
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new dendrimer series: synthesis, free radical scavenging and protein binding studies†
Dhaval Makawana and
Man Singh *
*
School of Chemical Sciences, Central University of Gujarat, Gandhinagar-382030, India. E-mail: dhavalmak@cug.ac.in; mansingh50@hotmail.com
First published on 8th June 2020
Abstract
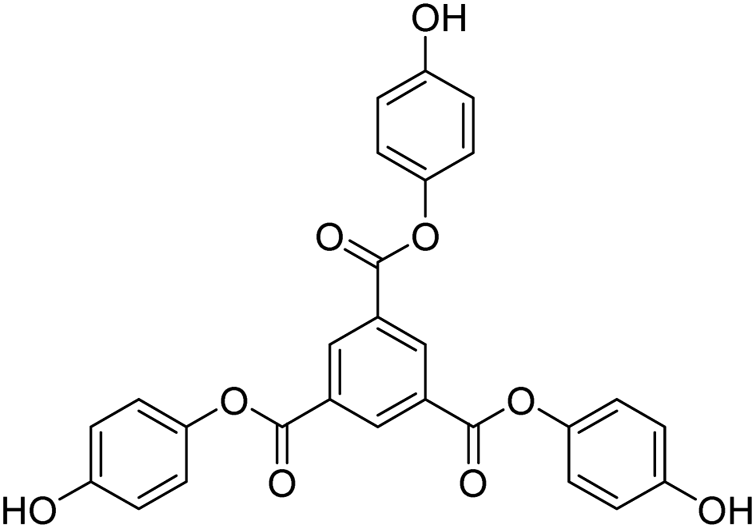
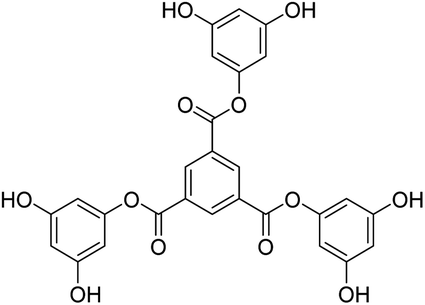
Tri-o-tolyl benzene-1,3,5-tricarboxylate (TOBT (T0)), tri-4-hydroxyphenyl benzene-1,3,5-tricarboxylate (THBT (T1)), and tri-3,5-dihydroxyphenyl benzene-1,3,5-tricarboxylate (TDBT (T2)), a series of 1st tier dendrimers with a common 1,3,5-benzenetricarbonyl trichloride/trimesoyl chloride (TMC) core, are reported. T0 does not have any replaceable H+ on its terminal phenyl group, acting as a branch. T1 has one phenolic –OH at the para position and T2 has two phenolic –OH groups at the 3 and 5 positions of each terminal phenyl group. During synthesis, these –OH groups at the terminal phenyl groups were protected through tert-butyldimethylsilyl chloride (TBDMSCl) assisted with t-BuOK in DCM, THF, indazole, 4-dimethylaminopyridine (DMAP), and tertiary-n-butyl ammonium fluoride (TBAF). MTBDMSP (mono-tertiary butyl dimethylsilane phloroglucinol), DTBDMSP (di-tertiary butyl dimethylsilane phloroglucinol), and TTBDMSP (tri-tertiary butyl dimethylsilane phloroglucinol) were obtained with >90% yield, and TTBDMSP phenolic derivatives (PDs) were developed to synthesize T0, T1, and T2 dendrimers by deprotecting with TBAF. T0 showed superhydrophobic properties as it did not dissolve in methanol, contrary to T1 and T2, but dissolved in acetone. Their structures were determined using 1H and 13C NMR spectroscopies, and mass spectrometry. Their scavenging activities were studied using UV-Vis spectrophotometry compared with ascorbic acid and protein binding was studied with bovine serum albumin (BSA) and lysozyme (lyso). T0 exhibited exceptional optical activity contrary to T1 and T2, which acted as antioxidants to scavenge free radicals.
1. Introduction
Generally, webdrimers like PAMAM (poly(amidoamine)) or starburst dendrimers with repeated branched subunits of amide and amine functionality, like TTDMM (trimesyol-1,3,5-trimethyl malonate), TSMM (trisurfactantomethylol melamine), and others do not have replaceable hydrogen atoms. Therefore, they are only capable of binding bionanoparticles, transporting them, and releasing them at desired sites as they have adequate steric activities, which are generated by their branches to encapsulate the drugs or genes safely. The branching induces the required rotational, vibrational and translational motions without altering the chemical activities of their void spaces, which is supportive of safer encapsulation. Thus, these webdrimers act as ideal gene delivery vehicles. To date, no webdrimer has been reported that can liberate H+ ions or protons, while also behaving as a drug or gene delivery vehicle. Such dendrimers behave differently in acidic and alkaline chemical environments, so they are medium selective moieties. Also, these webdrimers liberate protons when there is spontaneity for them, otherwise they do not liberate or furnish protons. Dendrimers remain monodispersed in the desired solvents and do not strongly bind or aggregate together and they maintain higher surface area, which is a most desired characteristic for molecular sensors. Dendrimers as multifunctional nanomaterials are compatible in nature so their physicochemical properties are a thrust area of current research. Dendrimers do not have structural constituents to support electronic transitions from the HOMO to LUMO state apart from scavenging activities. Detection of the scavenging activities of free radicals (FRs) is a major challenge in the food industry, biochemical reactions, and bioengineering due to the thermodynamically and kinetically active chemical moieties.1 FR scavenging has become an inevitable task to prevent the damage of valuable chemical processes of biomolecules and their combinations by FRs.1–3 FRs have lone pair electrons (LPEs), π-conjugation, sigma bonds, and functional groups to intensify the FR activities. Sensors may attract FRs to void spaces to immobilize them through van der Waals forces.4 Antioxidants are extensively used to prevent the threat of chronic diseases such as cancer and cardiovascular diseases, along with other irregular functions in cells.5,6 For the last few decades, synthetic molecules have been used to prevent redox, but they have a lower number of phenolic –OH.7 Flavonoids with large –OH numbers to release protons for scavenging are being synthesized. Vitamin C, beta-carotene, and polyphenolic flavonoids are used as a source of antioxidants in the diet. Flavonoids provide labile H+ to sense FRs, and accommodate the pH and potential energies of biologically and biochemically active liquid mixtures against oxidative deformation of molecular structures.1,4,8–10 The solubility of polyphenols in aqueous systems is negligible. Antioxidant effectiveness in vitro measures the radical scavenging activity (RSA) against FRs: singlet oxygen (1O2), superoxide radical anions (O2˙−), hydroxyl radicals (˙OH) and hydrogen peroxide.5,7,11 T1 and T2 have –OH and functional void spaces with π-conjugation favorable to release H+ in steps and scavenge FRs. No such dendrimer antioxidants have been reported yet, where phenolic molecules are used as the core as well as branches. These have higher stability with void spaces, and steric ability.12–14 These induce rotational, translational, vibrational, oscillatory and electronic motions to encapsulate a drug.10,14–16 Liu and co-workers have used DPPH radicals with stronger scavenging of natural antioxidants in combination than individual antioxidants.17 More phenolic –OH groups in a molecule act as effective antioxidants due to intramolecular synergistic activities with certain tentropy among them.18 Nishiyama and coworkers have investigated intramolecular synergistic activities between chromanol and thiopropionate groups of a series of antioxidants in tetralin oxidation.19,20 Dendrimers with a web-like structure are a class of functional webdrimer molecules that have functional void spaces for drug or gene binding, transport and release. They do not affect the chemical environment, like pH, ionic strength, temperature or aggregation of biochemical, biological, and biophysical processes as they do not undergo dissociation, ionization, or dimerization. The webdrimers remain monodispersed with larger surface area and surface activities with comparatively higher entropy than ordinary molecules. So, they have extraordinarily stronger drug or nanoparticle binding and transporting abilities in media of wider pH, as they are least affected by the pH variations of a process. The novelty of these molecules is that they do not undergo any structural changes within the infinitesimal thermodynamic conditions as they have a unique structure, including a core and branching, which mutually stabilizes and chemically balances their chemical activities, allowing them to remain stabilized.2. Experimental and methods
2.1 Materials for conducting the reaction
TMC procured from Sigma Aldrich was used as received; o-cresol, hydroquinone, phloroglucinol, and potassium tert-butoxide were obtained from Sisco Research Laboratories Pvt. Ltd. India. Structures were determined with 1H NMR and 13C NMR spectra recorded in CDCl3 and DMSO4 (NMR grade, 99.99% yield) with a Bruker Biospin Avance-III 500 MHz FT-NMR. TMS was used as an internal standard due to its highest shielding state. 99% pure solvents procured from Rankem were used. Analytical thin-layer chromatography (TLC) was performed with pre-coated silica gel plates (200 μm) with fluorescent indicator acquired from Sigma Aldrich. For flash column chromatography, silica of 200–400 mesh size procured from SRL was used. Imidazole and t-BuOK maintained a basic medium in protecting the phenolic groups with TBCDMS and deprotection with TBAF during synthesis, which was catalyzed by DMAP. For clarity of their participation in the chemical processes, the chemical structures are given in (Scheme 1).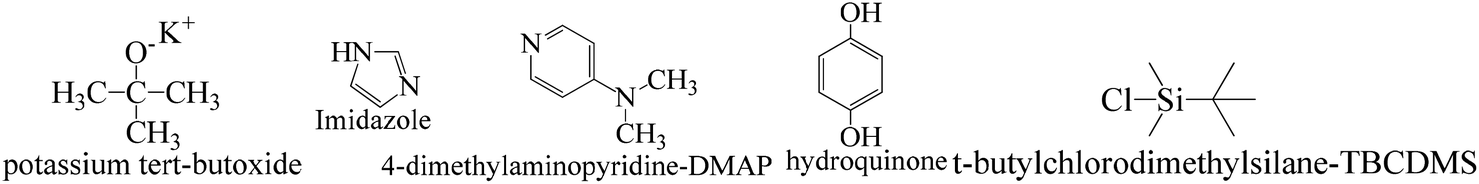 | ||
| Scheme 1 Functional structures of the molecules used as catalysts and protection and deprotection agents. | ||
2.2 Protection and deprotection of phenolic derivatives
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, which gave a colorless solid (55%).33 DTBDMSH (di-tertiary butyl dimethyl silane hydroquinone) and MTBDMSH (mono-tertiary butyl dimethyl silane hydroquinone) were obtained in 60 and 40% yield, respectively (Scheme 2).
:1 ratio, which gave a colorless solid (55%).33 DTBDMSH (di-tertiary butyl dimethyl silane hydroquinone) and MTBDMSH (mono-tertiary butyl dimethyl silane hydroquinone) were obtained in 60 and 40% yield, respectively (Scheme 2).
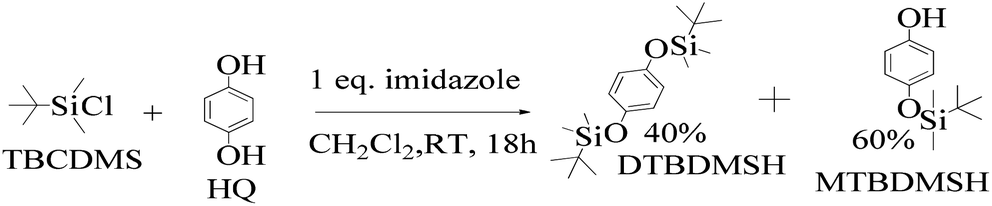 | ||
| Scheme 2 The mechanism of the reaction in the prescribed medium and experimental conditions. Imidazole is a base and attracts H+ from HQ and gains a positive charge by transferring its LPE to a H+ ion, which attracts Cl− from TBCDMS, and it catalyzes the reaction as shown in Scheme 3. | ||
 | ||
| Scheme 3 The charge orientation mechanism to develop nucleophilic and electrophilic centers to conduct the reaction in the prescribed medium and experimental conditions. | ||
HQ releases H+ in a basic chemical environment induced by imidazole, a planar five membered ring with vibrant electronic shifts, because hydrogen binds to one of its nitrogen atoms (Scheme 4). Imidazole with 3.67 debye is water soluble.
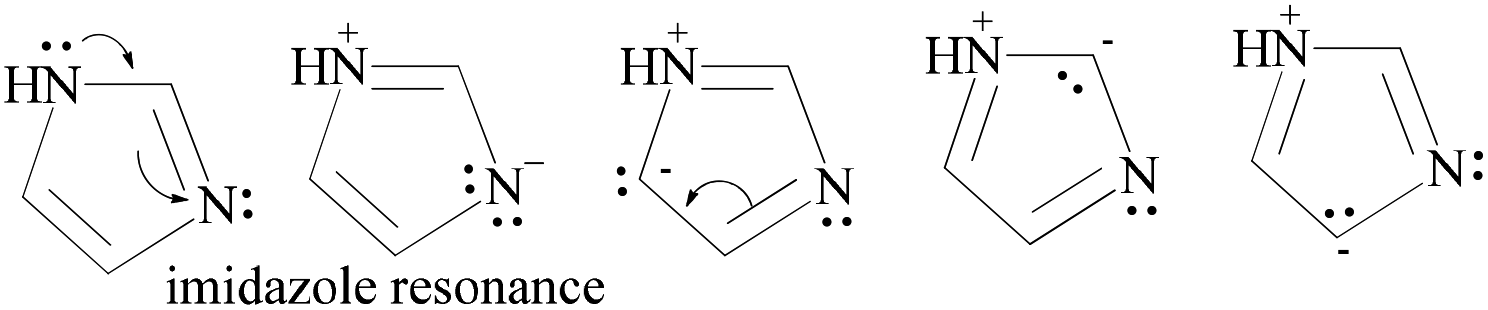 | ||
| Scheme 4 The charge orientation mechanism of the imidazole structure to develop nucleophilic and electrophilic centers to conduct the reaction in the prescribed medium. | ||
This mechanism leads to covalent bond formation, which is further broken by hydrolysis.
