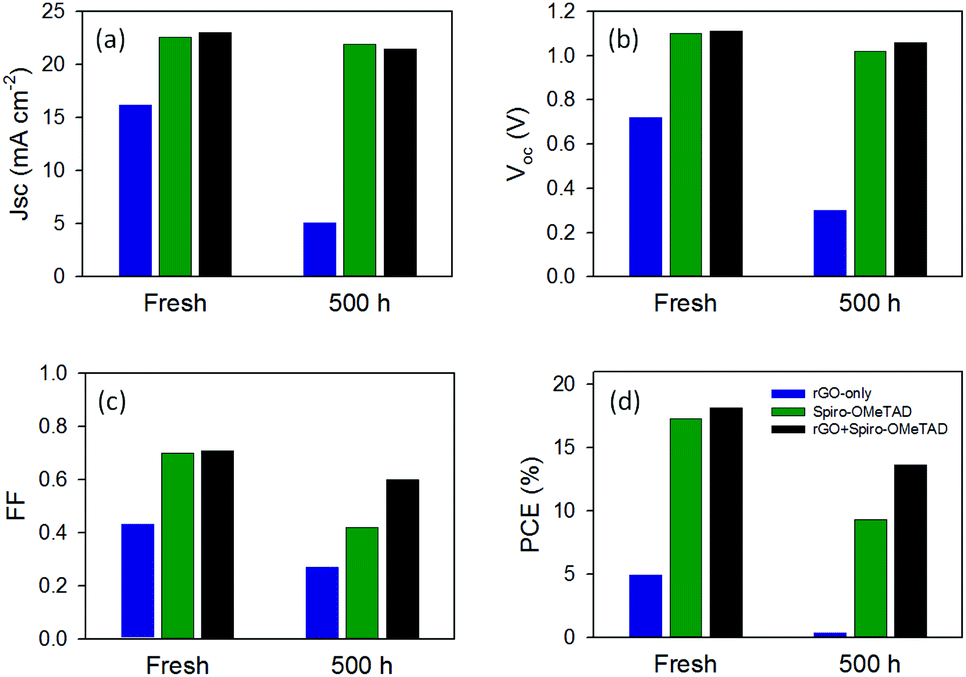 | ||
| Fig. 5 Stability of PV parameters of rGO-only, Spiro-OMeTAD and rGO + Spiro-OMeTAD based PSCs. | ||
This work demonstrates that solution processed rGO can be prepared from naturally abundant source in Mongolian mine and is an effective HTM candidate for improving PSC stability while achieving high efficiency. In addition, graphene derivatives are promising candidates for enhancing the efficiency of solar cells by incorporating them into the electron transporting layers.35 In this regard, composite materials based on our rGO and functional metal oxides53 are expected to significantly increase the PSC efficiency. Future studies should also explore the functionalization (heteroatom doping, creating heterostructure and so on) of our rGO nanosheets and their potential applications including batteries, catalysis and sensors.
Conclusions
2D graphene derivatives, rGO nanosheets, were successfully prepared from naturally abundant graphite flakes (obtained from a Mongolian mine) via chemical oxidation and thermal reduction methods. We also demonstrated the successful application of solution processed rGO as an effective HTM for PSCs. We found that the use of rGO in the HTL significantly improves the device stability, while also enhancing the PV efficiency. A PCE of 18.13% is achieved for the rGO incorporated Spiro-OMeTAD based PSC, while the reference cell with only Spiro-OMeTAD delivered an efficiency of 17.26%. The rGO helps to preserve 75% of the initial device performance after storing 500 h in ambient condition. This work provides an opportunity to address significant issues of PSCs using rGO nanosheets prepared from naturally abundant graphite sources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially supported by Fellow research grant of National University of Mongolia (P2019-3648). The support of the Australian Research Council Discovery Program (DP160101301) is gratefully acknowledged. The authors thank research groups of G. Sevjidsuren from Institute of Physics and Technology, Mongolian Academy of Sciences, Mongolia and B. Munkhjargal at the National University of Mongolia, Mongolia for their facility supports. We also greatly acknowledge the use of the Queensland node of the Australian National Fabrication Facility (ANFF-Q) and Centre for Microscopy and Microanalysis (CMM) at the University of Queensland, Australia.Notes and references
- M. Batmunkh, M. J. Biggs and J. G. Shapter, Small, 2015, 11, 2963–2989 CrossRef CAS PubMed.
- P. Peumans, S. Uchida and S. R. Forrest, Nature, 2003, 425, 158–162 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- L. Yu, A. S. R. Bati, T. S. L. Grace, M. Batmunkh and J. G. Shapter, Adv. Energy Mater., 2019, 9, 1901063 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- M. Chen and L.-L. Shao, Chem. Eng. J., 2016, 304, 629–645 CrossRef CAS.
