
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysical gels of poly(vinylamine) by thermal curing†
Thorsten Fischer a,
Jens Köhlera,
Martin Möller*ab and
Smriti Singh*a
a,
Jens Köhlera,
Martin Möller*ab and
Smriti Singh*a
aDWI-Leibniz-Institute for Interactive Materials, e.V., RWTH Aachen University, Forckenbeckstraße 50, D-52074 Aachen, Germany. E-mail: singh@dwi.rwth-aachen.de
bA. N. Nesemeyanov Institute of Organoelement Compounds of Russian Academy of Sciences (INEOS RAS), Vavilova 28, 119991 Moscow, Russia
First published on 9th June 2020
Abstract
Physical gels are a versatile class of materials which can find application in sensors, electrochemistry, biomedicine or rheological modifiers. Herein, we present a hydrogen-bonded physical gel which is based on the interaction between phenylcarbonate telechelic poly(ethylene glycol) (PEG-PC) and poly(vinyl amine-co-acetamide) (p(VAm-co-VAA)). The critical gelation concentration was found to be 10 wt% by rheology and NMR. UV-vis spectroscopy and dynamic light scattering reveal the formation of aggregates in the gel. Rheology and differential scanning calorimetry (DSC) was used to show the effect of thermal curing on the mechanical properties of the physical gel.
1. Introduction
Physically crosslinked hydrogels have unique self-healing properties and are therefore a powerful material class with a wide range of applications. The variety of non-covalent bonds including ionic interactions,1 hydrogen bonding,2,3 π–π-stacking,4 host–guest interactions,5 and the hydrophobic effect6 provides an ideal platform for the fabrication of physical gels with tailored strength and properties. Due to their associative and transient nature, they have inherently unique features.7,8The variety and versatility of the interactions are also reflected by the variety of manufacturing methods.9,10 Gelation induced by mixing two components in a good solvent is in this context a simple and straightforward pathway. In this case, the attraction of the first component to the second component has to be higher than attraction to the the solvent. Among the different physical interactions, hydrogen bonding is one of the best investigated.2,3,11–13 In this context poly(vinylamine) (PVAm), a waterborne polymer is an attractive candidate for formation of physical gels as it can facilitate hydrogen bonding with its high density of primary amines.14,15 Few papers published in the last decade focused on the use of PVAm for chemically crosslinked microgels,16–19 or hydrogel capsules.20 While very few literatures report on the fabrication of a physical gel based on PVAm, this is only by exploiting its cationic behavior, for example with carboxymethyl cellulose or copper.21,22 Alternative to ionic interaction for the formation of a physical gel, PVAm can be easily formulated exploiting hydrogen bonding interaction. Since PVAm can, if protonated, act as a hydrogen donor, addition of a second component acting as hydrogen acceptor will facilitate gel formation. Organic carbonates have shown to be strong hydrogen bond acceptors.23 To the best of our knowledge, there are no physical gels reported using organic carbonate compounds.
However, physical gels including those which are formed by hydrogen bonds can be kinetically trapped in a meta-stable state during gelation. Thermal curing, which is composed of a heating and a cooling step, can drive the structure towards the thermodynamical minimum. On heating hydrogen bonds are disrupted, while on cooling the bonds will be reformed and the elastic properties recover. Nevertheless the effect of thermal curing is not well investigated. Recently, Fuentes-Caparrós et al. investigated the effect of thermal curing on low molecular weight gelator peptide gels. They found out, that their gels are kinetically trapped and that thermal curing can change the microstructure of the gels leading to significantly different properties compared to the as-prepared gels.24
Taking this into account, in this work we report the formation of physically crosslinked hydrogels using PVAm and phenylcarbonate-telechelic-poly(ethylene glycol) (PEG-PC) and show the effect of thermal curing on the mechanical properties of the gel. We used 1H NMR, rheology, and UV-vis spectroscopy to analyze the predominant physical interaction responsible for the formation of the gel.25,26 The critical gelation concentration for the formation of the gel was determined by measuring the storage modulus G′ and the loss modulus G′′ in dependence on the time, with different wt% of p(VAm-co-VAA). The frequency response of G′ and G′′ of the physical gel was measured by small-amplitude oscillation shear (SAOS) experiments to investigate, whether the bonds have a transient physical or permanent chemical nature. To show the effect of thermal curing on the change in the mechanical properties of the gels, the gels were subjected to repeated heating–cooling cycles. The versatility of the system also comes from the fact that these hydrogels can be further chemically crosslinked via carbamate linkages by the reaction of phenylcarbonate and primary amine in the presence of a base as shown by us previously.27 The p(VAm-co-VAA) was obtained by selective hydrolysis of formamide to primary amines from the statistical copolymer of poly(vinyl formamide-co-acetamide). Under the reaction conditions used acetamide moiety remained intact.27
2. Materials and methods
2.1 Materials
N-Vinylformamide (NVF), phenylchloroformate (PCF), and poly(ethylene glycol) (PEG) (Mn = 400 Da) were purchased from Sigma Aldrich. N-Vinylacetamide (NVA) was purchased from ABCR. 2,2′-Azobis[2-(2-imidazolin-2-yl)propane]dihydro-chloride was from TCI. Methanol (MeOH) and triethylamine (TEA) were purchased from Sigma Aldrich. Deuterated methanol and water were purchased from Deutero. All chemicals were used as received without any purification.2.2 Methods
A typical set of breakage-thermal curing cycles consists of 5 heating cooling cycles, a frequency sweep, and a subsequent amplitude sweep as shown in Fig. 1.