Imidazole being amphoteric acts as an acid and a base, where an acidic proton is bound to the nitrogen, which deprotonates to form an imidazole anion. The basic site is the nitrogen with a lone pair (and not bound to hydrogen). Its protonation produces the imidazolium cation (Scheme 5). DMAP being basic acts as a nucleophilic catalyst for hydrosilylations with efficient kinetic resolution for phenolic –OH groups (Scheme 5).
 | ||
| Scheme 5 The proton shift from dihydroxybenzene to imidazole to sustain a reaction in collaboration with DMAP for the protection mechanism via nucleophilic and electrophilic centers in the prescribed medium and experimental conditions. | ||
Structure: 1H-NMR (500 MHz, CDCl3) δ: 0.19 (s, 6H), 1.00 (s, 9H), 6.72 (m, 4H). 13C-NMR (126 MHz, CDCl3) δ: −4.41, 18.27, 25.82, 116.22, 120.97, 149.26, 149.84.
 | ||
| Scheme 6 DTBDMSH deprotection mechanism to MTBDMSH and the dehydration mechanism of n-Bu4NF·3H2O to assist MTBDMSH formation. | ||
The mixture was evaporated, and the residue was collected in ethyl acetate and washed with saturated NaHCO3 and finally with brine. The residue was purified by flash chromatography with hexane and ethyl acetate in a 4:1 ratio to get a colorless solid. TBAF maintains solubilization of the reacting species. The 3H2O of n-Bu4NF·3H2O seems to facilitate the hydrolysis of these silyl ethers through bifluoride (HF2− or H+) and hydroxide (OH−), as well as fluoride. Silyl ethers are hydrolyzed in THF, an aprotic solvent with 7.6 debye. It moderately dissolves n-Bu4NF·3H2O, DTBDMSH, and MTBDMSH. It is water-miscible for hydrolysis of both DTBDMSH and MTBDMSH for better hydrolysis at RT. The structure to understand the role of TBAF in the reaction mechanism is given below.
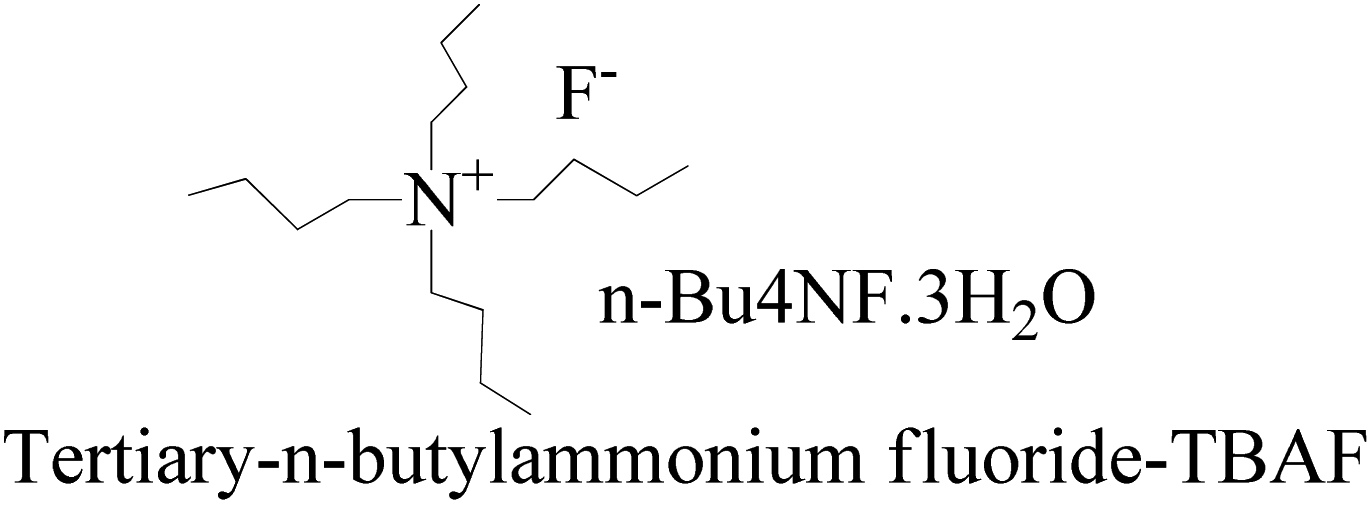
Silyl ethers protect groups from alcoholic or phenolic OH in organic synthesis as they combine with different groups for a number of silyl ethers. Ether groups provide selectivity for protecting group chemistry. Trimethylsilyl (TMS), tert-butyldiphenylsilyl (TBDPS) and tert-butyldimethylsilyl (TBDMS) are silyl ethers which are installed and removed very selectively under mild conditions.
:1 ratio and a colorless solid (61%) was collected. Imidazole and DMAP assist safer protection of the phloroglucinol with silyl ethers. MTBDMSP (mono-tertiary butyl dimethyl silane phloroglucinol), DTBDMSP (di-tertiary butyl dimethyl silane phloroglucinol) and TTBDMSP (tri-tertiary butyl dimethyl silane phloroglucinol) were obtained with >90% TTBDMSP.
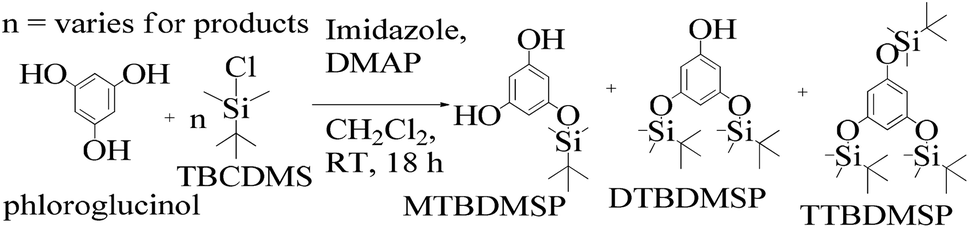 | ||
| Scheme 7 The phloroglucinol and TBCDMS mechanism in DMAP and CH2Cl2 medium at RT for preparing MTBDMSP and other species in small amounts. | ||
Therefore, a unique mathematical representation of the participating molecules could be presented as follows:
| P + nTB = (n − 1)MT + (n − 2)DT + (n − 3)TT |
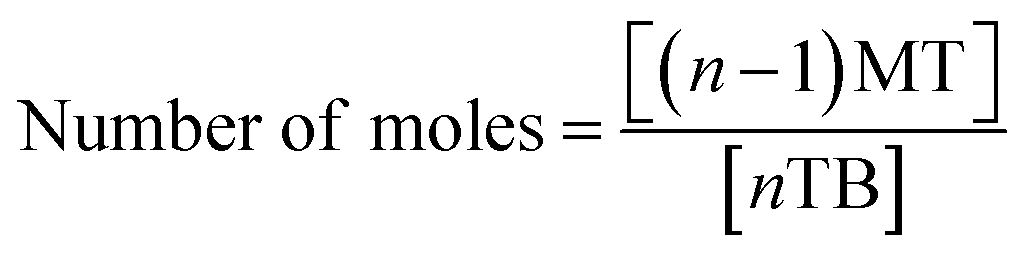
Since TBCDMS and MTBDMSP interact in a 1:1 ratio, the stoichiometry for MTBDMSP could be considered as:
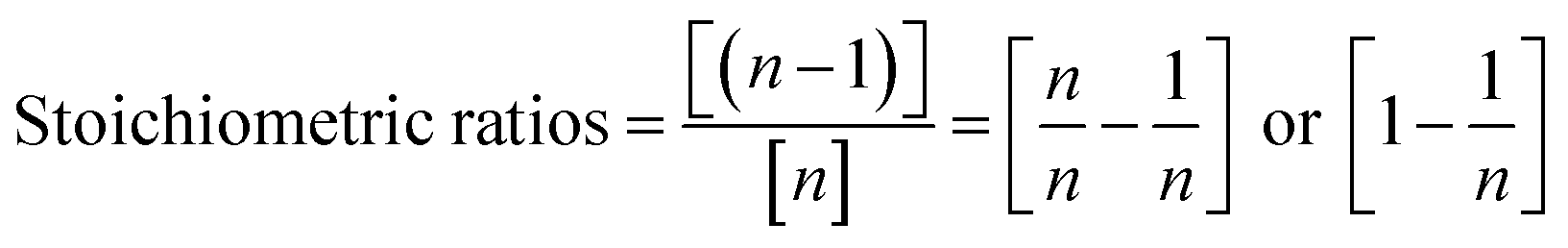
Similarly, the number of molecules calculated for DTBDMSP and TTBDMSP is:
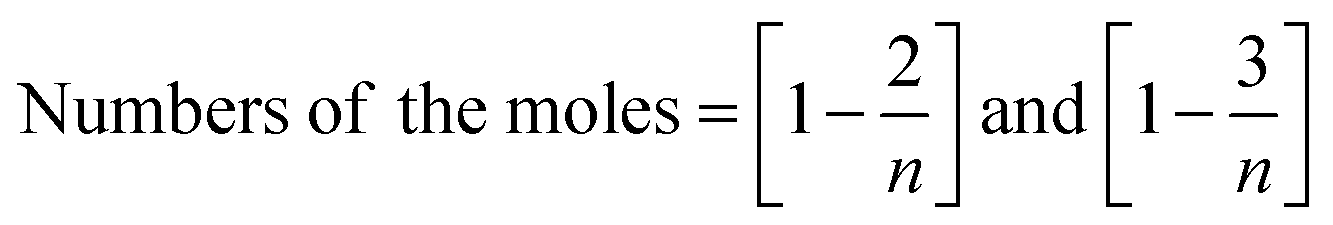
Structure: 1H NMR (500 MHz, CDCl3) δ: 0.18 (s, 6H), 0.96 (s, 9H), 4.73 (s, 1H), 5.94 (s, H) 5.98 (s, 2H). 13C NMR (126 MHz, CDCl3) δ: −4.28, 18.34, 25.79, 101.32, 105.24, 156.95, 157.41. HR-MS (CH3CN) + (m/z) found: 354.3447; calc value for C18H34O3Si2: 354.2046.
2.3 DTBDMSP deprotection
As shown in Scheme 8, a mixture of the silyl ether of TTBDMSP (tri-tertiary butyl dimethyl silane hydroquinone) (1 mM) and n-Bu4NF·3H2O (0.3 mM) in dry THF was stirred @700 rpm for 1 h at 0 °C. The n-Bu4NF·3H2O catalyzed the hydrolysis of TTBDMSP to get DTBDMSP and MTBDMSP. The solvent is evaporated, and the residue was collected in ethyl acetate. It was washed with dilute HCl, then saturated NaHCO3 and finally with brine. The residue was purified by flash chromatography using hexane and ethyl acetate in a 4:1 ratio for the solid product. TBAF catalyzed the hydrolysis of the silyl ethers at RT in a sequential manner. Three times tert-butyldimethylsilyl hydroxide was also obtained as a byproduct.
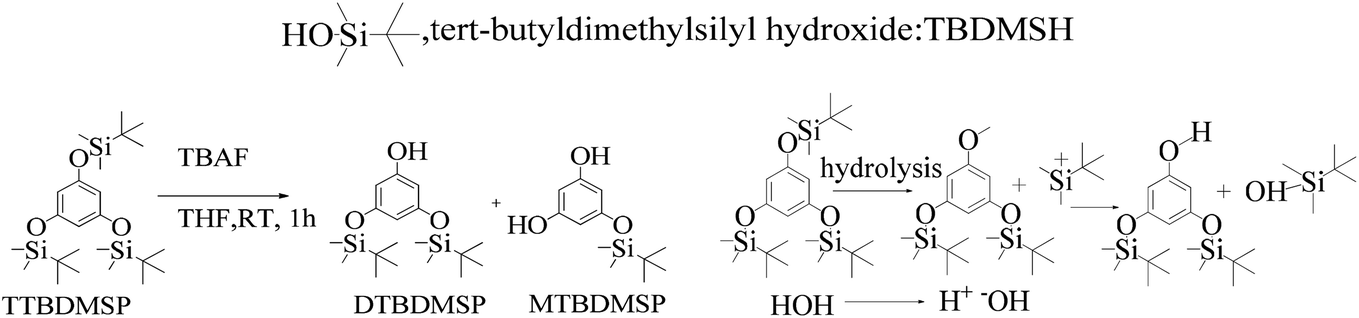 | ||
| Scheme 8 The TTBDMSP deprotection mechanism in the prescribed medium to prepare MTBDMSP and other species in small amounts at RT. | ||
2.4 1st generation dendrimer preparation
A 1 mM solution of MTBDMSP, DTBDMSP or TTBDMSP (3.2 equiv.) phenolic derivative and 2-methylphenol separately and t-BuOK base (3.1 equiv.) in dry THF (10 ml) were prepared. 1 equiv. TMC solution in THF was also prepared and added to the PD solutions separately at RT. The contents were vigorously stirred @700 rpm and the reaction was monitored with TLC. The brief mechanism is depicted in Scheme 9.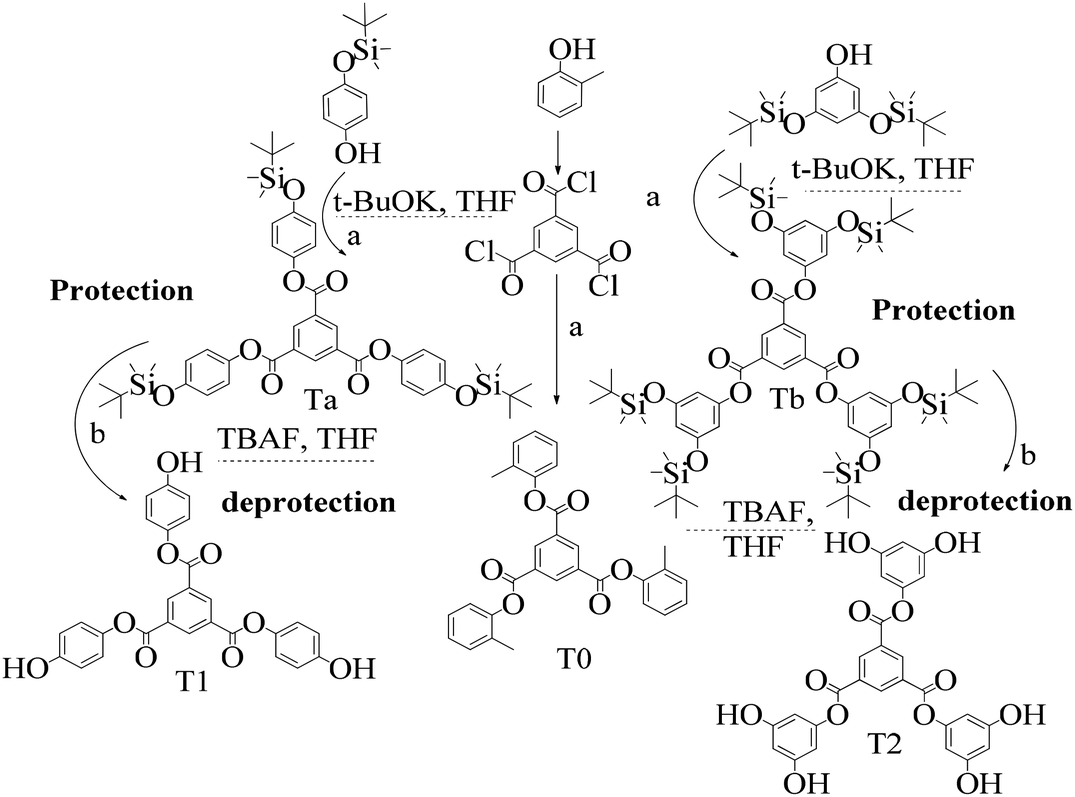 | ||
| Scheme 9 Brief illustration of 1st tier dendrimer synthesis: (a) t-BuOK, THF; (b) TBAF, THF. | ||
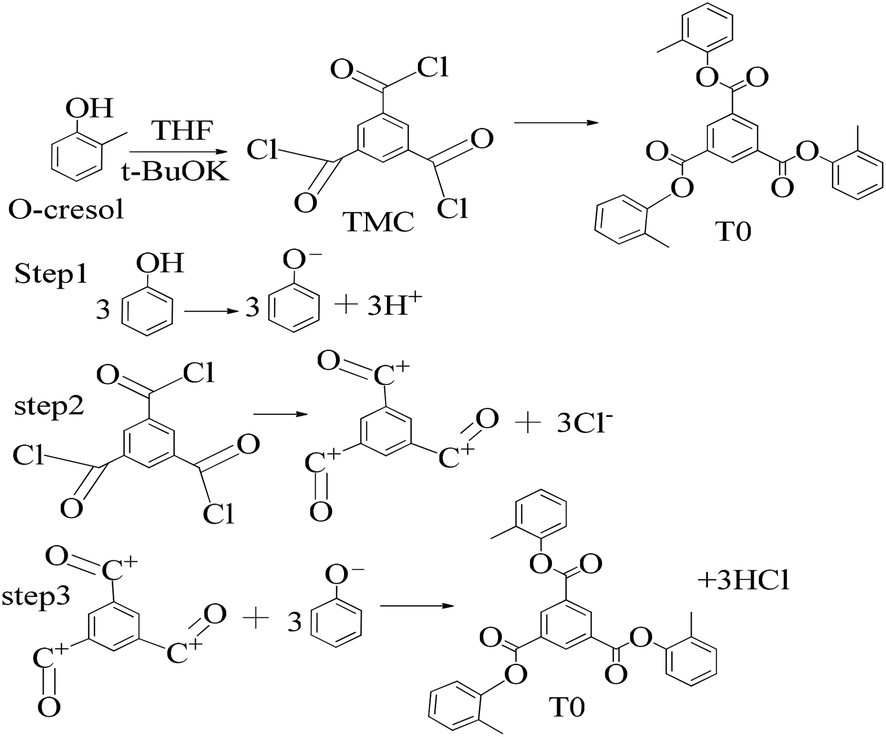 | ||
| Scheme 10 Synthesis of tri-o-tolyl benzene-1,3,5-tricarboxylate (T0) in the prescribed medium in three steps at RT. | ||
Structure: Rf 0.75 (25:75: EtOAc:n-hexane); 1H NMR (500 MHz, CDCl3): δ 9.29 (s, 3H), 7.25 (m, 13H), 2.28 (s, 9H) ppm; 13C NMR (125 MHz, CDCl3): δ 163.29, 149.38, 136.27, 131.59, 131.38, 130.33, 127.39, 126.77, 121.95, 16.53. HR-MS (CH3CN) + (m/z) found: 480.9243; calc. value for C30H24O6: 480.1573.