- M. Chen, G.-C. Wang, L.-L. Shao, Z.-Y. Yuan, X. Qian, Q.-S. Jing, Z.-Y. Huang, D.-L. Xu and S.-X. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 31208–31224 CrossRef CAS PubMed.
- M. Chen, G.-C. Wang, W.-Q. Yang, Z.-Y. Yuan, X. Qian, J.-Q. Xu, Z.-Y. Huang and A.-X. Ding, ACS Appl. Mater. Interfaces, 2019, 11, 42156–42171 CrossRef CAS PubMed.
- M. Chen, G. Zhao, L.-L. Shao, Z.-Y. Yuan, Q.-S. Jing, K.-J. Huang, Z.-Y. Huang, X.-H. Zhao and G.-D. Zou, Chem. Mater., 2017, 29, 9680–9694 CrossRef CAS.
- M. Chen, L.-L. Shao, Z.-Y. Yuan, Q.-S. Jing, K.-J. Huang, Z.-Y. Huang, X.-H. Zhao and G.-D. Zou, ACS Appl. Mater. Interfaces, 2017, 9, 17949–17960 CrossRef CAS PubMed.
- H. K. H. Lee, J. Wu, J. Barbé, S. M. Jain, S. Wood, E. M. Speller, Z. Li, F. A. Castro, J. R. Durrant and W. C. Tsoi, J. Mater. Chem. A, 2018, 6, 5618–5626 RSC.
- M. Chen, L. L. Shao, Y. Xia, Z.-Y. Huang, D.-L. Xu, Z.-W. Zhang, Z.-X. Chang and W.-J. Pei, ACS Appl. Mater. Interfaces, 2016, 8, 26030–26040 CrossRef CAS PubMed.
- M. Chen, L.-L. Shao, Y.-X. Guo and X.-Q. Cao, Chem. Eng. J., 2016, 304, 303–312 CrossRef CAS.
- Z. Wu, P. Li, Y. Zhang and Z. Zheng, Small Methods, 2018, 2, 1800031 CrossRef.
- M. Batmunkh, T. J. Macdonald, C. J. Shearer, M. Bat-Erdene, Y. Wang, M. J. Biggs, I. P. Parkin, T. Nann and J. G. Shapter, Adv. Sci., 2017, 4, 1600504 CrossRef.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- S. Yang, W. Fu, Z. Zhang, H. Chen and C.-Z. Li, J. Mater. Chem. A, 2017, 5, 11462–11482 RSC.
- J. Duan, H. Xu, W. E. I. Sha, Y. Zhao, Y. Wang, X. Yang and Q. Tang, J. Mater. Chem. A, 2019, 7, 21036–21068 RSC.
- H. Wang, X. Liang, J. Wang, S. Jiao and D. Xue, Nanoscale, 2020, 12, 14–42 RSC.
- Q. Guo, F. Yuan, B. Zhang, S. Zhou, J. Zhang, Y. Bai, L. Fan, T. Hayat, A. Alsaedi and Z. a. Tan, Nanoscale, 2019, 11, 115–124 RSC.
- M. Batmunkh, K. Vimalanathan, C. Wu, A. S. R. Bati, L. Yu, S. A. Tawfik, M. J. Ford, T. J. Macdonald, C. L. Raston, S. Priya, C. T. Gibson and J. G. Shapter, Small Methods, 2019, 3, 1800521 CrossRef.
- J.-D. Chen, T.-Y. Jin, Y.-Q. Li and J.-X. Tang, Nanoscale, 2019, 11, 18517–18536 RSC.
- Z. Yu and L. Sun, Adv. Energy Mater., 2015, 5, 1500213 CrossRef.
- X. Liu, X. Zheng, Y. Wang, Z. Chen, F. Yao, Q. Zhang, G. Fang, Z.-K. Chen, W. Huang and Z.-X. Xu, ChemSusChem, 2017, 10, 2833–2838 CrossRef CAS PubMed.
- J. Urieta-Mora, I. García-Benito, A. Molina-Ontoria and N. Martín, Chem. Soc. Rev., 2018, 47, 8541–8571 RSC.
- H. Kwon, J. W. Lim, J. Han, L. N. Quan, D. Kim, E.-S. Shin, E. Kim, D.-W. Kim, Y.-Y. Noh, I. Chung and D. H. Kim, Nanoscale, 2019, 11, 19586–19594 RSC.
- S. N. Habisreutinger, T. Leijtens, G. E. Eperon, S. D. Stranks, R. J. Nicholas and H. J. Snaith, J. Phys. Chem. Lett., 2014, 5, 4207–4212 CrossRef CAS PubMed.
- S. N. Habisreutinger, T. Leijtens, G. E. Eperon, S. D. Stranks, R. J. Nicholas and H. J. Snaith, Nano Lett., 2014, 14, 5561–5568 CrossRef CAS PubMed.
