
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn inkjet-printed polysaccharide matrix for on-chip sample preparation in point-of-care cell counting chambers†
Xichen Zhanga,
Dorothee Wasserberga,
Christian Breukersa,
Bridgette J. Connellb,
Pauline J. Schipperb,
Joost van Daluma,
Ellen Baetenc,
Dorine van den Blinkc,
Andries C. Bloemc,
Monique Nijhuisb,
Annemarie M. J. Wensingb,
Leon W. M. M. Terstappen a and
Markus Beck*a
a and
Markus Beck*a
aMedical Cell Biophysics, MIRA Institute for Biomedical Technology and Technical Medicine, Faculty of Science and Technology, University of Twente, PO Box 217, Enschede, 7500 AE, The Netherlands. E-mail: m.beck@utwente.nl
bUniversity Medical Center Utrecht, Department of Medical Microbiology, Virology, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands
cUniversity Medical Center Utrecht, Laboratory of Translational Immunology, Section Diagnostics, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands
First published on 12th May 2020
Abstract
On-chip sample preparation in self-contained microfluidic devices is a key element to realize simple, low-cost, yet reliable in vitro diagnostics that can be carried out at the point-of-care (POC) with minimal training requirements by unskilled users. To address this largely unmet POC medical need, we have developed an optimized polysaccharide matrix containing the reagents which substantially improves our fully printed POC CD4 counting chambers for the monitoring of HIV patients. The simply designed counting chambers allow for capillary-driven filling with unprocessed whole blood. We carefully tailored a gellan/trehalose matrix for deposition by inkjet printing, which preserves the viability of immunostains during a shelf life of at least 3 months and enables controlled antibody release for intense and homogeneous immunofluorescent cell staining throughout the complete 60 mm2 image area within 30 min. Excellent agreement between CD4 counts obtained from our fully printed CD4 counting chambers and the gold standard, flow cytometry, is demonstrated using samples both from healthy donors and HIV-infected patients.
Introduction
A point-of-care (POC) test is a standalone diagnostic test that is carried out near or by the patient and delivers “immediate” results equivalent to lab testing. Therefore, it needs to be reliable, robust and simple to operate, as these tests are typically carried out by nurses or by the patients themselves. This also means that any sample preparation must be carried out automatically without user-intervention.1,2Very recently, the need for simple and rapid diagnostics that delivers immediate results has become evident within the context of the COVID-19 pandemic. To effectively contain such a virus, rapid testing on a massive scale at home and in physicians' offices is of utmost importance, as the infected can be quarantined immediately.
Another example is on-site diagnostics in disaster areas or ambulances, where the transport of samples to laboratories is impractical, while rapid on-site test results can save lives.
Paper-based tests, such as lateral flow assays are an excellent example which unites many of the desired properties of a POC test. However, these tests are limited to a relatively small number of assays and reliable quantitation is difficult. In particular, cell counting assays relying on immunostaining cannot easily be converted into lateral-flow assays.3
Our goal is to develop a POC cytometry test that is as simple to use as a lateral flow assay but allows for precise cell counting of intact cells. As an example, we chose to demonstrate such a cell counting test based on immunostaining for HIV (human immunodeficiency virus) monitoring. As the virus attacks Helper T-cells, the concentration of these cells in the blood of a patient is a good indication of the state of the disease (AIDS).4 Especially in rural areas of resource-limited countries, e.g. in Sub-Saharan Africa, a simple, low-cost POC test for HIV monitoring is still needed, as in many countries neither the existing POC CD4 tests nor alternative solutions (viral load testing) are currently affordable. Many solutions have been proposed, however, none have been widely adopted as they either lack in simplicity, precision or affordability.5–9
Key requirements to enable POC testing are reagent storage and automated sample preparation within a disposable device. With the emergence of microfluidic technology, the concept of complete on-chip sample preparation has evolved.10–12 Reagents needed for the assays have to be integrated into microfluidic devices and optimal interaction between sample and reagents within the devices needs to be ensured. A variety of solutions have been developed to integrate dry reagents in microfluidic devices to accomplish well-controlled on-chip sample preparation after the introduction of the sample fluid.13
Conventional approaches focus on defined patterning of reagents in glass microfluidic devices with sophisticated designs of channel networks to control sample/reagent interactions.14,15 Controlled reagent dissolution can be attained by directing sample fluid through intricate flow paths. However, the requirement of an external pump for flow control and complex channel structures impose difficulties on low-cost fabrication, robustness, and reliability. Storing reagents in paper-based microfluidic devices is an alternative approach to achieve the desired dissolution enabled by capillary-driven sample flow in lateral flow tests.16,17 However, as mentioned above, the realization of cell counting assays is difficult, as the flow of plasma and cells through the paper will generally differ.
