
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePt/Al2O3 coated with N-doped carbon as a highly selective and stable catalyst for catalytic hydrogenation of p-chloronitrobenzene to p-chloroaniline†
Donghong Zhu,
Xin Weng,
Yuqiong Tang,
Jingya Sun *,
Shourong Zheng and
Zhaoyi Xu
*,
Shourong Zheng and
Zhaoyi Xu
State Key Laboratory of Pollution Control and Resource Reuse, School of the Environment, Nanjing University, Nanjing 210046, China. E-mail: sunsun1118@126.com; Fax: +86-25-89680596; Tel: +86-25-89680373
First published on 7th April 2020
Abstract
Selective catalytic hydrogenation of p-chloronitrobenzene on Pt-based catalysts is a green and high-efficient way for p-chloroaniline production. However, supported monometallic Pt catalysts often exhibit undesirable p-chloroaniline selectivity. We herein reported supported Pt catalysts with N-doped carbon (NC) as an overcoating (Pt/Al2O3@NC) to overcome the disadvantage. Three Pt/Al2O3@NC catalysts with different NC coating amounts were prepared by in situ carbonization of an ionic liquid. For comparison, Al2O3 coated by NC and Pt/Al2O3 coated by SiO2 were also prepared. A combination characterization confirmed that the NC overcoating was successfully formed on Pt/Al2O3 surface and Pt particles were completely coated by NC layers when ion liquid amount increased to 25 μl per g catalyst. Due to the intimate contact of NC layers and Pt particles Pt-NC heterojunctions were effectively formed on the catalyst surface. For the catalytic hydrogenation of p-chloronitrobenzene, Pt/Al2O3@NC with 25 μl ionic liquid as the NC precursor exhibited 100% selectivity to p-chloroaniline at 100% conversion of p-chloronitrobenzene. A lower ionic liquid amount led to decreased selectivity to p-chloroaniline. Furthermore, no deactivation was observed on Pt/Al2O3@NC during 5 catalytic cycles. The findings in the study demonstrate that coating noble metal catalysts by N-doped carbon is a promising method to enhance the selectivity and stability for catalytic hydrogenation of p-chloronitrobenzene.
1. Introduction
As important chemical raw materials, chloroanilines (CANs) are widely used as intermediates in pharmaceuticals, herbicides, pigments and dyes.1–3 Conventionally, CANs could be produced via reduction of corresponding chloronitrobenzenes (CNBs) using stoichiometric reductants such as potassium borohydride and hydrazine hydrate.2–4 However, the use of stoichiometric reductants would bring large amounts of harmful waste and severe pollution to the environment. Alternatively, selective catalytic hydrogenation reduction is an eco-friendly and cost-effective method for the conversion of CNBs to CANs.For catalytic hydrogenation reduction, noble metals such as Pd and Pt are highly active because of their strong abilities for H2 activation under mild conditions.5,6 However, supported noble metal catalysts exhibited low selectivity because dechlorinated byproducts were inevitably formed due to their strong hydrodechlorination capabilities.5,7 For example, supported Pt catalysts were considered as the most selective ones among supported noble metals for the catalytic conversion of CNBs to CANs,3,8,9 however, Pt/C catalyst only displayed 75.2% selectivity to p-CAN at 100% conversion of p-CNB.10 Additionally, catalyst deactivation often occurs as a result of contamination of metal surface or/and metal leaching during the reaction processes.11,12 For example, Mahata et al.11 found that Pt/CNT deactivated drastically in the catalytic hydrogenation of o-CNB during the second and subsequent reaction runs. Hence, exploring effective strategies to improve the catalytic selectivity and stability of supported noble metal catalysts for hydrogenation of CNBs to corresponding CANs is of significant importance.
Many attempts such as addition of a promoting metal or decorating metal particles with organic modifiers have been made to improve the catalytic selectivity for p-CNB hydrogenation.13–17 For example, Zhu et al.18 prepared a core–shell structural bimetallic Pt@Cu/TiO2 catalyst with monolayer Cu as the shell for catalytic hydrogenation of p-CNB and obtained 100% selectivity to p-CAN. Very recently, coating supported catalysts have been proved to be an effective approach to enhance catalytic selectivity. Chen et al.14 prepared a Pt-nanowire catalyst chelated by ethylenediamine on catalyst surface, which exhibited prominent selectivity for the catalytic production of N-hydroxylanilines from nitrobenzene. Leng et al.19 reported that controlled and chemoselective hydrogenation of nitrobenzene was achieved on a Ru@C60 catalyst. Notably, coating supported catalysts was also reported as a prominent strategy to enhance catalytic stability through protecting metal particles from leaching and poisoning. For example, Fu et al.20 illustrated that a Ni/Al2O3 catalyst coated with carbon nitride layer provided an exceptional stability for the catalytic hydrogenation of nitrobenzene. Li et al.21 developed a Pt catalyst with Pt particles embedded in N-doped carbon rods and observed a superior catalytic stability in the liquid-phase catalytic hydrogenation of bromate. Recent years, N-doped carbon (NC) materials have attracted tremendous attentions because of their unique and versatile properties. For example, N-doped carbon could transfer electrons to metal particles or vice versa via metal–NC heterojunctions and thus serve as catalytically active sites for the adsorption and activation of H2 and reactants.20–22 Inspired by recent advances, it is reasonable to speculate that coating supported noble metal catalysts with N-doped carbon may probably improve the catalytic selectivity and stability for hydrogenation of CNBs. However, few studies have been conducted for the selective hydrogenation of p-CNB on coated noble metal catalysts thus far.
The main objective of the present study is to develop highly selective and stable supported Pt catalysts for the catalytic hydrogenation of CNBs to CANs. We prepared three Pt/Al2O3 catalysts coated by NC layer with different NC amounts by in situ carbonization of ion liquid (1-ethyl-3-methylimidazolium dicyanamide, EMIm-dca). For comparison, Al2O3 coated with NC and Pt/Al2O3 coated with SiO2 were also prepared. The structural properties of the synthesized catalysts were characterized by a series of techniques and the catalytic performances of the catalysts for hydrogenation of p-CNB were examined.
2. Experimental
2.1 Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in a three-necked round flask under vigorous stirring at 80 °C for controlled hydrolysis of aluminum isopropoxide. After refluxing at 80 °C for 1 h, the mixture was further evaporated at 90 °C for 1 h to remove formed alcohol. The pH of the mixture was then adjusted to 3.0 using 10% HNO3 solution. Aluminum sol was obtained after the mixture further refluxed at 80 °C for 24 h.