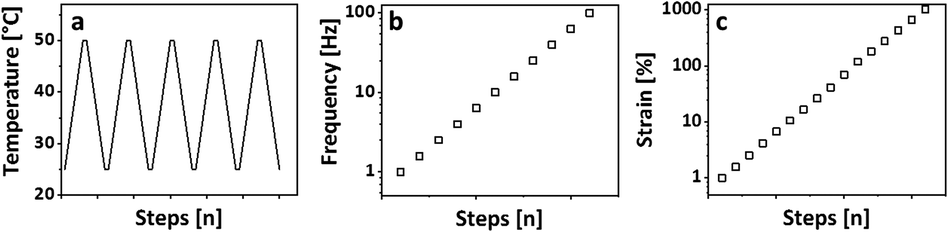 | ||
| Fig. 1 Breakage-thermal curing set, which consists of (a) 5 heating/cooling cycles with 5 °C min−1, at a frequency of 1 Hz and an amplitude of 1%; (b) frequency sweep from 1–100 Hz at 25 °C with an amplitude of 1% strain, and (c) an amplitude sweep from 1–1000% strain at 25 °C and a frequency of 1 Hz. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1
:1
| Compound 1 | Wt% | Compound 2 | Wt% | Formation of physical gel | |
|---|---|---|---|---|---|
| a | PEG-PC | 34 | — | — | − |
| b | p(VAm-co-VAA) | 10 | — | — | − |
| c | p(VAm-co-VAA) | 5 | PEG-PC | 20 | − |
| d | p(VAm-co-VAA) | 10 | PEG-PC | 34 | + |
2.3 Synthesis
1H NMR (400 MHz in D2O, δ in ppm): 1.35–1.75 (–CH2 backbone), 1.75–1.95 (–CH3), 3.0–4.0 (–CH backbone), 7.5–8 (–CHO).
SEC: Mn = 5.6 × 104 g mol−1, Mw = 1.2 × 105 g mol−1, PDI = 2.2.
1H-NMR (400 MHz in D2O, δ in ppm): 1.2–2.1 (–CH2 backbone, –CH3), 2.8–4.1 (–CH backbone).
SEC: Mn = 5.1 × 104 g mol−1, Mw = 1.1 × 105 g mol−1, PDI = 2.1.
1H NMR (400 MHz in MeOD, δ in ppm): 3.3–3.5 (–CH2, backbone), 3.55–3.65 (–CH2), 4.2–4.3 (–CH2), 6.65–6.75 (unknown side product), 7.0–7.1 (–CH, aryl, para), 7.1–7.2 (–CH, aryl, para), 7.2–7.35 (–CH, aryl, meta).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1. p(VAm-co-VAA) (50 mg, 394 μmol amine groups) was dissolved in methanol (450 mg, 570 μL) and PEG-PC (252 mg, 394 μmol phenylcarbonate groups) was added.