Structure: Rf 0.75 with an EtOAc and hexane mixture in 20:80 ratio. Ta was used to synthesize T1 as such (Scheme 9). 1H NMR (500 MHz, CDCl3) δ 9.13 (s, 3H), 7.12 (d, J = 8.4 Hz, 6H), 6.90 (d, J = 8.5 Hz, 6H), 1.00 (s, 27H), 0.22 (s, 18H). 13C NMR (126 MHz, CDCl3): δ 163.5, 153.6, 144.4, 135.9, 131.3, 122.1, 120.7, 25.6, and 4.4.
Structure: Rf 0.75 1H NMR (500 MHz, CDCl3) δ 9.13 (s, 3H), 6.40 (s, 6H), 6.29 (s, 3H), 0.98 (s, 54H), 0.23 (s, 36H); 13C NMR (126 MHz, CDCl3) δ 4.29, 25.76, 29.85, 107.28, 110.33, 131.25, 136.23, 151.64, 157.12, 163.19.
:0.5 ratio to get a white solid (90%).
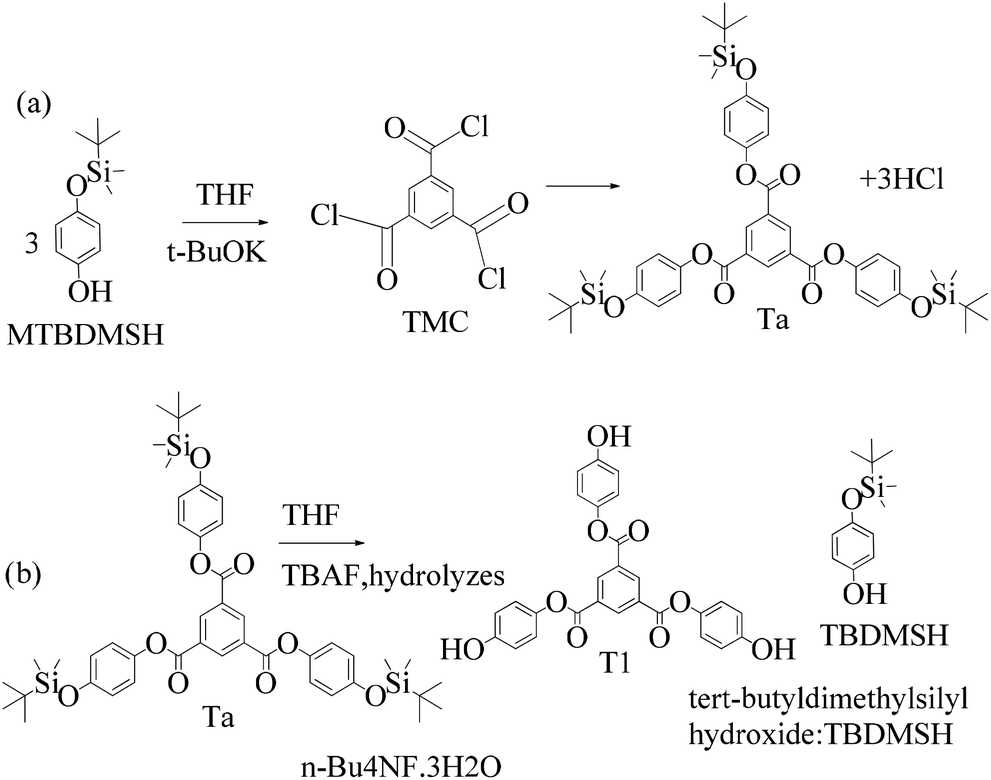 | ||
| Scheme 11 (a) and (b) Synthesis of tris(4-hydroxyphenyl) benzene-1,3,5-tricarboxylate (T1) in the prescribed medium at RT. | ||
Structure: 1H NMR (500 MHz, DMSO) δ 9.61 (s, 3H), 8.84 (s, 3H), 7.13 (d, J = 8.0 Hz, 6H), 6.82 (d, J = 8.0 Hz, 6H); 13C NMR (125 MHz, DMSO): δ 163.63, 155.48, 142.54, 134.63, 130.67, 122.60, and 115.76. HR-MS (CH3CN) + (m/z) found: 487.1025; calc. value for C27H18O9: 487.0951.
:1 ratio.
 | ||
| Scheme 12 (a) and (b) Synthesis of tris(3,5-dihydroxyphenyl)benzene-1,3,5-tricarboxylate (T2) in the prescribed medium at RT. | ||
Structure: 1H NMR (500 MHz, DMSO) δ 9.69 (s, 6H), 8.81 (s, 3H), 6.15 (s, 6H), 5.76 (s, 3H). 13C NMR (126 MHz, DMSO): δ 158.99, 151.88, 144.15, 134.78, 131.76, 100.49, 100.17. HR-MS (CH3CN) + (m/z) found: 535.0872; calc. value for C27H18O12: 535.0798.
Efficient and facile methods for the synthesis of trinuclear Schiff base ligands derived from chiral aminoalcohols were reported elsewhere using vanadium catalysts, but these are altogether different studies. Our study is focused on using free –OH groups to release H+ ions to scavenge free radicals in a safer manner. Thereby, the protection and deprotection routes were adopted in very simple chemical processes without using any metallic catalyst. The rate of release of H+ is significant to scavenge free radicals. In this context, T1 and T2 dendrimers have been proven very effective within the standard norms of antioxidant activities.21
3. Results and discussion
3.1 Characterization
The Ta and Tb were deprotected and the peaks for silyl ether were absent in T1 and T2, predicting the structures. The mass spectra of T0, T1, and T2 implied the structures. T0 with larger pi conjugation and effective delocalization of the terminal phenyl groups, core, and LPE of the O atoms of its ester groups is optically active compared to T1 and T2. The dendrimers are insoluble in water and their solutions were prepared in organic solvents. T0 was dissolved in acetone, while T1 and T2 were dissolved in methanol for the optical activities study. They were mixed with DPPH free radicals. T1 and T2 were also mixed with aqueous solutions of BSA and lyso at RT. The DPPH solution was prepared in methanol. T0 did not scavenge free radicals, while T1 and T2 both scavenged them. Such experiments are of significance as free radicals initiate undesired activities in biological and biochemical processes. The radicals are highly energetic and interact with proteins, enzymes, and other sensitive biomolecules in cells. T1 and T2 acted as biocompatible sensors as they scavenged free radicals and bound the proteins.
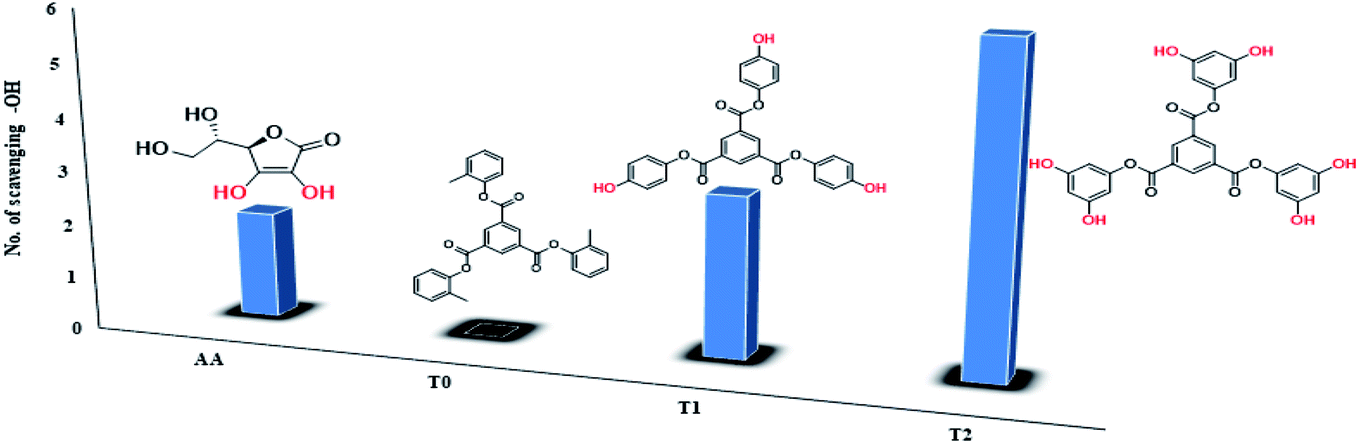
T1 and T2 with 3 and 6-OH acted as flavonoids to detect DPPH (1,1-diphenyl-2-picrylhydrazyl) free radicals and scavenge them. T1 and T2 have aggregated BSA and lyso in their void spaces, which decreases their antioxidant activities compared to ascorbic acid as a standard antioxidant. The free radical scavenging kinetics for 25 μM (micromol L−1) to 1 mM (millimol L−1) T1 and T2 were investigated separately with DPPH at RT using UV-Vis spectrophotometry. This determined the efficient concentration (EC50) of T1 and T2 for DPPH radical scavenging calculated from UV-Vis absorption. T1 and T2 have 0.424 and 0.04312 mM EC50 values, respectively, and quickly scavenged radicals. T2 induced comparatively stronger electronic activities than T1 by acquiring more activation energy. The BSA and lyso concentrations were studied separately and found to be 0.424 and 0.04312 mM for T1 and T2, respectively. The binding of more BSA than lyso may be due to the fact that the BSA preferred entering the void spaces due to the hydrophobic environment of the void spaces.
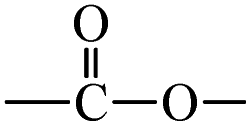 ) constituents as the linker, phenyl as the branches, and –CH3 with T0, 1-OH with T1 and 2-OH with T2, respectively, induced hydrophobicity in the structures, as shown in Fig. 1. The –OH groups shown in red have H+ releasing ability.