:1 in a three-necked round flask under vigorous stirring at 80 °C for controlled hydrolysis of aluminum isopropoxide. After refluxing at 80 °C for 1 h, the mixture was further evaporated at 90 °C for 1 h to remove formed alcohol. The pH of the mixture was then adjusted to 3.0 using 10% HNO3 solution. Aluminum sol was obtained after the mixture further refluxed at 80 °C for 24 h.2.2 Catalyst characterization
Pt contents in the samples were precisely determined by an inductively coupled plasma-atomic emission spectrometer (ICP-AES) (J-A1100, Jarrell-Ash, USA). The XRD patterns of the samples were collected on a Rigaku D/max-RA powder diffraction-meter using a Cu Kα radiation (Rigaku, Tokyo, Japan). The carbon and nitrogen contents in the catalysts were determined using an elementary analyzer CHN–O-rapid apparatus (Heraeus, Germany). The Brunauer–Emmett–Teller (BET) surface areas of the catalysts were measured using a Micromeritics ASAP 2200 instrument (Micromeritics Instrument Co., Norcross, GA). High-angle annular dark field-scanning transmission electron microscope (HAADF-STEM) and elemental mapping images were acquired using an ARM-200F transmission electron microscope (JEOL Co., Japan.) equipped with an X-MAX energy dispersive spectrometer (Oxford, UK). X-ray photoelectron spectroscopy (XPS) spectra were recorded on a PHI5000 VersaProbe with a monochromatized Al Kα X-ray source (hν = 1486.6 eV) (ULVAC-PHI, Japan). The binding energy was calibrated by measuring the C 1s peak at 284.6 eV.Temperature programmed oxidation (TPO) was conducted on an AMI-300 setup (Altamira Instruments, USA) with a gas analysis system (Thermo Star, Germany). Typically, 50 mg of catalyst was loaded in a U-shaped quartz tube, which was pretreated at 120 °C for 1 h using a 25 ml min−1 of He flow in a heating furnace. After cooling down, the catalyst was in situ oxidized with a 5% O2/He flow (25 ml min−1) from ambient temperature to 600 °C at a rate of 5 °C min−1. The evolved gases were monitored with intensities of m/z 17 (NH3), 28 (CO), 30 (NO), 44 (CO2) and 46 (NO2), respectively.
2.3 Catalytic hydrogenation of p-chloronitrobenzene
The catalytic hydrogenation of p-CNB over the catalysts was conducted in a high pressure stainless steel autoclave with a controlling system. Briefly, 20 mg catalyst, 100 mg p-CNB and 20 ml ethanol was added to the autoclave. Before hydrogenation reaction, the autoclave was flushed with N2 for 5 times to replace air in the reactor under ambient condition. The reaction was conducted under 1 MPa pressure of H2 at 80 °C with stirring rate fixing at 1000 rpm. Samples were taken periodically and detected by a 6980 gas chromatograph equipped with a FID detector and a DB-5 capillary column (Agilent Technologies, US), and n-hexane was used as the inner standard substance. For catalyst reuse, the used catalyst was prepared under the same reaction conditions but without sampling. After reaction for 180 min, the catalyst was collected by filtration, rinsing with ethanol and distilled water and drying at 60 °C for 12 h. The catalyst was then subjected to the subsequent reaction test.The conversion of p-CNB and selectivity to p-CAN and aniline were calculated following equations below:
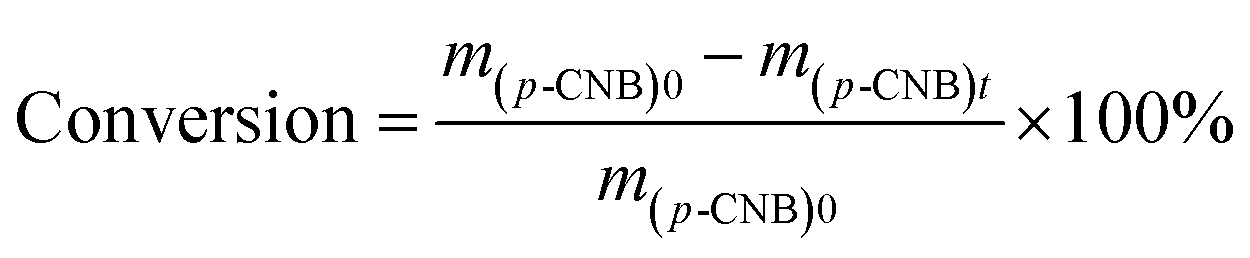 | (1) |
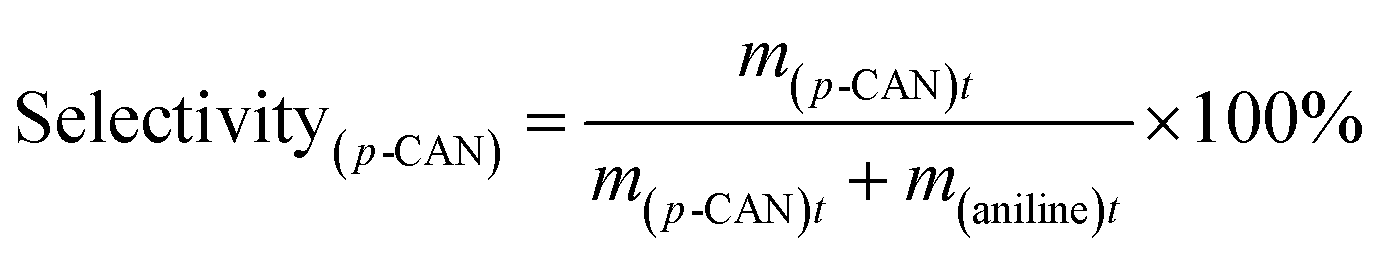 | (2) |
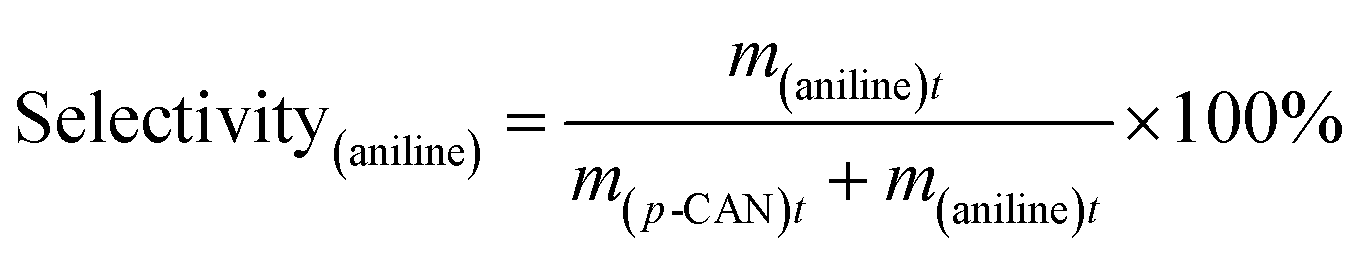 | (3) |
3. Results and discussion
3.1 Catalyst characterization
Table 1 shows Pt content of the catalysts determined by ICP-AES. For Pt/Al2O3, Pt content was determined to be 0.96 wt%. As for Pt/Al2O3@NC, Pt content were in the range of 0.71–0.78 wt%, which were lower than that of Pt/Al2O3 due to successful loading of NC overcoating.| Catalysts | Pt contenta (wt%) | C contentb (wt%) | N contentb (wt%) | Surface concentration of Nc (at%) | N1/N2c | BET surface aread (m2 g−1) | d1e (nm) | CO adsorption amountf (μmol g−1) | Coverage of Pt surfacef (%) | d2f (nm) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by ICP.b Determined by elemental analysis.c Determined by XPS, N1/N2 refers to the ratio of pyridinic N amount to pyrrole N amount on catalyst surface.d Calculated from BET.e Calculated from HAADF-STEM.f Calculated by CO chemisorption.g Below determine limit. | ||||||||||
| Pt/Al2O3 | 0.96 | BDL | BDL | BDL | BDL | 103 | 4.0 | 12.0 | — | 4.3 |
| Pt/Al2O3@NC-10 | 0.78 | 0.33 | 0.17 | 0.49 | BDL | 100 | 3.9 | 3.7 | 69% | — |
| Pt/Al2O3@NC-15 | 0.71 | 0.63 | 0.30 | 0.65 | 0.92 | 101 | 4.0 | 0.5 | 96% | — |
| Pt/Al2O3@NC-25 | 0.78 | 0.76 | 0.39 | 0.79 | 0.71 | 99 | 4.1 | 0 | 100% | — |
| Al2O3@NC-25 | BDLg | 0.58 | 0.24 | 0.61 | 0.47 | 110 | — | BDL | — | — |