:1. p(VAm-co-VAA) (50 mg, 394 μmol amine groups) was dissolved in methanol (450 mg, 570 μL) and PEG-PC (252 mg, 394 μmol phenylcarbonate groups) was added.3. Results and discussion
The physical gels were obtained by dissolving 10 wt% of p(VAm-co-VAA) in methanol with subsequent addition of PEG-PC (Table 1). This leads to a phase separation with precipitation of a colourless to yellowish gel. The methanol content of the physical gel was determined by removing the supernatant and subsequent drying of the gel fraction and found to be 45.5% ± 1.5%. Attempts to swell the gel, either in methanol or in water, failed as the gel dissolves in an excess of solvent suggesting physical crosslinks. In order to estimate the critical concentration at which gelation occurs, we performed rheological measurements. Three samples of p(VAm-co-VAA) (5 wt%, 7 wt%, and 10 wt%) in methanol were mixed with PEG-PC (amine-to-carbonate molar ratio 1:1). G′ and G′′ were monitored (1 Hz, 1%) in dependence on the time. From these values, the dissipation factor tanδ was calculated (G′′/G′) and plotted against the time (Fig. S1 ESI†).
The lower the tanδ value the more elastic is the response, i.e. at a tanδ of 1 and/or lower, the elastic response is dominating. At 10 wt%, the tanδ approximates 1 (Fig. S1 ESI†); this corresponds to the critical gel concentration. To validate the transient nature of the bonds, the gels were measured with a SAOS experiment measuring G′ and G′′ (pre-conditioning time: 3 h, amplitude: 0.1%). As can be seen in Fig. 2 G′ and G′′ increases linearly with the frequency, which is common for physical gels.28 Furthermore, there is a crossover point, where G′ exceeds G′′, at around 0.5 Hz, which is related to the average lifetime of the bond.29 From these observations, we conclude that the gel is physically crosslinked. This hypothesis is supported by concentration dependent 1H NMR. Gradual shifts of the aromatic protons and the protons nearby the carbonate group of the PEG-PC indicate an intermolecular interaction as this is not seen in the concentration dependent 1H NMR of the pure PEG-PC (Fig. S2 and S3 ESI†). Peak 1 and 2 are not visible because of their short relaxation time in the 1H NMR (backbone signals of the polymer). Furthermore, the absence of phenol in the spectrum rules out any chemical crosslinking (Scheme S1 ESI†).27
 | ||
| Fig. 2 Frequency dependence of the physical gel with an amplitude of 0.1%. The frequency dependence indicates the presence of physical crosslinks. | ||
The 1H NMR spectrum of the supernatant reveals, that it consists mainly of PEG-PC, although the presence of p(VAm-co-VAA) cannot be ruled out because of its weak signals (Fig. S4 ESI†).
To further investigate the nature of the physical crosslinks like hydrogen bonding and π–π-stacking, which can be affected by the change in temperature, temperature dependent UV-vis spectroscopy was performed. The UV-vis spectra was recorded from 250 nm to 450 nm. The temperature range was kept between 50 °C to 10 °C with a step of 5 °C, keeping the temperature constant during each measurement (Fig. 3). Four sets of measurements were done using (a) only PEG-PC and (b) only p(VAm-co-VAA) in the concentrations which resulted into the formation of physical gels and a combination of PEG-PC and p(VAm-co-VAA) which did not (c) and did (d) resulted into the corresponding physical gel. The respective wt% is mentioned in Table 1. On decreasing the temperature, UV spectrum of PEG-PC (Fig. 3a) reveals two effects. The main maximum at 290 nm, which is due to the π → π* transition of the carbonyl-group,30 experiences a hypochromic shift and a shoulder appears at ∼310 nm, which is due to the π → π* transition of the phenyl-group.