For cell counting, we envision a simplistic on-chip sample preparation approach, of which we demonstrated a proof-of-concept previously.18 Reagents are contained in a simple microfluidic chamber with just two openings. Sample fluid flows into the chamber via capillary force and stops when the chamber is filled. During inflow, reagents are mixed with sample fluid to initialize biochemical reactions. However, uniform mixing of reagents and sample fluid, which is critical for most analytical readout, is challenging to realize in a microfluidic chamber with such a stopped-flow configuration. Reagents are washed-off during sample inflow, thus resulting in a laterally inhomogeneous distribution, which leads to inhomogeneous mixing and staining. To achieve uniform mixing, embedding reagents in a matrix inside a microfluidic chamber to delay reagent release during sample inflow is an effective solution.18–23 On the one hand, the release has to be delayed during sample inflow. On the other hand, the reagents should be available as quickly as possible after the inflow has stopped, to allow for optimized staining within minimal incubation time. Therefore, a careful choice and tuning of the release matrix in our microfluidic chambers are required in our approach. Fig. 1 illustrates the effect of the release matrix on cell staining. The absence of a release matrix would result in reagent wash-off and hence inhomogeneous mixing (Fig. 1, left chamber), whereas optimized release enables homogeneous mixing (Fig. 1, right chamber) after sample filling.
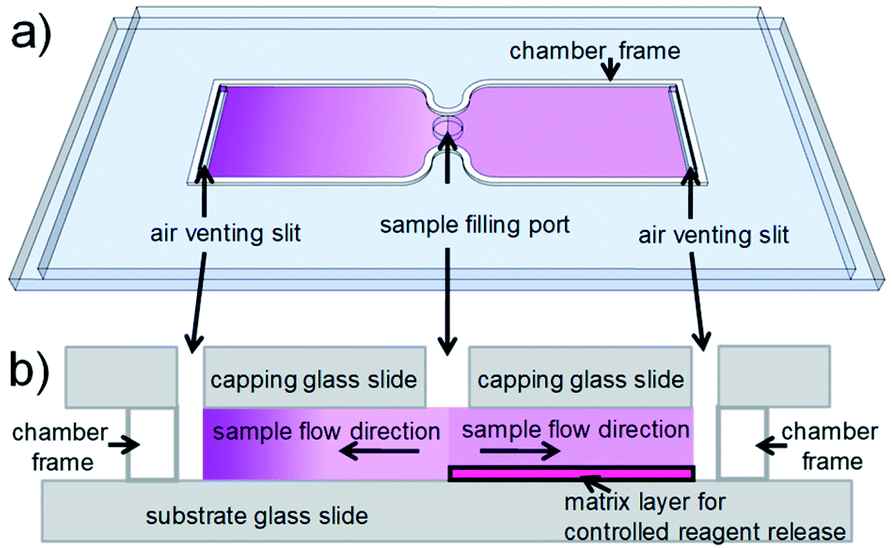 | ||
| Fig. 1 Illustration of the on-chip sample preparation approach. (a) Perspective view of a simple glass chip with two chambers. Each chamber (some tens of μm high) has two openings to enable capillary flow. Purple shading indicates reagent concentration. (Left) In a (hypothetical) chamber with reagents deposited without matrix, reagents would be washed off toward the end of the chamber during inflow. (Right) In a chamber with reagents embedded in a matrix, reagents are homogeneously distributed, as their release is delayed during inflow. (b) (Side view) (Left) The chamber without matrix shows strong reagent wash-off. (Right) The chamber with matrix shows uniform reagent distribution. Purple shading indicates reagent concentration. | ||
Hydrogel matrices, widely used in drug delivery,24 have the potential to enable such controlled reagent release. In a previous approach, we used actively temperature-controlled release from a gelatin matrix,25 which does allow for excellent staining homogeneity due to the external control of the antibody release. However, the need to activate the release by heating the device requires additional electric power, and the additional sample preparation step reduces the simplicity and robustness of the POC test. Furthermore, a gradual change of the properties of gelatin, especially after inkjet printing,25,26 limited the shelf life to a maximum of a few weeks. To further simplify the procedure and reduce power consumption, we set out to tailor a suitable matrix, capable of passive reagent release with high reproducibility over a long storage time, not requiring external stimuli.