The compositions of C and N elements in the catalysts were determined by elemental analysis and the results are summarized in Table 1. For Pt/Al2O3@NC-10, Pt/Al2O3@NC-15 and Pt/Al2O3@NC-25, the C contents were 0.33%, 0.63% and 0.76% respectively, exhibiting increased C content with the increase of ion liquid loading amount from 10 μl to 25 μl. Consistently, N contents in Pt/Al2O3@NC catalysts increased from 0.17% to 0.39% with ion liquid loading. For Al2O3@NC-25, the C and N content were determined to be 0.58% and 0.24% respectively, lower than those of Pt/Al2O3@NC-25, probably because the presence of Pt particles on Al2O3 surface stabilized the NC matrix formed by pyrolysis of ion liquid via evoking interactions with NC matrix. The molar N/C ratios in the catalysts were in the range of 0.35–0.44 which was lower than 0.63 of ion liquid, this can be ascribed to more prominent loss of unstable N species upon carbonization at high temperatures.24,25
Fig. 1 shows the XRD patterns of the catalysts in the range of 20–80°. Diffraction peaks centered at 37.3°, 39.5°, 45.7°, and 67.1° indexed to the γ-Al2O3 phase, were clearly observed on all the catalysts. No diffraction peaks characteristic of Pt were found in the XRD patterns probably due to low metal contents of the catalysts.26 Notably, in the range of 15–25°, a weak and broad peak centered at 19.5° were observed on Pt/Al2O3@NC catalysts. The very weak peak was ascribed to amorphous N-doped carbon, which was lower than that of amorphous carbon (23.5°) due to the increase of inter-layer distance of carbon upon N doping.27 As for the XRD pattern of Pt/Al2O3@SiO2, the broad peak centered at 23.1° was assigned to amorphous silica. In comparison with Pt/Al2O3@NC, Pt/Al2O3@SiO2 had markedly weakened intensities of peaks of γ-Al2O3 phase, likely resulting from the thicker SiO2 overcoating than NC overcoating.
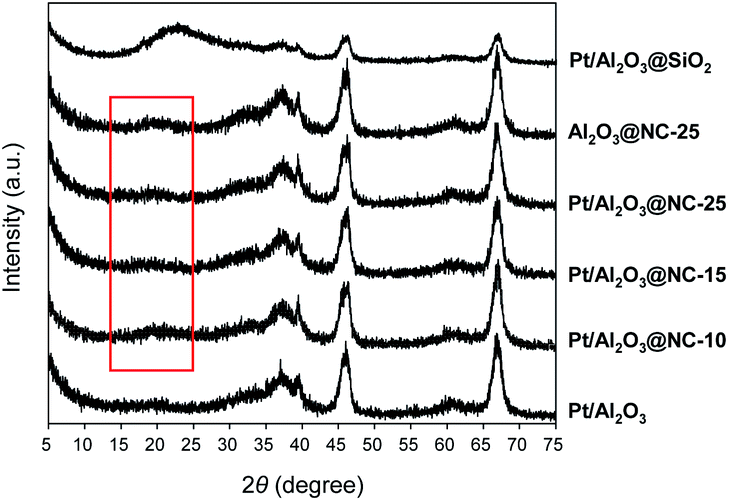 | ||
| Fig. 1 XRD patterns of the catalysts. | ||
Fig. S1, ESI† shows the HAADF-STEM images of the catalysts. Al2O3 had uniform rhombus morphology with diameter in the range of 30–60 nm. The TEM images showed that Pt particles were spherical and were well dispersed on Al2O3 surface. The average Pt particle diameter was measured based on the surface area weighted diameter:28,29
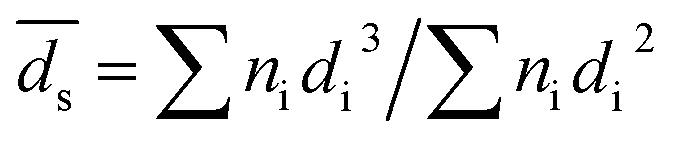 | (4) |
Results are listed in Table 1. The average sizes of Pt particles of Pt/Al2O3, Pt/Al2O3@NC-10, Pt/Al2O3@NC-15 and Pt/Al2O3@NC-25 were 4.0, 3.9, 4.0 and 4.1 nm, respectively, indicating that NC coating did not affect the structure of Pt/Al2O3. However, NC overcoating layer was not clearly visible in TEM images, probably because of the very low NC contents (as indicated by elemental analysis results). To verify the presence of NC overcoating on the catalysts, EDS mapping analysis was conducted on Pt/Al2O3@NC-25 and the images are presented in Fig. 2. In Fig. 2a light particles with diameter around 5–7 nm could be clearly observed as marked by red arrows. Consistently, Pt signal was very dense in the same area in the mapping image in Fig. 2c, reflecting that the lightness variation was due to the presence of Pt particles on Al2O3 support. The EDS mapping of elemental C and N in Pt/Al2O3@NC-25 (see Fig. 2d and e) showed that elemental C and N were evenly distributed on Al2O3 surface. The clear spatial correlation among elemental Al, C and N was indicative of a close contact of NC with Al2O3 support, verifying the presence of NC overcoating on Al2O3 surface.
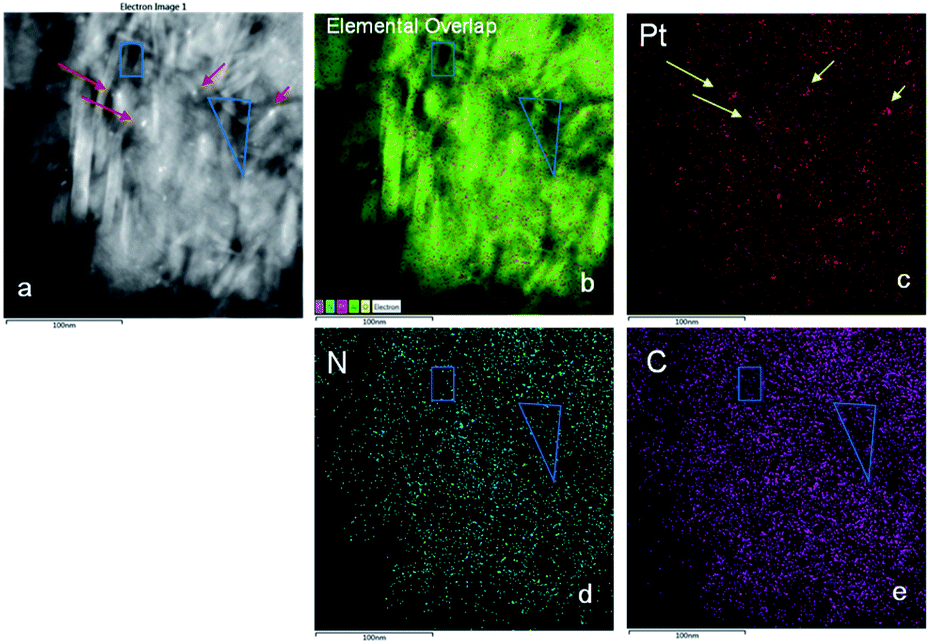 | ||
| Fig. 2 (a) HAADF-STEM image of Pt/Al2O3@NC-25 and the corresponding EDS elemental mapping of (b) the overlapping of Pt, C, and N element, (c) Pt element (red), (d) N element (blue), and (e) C element (purple). | ||
To further investigate the composition of Pt/Al2O3@NC-25, temperature-programed oxidation of Pt/Al2O3@NC-25 was conducted, and the dependence of gas release on oxidation temperature are shown in Fig. 3. In Fig. 3, CO and CO2 exhibited very similar evolving profiles, in which a strong and broad peak with two shoulders presented in the range of 200–550 °C. In parallel, marked release of NH3 and NO was also observed in the range of 180–530 °C. The presence of CO2, CO, NH3 and NO2 could be ascribed to the oxidation of C and N containing species, confirming the presence of NC overcoating on the catalyst surface.30 Notably, the oxidation temperature of N species was slightly lower than that of C species, reflecting that N species were more prone to lose than C species upon high temperature, in line with elemental analysis results.