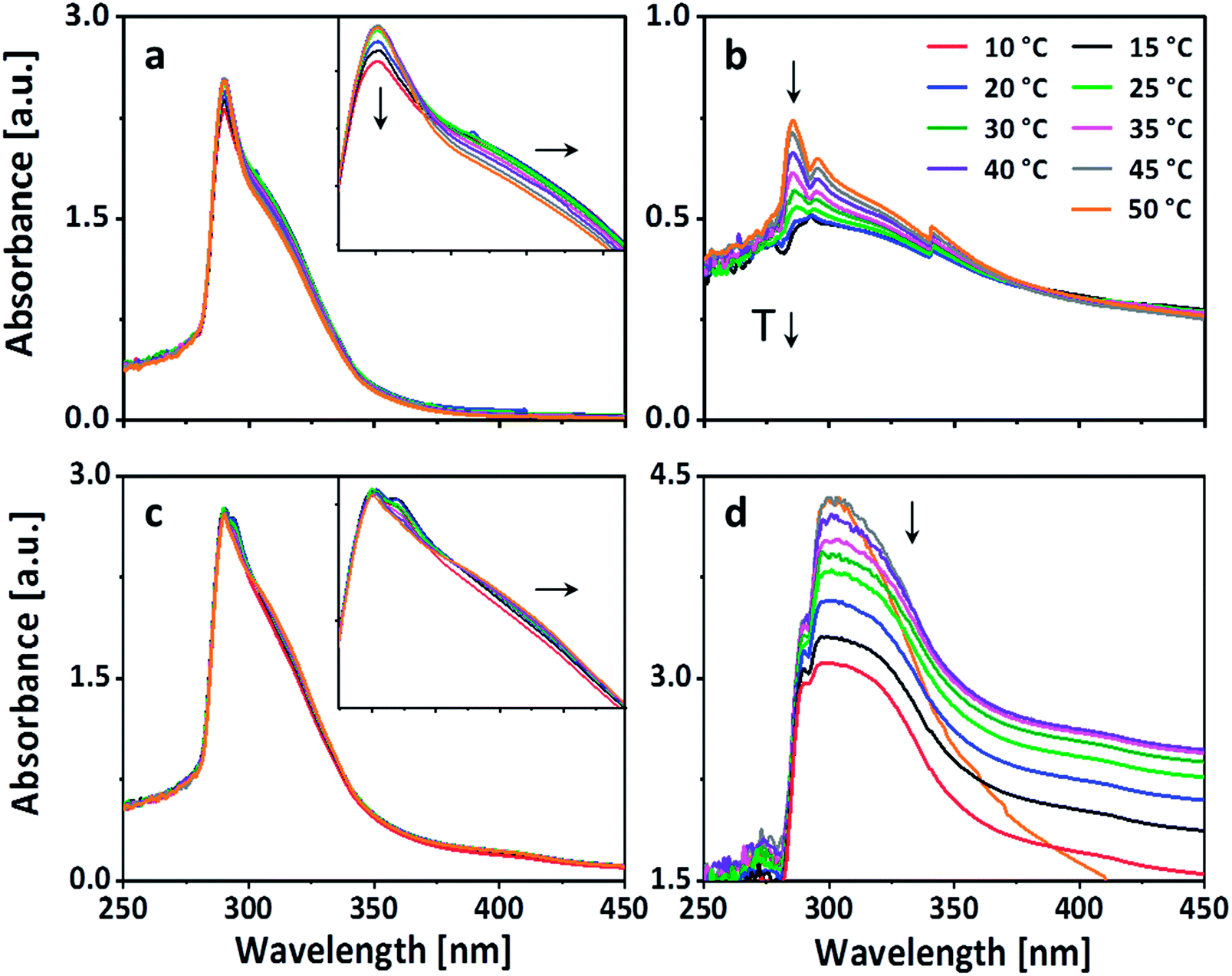 | ||
| Fig. 3 UV-vis spectrum of (a) PEG-PC in methanol, (b) p(VAm-co-VAA) in methanol, (c) PEG-PC + 5 wt% p(VAm-co-VAA) in methanol, and (d) PEG-PC + 10 wt% p(VAm-co-VAA) in methanol in dependence on the temperature. The arrows indicate the shift of the absorbance with decreasing temperature. PEG-PC exhibits due to π–π stacking a bathochromic shift upon decreasing temperature, which is also visible in a combination with 5 wt% of p(VAm-co-VAA). The hypochromic shifts in (a, b, and d) are caused by aggregation. | ||
The shoulder at 310 nm indicates π–π-stacking.31 With decreasing temperature, the π–π-stacking is enhanced because the energy gap between the π and the π* orbital is decreased. Therefore, less energy is needed for the transition, this results in a bathochromic shift towards a longer wavelength. The hypochromic shift, which appears along with the bathochromic shift, supports this hypothesis. Generally, hypochromic shifts are associated with conformational changes in proteins and DNA. The unstacking of the bases in case of DNA at elevated temperature induces a conformational change in the structure, which in turn increases the number of exposed chromophores and consequently the absorbance.32 Taking this into account, the UV-vis spectrum of PEG-PC suggests the presence of aggregates at room temperature, which are stabilized by π-stacking. The spectrum of p(VAm-co-VAA) (Fig. 3b) is dominated by a hypochromic shift of the main maximum at 285 nm, which is due to the n → π* transition of the acetamide group.33 This shift also indicates the presence of aggregates, which was verified by DLS (Fig. S5 and S6 ESI†). This aggregation is caused by either hydrogen bonding of the amine and carbonyl-group, or by hydrophobic interaction of the acetamide. Furthermore, the UV-vis spectrum shows a gradual hypsochromic shift of the maximum. This shift is related to hydrogen bonding either with the solvent or the polymer itself.32
However, mixing 5 wt% of p(VAm-co-VAA) with PEG-PC (no gelation), the UV-vis spectrum is dominated by the absorption of PEG-PC (Fig. 3c). The bathochromic shift is still visible indicating π–π-stacking. Furthermore, there is a hypsochromic shift of the maximum indicating hydrogen bonding. Since this was not visible without the p(VAm-co-VAA), it is caused by hydrogen bonding between protonated amine groups of p(VAm-co-VAA) and the carbonate groups of the PEG-PC. With increasing the concentration to 10 wt% the spectrum changes dramatically (Fig. 3d).