Polysaccharide based hydrogels represent a promising class of materials to act as such release matrices in microfluidic devices. Such hydrogels are well-suited to our on-chip sample preparation approach for a number of reasons: first, many of these materials exhibit good compatibility with a variety of biological reagents27 e.g. proteins. Particularly, if these hydrogel based matrices contain disaccharides, like trehalose, the secondary conformation of embedded proteins is largely preserved even after dehydration.28 Second, their use is well-documented in controlled release studies for diverse drug delivery applications.29 Third, polysaccharides are hydrophilic materials due to their high polarity. Hydrophilicity is paramount for the capillary filling of microfluidic devices by enhancing the wetting of the surface with aqueous sample fluids.30,31 Fourth, no significant structural change is found in polysaccharide matrices stored under dry and cool conditions,32,33 suggesting polysaccharides are stable for months with negligible ageing issues. Such long-term stability would ensure the consistency of the desired release kinetics and reliable sample preparation and, therefore, excellent shelf life. Last, polysaccharides are abundant, non-toxic materials, available at low (processing) cost.34,35
There were about 38 million HIV-infected people worldwide in 2018, most of which reside in resource-limited regions, e.g. sub-Saharan Africa.36 The progression of AIDS needs to be monitored regularly, both to determine start of treatment and monitor treatment effectiveness. There are two widely used monitoring methods, CD4 counting, and viral load testing.37 The CD4 count is the number of CD4 positive (CD4+) T-cells per microliter of whole blood, while viral load is quantified as the copy number of viral RNA per milliliter of whole blood. Despite new World Health Organization (WHO) guidelines38 that recommend to treat patients irrespective of their CD4 count, CD4 counting is still commonly used where viral load testing is not affordable. Our simple and low-cost on-chip sample preparation concept fulfills this need perfectly.
Here, we demonstrate a successful CD4 counting assay with our simplistic on-chip sample preparation approach18 using recently developed fully printed microfluidic chambers26 containing a new polysaccharide matrix material with embedded fluorophore labeled antibody (Ab). In order to optimize controlled on-chip reagent release for uniform and intensive cell staining in the chamber, the reagent release matrix in the chamber was selected after screening a number of polysaccharide materials according to 4 criteria in the following order: smooth topography, homogeneous Ab distribution, high staining intensity, and long-term stability. After these 4 initial screening steps, gellan proved to be the superior material and therefore, the procedure for its deposition in counting chambers was optimized.
Chambers were fabricated by printing techniques in an automated manner. Both, gellan layers with embedded Abs as well as chamber frames were deposited via (inkjet) printing. A validation of our CD4 counting assay, using fully printed counting chambers, was also carried out. Our custom-built image cytometer18 was used to analyze patient samples in the fully printed chambers and the obtained cell counts showed excellent correlation with the results from standard flow cytometry analysis used in the clinical lab.
Furthermore, the printed chambers proved to have a shelf life of at least 3 months without any noticeable effect on the accuracy in dry, cool storage and to be still functional for 1 week, when stored under less favorable conditions (high temperature and humidity).