) constituents as the linker, phenyl as the branches, and –CH3 with T0, 1-OH with T1 and 2-OH with T2, respectively, induced hydrophobicity in the structures, as shown in Fig. 1. The –OH groups shown in red have H+ releasing ability.
 | ||
| Fig. 1 Structures of DPPH, AA, T0, T1, and T2. | ||
T0, T1, and T2 can bind greasy hydrophobic waste materials due to hydrophobic interactions. T0 with electron releasing 3-CH3 attached to its terminal phenyl group acted as a superhydrophobe as it is not soluble in methanol (CH3OH) but is soluble in acetone (CH3)2CO. T1 and T2 were dissolved in methanol, which indicated interaction of phenolic –OH and alcoholic –OH groups. T0 initiated hyperconjugation due to its sigma bond activities of the H atom of the –CH3 with the pi bond of the terminal phenyl. It adsorbed more UV/Vis light than T1 and T2. T0, T1, and T2 have H+ releasing 0, 3, and 6-OH respectively (Fig. 2) with scavenging activities.
 | ||
| Fig. 2 Number of –OH groups and DPPH free radical scavenging activities. | ||
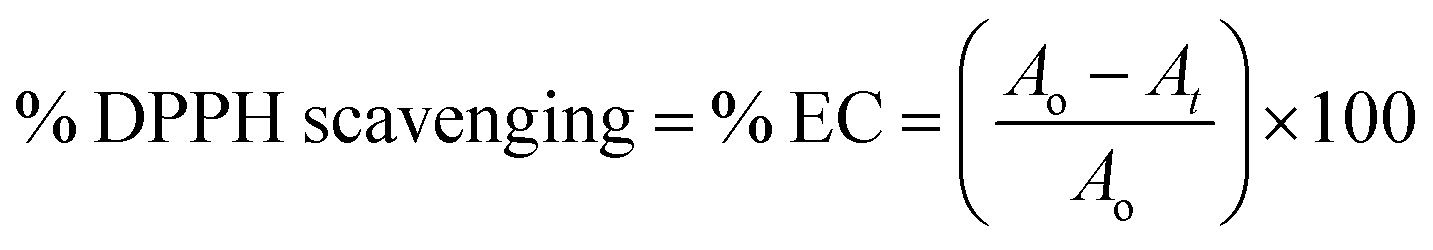 | (1) |
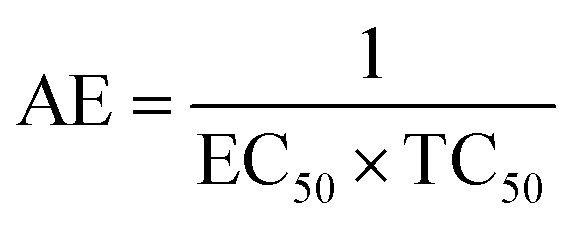 | (2) |
The scavenging capacities of T1 and T2 compared to ascorbic acid as a standard are evaluated.
|
[A] = [A]0e−kt
| (3) |
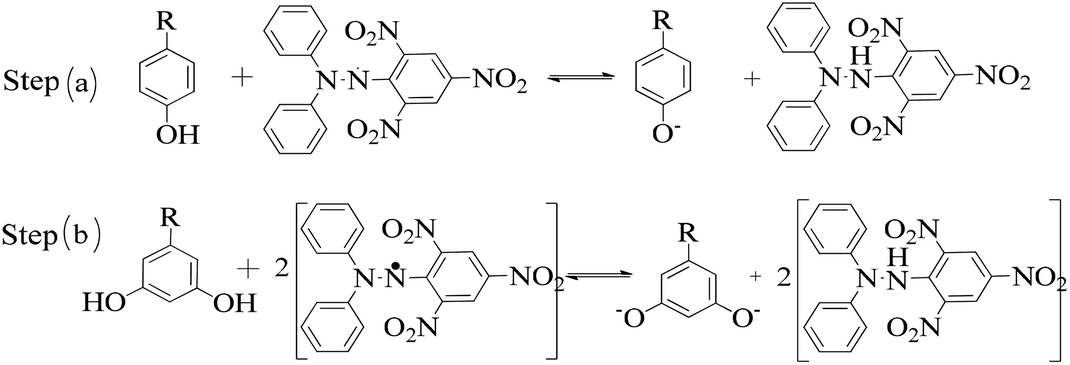 | ||
| Fig. 3 (a, b) Interacting activities of the dendrimers with DPPH radicals in two steps. Step (a) occurs when the ratio of dendrimer to DPPH is 1:1, while step (b) occurs with a 1:2 ratio according to the H+ holding ability of the PDs to decide the rate constant. | ||
The k values are calculated from the slopes of the UV/vis abs versus time plot. The DPPH concentration [A] left after time t s is determined from UV abs. The initial DPPH [A]0 concentration is known. The scavenging kinetics of DPPH with time ascertained its order. The Gibbs energy (ΔG, J mol−1) is determined for the scavenging activities of T0, T1 and T2 at RT. ΔG is calculated using the equation:32
|
ΔG = −2.303RTlog(A/A0)
| (4) |
3.2 Scavenging activities and kinetics of the dendrimers
The lowering of the UV/Vis absorption with DPPH on increasing the dendrimer concentration implies a significant DPPH scavenging (Fig. 5). The EC50 for the dendrimers is determined compared to the standard AA. Increasing the dendrimer concentration increased the scavenging activities depending on the individual amount of T0, T1, and T2. DPPH scavenging depends on the H+ releasing ability of the dendrimers. A H+ ion accepts the extra electron of DPPH and develops a –N–H covalent bond by reduction. The scavenging ability increases on increasing their concentrations because the number of –OH also increases, but despite the release of H+, their pH remains constant. T1 and T2 act as a buffer with adequate DPPH amount as the released H+ is directly engaged in FR scavenging without affecting the pH. This is a novelty of T0, T1 and T2. The moment they release H+, it is used by an unshared electron on the N atom of DPPH. The H+ release is balanced by a delocalization of the phenyl or benzene ring. T0 has 3 terminal phenyl groups containing one –CH3 each. T0 does not have –OH so no H+ is released and no free radical scavenging occurred. T0 is similar to TTDMM reported in our previous work, but it has delocalization in the branching contrary to T0, so it can encapsulate the drug silibinin. T1 with 3-OH and T2 with 6-OH scavenged DPPH by 94 and 92%, respectively, at 0.424 and 0.4312 mM respectively. T1 and T2 release 3 and 6H+, respectively, but T1 with 50% less H+ scavenged FRs by 2% more than T2. The H+ releasing action of T1 is stronger than that of T2, due to an interruption as 2-OH are placed at the meta position of benzene. Such interference occurs due to electron–electron interactions among their LPEs, which is missing in T1. T0 contains a H atom in TMC as well as in the terminal benzene rings and –CH3, but no H+ was released. T1 and T2 have detected H+ release. T1 and T2 have H+ releasing –OH so they tune their electron delocalization. The H+ releasing activities of T1 and T2 are controlled by their extended delocalization. T1 produced 94 and T2 produced 92% scavenging. The rate of release and use of H+ should have been equal but release required some activation energy due to interrupted delocalization as is noted in Fig. 4. The –OH with 2 LPEs on the O atoms initiate electron withdrawing activities from the benzene rings of T1 and T2, where T1 facilitates expeditious H+ release without any interruption. So T1 does not undergo any electronic reorientation contrary to T2. It faces interruption due to another –OH as it also initiates extended conjugation. It needs comparatively more activation energy as it has a comparatively higher potential energy (Fig. 4). The activation energies of T1 and T2 are T2 > T1 with frequency factor A being T1 > T2. Abs is a function of time, min, so these data are calculated using the Arrhenius eqn (5).
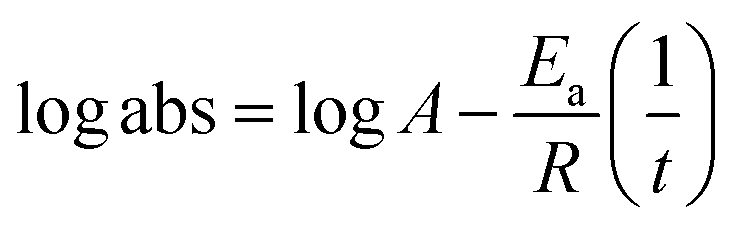 | (5) |
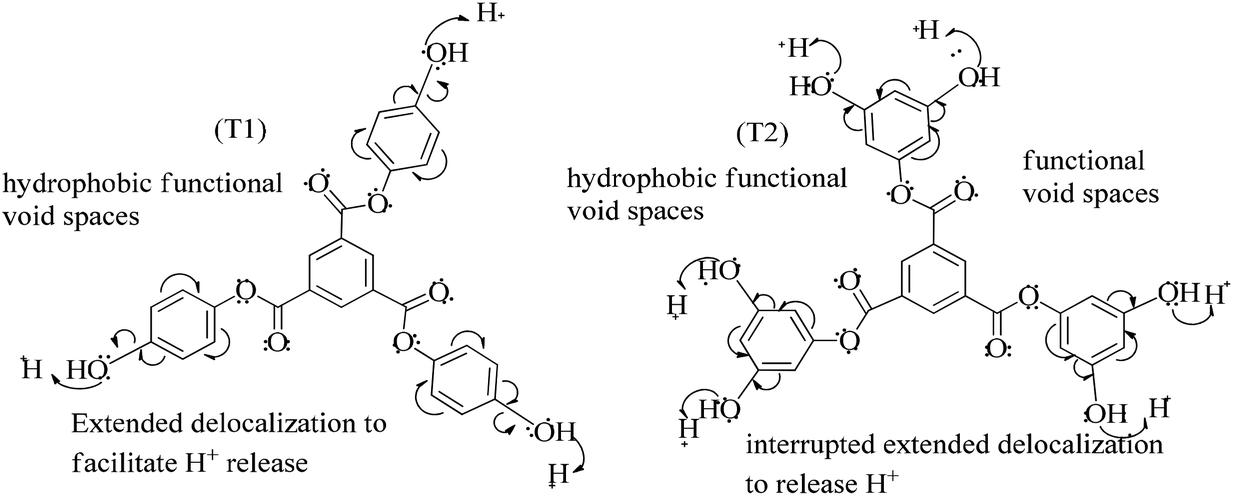 | ||
| Fig. 4 H+ releasing activities of T1 and T2 to scavenge DPPH radicals. | ||
The higher activation energy for T2 than T1 implies stronger T1 spontaneity in releasing H+ compared to T2. This is supported by the higher frequency factor A for T1. Regression analysis of the data is given in the equation depicted in Fig. 5 along with the respective plots. The 2-OH of T2 restricts its number of collisions compared to T1 because of a localized competitive extended delocalization of both the –OH groups. A weaker H+ releasing spontaneity is observed for T2 that required higher activation energy, which could be possible as both the –OH groups may also develop hydrogen bonding. T2 scavenged DPPH by 92% compared to 94% for T1, so the numbers of –OH do not matter to scavenge FRs but their position on the host unit decides their scavenging activities.
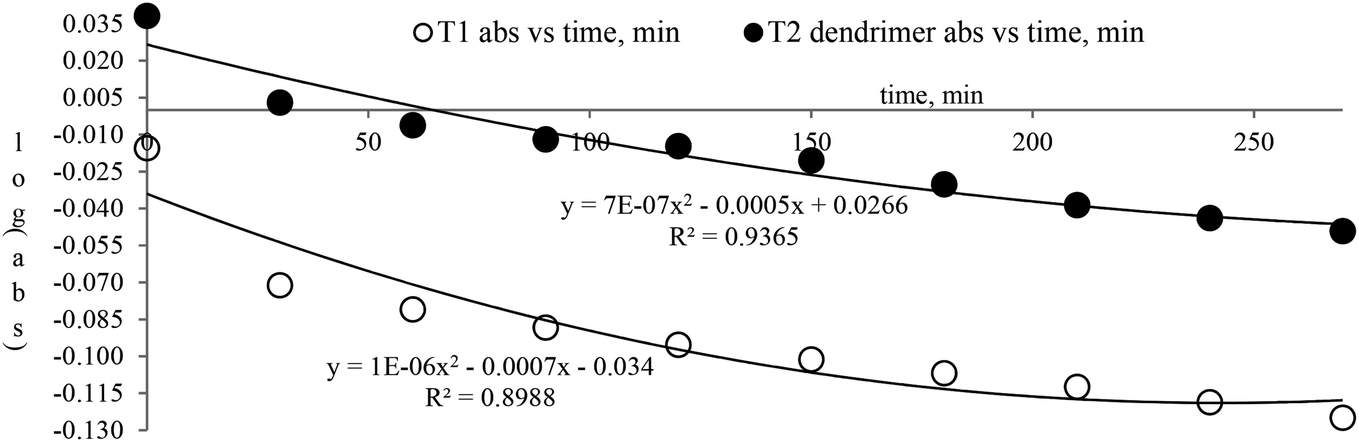 | ||
| Fig. 5 Abs vs. time of T1 and T2. | ||
Uninterrupted delocalization of T1 due to 3H+ and the presence of 6H+ with T2, respectively, occurs through electron or delocalization activities. Thus initiating H+ release as well as scavenging requires energy. On getting the required activation energy, their reorientations tune the nucleophilic and electrophilic sites as per sp2 to sp3 electronic transitions through the HOMO (highly occupied molecular orbital) and LUMO (lowest occupied molecular orbital) of T0, T1, and T2. The activation energy requiring activities of these two sites are not equal for T0, T1, and T2 and create a hysteresis in their optical activities vs. scavenging potential.
T0 among AA, T0, T1, and T2, has higher abs due to hyperconjugation as it has electron releasing –CH3 (Fig. 6). A sigma bond of –CH3 and pi bond of phenyl initiate hyperconjugation, which detains a larger amount of UV/Vis light. Fig. 6 shows the higher abs with DPPH when AA was not added so a larger population of radicals absorb much UV/vis light. On increasing the AA concentration in the DPPH solution, the H+ release simultaneously decreased abs. A single or unpaired electron developed intensified cycles of h+ and e− holes, which engage UV light or photons. The H+ neutralized an unpaired single electron of DPPH and the lowest number of redox cycles occurs. On using H+, the UV/Vis abs is decreased. The H+ consumption occurred at a similar λmax. No transitions occur in the structures except for the release of H+ from T0, T1, and T2 and the consumption of the released H+ by DPPH. Experiments at different pH, and the scavenging activities of T0, T1, and T2 are being pursued and will soon be communicated. The H releasing mechanisms of T1 and T2 are controlled by their extended delocalization, which induces faster sp2 to sp3 electronic transitions on receiving UV/Vis light. T1 predicted 94% and T2 92% scavenging. The rates of release and use of H+ should have been equal or more for T2 as it has 2 times more H+ in its structure. Probably H+ release requires more activation energy for reorientation of molecules to match the nucleophilic and electrophilic sites, which requires time. These two events are not equal so a difference occurs in H+ release and FR scavenging. A linear relationship between abs vs. AA concentration indicates direct scavenging. A 0.5 mM AA stock solution was prepared in MeOH and from 5.0 to 200 μM scavenged DPPH radicals. The AA from 5.0 to 55 μM had a reduced transition state due to protonation of DPPH radicals. But 55 μM AA stopped responding to UV abs at 520 nm due to complete AA oxidization by DPPH radicals getting reduced themselves. The decrease in abs is almost constant on increasing the AA concentration (Fig. 7 and 8). The EC50 value calculated from the abs vs. concentration plot is 32.61 μM. T0 was dissolved in acetone from 10.0 to 400.0 μM and the abs for T0 is plotted in Fig. 7. The T0 undergoes aggregation on increasing its concentration from the initial 10.0 μM. These increments in abs on increasing T0 concentration largely occur as it does not have electron donating –OH or H+ releasing groups, so it did not scavenge, rather it gets monodisperse and absorbs visible light at 520 nm from 20.0 to 300.0 μM. T0 has 3 phenyl groups each with one –CH3, which induce adequate electronic transitions with more UV/Vis light abs. However, after this concentration range with DPPH its abs increased drastically (Fig. 7(A–E)). This shows stability in abs due to saturation in the electronic transitions from HOMO to LUMO. The abs is increased at a certain concentration with a 2nd order mechanism compared to the 1st order interaction of T1 and T2. T0 activates DPPH to absorb visible light from 10.0 to 300.0 μM with a minor increment. This is not due to radical scavenging but due to π–π interactions. Visible light absorption by T0 implies activities of π-conjugation of phenyl groups, LPEs of O atoms, and electronic activities of its –CH3. These electronic activities imply its structural stability and active optical activities.