The absorption between 280–310 nm decreases showing a predominant hypochromic shift with decreasing temperature, while there is no bathochromic shift visible anymore. This indicates that the hydrogen bonds are dominating the formation of the hydrogel as the number of exposed chromophores decreases with decreasing temperature implying that the carbonate groups of the PEG-PC are strongly hydrogen-bonded to the protonated amine groups of p(VAm-co-VAA) restricting the mobility of the phenylcarbonate groups. Based on these results, we postulate a physical crosslinked structure as shown in Scheme 1.
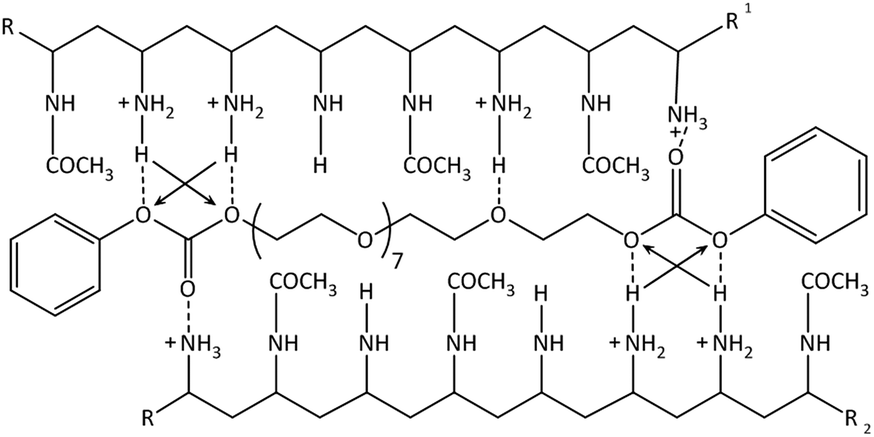 | ||
| Scheme 1 Suggested structure of the physical gel: hydrogen bonds are shown as the dotted lines. The arrows indicate secondary attractive forces. π–π-stacking not shown. | ||
The strong intermolecular interaction resulting in the formation of physical gels is likely due to the formation of hydrogen bonds between the carbonate group of PEG-PC as a strong hydrogen acceptor and protonated amines of p(VAm-co-VAA) as hydrogen donor, although the possibility of π–π-stacking between the phenyl-groups of PEG-PC cannot be ruled out. From titration curves of p(VAm-VAA) in water27 and due to the fact that the p(VAm-co-VAA) was neutralized to pH = 7 after hydrolysis, it can be estimated that around 50% of the amine groups were protonated and available for hydrogen bonding. Due to the transient nature of the hydrogen bonds, the gel should exhibit a temperature sensitive behaviour. As it is well known from literature hydrogen bonds can break with increasing temperature.34 To investigate the temperature responsive behaviour of the gels, rheological measurements were conducted with a temperature ramp. The temperature was increased to 50 °C with subsequent cooling to 25 °C (1 Hz, 1%, 5 °C min−1, Fig. 4a). In total 5 of these heating/cooling cycles were performed subsequently. It can be seen from Fig. 4a, that instead of an expected decrease in G′, the values drastically increases from ∼50 Pa to ∼7 kPa. We postulate that this increase in modulus is caused by thermal annealing of the gel resulting in a change in gel microstructure. This means that the increase of the temperature reduces the aggregates formed due to fast gelation and leads to better mixing of the gel (schematic illustration Fig. S7 ESI†). Polymer mixing is an enthalpic (endothermic) process that can be monitored by differential scanning calorimetry (DSC). DSC was performed in sealed stainless steel crucibles to ensure that no solvent evaporations happen and for at least 10 heating/cooling cycles with 5 °C min−1. p(VAm-co-VAA), PEG-PC and the physical gel were measured. In the latter case, the gel and the supernatant were analyzed. For p(VAm-co-VAA) and PEG-PC no signal was observed (Fig. S8 and S9 ESI†). The supernatant (Fig. S10 ESI†) reveals a prominent endothermic signal at 40 °C, whereas the physical gel (Fig. S11 ESI†) has a weak signal at 55 °C. These signals are attributed to mixing, which means the homogenization process mentioned above. This homogenization temperature is decreased for the supernatant. The supernatant is in a liquid state, so it is more mobile and the energy barrier to break the hydrogen bonds is decreased. Furthermore, the signal is more prominent than in the strongly bound physical gel and anneals after the 10 heating/cooling cycles.