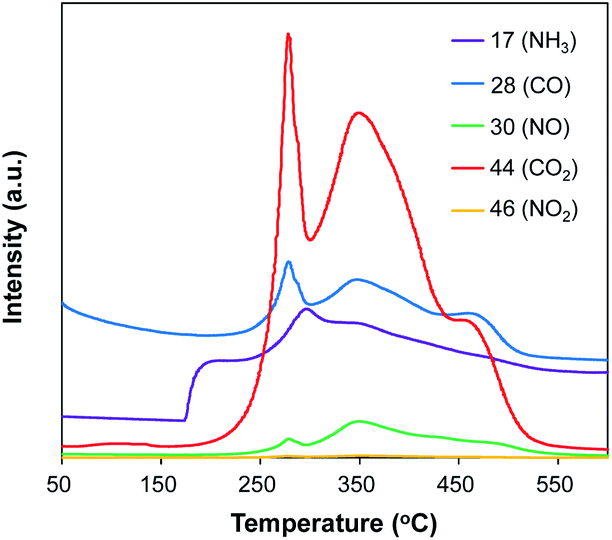 | ||
| Fig. 3 Temperature programmed oxidation profile of Pt/Al2O3@NC-25. | ||
The effective decoration of NC overcoating on Pt particle could be further verified by CO chemisorption (see Table 1). The CO adsorption amount on Pt/Al2O3 was determined to be 12.0 μmol g−1. Accordingly, the average diameter of Pt particles of Pt/Al2O3 could be calculated by CO chemisorption amount with a chemisorption stoichiometry of CO/Pt = 1. The diameter was calculated to be 4.3 nm, in good agreement with that measured by TEM. After NC coating, the CO adsorption amount markedly decreased. For example, CO adsorption amount of Pt/Al2O3@NC-10 and Pt/Al2O3@NC-15 was 3.7 μmol g−1 and 0.5 μmol g−1 respectively, much lower than that of Pt/Al2O3. Because CO could chemically adsorb only on exposed Pt sites of the catalyst, the decreased chemisorption amount of CO clearly reflected lowered exposure of Pt sites of the catalysts as a result of NC overcoating.31 Further increasing NC coating amount even led to the absence of CO adsorption on Pt/Al2O3@NC-25, indicating that Pt sites were completely blocked by NC overcoating in the catalyst. For Pt/Al2O3@NC-25 calcined at 500 °C in air, the CO chemisorption amount increased to 7.0 μmol g−1 (see Table 3). Because calcination led to the oxidation of NC matrix of the catalyst and thus resulted in the exposure of blocked Pt sites for CO chemisorption. Notably, the CO chemisorption amount of Pt/Al2O3@NC-25-calcined was lower than that of Pt/Al2O3, probably due to incomplete oxidation of NC overcoating upon calcination.
Fig. 4 shows the XPS spectra of the catalysts. Because Pt 4f region was overlapped by the strong Al 3p peak, the surface Pt compositions of the catalysts were analyzed by XPS spectra in Pt 4d region. In Fig. 4a, two peaks around 315.2 and 332.7 eV were found for Pt/Al2O3, characteristic of Pt 4d5/2 and 4d3/2, respectively (see Fig. 4a).32,33 Upon NC coating, slightly lowered binding energies of Pt 4d were observed. For example, the binding energies of Pt 4d5/2 was 315.0, 314.8 and 314.7 eV for Pt/Al2O3@NC-10, Pt/Al2O3@NC-15 and Pt/Al2O3@NC-25 respectively. The gradual red shift of the binding energy of Pt 4d5/2 with NC coating amount in the catalysts may be ascribed to the electron transfer from electronic-enriched N sites to Pt due to their strong interactions.34,35 Fig. 4b shows XPS spectra of the catalysts in the N 1s region. Deconvolution of the N 1s peak gave rise to two peaks located at 398.6 and 400.2 eV, characteristic of pyridinic N and pyrrole N respectively, further confirming the successful formation of NC overcoating layers. The ratio of pyridinic N to pyrrole N of the catalysts were calculated and the results are summarized in Table 1. Notably, the ratio of pyridinic N (N1) to pyrrole N (N2) markedly differed with samples. For Al2O3@NC-25, the ratio of N1/N2 was 0.47, which was much lower than that of Pt/Al2O3@NC-25 (0.71). In comparison with Pt/Al2O3@NC-25, Pt/Al2O3@NC-15 with a lower NC content has a higher N1/N2 ratio of 0.92. Notably, binding energy in N 1s region characteristic of covalently bound N–Pt species was found to be 398.6 eV, which was identical to the binding energy of pyridinic N.36 Hence, the much higher N1/N2 ratio in Pt/Al2O3@NC-25 than that of Al2O3@NC-25 could be attributed to the formation of additional N–Pt species when ion liquid was pyrolyzed on catalyst surface. The results clearly confirmed strong interactions between Pt and N atoms in NC matrix as also indicated by elemental analysis results. Consistently, the higher N1/N2 ratio of Pt/Al2O3@NC-15 can be explained in terms of the formation of more Pt–N species as a result of the higher Pt:N ratio in Pt/Al2O3@NC-15 than that of Pt/Al2O3@NC-25 assuming identical Pt sites in Pt/Al2O3@NC-15 to those in Pt/Al2O3@NC-25.