- M. Cai, V. T. Tiong, T. Hreid, J. Bell and H. Wang, J. Mater. Chem. A, 2015, 3, 2784–2793 RSC.
- A. Capasso, F. Matteocci, L. Najafi, M. Prato, J. Buha, L. Cinà, V. Pellegrini, A. D. Carlo and F. Bonaccorso, Adv. Energy Mater., 2016, 6, 1600920 CrossRef.
- M. Batmunkh, C. J. Shearer, M. J. Biggs and J. G. Shapter, J. Mater. Chem. A, 2015, 3, 9020–9031 RSC.
- D. Selvakumar, G. Murugadoss, A. Alsalme, A. M. Alkathiri and R. Jayavel, Sol. Energy, 2018, 163, 564–569 CrossRef CAS.
- G.-H. Kim, H. Jang, Y. J. Yoon, J. Jeong, S. Y. Park, B. Walker, I.-Y. Jeon, Y. Jo, H. Yoon, M. Kim, J.-B. Baek, D. S. Kim and J. Y. Kim, Nano Lett., 2017, 17, 6385–6390 CrossRef CAS PubMed.
- R. Ishikawa, S. Watanabe, S. Yamazaki, T. Oya and N. Tsuboi, ACS Appl. Energy Mater., 2019, 2, 171–175 CrossRef CAS.
- M. Batmunkh, C. J. Shearer, M. J. Biggs and J. G. Shapter, J. Mater. Chem. A, 2016, 4, 2605–2616 RSC.
- M. Acik and S. B. Darling, J. Mater. Chem. A, 2016, 4, 6185–6235 RSC.
- K. Yan, Z. Wei, J. Li, H. Chen, Y. Yi, X. Zheng, X. Long, Z. Wang, J. Wang, J. Xu and S. Yang, Small, 2015, 11, 2269–2274 CrossRef CAS PubMed.
- F. Wang, M. Endo, S. Mouri, Y. Miyauchi, Y. Ohno, A. Wakamiya, Y. Murata and K. Matsuda, Nanoscale, 2016, 8, 11882–11888 RSC.
- M. Chen, R.-H. Zha, Z.-Y. Yuan, Q.-S. Jing, Z.-Y. Huang, X.-K. Yang, S.-M. Yang, X.-H. Zhao, D.-L. Xu and G.-D. Zou, Chem. Eng. J., 2017, 313, 791–800 CrossRef CAS.
- A. L. Palma, L. Cinà, S. Pescetelli, A. Agresti, M. Raggio, R. Paolesse, F. Bonaccorso and A. Di Carlo, Nano Energy, 2016, 22, 349–360 CrossRef CAS.
- Q. Luo, Y. Zhang, C. Liu, J. Li, N. Wang and H. Lin, J. Mater. Chem. A, 2015, 3, 15996–16004 RSC.
- K. T. Cho, G. Grancini, Y. Lee, D. Konios, S. Paek, E. Kymakis and M. K. Nazeeruddin, ChemSusChem, 2016, 9, 3040–3044 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- Z. Q. Li, C. J. Lu, Z. P. Xia, Y. Zhou and Z. Luo, Carbon, 2007, 45, 1686–1695 CrossRef CAS.
- S. Some, Y. Kim, E. Hwang, H. Yoo and H. Lee, Chem. Commun., 2012, 48, 7732–7734 RSC.
- J. Guo, K. Song, B. Wu, X. Zhu, B. Zhang and Y. Shi, RSC Adv., 2017, 7, 22875–22881 RSC.
- D. H. Seo, M. Batmunkh, J. Fang, A. T. Murdock, S. Yick, Z. Han, C. J. Shearer, T. J. Macdonald, M. Lawn, A. Bendavid, J. G. Shapter and K. Ostrikov, FlatChem, 2018, 8, 1–8 CrossRef CAS.
- M. Chen, L.-L. Shao, J.-J. Li, W.-J. Pei, M.-K. Chen and X.-H. Xie, RSC Adv., 2016, 6, 35228–35238 RSC.
- M. Chen, Q.-S. Jing, H.-B. Sun, J.-Q. Xu, Z.-Y. Yuan, J.-T. Ren, A.-X. Ding, Z.-Y. Huang and M.-Y. Dong, Langmuir, 2019, 35, 6321–6332 CrossRef CAS PubMed.
- S. Fang, D. Huang, R. Lv, Y. Bai, Z.-H. Huang, J. Gu and F. Kang, RSC Adv., 2017, 7, 25773–25779 RSC.
- J. Wang, Y. Liu, X. Chen, C. Chen, P. Chen, Z. Wang and Y. Duan, ChemPhysChem, 2019, 20, 2580–2586 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01423k |
| This journal is © The Royal Society of Chemistry 2020 |