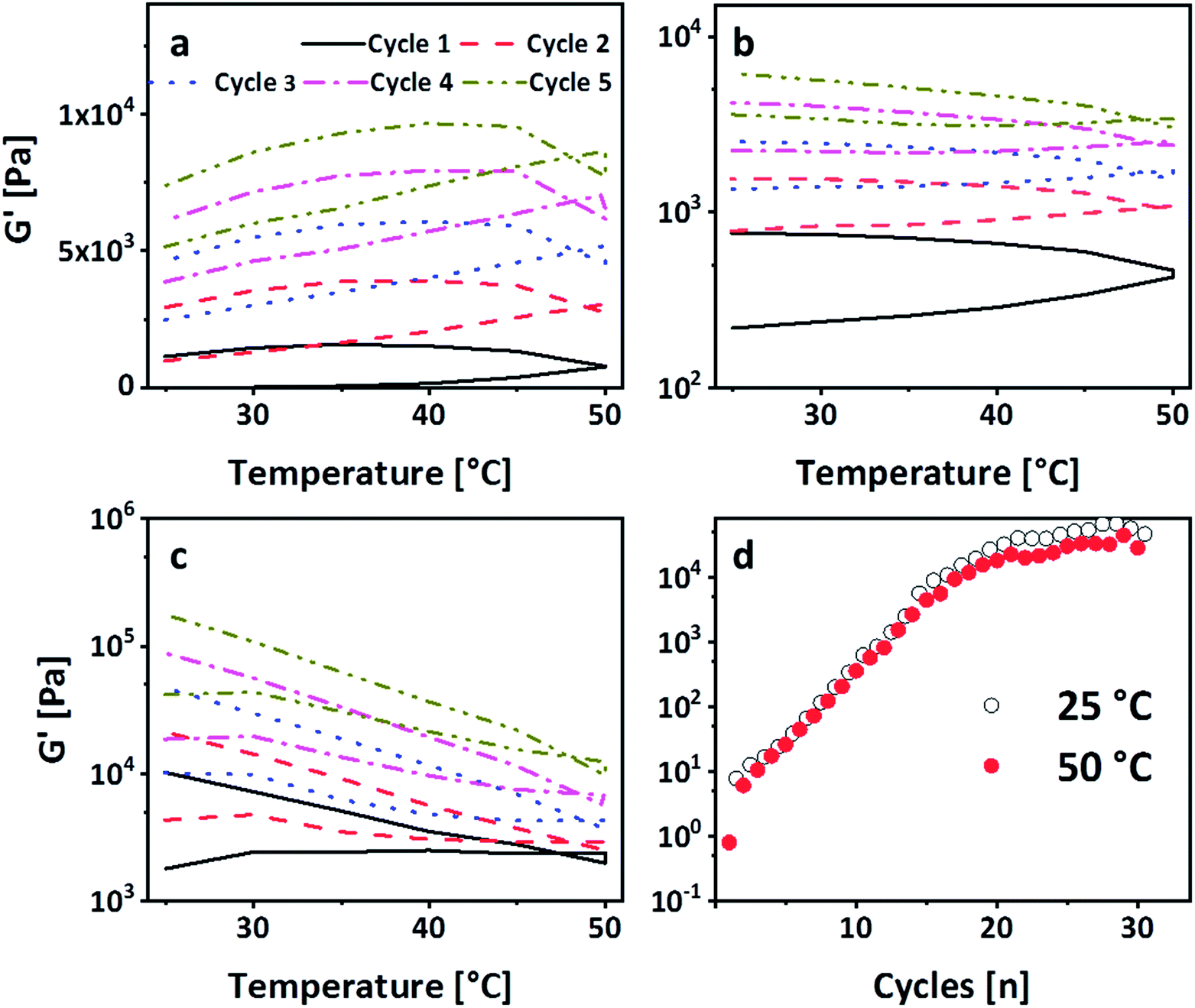 | ||
| Fig. 4 (a) G′ dependence on the temperature performing five subsequent heating–cooling cycles with 5 °C min−1. G′ increases with each cycle indicating a homogenization. (b) 30 heating/cooling cycles with a frequency of 1 Hz, an amplitude of 1%, and a heating rate of 5 °C min−1. A plateau is reached after 20 cycles. (c) 2nd set of heating/cooling cycles after the amplitude sweep of the 1st set. The storage modulus is regained after these cycles, so the gel can be cured by the temperature cycles and (d) 3rd set of heating/cooling cycles after the amplitude sweep of the 2nd set. It is visible that the shape of G′ as a function of the temperature changes with the number of sets. | ||
To verify that the gel is still only physically bound after temperature treatment, the gelation in the NMR tube was performed. The tubes were heated up to 50 °C and 80 °C respectively for 4 h (Fig. S12 ESI†). Even after the treatment at 80 °C, no phenol formation was observed.27 In accordance with these results, a subsequent SAOS measurement after the heating/cooling cycle exhibited a dependency of G′ and G′′ on the frequency, confirming the physical nature of crosslinks (Fig. S13 ESI†). Thus, these heating/cooling cycles induce a thermo-curing of the gel with an increase of G′ by more than two orders of magnitude. To the best of our knowledge, this behaviour has not been reported so far. In general, physical gels have an inherent self-healing behaviour due to the transient nature of the bonds. To investigate whether the gel shows this kind of behaviour and can return to the enhanced modulus, the gel was broken in an amplitude sweep, i.e. the amplitude was subsequently increased to 1000% (1 Hz, Fig. S14 ESI†). Consequently, G′ drops down to ∼200 Pa. Immediately after the amplitude sweep, the gel was subjected to another five heating/cooling cycles. It is visible from Fig. 