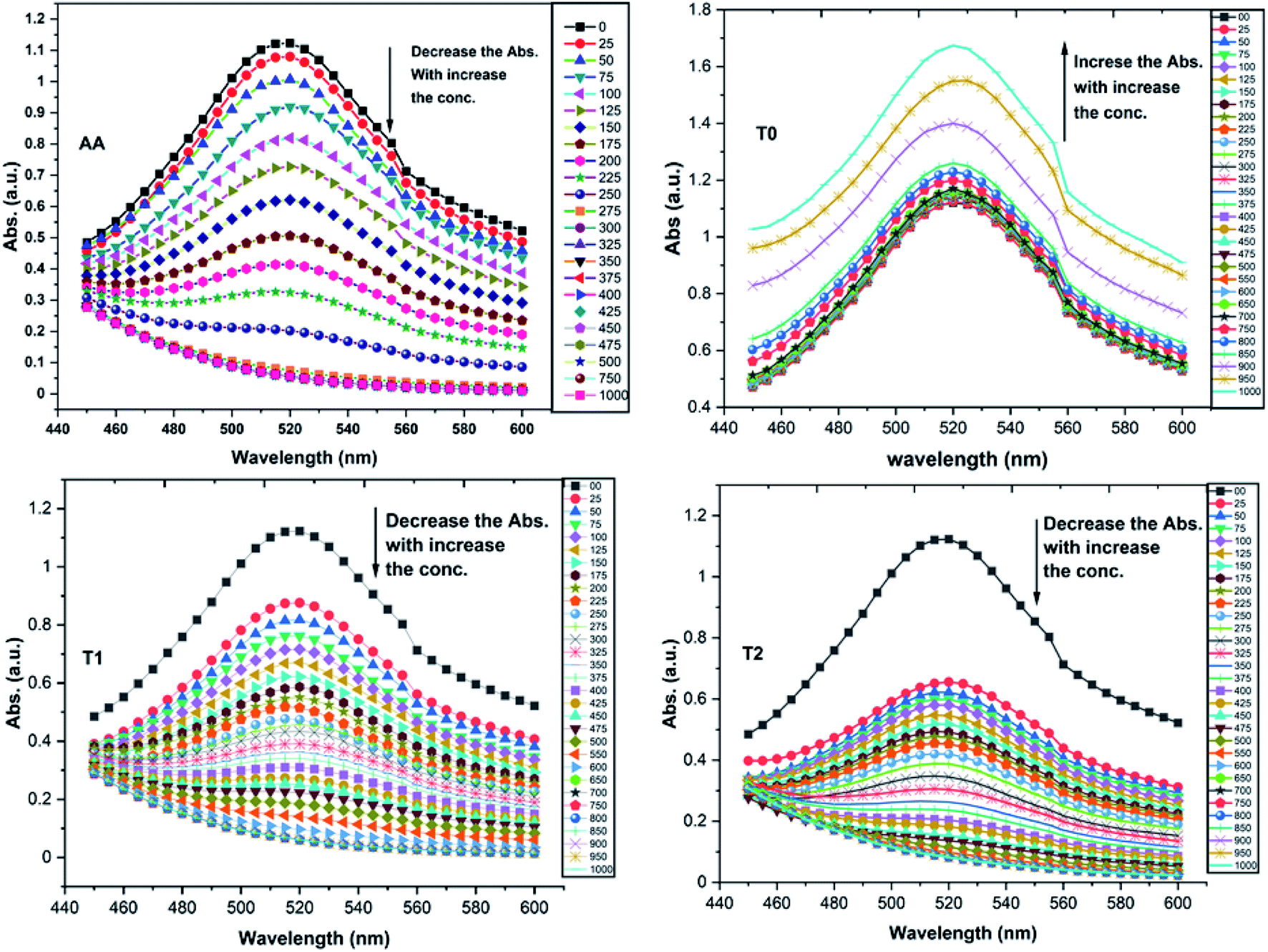 | ||
| Fig. 6 UV-Vis abs spectra for DPPH radicals with AA (ascorbic acid), T0, T1, and T2. | ||
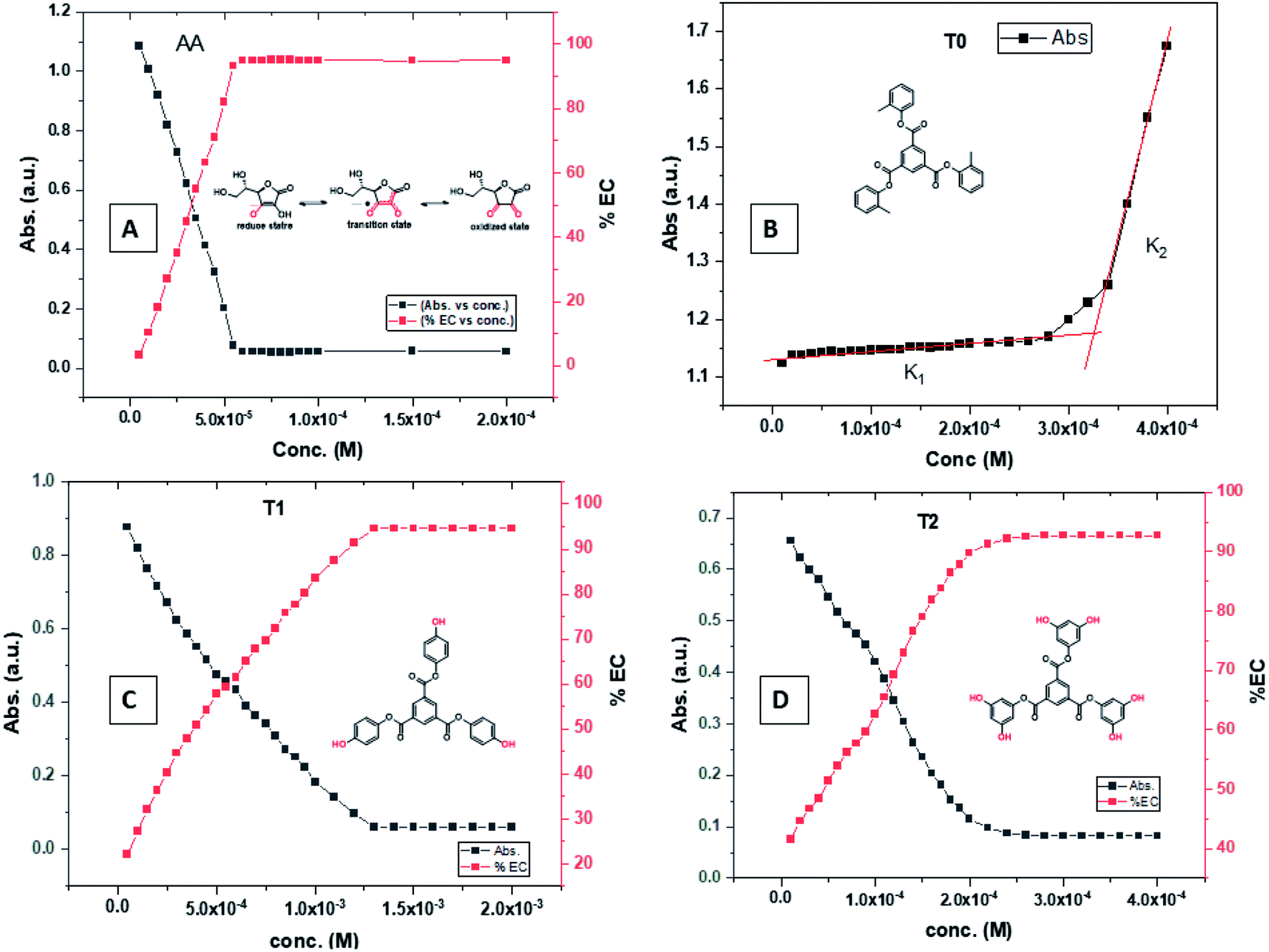 | ||
| Fig. 7 (A) Abs vs. conc. and abs vs. % EC of AA, (B) abs vs. conc. of T0, (C) abs vs. conc. and abs vs. % EC of T1, (D) abs vs. conc. and abs vs. % EC of T2. | ||
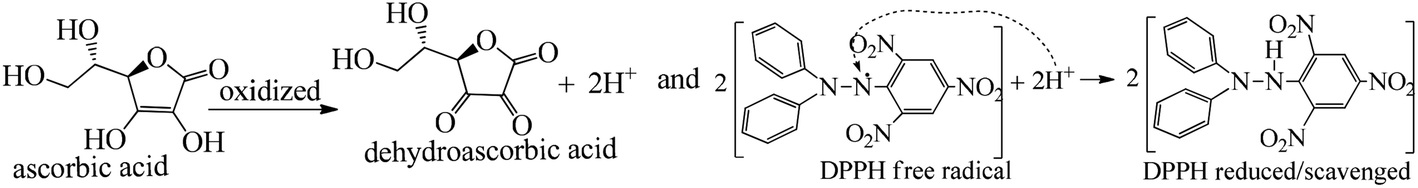 | ||
| Fig. 8 DPPH scavenging mechanism of ascorbic acid to dehydroascorbic acid. | ||
The electron releasing –CH3 and benzene ring induce coulombic interaction with T0 due to intramolecular electronic orientations. Delocalization of the π bond of the terminal phenyl along with ERG develops a negative charge at the center of the phenyl, which may repel DPPH radicals to other energy states. Electron–electron repulsions cause aggregation of T0, rather than opting for monodispersion; however, the medium plays a critical role. Acetone tends towards DPPH so after a certain concentration, T0 undergoes self-aggregation, and UV/Vis implies it as the critical aggregation concentration. Contrary to that, T0 showed increased abs with concentration, and T1 and T2 both showed decreased abs at 520 nm. The decreasing abs rate from T1 and T2 changes with concentration due to 3 and 6-OH with T1 and T2, respectively. Abs vs. concentration curves of AA, T0, T1, and T2 are depicted in Fig. 7(A–D); AA shows a sharper transition in DPPH scavenging, while T0 with more UV/Vis abs produces a smooth transition in optical activities at 3.3 × 10−4 M. A higher abs by T0 occurs due to electron releasing 3-CH3, due to stronger transition as no part of the abs is lost contrary to T1 and T2 which participate in releasing H+ on absorbing UV/Vis light. T0 is a robust UV/Vis light absorbent, and it has photocatalyzed the conversion of fluorescent dye methylene blue to colorless leuco methylene blue (Fig. 10A).