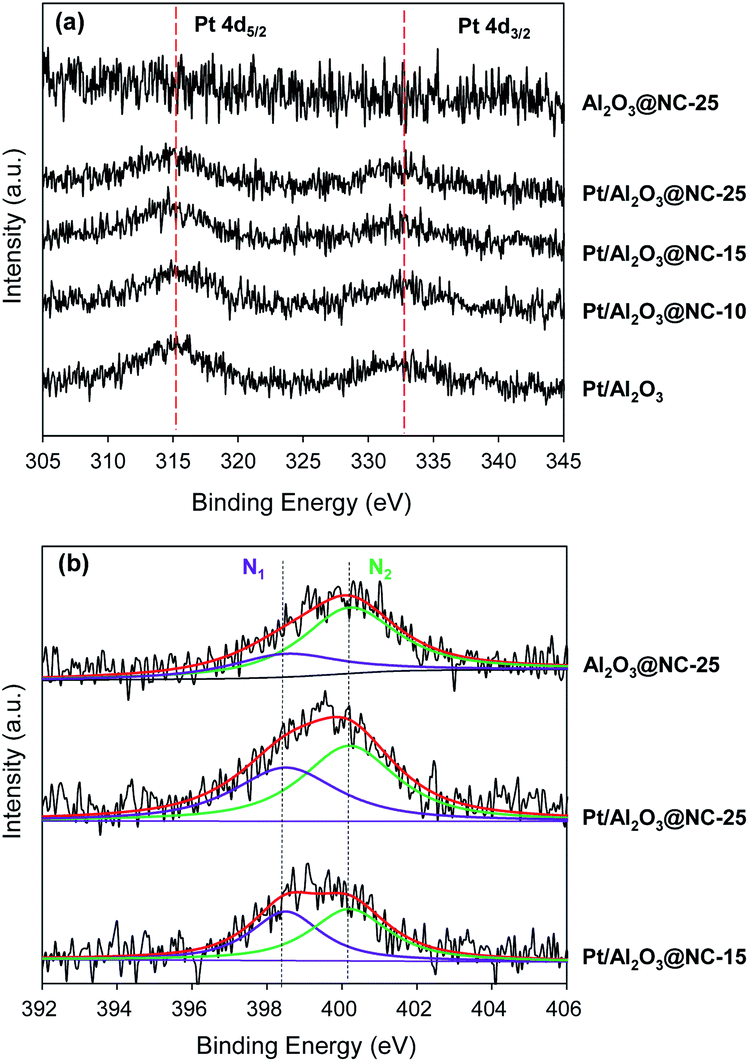 | ||
| Fig. 4 XPS spectra of catalysts in the regions of (a) Pt 4d and (b) N 1s. | ||
3.2 Catalytic hydrogenation of p-CNB
The catalytic performances of the catalysts for hydrogenation of p-CNB are presented in Fig. 5 and the reaction constants are compared in Table 2. Aniline and p-CAN were identified as products in all the reactions. Control experiment showed that Al2O3@NC-25 exhibited very low activity for catalytic hydrogenation of p-CNB with p-CNB conversion of 0.6% within 180 min of reaction time, reflecting the very weak capability of NC overcoating and Al2O3 for H2 activation under reaction conditions. In contrast, Pt/Al2O3 exhibited a much higher catalytic activity with 100% of p-CNB conversion within 30 min indicating the high activity of Pt particles for H2 activation and p-CNB reduction. Upon NC coating, the conversion of p-CNB over Pt/Al2O3@NC gradually decreased with NC coating amount. For example, p-CNB was completely converted at 50, 90, and 150 min on Pt/Al2O3@NC-10, Pt/Al2O3@NC-15 and Pt/Al2O3@NC-25 respectively (see Table 2). The deteriorated catalytic activity of Pt/Al2O3@NC was likely due to blocking Pt sites by NC overcoating from p-CNB as evidenced by CO chemisorption results. Accordingly, decreased catalytic activity as a result of coating catalyst surface had been reported previously.37,38 Notably, effective hydrogenation of p-CNB was found on Pt/Al2O3@NC-25 although Pt sites were completely blocked by NC matrix (see CO chemisorption results). This results suggested that H2 was capable of being activated and p-CNB could be reduced on NC matrix on catalyst surface. Considering the negligible catalytic activity of Al2O3@NC-25, a synergistic effect between Pt and NC overcoating could be deduced. Previous reports proved that supported metal catalysts with carbonaceous materials as the overcoating could catalyze hydrogenation reactions.20,21,37 It was proposed that intimate contact between metal and carbon matrix would cause a metal–carbon matrix heterojunction, through which H2 could be adsorbed and activated and electrons could be transferred to reactants. Accordingly, the mechanism well explained the hydrogenation of p-CNB on Pt/Al2O3@NC-25. XPS results confirmed strong interactions between Pt particles and NC matrix, forming Pt-NC heterojunction. Furthermore, the catalytic performance of the catalyst with non-conductive overcoating also confirmed this mechanism. For example, Pt/Al2O3 coated by SiO2 exhibited negligible catalytic activity for hydrogenation of p-CNB (1.1% p-CNB conversion in 180 min reaction time) (see Table 2). This was because that SiO2 is an inert insulator, on which electron transfer was substantially suppressed, thus resulting in a negligible catalytic activity than that of Pt/Al2O3@NC-25. Similarly, Li et al.12 observed a much higher catalytic activity of Pd/CNT with N-doped carbon as overcoating than that of SiO2 coated Pd/CNT for catalytic reduction of Cr (VI) in liquid phase.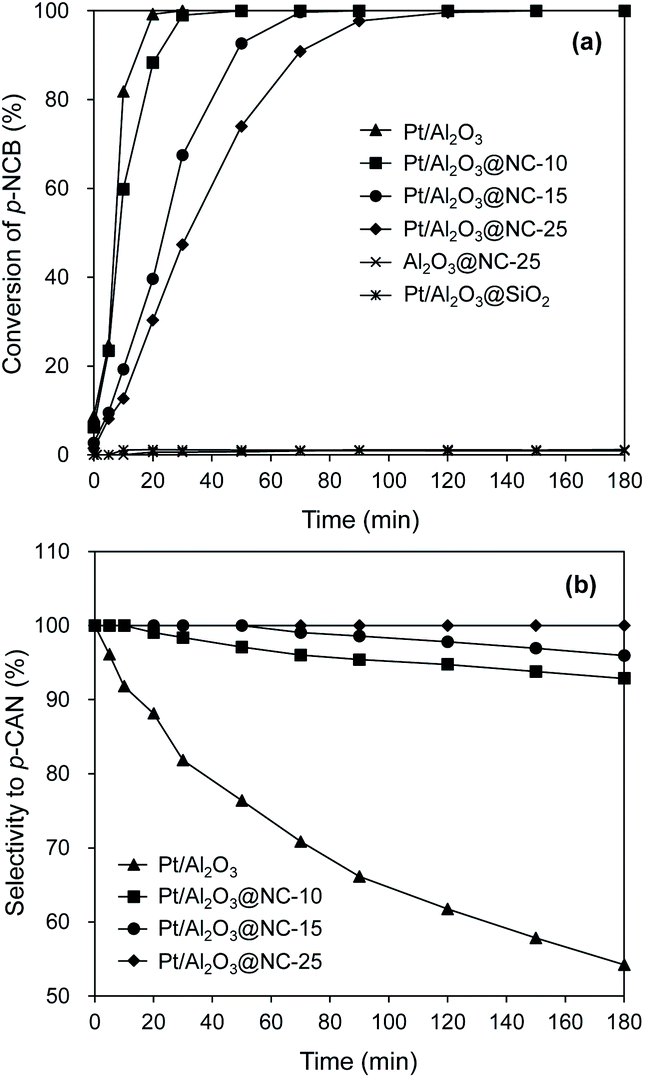 | ||
| Fig. 5 Catalytic hydrogenation of p-CNB on the catalysts. Reaction conditions: catalyst dosage: 20 mg, alcohol: 20 ml, p-NCB amount: 100 mg, reaction temperature: 80 °C, H2 pressure: 1 MPa. | ||
| Catalysts | Reaction time (min) | Conversion (%) | p-CAN selectivity (%) | Aniline selectivity (%) |
|---|---|---|---|---|
| a Reaction conditions: catalyst dosage: 20 mg, alcohol: 20 ml, p-nitrochlorobenzene: 100 mg, reaction temperature: 80 °C, reaction time: 60 min; H2 pressure: 1 MPa. | ||||
| Pt/Al2O3 | 30 | 100.0 | 81.8 | 18.2 |
| Pt/Al2O3@NC-10 | 50 | 100.0 | 97.1 | 2.9 |
| Pt/Al2O3@NC-15 | 90 | 100.0 | 98.6 | 1.4 |
| Pt/Al2O3@NC-25 | 150 | 100.0 | 100.0 | 0.0 |
| Al2O3@NC-25 | 180 | 0.6 | 100.0 | 0.0 |
| Pt/Al2O3@SiO2 | 180 | 1.1 | 100.0 | 0.0 |