4b that G′ recovers after these 5 cycles (G′ ∼ 7 kPa). This explains the transient nature of the physical bonds as they are re-formed after breakage. Repeating this breakage and thermal curing sets a further increase of G′ was observed, which was recorded up to 20 kPa (Fig. 4c). We deduce this increase in modulus to further homogenization. In order to decouple the increase of the modulus from the amplitude sweep, another gel sample was subjected to 30 heating/cooling cycles (Fig. 4d). It is visible that after 20 cycles a plateau is reached with a G′ of 50 kPa confirming the previous results. Moreover, it is visible from Fig. 4d that after reaching the plateau the G′ at 25 °C is always higher than 50 °C. This shows that higher temperature leads to a breakage of the hydrogen bonds but only after complete homogenization we see the effect. However, in the stage of homogenization (most prominent between cycles 5 and 15), there is a linear increase on the logarithmic scale. To understand this behaviour the single sets of the breakage-thermal curing cycles were analyzed further (Fig. 4a–c). It is visible that the “shape”, which means the slope of G′ during heating and cooling, of the temperature cycles changes: at the first two sets G′ increases with increasing temperature (Fig. 4a and b), whereas this is not the case for the 3rd and the following sets (Fig. 4d). The increase of the temperature leads to a breakage of the hydrogen bonds on the one hand, but on the other also results in homogenization, which overcompensates the reduction of G′ due to breakage of comparably few hydrogen bonds. However, in the 3rd set the gel is homogenized (which is also visible by the increase of G′ from 7 kPa after the 2nd set to 20 kPa after the 3rd set) to an extent that further homogenization does not overcompensate the breakage of the hydrogen bonds leading therefore to a decrease of G′ with increasing temperature.