Experimental
Preparation of polysaccharide/antibody (Ab) solutions
All materials were purchased from Sigma-Aldrich unless stated otherwise. Six different types of polysaccharides: chitosan (chi, viscosity 20–300 cps 1% solution in 1% acetic acid, cat. no. 448869), gellan (gell, MW ∼ 1000 kD, cat. no. G1910), xanthan (xan, viscosity 800–1200 cps 1% solution in water, cat. no. G1253), carrageenan (carr, cat. no. C1013), alginate (alg, viscosity 15–25 cps 1% solution in water, cat. no. 180947) and pectin (pec, cat. no. P9135), were received as powders. Chi was dissolved in acetic acid (0.4% v/v) at room temperature (RT, ∼20 °C), while stirring for 1 h. The other polysaccharide materials were dissolved in Milli-Q® water at 80 °C, while stirring for 1 h, and then left to cool to RT. All the solutions were filtered gravitationally using a membrane filter (CellTrics®, mesh size 20 μm, Partec) before use.Polysaccharide materials tested in this paper were: pure polysaccharides (chi, gell, xan, and carr), polysaccharide blends (gell/chi, xan/chi, carr/chi, alg/chi, and pec/chi) and ionically crosslinked polysaccharides (alg/Ca2+ and pec/Ca2+). Pure polysaccharide solutions (0.1% w/v) were prepared in water. Solutions of the polysaccharide blends were each prepared by mixing an aqueous solution of polycationic chi with one of the five polyanionic polysaccharides (gell, xan, carr, alg, and pec) solutions. The blends were equimolar mixtures with regard to the amine groups (–NH2) of chi and the carboxylic acid groups (–COOH) of its respective counterpart. Ionically crosslinked polysaccharides were prepared by adding CaCl2 to alg or pec solutions. The added Ca2+ amounted to half a molar equivalent of the carboxylic acid groups of alg or pec, in order to achieve the highest degree of crosslinking, aiming for the so-called egg-box conformation.39,40 The total concentration of polysaccharides in blends and ionically crosslinked polysaccharides was kept at only 0.02% w/v to prevent precipitation. The concentrations of different polysaccharide solutions differ due to their solubility. By adjusting the deposited volume, the layer thicknesses stay comparable in each screening step.
Casting solutions were prepared by adding the supernatant of APC-αCD3 (clone SK7, 260 kDa, BD) and, where necessary, of PerCP-αCD4 (clone MEM241, 308 kDa, Exbio) solutions, after centrifugation (21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g for 3 min), to each of the above polysaccharide solutions to yield final concentrations of 2.5 or 0.5 μg mL−1 APC-αCD3 and 0.5 or 0.1 μg mL−1 PerCP-αCD4 in 0.1 or 0.02% w/v polysaccharide solutions, respectively.
000×g for 3 min), to each of the above polysaccharide solutions to yield final concentrations of 2.5 or 0.5 μg mL−1 APC-αCD3 and 0.5 or 0.1 μg mL−1 PerCP-αCD4 in 0.1 or 0.02% w/v polysaccharide solutions, respectively.
Fabrication of chambers with cast polysaccharide/Ab layers
Chambers with cast layers were fabricated as illustrated in Fig. S1.† Precut laminating adhesive (nominal thickness 25.4 μm, 3 M) was attached to a glass substrate (Menzel, 76 mm × 26 mm × 1 mm) to create a confined casting area (15 mm × 9 mm). Into this area, pure polysaccharide (24 μL, 0.1% w/v), or polysaccharide blend (120 μL, 0.02% w/v) or ionically crosslinked polysaccharide (120 μL, 0.02% w/v), was cast and left to dry under ambient conditions to form a layer of ∼50 nm thickness (verified by white light interferometry as described below). The cast layers used in the evaluation of staining intensities contained 10 ng cm−2 APC-αCD3 and 2 ng cm−2 PerCP-αCD4 in the image area. In the other steps of the screening, cast layers only contained APC-αCD3 (10 ng cm−2 in the image area). The antibody concentrations were chosen to match the conditions previously determined for immunostaining in our application.18,25,26 After the cast layers had dried, an 11 mm wide capping glass slide (Menzel) was attached as a cover to create a counting chamber. The chambers with cast layers were stored over dry silica gel in the dark, which results in ∼10% relative humidity (RH). Containers were kept at 4 °C, 20 °C or 40 °C to test the dependence of Ab release on the storage temperature in time.Fabrication of counting chambers with printed polysaccharide/Ab layers and frames