Besides catalytic activity coating Pt/Al2O3 substantially impacted the selectivity in catalytic hydrogenation of the p-CNB. For example, Pt/Al2O3 displayed a catalytic selectivity of 81.8% to p-CAN at 100% of p-CNB conversion, suggesting that Pt sites were active for both reduction of NO2 group and activation of C–Cl bond in p-CNB. In contrast, Pt/Al2O3@NC exhibited significantly improved p-CAN selectivity, and the selectivity to p-CAN was strongly dependent on NC overcoating amount. Specifically, increasing ion liquid amount to 25 μl per g catalyst resulted in p-CAN selectivity increasing monotonically from 81.8% to 100% at 100% conversion of p-CNB, indicating effective inhibition of hydrodechlorination owing to NC coated on the surface of Pt particles. Notably, because Cl group in p-CNB would increase the electron density of NO2 group, the reduction of NO2 group in p-CNB was favored by the presence of Cl group.3,39 Accordingly, p-CAN is the main production in the hydrogenation of p-CNB. After p-CNB was 100% converted, however, the catalytic selectivity to p-CAN markedly decreased on all catalysts except Pt/Al2O3@NC-25. For example, the selectivity to p-CAN on Pt/Al2O3, Pt/Al2O3@NC-10 and Pt/Al2O3@NC-15 decreased to 54.2%, 92.8% and 95.9% after 180 min of reaction time, respectively. In contrast, Pt/Al2O3@NC-25 maintained 100% selectivity to p-CAN throughout the catalytic hydrogenation process even when only p-CAN left in the reaction system, clearly indicating that the catalytic dechlorination of p-CAN was completely inhibited on Pt/Al2O3@NC-25. In parallel, CO chemisorption showed that the coverage of Pt surface were 69%, 96% and 100% for Pt/Al2O3@NC-10, Pt/Al2O3@NC-15 and Pt/Al2O3@NC-25 respectively (see Table 1), confirming that C–Cl bond could only be cleaved on exposed Pt sites and NC overcoating was incapable of catalytic hydrodechlorination. Accordingly, the catalytic hydrogenation of p-CNB on Pt/Al2O3 and Pt/Al2O3@NC-25 were depicted in Scheme 1.
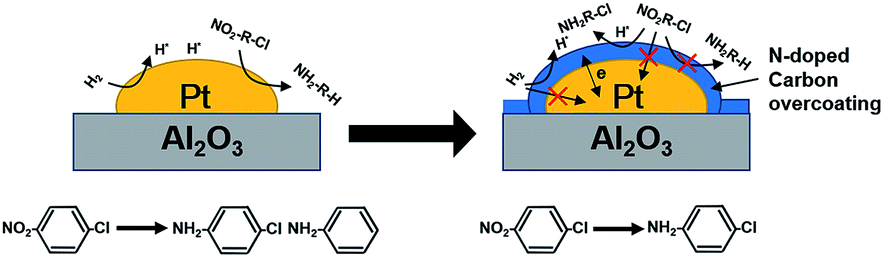 | ||
| Scheme 1 Schematic illustration of catalytic hydrogenation of p-CNB on Pt/Al2O3 and Pt/Al2O3@NC-25. | ||
To further explore the effect of NC overcoating on the catalytic performances of the coated catalysts. Pt/Al2O3@NC-25 was calcined at 500 °C in air for 2 h to remove NC coating and the catalytic hydrogenation of p-CNB was conducted on the calcined catalyst. The results are presented in Fig. 6. In comparison with Pt/Al2O3@NC-25, Pt/Al2O3@NC-25-calcined exhibited a significantly enhanced catalytic activity, which was very close to that of Pt/Al2O3. The increased catalytic activity of Pt/Al2O3@NC-25-calcined can be explained by the removal of NC overcoating on catalyst surface via calcination as reflected by TPO and CO chemisorption results. However, the catalytic selectivity of Pt/Al2O3@NC-25-calcined was still much higher than that of Pt/Al2O3, with a slightly decreased selectivity of 97.9% after 180 min reaction time in comparison with Pt/Al2O3@NC-25. CO chemisorption suggested that partial NC matrix remained on Pt/Al2O3@NC-25-calcined surface. Hence, the high selectivity of Pt/Al2O3@NC-25-calcined was probably related with NC matrix left on catalyst surface. On one hand, the remaining NC matrix would block partial Pt sites from catalytic hydrodechlorination. On the other hand, the interactions between Pt sites and residual NC matrix impacted the catalytic selectivity of Pt/Al2O3@NC-25-calcined. XPS results suggested that strong interactions and electron transfer existed between NC overcoating and Pt, causing negatively charged Pt sites. Because NH2 groups in p-CAN tended to donate electron to Cl groups, which would give rise to electrostatic repulsion between Cl group and negatively charged Pt sites and thus suppressed the cleavage of C–Cl bond.10,40,41 Similarly, Shi et al.35 reported that Pt supported on N-doped carbon nanotube exhibited high selectivities to haloanilines for catalytic hydrogenation of halonitrobenzenes because of the electron-rich chemical state of Pt particles.