4 Conclusions
In conclusion, physical gels were successfully prepared from p(VAm-co-VAA) and PEG-PC. Rheology, NMR, and UV-vis spectroscopy were used to determine the structure of the gel revealing the formation of aggregates. Heating/cooling cycles within the rheometer can homogenize the structure resulting in a tough physical gel with storage moduli above 50 kPa. Furthermore, the gel is fully self-healable. These experiments highlight the possibility to tune the modulus and to anneal inhomogeneities in physical gels.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the SFB 985 – “Functional Microgels and Microgel Systems” of the Deutsche Forschungsgemeinschaft and was performed in part at the Center for Chemical Polymer Technology, supported by the EU and North Rhine-Westphalia (EFRE 30 00883 02). One of the authors (M. M.) gratefully acknowledges the support of an Advanced Grant from the European Research Council (ERC), Jellyclock 695716. This work was also supported by the Ministry of Science and Higher Education of the Russian Federation (Grant of the Government of the Russian Federation No. 14.W03.31.0018).Notes and references
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Agasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- X. Dai, Y. Zhang, L. Gao, T. Bai, W. Wang, Y. Cui and W. Liu, Adv. Mater., 2015, 27, 3566–3571 CrossRef CAS PubMed.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- L. Li, B. Yan, J. Yang, L. Chen and H. Zeng, Adv. Mater., 2015, 27, 1294–1299 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima and A. Harada, Macromol. Rapid Commun., 2016, 37(1), 86–92 CrossRef CAS PubMed.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- L. Voorhaar and R. Hoogenboom, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- S. M. Mantooth, B. G. Munoz-Robles and M. J. Webber, Macromol. Biosci., 2018, e1800281, DOI:10.1002/mabi.201800281.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- A. Noro, M. Hayashi, A. Ohshika and Y. Matsushita, Soft Matter, 2011, 7, 1667–1670 RSC.
- W. Edwards and D. K. Smith, J. Am. Chem. Soc., 2013, 135, 5911–5920 CrossRef CAS PubMed.
- X. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS PubMed.
- R. K. J. Pinschmidt, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2257–2283 CrossRef CAS.
- R. Pelton, Langmuir, 2014, 30, 15373–15382 CrossRef CAS PubMed.
- Q. Chen, K. Xu, W. Zhang, C. Song and P. Wang, Colloid Polym. Sci., 2009, 287, 1339–1346 CrossRef CAS.
- J. Xu, A. Barros Timmons and R. Pelton, Colloid Polym. Sci., 2004, 282, 256–263 CrossRef CAS.
- K. Xu, Y. Tan, Q. Chen, H. An, W. Li, L. Dong and P. Wang, J. Colloid Interface Sci., 2010, 345, 360–368 CrossRef CAS PubMed.
- M. Yue, K. Imai, Y. Miura and Y. Hoshino, Polym. J., 2017, 49, 601–606 CrossRef CAS.
- J. Kim, H. J. Lim, Y. K. Hwang, H. Woo, J. W. Kim and K. Char, Langmuir, 2012, 28, 11899–11905 CrossRef CAS PubMed.
- X. Feng and P. Robert, Macromolecules, 2007, 40, 1624–1630 CrossRef CAS.
- J. G. Mendoza-Payan, S. Flores-Gallardo and A. Marquez-Lucero, J. Appl. Polym. Sci., 2010, 115, 790–801 CrossRef CAS.
- R. Alcalde, M. Atilhan, J. L. Trenzado and S. Aparicio, J. Phys. Chem. B, 2016, 120, 5015–5028 CrossRef CAS PubMed.
- A. M. Fuentes-Caparrós, F. de Paula Gomez-Franco, B. Dietrich, C. Wilson, C. Brasnett, A. Seddon and D. J. Adams, Nanoscale, 2019, 11, 3275–3280 RSC.
- S. N. Volkov, Int. J. Quantum Chem., 1979, 16, 119–132 CrossRef CAS.
- C. Rest, M. J. Mayoral, K. Fucke, J. Schellheimer, V. Stepanenko and G. Fernandez, Angew. Chem., Int. Ed., 2014, 53, 700–705 CrossRef CAS PubMed.
- T. Fischer, J. Köhler, H. Keul, S. Singh and M. Möller, Macromol. Chem. Phys., 2018, 219, 1800399–1800408 CrossRef.
- J. Zhao, K. Mayumi, C. Creton and T. Narita, J. Rheol., 2017, 61, 1371–1383 CrossRef CAS.
- T. Indei and J. Takimoto, J. Chem. Phys., 2010, 133, 194902 CrossRef PubMed.
- H. M. Zidan, J. Appl. Polym. Sci., 2003, 88, 104–111 CrossRef CAS.
- K.-Y. Wang, C. Chen, J.-F. Liu, Q. Wang, J. Chang, H.-J. Zhu and C. Li, Org. Biomol. Chem., 2012, 10, 6693–6704 RSC.
- N. A. Nicola and S. J. Leach, Int. J. Pept. Protein Res., 1976, 8, 393–415 CrossRef CAS PubMed.
- M. B. Robin, Higher Excited States of Polyatomic Molecules, Academic Press, New York, San Francisco, London, 1975 Search PubMed.
- S.-W. Kuo, J. Polym. Res., 2008, 15, 459–486 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01607a |
| This journal is © The Royal Society of Chemistry 2020 |