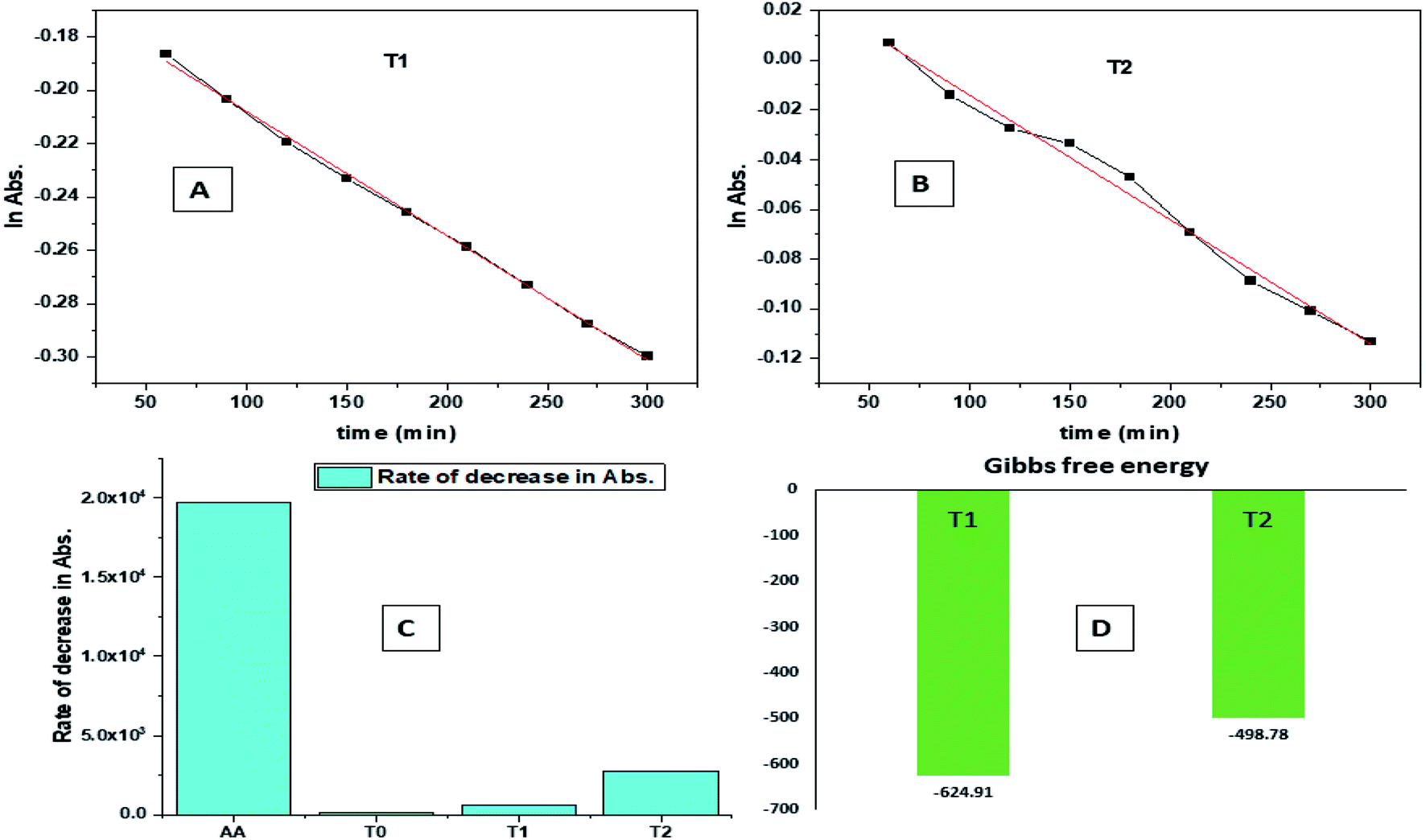 | ||
| Fig. 9 (A) Time vs. ln abs of T1. (B) Time vs. ln abs of T2. (C) Rate of decrease in abs. (D) Gibbs free energy. | ||
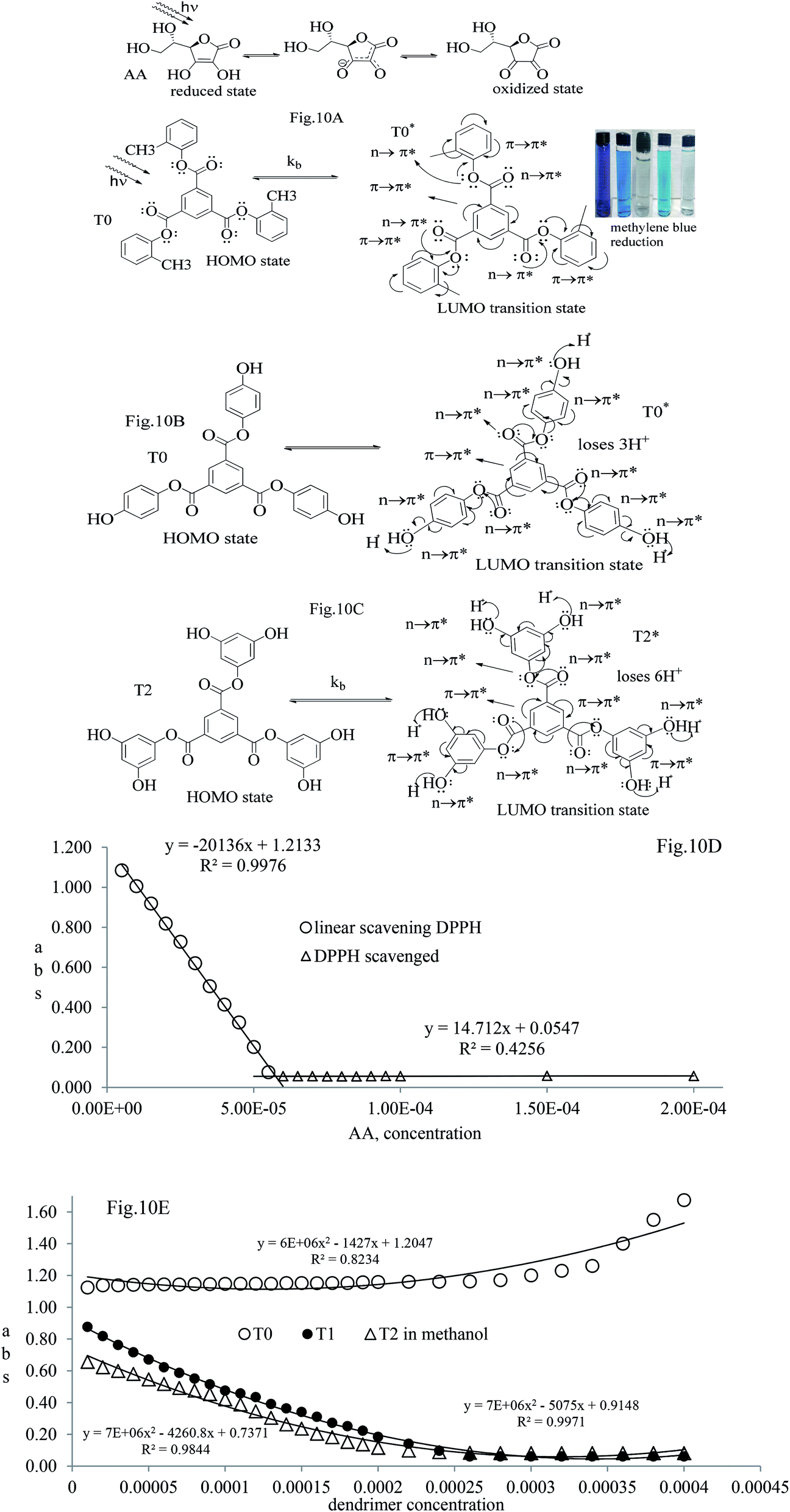 | ||
| Fig. 10 (A–C) Optical activities from the HOMO to LUMO to scavenge DPPH radicals. (D) Abs vs. AA composition with 1st order scavenging of DPPH free radicals. (E) Abs vs. dendrimer composition to scavenge DPPH free radicals. | ||
The H+ releasing activity of ascorbic acid is active as its 2-OH undergoes electronic transition to release H expeditiously, contrary to the dendrimers. T0 did not produce a sharper absorption point on increasing the concentration, but T1 and T2 produced a smooth saturation point of abs (Fig. 7A–D). One ascorbic acid reduced 2 DPPH radicals (Fig. 8). Also T1 and T2 each have reduced 3 and 6 DPPH radicals, respectively. Fig. 9(A–D) imply free radical reducing kinetics from 0 to 300 min for fixed compositions of radical and of antioxidants under similar experimental conditions. The H+ release and utilization is in a 1:1 ratio. The rates of abs by AA, T0, T1, and T2 decrease. This is supported by Gibbs free energy (Fig. 9A–D). T2 has higher Gibbs energy with 2-OH, which increases the stability.
The scavenging ability of T2 is 10 times higher than that of T1 so the MIC (minimum inhibition concentration) for T2 is ten times lower than that for T1. A single T2 releases 6H+ against 3H+ from T1 (Table 1). This denies a possibility of hydrogen bonding between the two species at a common benzene ring along with a constant effect of π conjugation. T1 and T2 follow a straight-line decrease in abs like AA, so no generation of a secondary chemical mechanism other than the scavenging activity occurs.
| Compound | Scavenging –OH | EC50, M (mol L−1) | Max, % EC | TC50 (min) | AE | Rate constant (M s−1) |
|---|---|---|---|---|---|---|
| T0 | 0 | — | — | — | — | — |
| T1 | 3 | 4.24 × 10−4 | 94.57 | 811.11 | 5.23 × 10−7 | 3.86 × 10−5 |
| T2 | 6 | 4.312 × 10−5 | 92.70 | 945.6 | 4.56 × 10−8 | 4.73 × 10−4 |
T1 may release H+ in three steps and scavenge FRs in three steps with three scavenging orders. The 1st H+ release and scavenging are faster as initially no H+ exists and no FR was scavenged so dispersion seems active. The 2nd H release could comparatively be lower and a 3rd H release could be lowest. For T2, the order to releasing H and scavenging FRs seems to be in 6 steps with subsequent lowering in the H release and scavenge steps. This is one reason that the T1 scavenging is 10 times faster than that of T2.
| T0 + hν ↔ T0*, | (6) |
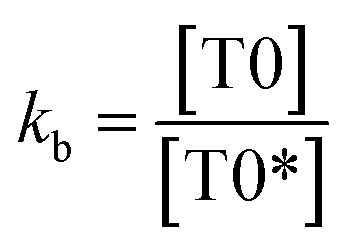 | (7) |
| NMR peak |
|
|
|
|||
|---|---|---|---|---|---|---|
| 1H | 13C | 1H | 13C | 1H | 13C | |
| a The –CH3 ERG at the p-position of the terminal benzene deshields the H of the benzene ring in the core. | ||||||
| Core benzene | δ 9.29 (s, 3H) | 136.27, 130.33 | δ 8.84 (s, 3H) | 134.63, 130.67 | δ 8.81 (s, 3H) | 134.78, 131.76 |
| Peripheral benzene | δ 7.25 (m, 12H) | 149.38, 131.59, 131.38, 127.39, 126.77, 121.95 | δ 7.13 (d, 6H), δ 6.82 (d, 6H) | 142.54, 155.48, 122.60, 115.76 | δ 6.15 (s, 6H), δ 5.76 (s, 3H) | 151.88, 144.15, 100.49, 100.17 |
| Ester bond | — | 163.29 | — | 163.63 | — | 158.99 |
| –CH3 | δ 2.28 (s, 9H) | 16.53 | — | — | — | — |
| –OH | — | — | δ 9.61 (s, 3H) | — | δ 9.69 (s, 6H) | — |
The kb predicts a transition state but after a certain concentration of AA, it is stabilized by developing an extreme transition state. Due to the –CH3, the T0 detains maximum UV/Vis light but it saturates at 2.0 × 10−4 μM as the abs starts increasing with its concentration. The AA did not release H+ causing electronic vibrations and oscillations which get saturated as the AA–AA–DPPH interactions might have aggregated the DPPH radicals. The abs with T0, T1, and T2 distinguishes the H+ releasing energy (Table 3).
| System | lnA |
S | S′ | A | Ea | kb | dG |
|---|---|---|---|---|---|---|---|
| AA | 8.46 × 103 | −0.1823 | 7.00 × 10−7 | 0 | 1.5156 | 1.2133 | −208.142 |
| T0 | 9.72 × 10−2 | −2.00 × 10−6 | 2.00 × 10−11 | 1.2508 | 1.66 × 10−5 | 1.2047 | −200.484 |
| T1 | −1.01 × 10 | 0.0002 | −1.00 × 10−8 | 10.3398 | 1.66 × 10−3 | 0.9148 | 95.86546 |
| T2 | −1.04 × 10 | 4.00 × 10−5 | −3.00 × 10−10 | 10.9724 | 3.33 × 10−4 | 0.7371 | 328.3781 |
Scavenging efficiency was calculated with eqn (8), and A0 = 1.123 is the abs for DPPH in methanol, whereas the abs A values for T1 and T2 at maximum scavenged DPPH sample in methanol.
 | (8) |
Abs values where maximum DPPH was scavenged were used from Fig. 10D and E. The AA, T1, and T2 scavenged 95.13, 93.32, and 94.21% DPPH, respectively.
Activation energies and frequency factor and DPPH radical scavenging were calculated fitting abs with Arrhenius eqn (5). The binding or scavenging constant was determined by analyzing the rate of radical scavenging on increasing the dendrimer composition. These are fitted using the Nernst equation for Gibbs energy determination (Table 3).
|
dG = −nRTlnkb
| (9) |
The activation energy and scavenging dG are correlated with frequency factor. T2 needs higher activation energy as it has 2-OH, which hinders extending the delocalization of the terminal benzene ring (Fig. 11A). The Ea values listed as AA > T1 > T2 > T0 imply that AA requires the least energy for releasing 2H+, whereas T0 needs higher activation energy to reach the LUMO from the HOMO (Fig. 11A). T2 compared to T1 needs more activation energy than T1 in releasing H+.