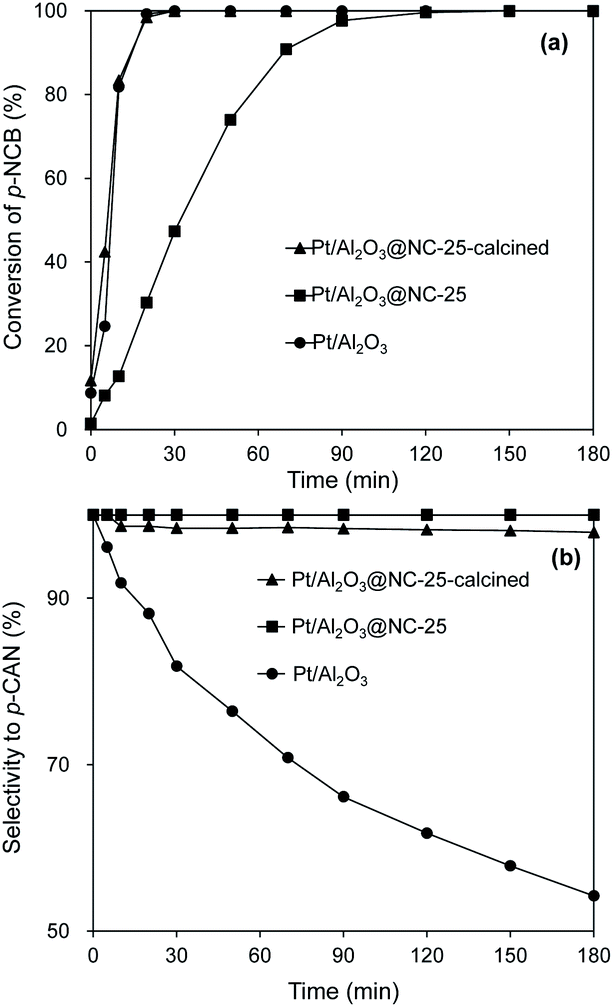 | ||
| Fig. 6 Catalytic hydrogenation of p-CNB on Pt/Al2O3@NC-25-calcined in comparison with Pt/Al2O3@NC-25 and Pt/Al2O3. Reaction conditions: catalyst dosage: 20 mg, alcohol: 20 ml, p-NCB amount: 100 mg, reaction temperature: 80 °C, H2 pressure: 1 MPa. | ||
The stability of catalyst is of important significance for supported noble metal catalysts. For stability test, the consecutive catalytic hydrogenation of p-CNB was examined on Pt/Al2O3@NC-25 and results are shown in Fig. 7. It was very interesting to find that in the initial two cycles markedly increased activity of Pt/Al2O3@NC-25 from 47.3% to 100% at 30 min reaction time was observed, likely due to the presence of exposed Pt sites as a result of partial elimination of NC overcoating via washing catalyst surface by reaction solution. Accordingly, previous studies indicated that surface coke of used catalysts could be partially removed via washing by organic solvents.42 Consistently, characterization results showed that CO chemisorption amount of Pt/Al2O3@NC-25 was 4.3 μmol g−1 after 5 catalytic cycles (see Table 3), reflecting a portion of Pt sites were again exposed. After 5 catalytic cycles, the catalytic activities remained constant and no catalyst deactivation was observed indicative of a very high catalytic stability of Pt/Al2O3@NC-25. HAADF-STEM and ICP results showed that the particle size of Pt and Pt content of Pt/Al2O3@NC-25-cycled were very similar to those of the fresh Pt/Al2O3@NC-25, further confirming the high stability of Pt/Al2O3@NC-25 (see Table 3). Similarly, enhanced stability of supported metal catalyst via carbon coating was also reported previously.43 Notably, although enhanced catalytic activity of Pt/Al2O3@NC-25 was observed and partial Pt sites were exposed on the catalyst surface after 2 reaction cycles, the catalytic selectivities to p-CAN were not accordingly decreased and remained 100% after 180 min reaction time in 5 reaction cycles, confirming that the electronic effect between Pt and NC overcoating played an important role in the high selectivity of the used Pt/Al2O3@NC-25.
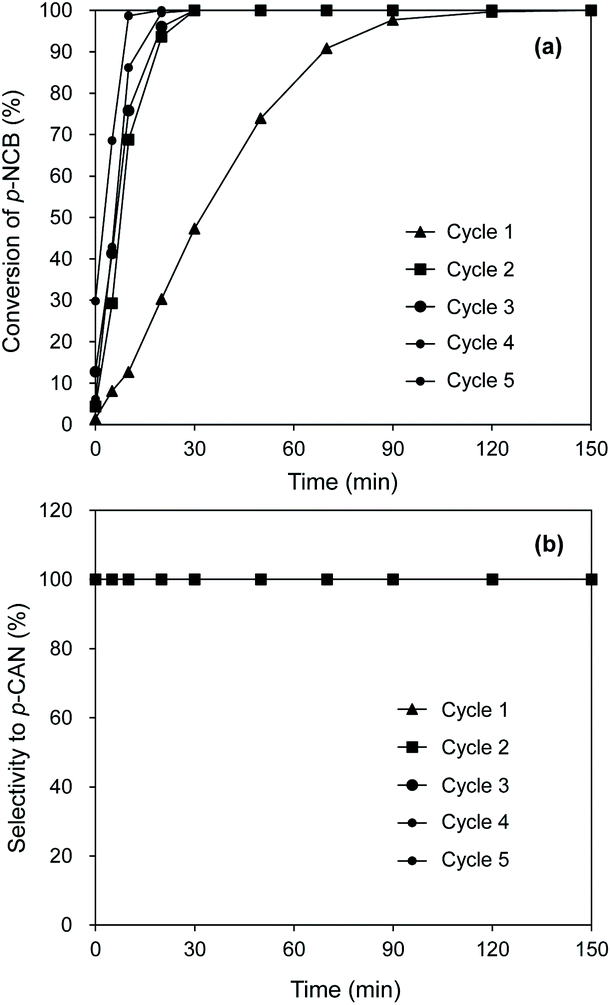 | ||
| Fig. 7 The reuse of Pt/Al2O3@NC-25 for catalytic hydrogenation of p-CNB. Reaction conditions: catalyst dosage: 20 mg, alcohol: 20 ml, p-NCB amount: 100 mg, reaction temperature: 80 °C, H2 pressure: 1 MPa. | ||
4. Conclusions
In the present work, we prepared a series of Pt/Al2O3@NC catalysts with different ion liquid amount and investigated selective catalytic hydrogenation of p-CNB on the catalysts. Characterization results showed that NC matrix was formed as overcoating on Pt/Al2O3 surface, which blocked exposed Pt sites effectively. Furthermore, strong interactions existed between Pt sites and N atoms in NC matrix and their interactions led to electron-enrichment of Pt atoms. For the selective catalytic hydrogenation of p-CNB to p-CAN, Pt/Al2O3@NC-25 exhibited marked hydrogenation activity and 100% selectivity to p-CAN, owing to the electroconductivity of NC overcoating and the complete blockage of surface Pt sites from catalytic dechlorination. The cleavage of C–Cl bond was also suppressed on Pt/Al2O3@NC-25 after calcination treatment due the electrostatic repulsion between Cl groups and negatively charged Pt sites resulting from Pt–N interactions. Consecutive cycle test showed that Pt/Al2O3@NC-25 displayed a good catalytic stability. Findings in this work reveal that surface coating of supported noble metal by N-doped carbon is a promising strategy for fabricating highly selective and stable catalysts for catalytic hydrogenation reduction of p-CNB.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the National Natural Science Foundation of China (no. 21976086) is gratefully acknowledged.Notes and references
- A. M. Tafesh and J. Weiguny, Chem. Rev., 1996, 96, 2035–2052 CrossRef CAS PubMed
.