For the fabrication of printed Ab release layers, inks were prepared as follows. Trehalose (Merck KGaA) was dissolved in Milli-Q® water at RT by extensive vortexing. Solutions were prepared by adding varying amounts of trehalose to a gell/Ab solution to reach fractions of trehalose of 0%, 9%, 17%, 33% and 50% (w/w) with regard to the mass of the dry layer. The final concentrations of gell, APC-αCD3, and PerCP-αCD4 in the ink solutions were kept constant at 0.3% w/v, 0.42 μg mL−1 and 0.14 μg mL−1, respectively.Fig. S2a† shows a schematic representation of a fully printed chamber. Inkjet printing was performed using an industrial inkjet printer (LP50, PiXDRO, Meyer Burger B.V.) with a Konica Minolta 512 MHX printhead (nozzle diameter ∼27 μm). Standard microscope glass slides (Menzel, 76 mm × 26 mm × 1 mm) were used as substrates for ink deposition. Droplets (∼18 pL) of ink solution were dispensed in 25 swaths of (360 dpi)2 resolution, each, and a delay of 84 s between them, resulting in a final resolution of (1800 dpi)2. These parameters were chosen in a process described earlier26 to minimize puddle formation, while maintaining sufficient throughput to keep nozzles jetting reliably. Resulting layers had an average thickness in the range from ∼370 to ∼560 nm after drying, for 0% (Fig. S2b†) and 50% trehalose, respectively. The resulting layers contained 3.8 ng cm−2 of APC-αCD3 and 1.3 ng cm−2 of PerCP-αCD4.
Chamber frames as shown in Fig. S2c† were printed as described previously.26 Chamber heights were determined individually for each chamber by a custom-built laser interferometry setup and were found to be 29.8 ± 1.8 μm (n = 232) for all chambers used for determining the CD4 counts of samples from HIV-infected patients.
Layer topography, matrix dissolution and Ab release of cast and printed layers were measured without capping slides, while the distribution of Ab after sample inflow and the characterization of staining intensity were performed using completed chambers.
Determination of layer topography
To quantify the surface roughness of the polysaccharide layers, the coefficient of variation (CV) of the thickness was determined from a representative region (2 mm2) for each material. The thickness of both, cast and printed layers were measured using a white light interferometer (smartWLImicroscope, GBS) after the material had been scratched off from some areas of the substrate for reference.Fluorescence imaging
Fluorescence images of both, cast and printed layers in (uncovered) chambers were taken using our custom-built laboratory fluorescence imaging system.18 Images of an area of 7.8 mm × 5.2 mm were captured with a resolution of 10 μm without scanning using one exposure with excitation light from a red LED (for APC) and one exposure with excitation light from a blue LED (for PerCP). The fluorescence intensity of APC-αCD3 or PerCP-αCD4 was recorded after background subtraction and analyzed to obtain a mean intensity, which can be used to evaluate the structural integrity of fluorophores. The CV of the fluorescence intensity of a dry layer is indicative of the homogeneity of the Ab distribution within its matrix.Determination of matrix dissolution and fractional Ab release – dipping experiments
To quantify matrix dissolution within a time period comparable to sample inflow (10 s), both cast and printed layers on substrates without capping slides were immersed in phosphate buffered saline (PBS, previously shown to be a suitable replacement for blood plasma23), for 10 s, while stirring. Following a gentle blow-drying step, the average layer thickness (d) of a specific region (2 mm2, next to a scratch used for alignment) was measured and compared with the layer thickness within the same area before dipping. The ratio dafter/dbefore yields the fraction of retained material during sample inflow.Furthermore, the fractional Ab release was measured at two characteristic time points (10 s: inflow period and 30 min: entire assay incubation time). First, fluorescence images (APC-αCD3 or PerCP-αCD4) of dry polysaccharide layers in uncovered chambers were recorded and the mean intensities after background subtraction (Ibefore) of 4.1 mm × 3.1 mm regions (adjacent to a scratch in the layer, used for alignment) were determined. Then, the layers were immersed in PBS, while stirring for 10 s. Following a gentle blow-drying step, the average intensities (I10s) of the exact same regions after background subtraction were determined. The resulting I10s/Ibefore is indicative of the mass (M) fractions of retained Ab in the layer during sample inflow. An additional 30 min immersion was carried out for all layers. Likewise, the mean intensities (I30min) of the exact same regions after background subtraction were determined again after 30 min immersion. The determined intensity ratio of the layer before and after immersion is inversely related to the fractional release of APC-αCD3 after 10 s (M10s/Mtotal) and after 30 min (M30min/Mtotal), where the intensity of the dry layers before immersion are proportional to Mtotal.