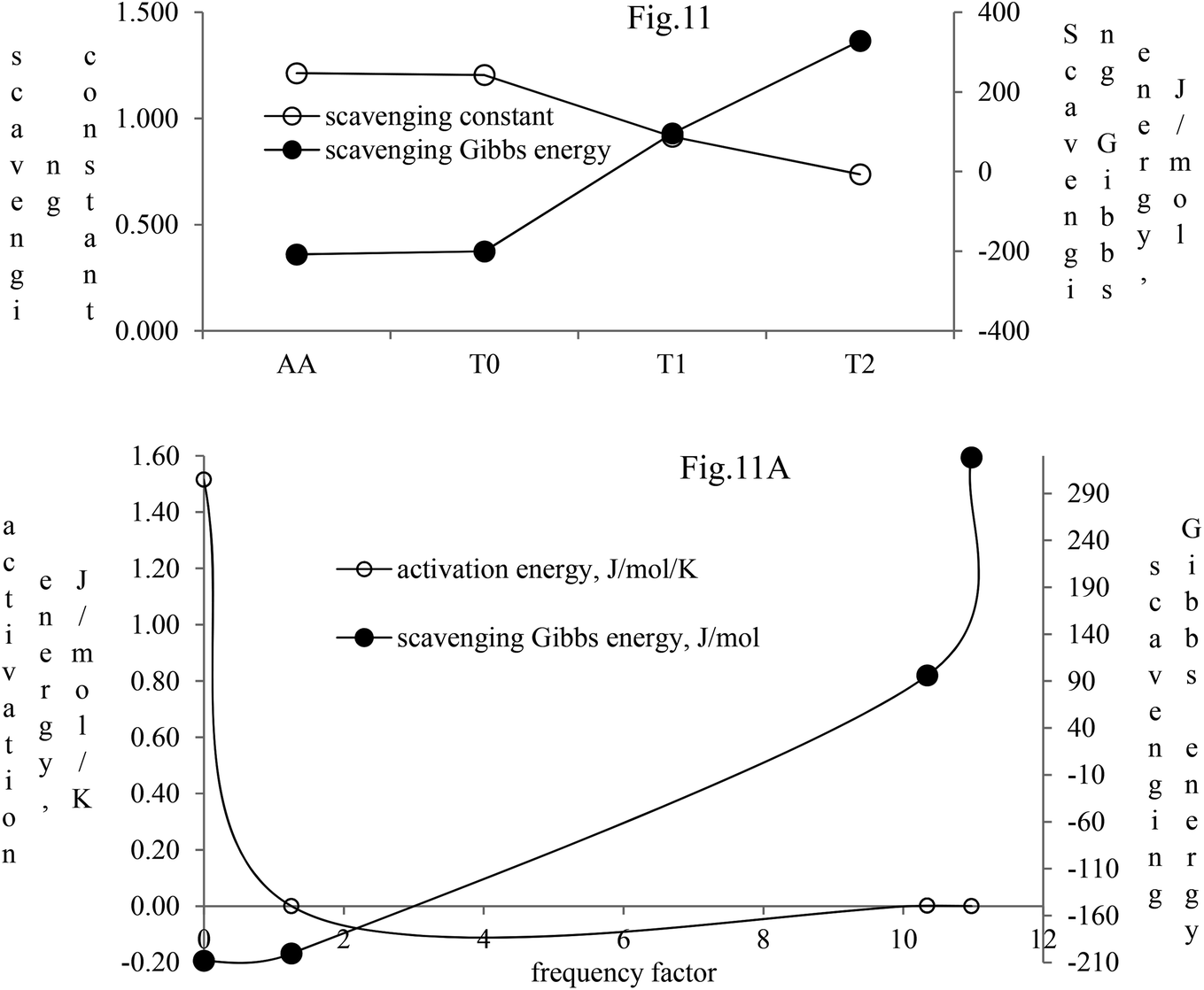 | ||
| Fig. 11 Scavenging constant and scavenging dG of the dendrimers with DPPH radicals. (A) Activation energy and scavenging dG with frequency factor. | ||
Net Gibbs energies were determined as follows:
| ΔH+releasingdG = ΔGT0 − ΔGT1 |
| ΔH+releasingdG = −200.484 − 95.866 J mol−1, −296.35 J mol−1 |
Similarly, for the energy needed to release 2H+ from T2:
| ΔH+releasingdG = ΔGT0 − ΔGT2 |
| ΔH+releasingdG = −200.484 − 328.378 J mol−1, −538.862 J mol−1 |
Net dG considered from T0 up to T2 is higher, partly because it is used in overcoming the hindrance being caused by the extended delocalization activities of its 2-OH on the terminal phenyl group. The activation energy, listed as AA > T1 > T2 > T0, induces a transition either to release H+ or to undergo a transition state. The abs with increasing concentration is absT0 > absT1 > absT2, on increasing n → π*, due to, –CH3, –OH and –2-OH species, respectively. Abs utilizes UV light to initiate HOMO to LUMO state through n → π*, and π → π*. The deshielding patterns of 3H of the core are T0 > T1 > T2 with 9.29 > 8.84 > 8.81 ppm chemical shifts, respectively. The –CH3 induces stronger deshielding than the others but when –OH are 2-OH exist with T1 and T2, respectively, then they weaken deshielding. This matches with the abs trends, but in a similar way deshielding of 13C is T2 > T1 > T0 with 131.76 > 130.67 > 130.33 ppm, respectively. This implies a high deshielding of T2 due to 2-OH. NMR and UV/Vis studies implied that the number and nature of the functional groups attached to the dendrimer deshield the core. Hence, a core contributes to the electronic populations. 1-OH to 2-OH of T1 and T2 enhances the shielding of H. This initiates H+ release from –OH and not from the benzene ring. The n → π* population stabilizes the H of the phenyl (Fig. 10A–B). The H of –OH of T1 and T2 are deshielded and release with lower abs. The C atom is shielded from T0 to T2. The kb and dG for antioxidants are depicted in Fig. 11A with higher values of kb implying faster H+ release activities denoted as:
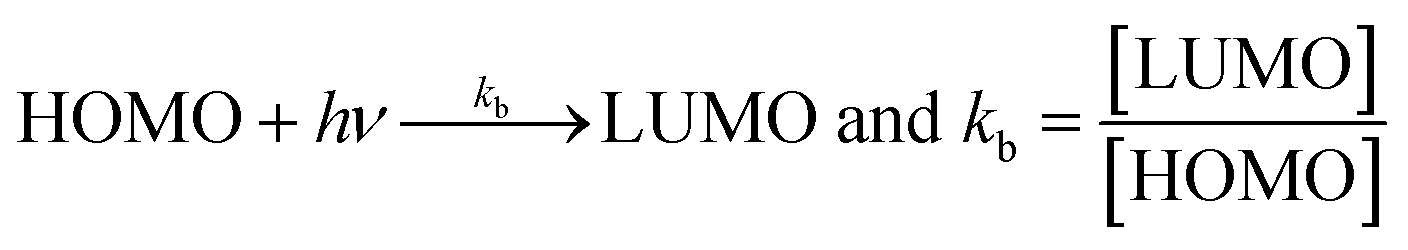
3.3 BSA and lysozyme
Dendrimers interact with proteins and lyso through their peptide bonds or electrostatic forces with synergetic effects on the EC50 of the dendrimers with variable concentration. 1, 3, and 5 mg mL−1 stock solutions of BSA and lyso were prepared with water and 100, 200, 300, 400, and 500 μl volumes were made up to 0.5 ml portions of both the BSA and lyso stock solutions, separately. These were added to DPPH solution separately containing the EC50 of AA, T1, and T2, which did not aggregate BSA and lyso due to their critically low concentrations.34,35 BSA was from 0.606 to 0.152 μM and the lyso was from 2.8 to 0.70 μM. With AA, on increasing the BSA concentration, the EC50 value is increased by 41.50% and later decreased to 25.30%. With T1, the EC50 is increased to 39.03% and decreased to −34.37%. With T2, the EC50 decreased by −39.74% and then increased to 155.86%. Fig. 12 and 13 show that AA and T1 both initially induce a dispersive effect, but on increasing the BSA concentration the dispersion is weakened. However, with T2, initially it synergized but later it is weakened due to opposing reorientations. After 3.03 μM BSA with T2, it was precipitated due to aggregation of the BSA within the void spaces of T2 as the solvent could not bind so the BSA opted to aggregate within the voids of T2. The proteins with lower additive concentrations ‘salt in’ but those with higher concentration ‘salt out’ due to aggregation. This did not occur with AA and T1 on adding BSA, which predicts a role of the two –OH of T2 in aggregating the BSA. Also the BSA has reduced scavenging limits contrary to AA. Thus, the BSA has entered the robust hydrophobic void spaces of both T1 and T2 (Fig. 5). T1 and T2 have both aggregated BSA and lyso, which has hampered their scavenging activities (Fig. 12). AA is quickly protonated forming H3O+ (hydronium) ions with water molecules, which catalyzed scavenging with BSA and lyso aqueous solutions (Fig. 13 and 14).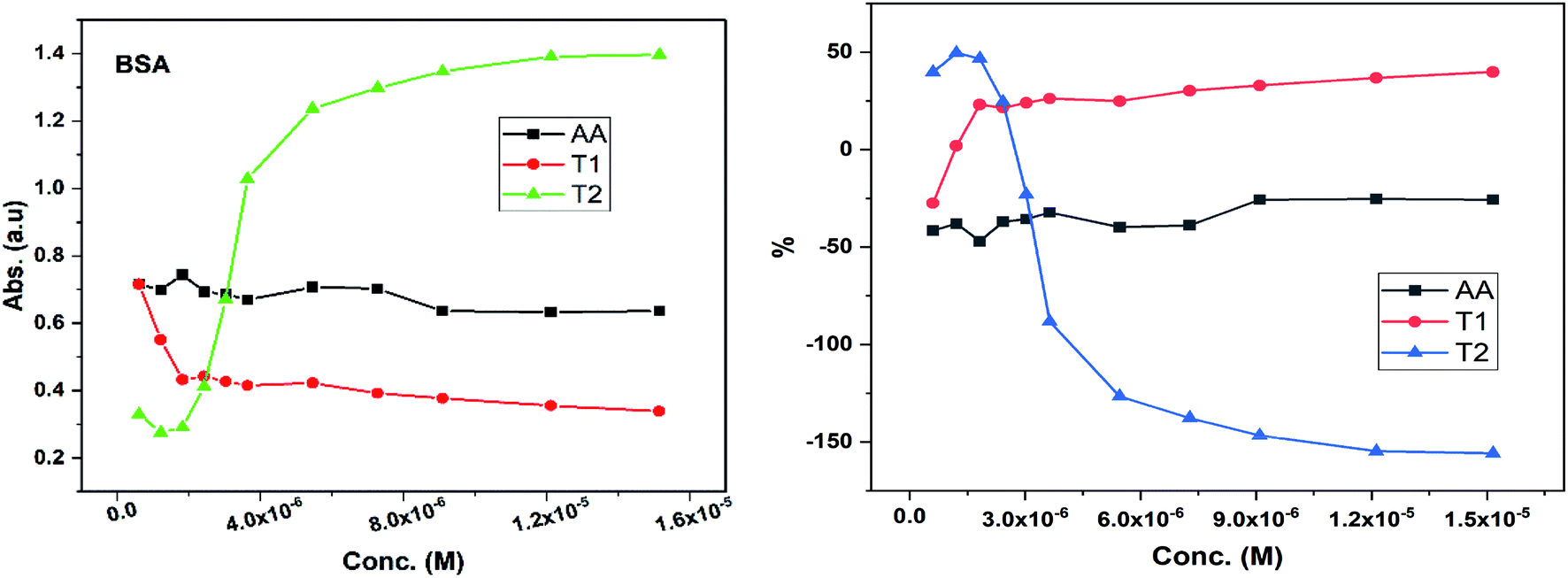 | ||
| Fig. 12 Abs vs. conc. and % EC50 vs. conc. of BSA with EC50 of AA, T1, and T2. | ||
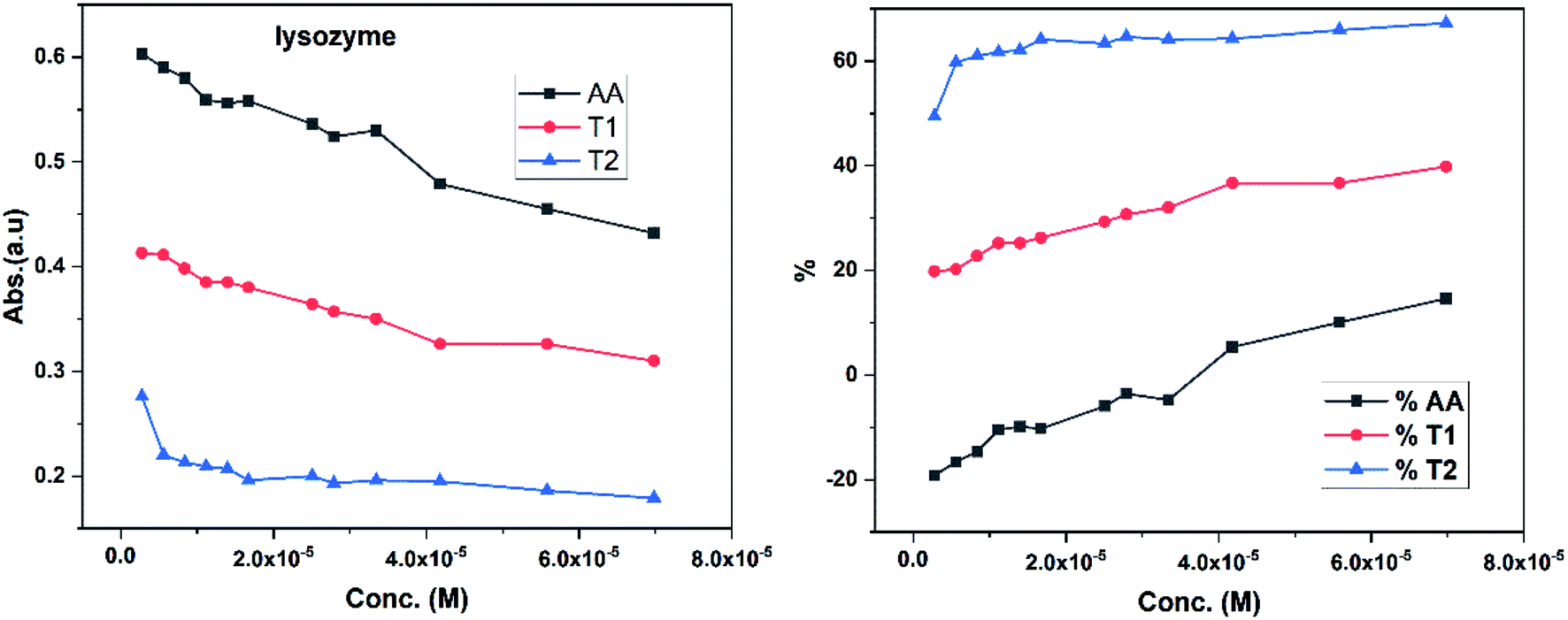 | ||
| Fig. 13 Abs vs. conc. and % EC vs. conc. graph of lyso with EC50 AA, T1, and T2. | ||
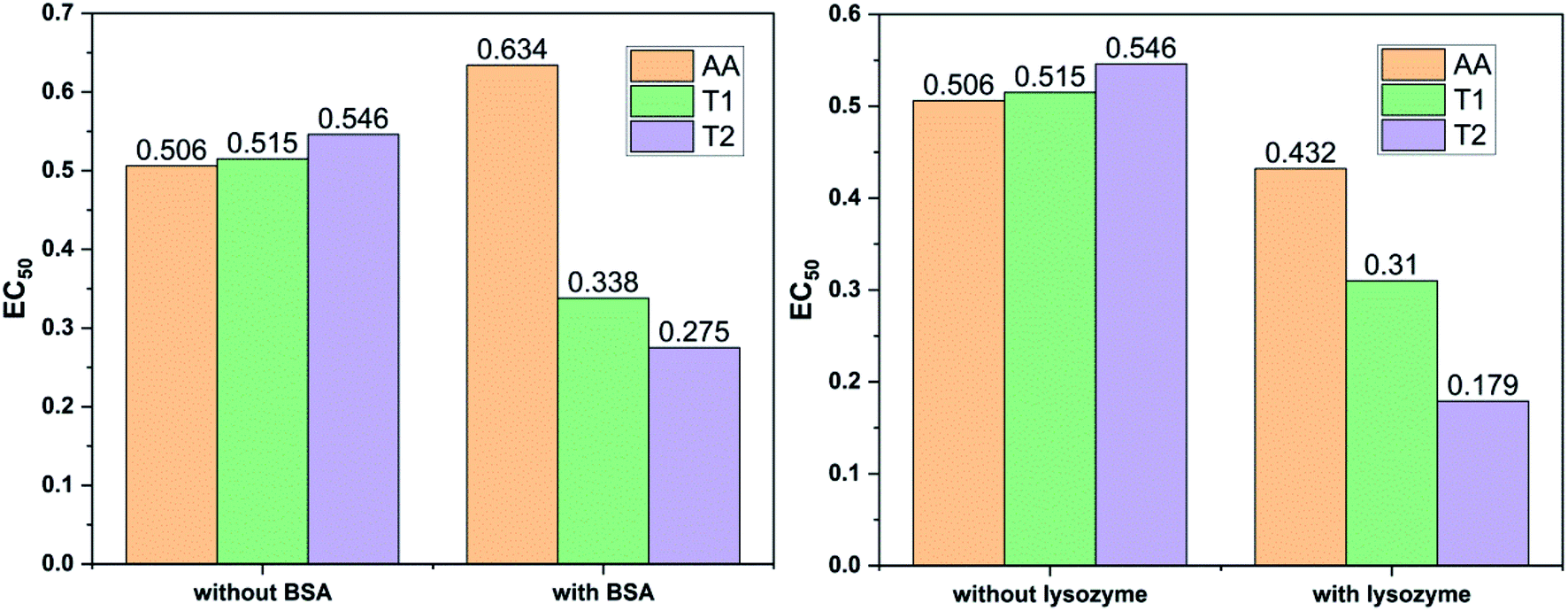 | ||
| Fig. 14 BSA and lysozyme inhibit free radical scavenging in aqueous menthol solutions. | ||
Abs for increasing BSA concentrations saturates at ∼6 μM in response to UV/Vis light and is diminished because BSA might have aggregated within the voids spaces of T2. Fig. 12 gives abs = 0.3 for blank and abs for ∼6 μM BSA = 1.3 with T2; putting these values in eqn (8), T2 aggregated 79.92% of BSA, contrary to 43.5% with T1. The BSA aggregating activities fractionate the scavenging activities of T1 and T2. Unlike BSA, the lyso with AA, T1, and T2 got aggregated in T2 > T1 > AA order. T2 aggregated ∼60% lyso compared to 79.92% BSA. Stronger binding of BSA and lyso with T2 than T1 occurred due its 2-OH.