- X. D. Wang, M. H. Liang, J. L. Zhang and Y. Wang, Curr. Org. Chem., 2007, 11, 299–314 CrossRef CAS
- M. Pietrowski, Curr. Org. Synth., 2012, 9, 470–487 CrossRef CAS
- A. Burawoy and J. P. Critchley, Tetrahedron, 1959, 5, 340–351 CrossRef CAS
- M. H. Liu, W. Y. Yu and H. F. Liu, J. Mol. Catal. A: Chem., 1999, 138, 295–303 CrossRef CAS
- A. B. Dongil, L. Pastor-Perez, J. L. G. Fierro, N. Escalona and A. Sepulveda-Escribano, Appl. Catal., A, 2016, 513, 89–97 CrossRef CAS
- Q. Wei, Y. S. Shi, K. Q. Sun and B. Q. Xu, Chem. Commun., 2016, 52, 3026–3029 RSC
- B. Coq and F. Figueras, Coord. Chem. Rev., 1998, 178, 1753–1783 CrossRef
- H. Chen, D. P. He, Q. Q. He, P. Jiang, G. B. Zhou and W. S. Fu, RSC Adv., 2017, 7, 29143–29148 RSC
- X. Li, Y. Wang, L. Q. Li, W. Q. Huang, Z. C. Xiao, P. F. Wu, W. B. Zhao, W. Guo, P. Jiang and M. H. Liang, J. Mater. Chem. A, 2017, 5, 11294–11300 RSC
- N. Mahata, O. S. G. P. Soares, I. Rodriguez-Ramos, M. F. R. Pereira, J. J. M. Orfao and J. L. Figueiredo, Appl. Catal., A, 2013, 464, 28–34 CrossRef
- M. H. Li, J. He, Y. Q. Tang, J. Y. Sun, H. Y. Fu, Y. Q. Wan, X. L. Qu, Z. Y. Xu and S. R. Zheng, Chemosphere, 2019, 217, 742–753 CrossRef CAS PubMed
- D. P. He, X. D. Jiao, P. Jiang, J. Wang and B. Q. Xu, Green Chem., 2012, 14, 111–116 RSC
- G. X. Chen, C. F. Xu, X. Q. Huang, J. Y. Ye, L. Gu, G. Li, Z. C. Tang, B. H. Wu, H. Y. Yang, Z. P. Zhao, Z. Y. Zhou, G. Fu and N. F. Zheng, Nat. Mater., 2016, 15, 564–569 CrossRef CAS PubMed
- A. Jalal and A. Uzun, J. Catal., 2017, 350, 86–96 CrossRef CAS
- C. S. Lu, H. K. Ji, Q. W. Zhu, X. J. Zhang, H. Wang, Y. B. Zhou, Q. Q. Liu, J. J. Nie, J. T. Ying and X. N. Li, J. Mater. Sci., 2019, 54, 10153–10167 CrossRef CAS
- H. Liu, K. Tao, C. Xiong and S. Zhou, Catal. Sci. Technol., 2015, 5, 405–414 RSC
- D. Zhu, L. Long, J. Sun, H. Wan and S. Zheng, Appl. Surf. Sci., 2020, 504, 144329 CrossRef
- F. Q. Leng, I. C. Gerber, P. Lecante, S. Moldovan, M. Girleanu, M. R. Axet and P. Serp, ACS Catal., 2016, 6, 6018–6024 CrossRef CAS
- T. Fu, M. Wang, W. M. Cai, Y. M. Cui, F. Gao, L. M. Peng, W. Chen and W.
P. Ding, ACS Catal., 2014, 4, 2536–2543 CrossRef CAS
- M. H. Li, Y. Hu, H. Y. Fu, X. L. Qu, Z. Y. Xu and S. R. Zheng, Chem. Commun., 2019, 55, 11786–11789 RSC
- T. Wang, Z. Dong, T. Fu, Y. C. Zhao, T. Wang, Y. Z. Wang, Y. Chen, B. H. Han and W. P. Ding, Chem. Commun., 2015, 51, 17712–17715 RSC
- Y. Y. Chu, J. Cao, Z. Dai and X. Y. Tan, J. Mater. Chem. A, 2014, 2, 4038–4044 RSC
- R. Arrigo, M. E. Schuster, Z. Xie, Y. Yi, G. Wowsnick, L. L. Sun, K. E. Hermann, M. Friedrich, P. Kast, M. Hävecker, A. Knop-Gericke and R. Schlögl, ACS Catal., 2015, 5, 2740–2753 CrossRef CAS
- R. F. Nie, H. Z. Jiang, X. H. Lu, D. Zhou and Q. H. Xia, Catal. Sci. Technol., 2016, 6, 1913–1920 RSC
- Y. B. Li, C. B. Zhang, J. Z. Ma, M. Chen, H. Deng and H. He, Appl. Catal., B, 2017, 217, 560–569 CrossRef CAS
- H. L. Peng, Z. Y. Mo, S. J. Liao, H. G. Liang, L. J. Yang, F. Luo, H. Y. Song, Y. L. Zhong and B. Q. Zhang, Sci. Rep., 2013, 3, 1765 CrossRef
- T. Paryjczak and J. A. Szymura, Z. Anorg. Allg. Chem., 1979, 449, 105–114 CrossRef CAS
- C. Amorim and M. A. Keane, J. Colloid Interface Sci., 2008, 322, 196–208 CrossRef CAS PubMed
- K. F. Ortega, R. Arrigo, B. Frank, R. Schlogl and A. Trunschke, Chem. Mater., 2016, 28, 6826–6839 CrossRef
- Y. X. Han, J. Zhou, W. J. Wang, H. Q. Wan, Z. Y. Xu, S. R. Zheng and D. Q. Zhu, Appl. Catal., B, 2012, 125, 172–179 CrossRef CAS
- J. Z. Shyu and K. Otto, Appl. Surf. Sci., 1988, 32, 246–252 CrossRef CAS
- J. C. Serrano-Ruiz, G. W. Huber, M. A. Sanchez-Castillo, J. A. Dumesic, F. Rodriguez-Reinoso and A. Sepulveda-Escribano, J. Catal., 2006, 241, 378–388 CrossRef CAS
- X. M. Ning, H. Yu, F. Peng and H. J. Wang, J. Catal., 2015, 325, 136–144 CrossRef CAS
- W. Shi, B. S. Zhang, Y. M. Lin, Q. Wang, Q. Zhang and D. S. Su, ACS Catal., 2016, 6, 7844–7854 CrossRef CAS
- J. P. Macquet, M. M. Millard and T. Theophanides, J. Am. Chem. Soc., 1978, 100, 4741–4746 CrossRef CAS
- H. N. Pham, A. E. Anderson, R. L. Johnson, T. J. Schwartz, B. J. O'Neill, P. Duan, K. Schmidt-Rohr, J. A. Dumesic and A. K. Datye, ACS Catal., 2015, 5, 4546–4555 CrossRef CAS
- B. Sun, T. J. Fu and H. C. Zeng, Chem. Commun., 2018, 54, 7030–7033 RSC
- Q. Xu, X. M. Liu, J. R. Chen, R. X. Li and X. J. Li, J. Mol. Catal. A: Chem., 2006, 260, 299–305 CrossRef CAS
- S. Iihama, S. Furukawa and T. Komatsu, ACS Catal., 2016, 6, 742–746 CrossRef CAS
- Y. Sheng, X. G. Wang, Z. K. Xing, X. B. Chen, X. J. Zou and X. G. Lu, ACS Sustainable Chem. Eng., 2019, 7, 8908–8916 CrossRef CAS
- M. R. Gray, Y. Zhao and C. M. McKnight, Fuel, 2000, 79, 285–294 CrossRef CAS
- M. Zhou, M. Li, C. Hou, Z. Li, Y. Wang, K. Xiang and X. Guo, Chin. Chem. Lett., 2018, 29, 787–790 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01578d |
| This journal is © The Royal Society of Chemistry 2020 |