The water which bound BSA and lyso in their aqueous solutions without mixing with AA is now available for AA molecules. Even mildly protic methanol molecules were in close chemical contact with AA, which is now engaged by a proton in the resultant sample. The AA got free and interacts with BSA, while on the other hand the T1 and T2 both aggregate BSA and lyso in the void spaces. Now the T1 and T2 are engaged with BSA and lyso in holding them in the void spaces, rather than releasing H+ that scavenges the DPPH radicals. Fig. 12 depicts abs with increasing BSA concentration with the EC50 of AA, T1, and T2. Abs with T2 for lower concentrations exponentially increases due to BSA dispersion but when its concentration continuously increases, then the BSA molecules tend to occupy the void spaces, which activates the electronic transitions. This could interrupt extended conjugations due to 2-OH with T2. Also the BSA molecules may partially move towards the void spaces and later get aggregated with variable orientational vibrations which absorb UV/Vis light (Fig. 13). The AA and T1 did not absorb much UV/Vis light on increasing the BSA concentration as these do not have complicated pi-conjugations. In particular, the T1 has only 1-OH so the extended conjugations are not interrupted and a single electronic chain from HOMO to LUMO occurred contrary to 2-OH of T2. Also the 2HOMO to 2LUMO for each –OH might be materialized simultaneously, which absorbs more UV/Vis light (Fig. 14). Abs is T2 > AA > T1; the AA has no pi-conjugation but releases two H+ which absorb more UV/Vis light than even the simple extended conjugation of T1, while T2 has 2-OH and absorbed more UV/Vis light.
Increasing concentrations of lyso with EC50 of AA, T1 and T2 have absorb UV/Vis light in the order of AA > T1 > T2. The lyso has less amino acids in its structure and strongly interacted with AA and absorbed more UV/Vis light, but T2 might have aggregated lyso within its void spaces. So it may not allow excitation as a stronger aggregation developed stronger potential energy, which defuncts and opposes electronic transitions. Lyso aggregation is as T2 > T1 > AA; T1 induced a synergetic effect with BSA, while AA produced a less synergetic effect, but T2 has an opposing effect with BSA. With lyso, the EC50 value of AA increased to 19.17% with increasing lyso concentration and later it decreased to −14.62%. The EC50 value with T1 is decreased to −19.80% and also decreased to −39.81% and with T2, the EC50 decreased to −49.45% and then decreased to −67.21%. Lyso is a competitive component to decrease the EC50 value of the dendrimers.
4. Conclusion
T0, T1 and T2 dendrimers as potential antioxidants have scavenged DPPH radicals studied spectrophotometrically. The scavenging abilities of T2 with 6-OH are 10 times higher than that of T1 with 3-OH. The AA showed a higher antioxidant activity than T1 and T2 due to its faster electronic reorientation to quickly release H+ with a higher resonance. Dendrimers acted as smart hydrophobes with hydrophobically functional void spaces. To reduce chemical waste and increase yield, branching units were synthesized via selective protection and deprotection routes. BSA and lysozyme both showed good interaction with the dendrimers compared to standard AA. Lyso acted as a promising protein to give a synergetic effect. These proteins increased the antioxidant activities of the dendrimers. The results significantly reveal a mechanism for effective biodegradable dendrimers, bioremediation, drug efficacy, and protein interacting capacity. T0, T1 and T2 dendrimers with 0, 3 and 6-OH groups have potential to further engage –OH groups to initiation condensation reactions in a suitable medium to develop macromolecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to the Central University of Gujarat for infrastructures. Dhaval Makawana acknowledges RGNF awarded (F1-17.1/2015-16/RGNF-2015-17-SC-GUJ-24172/(SA-III/Website)) for financial support.References
- R. K. Ameta and M. Singh, A thermodynamic in vitro antioxidant study of vitamins B (niacin and niacin amide) and C (ascorbic acid) with DPPH through UV spectrophotometric and physicochemical methods, J. Mol. Liq., 2014, 195, 40–46, DOI:10.1016/j.molliq.2014.01.029.
- N. C. Cook and S. Samman, Flavonoids—chemistry, metabolism, cardioprotective effects, and dietary sources, J. Nutr. Biochem., 1996, 7, 66–76, DOI:10.1016/S0955-2863(95)00168-9.
- O. I. Aruoma, Characterization of drugs as antioxidant prophylactics, Free Radic. Biol. Med., 1996, 20, 675–705, DOI:10.1016/0891-5849(95)02110-8.
- N. Vashistha, A. Chandra and M. Singh, Influence of rhodamine B on interaction behaviour of lanthanide nitrates with 1st tier dendrimer in aqueous DMSO: a physicochemical, critical aggregation concentration and antioxidant activity study, J. Mol. Liq., 2018, 260, 323–341, DOI:10.1016/j.molliq.2018.03.056.
- F. Shahidi and Y. Zhong, Measurement of antioxidant activity with .pdf, J. Funct. Foods, 2015, 18, 757–781, DOI:10.1016/j.jff.2015.01.047.
- B. N. Dar, S. Sharma and G. A. Nayik, Effect of storage period on physiochemical, total phenolic content and antioxidant properties of bran enriched snacks, J. Food Meas. Charact., 2016, 10, 755–761, DOI:10.1007/s11694-016-9360-x.
- B. Halliwell, Antioxidant characterization. Methodology and mechanism, Biochem. Pharmacol., 1995, 49, 1341–1348, DOI:10.1016/0006-2952(95)00088-H.
- H. M. Ali, A. Abo-Shady, H. A. Sharaf Eldeen, H. A. Soror, W. G. Shousha, O. A. Abdel-Barry and A. M. Saleh, Structural features, kinetics and SAR study of radical scavenging and antioxidant activities of phenolic and anilinic compounds, Chem. Cent. J., 2013, 7, 1–9, DOI:10.1186/1752-153X-7-53.
- P. Malik and M. Singh, Study of curcumin antioxidant activities in robust oil-water nanoemulsions, New J. Chem., 2017, 41, 12506–12519, 10.1039/c7nj02612a.
- R. A. Gossage, E. Munoz-Martinez, H. Frey, A. Burgath, M. Lutz, A. L. Spek and G. Van Koten, A novel phenol for use in convergent and divergent dendrimer synthesis: access to core functionalizable trifurcate carbosilane dendrimers-the X-ray crystal structure of [1,3,5-tris{4-(triallylsilyl)phenyl ester}benzene], Chem.–Eur. J., 1999, 5, 2191–2197, DOI:10.1002/(SICI)1521-3765(19990702)5:7<2191::AID-CHEM2191>3.0.CO;2-J.
- K. Esumi, H. Houdatsu and T. Yoshimura, Antioxidant action by gold-PAMAM dendrimer nanocomposites, Langmuir, 2004, 20, 2536–2538, DOI:10.1021/la036299r.
- C. Li, P. Sun, H. Yu, N. Zhang and J. Wang, Scavenging ability of dendritic PAMAM bridged hindered phenolic antioxidants towards DPPH˙ and ROO˙ free radicals, RSC Adv., 2017, 7, 1869–1876, 10.1039/c6ra26134e.
- C. Y. Lee, A. Sharma, J. E. Cheong and J. L. Nelson, Synthesis and antioxidant properties of dendritic polyphenols, Bioorg. Med. Chem. Lett., 2009, 19, 6326–6330, DOI:10.1016/j.bmcl.2009.09.088.
- J. Handique and D. Mahanta, Synthesis and Electrochemical Behavior of Some Dendritic Polyphenols as Antioxidants, Lett. Org., 2013, 2370210, 53–59 Search PubMed , http://www.ingentaconnect.com/content/ben/loc/2013/00000010/00000001/art00013.
- K. Rajavelu, M. Subaraja and P. Rajakumar, Synthesis, optical properties, and antioxidant and anticancer activity of benzoheterazole dendrimers with triazole bridging unit, New J. Chem., 2018, 42, 3282–3292, 10.1039/c7nj04060a.
- J. D. amou, K. Twibanire and T. B. Grindley, Polyester dendrimers, Polymers, 2012, 4, 794–879, DOI:10.3390/polym4010794.
- D. Liu, J. Shi, A. Colina Ibarra, Y. Kakuda and S. Jun Xue, The scavenging capacity and synergistic effects of lycopene, vitamin E, vitamin C, and β-carotene mixtures on the DPPH free radical, LWT - Food Sci. Technol., 2008, 41, 1344–1349, DOI:10.1016/j.lwt.2007.08.001.
- A. J. Young and G. M. Lowe, Antioxidant and Prooxidant Properties of Carotenoids, Arch. Biochem. Biophys., 2001, 385, 20–27, DOI:10.1006/abbi.2000.2149.
- T. Nishiyama, T. Sugimoto and Y. Andoh, Antioxidant activity of phenols in intramolecularly cooperating stabilizing systems, Polym. Degrad. Stab., 2001, 74, 189–193, DOI:10.1016/S0141-3910(01)00151-3.
- T. Nishiyama, Y. Andoh, T. Sugimoto and T. Okamoto, New bifunctional antioxidants—intramolecular synergistic effects between chromanol and thiopropionate groups, Polym. Degrad. Stab., 2003, 81, 409–413, DOI:10.1016/S0141-3910(03)00125-3.
- P. Suresh, S. Srimurugan, B. Babu and H. N. Pati, Asymmetric sulfoxidation of prochiral sulfides using aminoalcohol derived chiral C3-symmetric trinuclear vanadium Schiff base complexes, Tetrahedron: Asymmetry, 2007, 18, 2820–2827 CrossRef CAS.
- V. Thavasi, L. P. Leong and R. P. A. Bettens, Investigation of the influence of hydroxy groups on the radical scavenging ability of polyphenols, J. Phys. Chem. A, 2006, 110, 4918–4923, DOI:10.1021/jp057315r.
- E. N. Khimich, G. S. Buslaev, A. V Zdravkov, N. N. Khimich and M. G. Voronkov, Polysiloxane Structures Cross-Linked with Hydroquinone and Phloroglucinol, Russ. J. Appl. Chem., 2008, 81, 1070–4272, DOI:10.1134/S107042720808017X.
- W. Brand-Williams, M. E. Cuvelier and C. Berset, Use of a free radical method to evaluate antioxidant activity, LWT - Food Sci. Technol., 1995, 28, 25–30, DOI:10.1016/S0023-6438(95)80008-5.
- R. J. Elias, S. S. Kellerby and E. A. Decker, Antioxidant activity of proteins and peptides, Crit. Rev. Food Sci. Nutr., 2008, 48, 430–441, DOI:10.1080/10408390701425615.
- R. T. Dean, S. Fu, R. Stocker and M. J. Davies, Biochemistry and pathology of radical-mediated protein oxidation, Biochem. J., 1997, 324(1), 1–18, DOI:10.1042/bj3240001.
- M. P. Almajano and M. H. Gordon, Synergistic Effect of BSA on Antioxidant Activities in Model Food Emulsions, J. Am. Oil Chem. Soc., 2004, 81, 1–6, DOI:10.1007/s11746-004-0895-6.
- M. Yang, Y. Wu, J. Li, H. Zhou and X. Wang, Binding of curcumin with bovine serum albumin in the presence of ι-carrageenan and implications on the stability and antioxidant activity of curcumin, J. Agric. Food Chem., 2013, 61, 7150–7155, DOI:10.1021/jf401827x.
- M. Voicescu, G. Neacsu, A. Beteringhe, O. Craciunescu, R. Tatia and L. Moldovan, Antioxidant and cytotoxic properties of riboflavin in PEG/BSA systems, Chem. Pap., 2017, 71, 1107–1117, DOI:10.1007/s11696-016-0057-8.
- S. J. You, C. C. Udenigwe, R. E. Aluko and J. Wu, Multifunctional peptides from egg white lysozyme, Food Res. Int., 2010, 43, 848–855, DOI:10.1016/j.foodres.2009.12.004.
- H. Liu, Amelioration of oxidant stress by the defensin lysozyme, Am. J. Physiol. Endocrinol. Metabol., 2006, 290, E824–E832, DOI:10.1152/ajpendo.00349.2005.
- P. Sharma and R. P. Singh, Evaluation of antioxidant activity in foods with special reference to TEAC method, Am. J. Food Technol., 2013, 8, 83–101, DOI:10.3923/ajft.2013.83.101.
- A. K. Jangid, D. Pooja and H. Kulhari, Determination of solubility, stability and degradation kinetics of morin hydrate in physiological solutions, RSC Adv., 2018, 8, 28836–28842, 10.1039/c8ra04139c.
- A. Lo, P. Wentworth, F. Sieber, W. A. Metz and K. D. Janda, Soluble Polymer-Supported Chemoenzymatic Synthesis of the C21–C7 Fragment of the Bryostatins, J. Org. Chem., 2000, 65, 8527–8531, DOI:10.1021/jo005539u.
- G. Houen, K. Bechgaard, K. Bechgaard, J. Songstad, M. Leskelä, M. Polamo, M. N. Homsi, F. K. H. Kuske, M. Haugg, N. Trabesinger-Rüf and E. G. Weinhold, The Solubility of Proteins in Organic Solvents, Acta Chem. Scand., 1996, 50, 68–70, DOI:10.3891/acta.chem.scand.50-0068.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04102e |
| This journal is © The Royal Society of Chemistry 2020 |