Measurement of Ab distribution after sample inflow and on-chip immunostaining
To quantify the wash-off of Ab during sample inflow, fluorescence images before and after inflow were compared: first, a fluorescence image (APC-αCD3 readout) of each type of polysaccharide layer in completed chambers, before sample inflow, was recorded. Then, approximately 5 μL of whole blood was added to the counting chamber. Reference staining intensities were obtained from samples prestained with APC-αCD3 (0.83 μg mL−1) and PerCP-αCD4 (0.25 μg mL−1) solutions in chambers without Ab release layers. Immediately after sample inflow had ended, a second fluorescence image (APC-αCD3 readout) of each of the filled counting chambers was taken. Normalized images resulted from the ratio between background-corrected fluorescence images of the filled versus the unfilled chamber. These normalized images are assumed to represent the ratio between Ab concentration after and before sample inflow. From these normalized images, normalized intensity profiles were obtained by averaging over fluorescence intensities perpendicular to the inflow direction at each point along the chambers' inflow direction. After 30 min incubation at RT, a set of fluorescence images of both APC and PerCP readouts were recorded. Automated image analysis using ImageJ41 was executed to identify objects (2–9 pixels in diameter) with the fluorescence intensity of at least one of the readout signals exceeding the local background level significantly. Background inhomogeneity is caused by the granular structure of the sample, i.e. unstained cells (including red blood cells) displacing fluorophore labeled antibody in the blood plasma, in which the cells are suspended.26 However, the concentration of the fluorophore labeled antibody has been optimized so that this background inhomogeneity is small compared to the intensity of the labelled cells. Each object's (i.e. each identified cell's) emission was quantified as its integrated intensities for both fluorophores after background subtraction. Objects with above-threshold fluorescence intensities for both fluorophores were identified as CD4+ T-cells and counted with our automated image analysis software, reported previously.26,42 The CD4 count was obtained via dividing the number of identified CD4+ T-cells by the sample volume, which is determined by the image area and the chamber height.All experiments were performed in compliance with the Dutch Medical Research Involving Human Subjects Act (WMO), approved by the Medical Ethics Review Committee (METC) of the Medisch Spectrum Twente in Enschede. Blood samples from healthy volunteers were obtained anonymously via the TNW-ECTM-donor service at the University of Twente and all donors gave written informed consent before donating blood.
Shelf life testing of fully printed counting chambers
The fully printed chambers with Ab embedded in gell/trehalose matrices were stored in vacuum packs (vacuum condition; oxygen and moisture depleted), over silica gel (silica gel condition; ∼10% RH) or over saturated potassium chloride solutions (high humidity condition; ∼85% RH) each at 4 °C, 20 °C or 40 °C for shelf life testing. After 7, 28, 90 days, the average intensities of printed layers in 4 chambers, stored under each of the conditions mentioned above, were determined and compared with the results from the respective freshly prepared layer. The change in fluorescence intensity of the layers over time was used to assess the structural integrity of the fluorophores (APC and PerCP). The same counting chambers, subject to certain conditions for specific storage periods, were also used to carry out CD4 counting. The dependence of staining intensity and counting accuracy on storage conditions and time were analyzed.Evaluation of the CD4 count using patient samples
For the validation of the CD4 count, a custom-built prototype instrument was used. The instrument uses the same type of LED excitation and optical filters as used in the laboratory setup, but with a different lens (TC23009, 1× telecentric lens, OptoEngineering) and a different camera (MR655MU-BH, Ximea), resulting in an image area of 60 mm2.All experiments were performed in compliance with the Dutch Medical Research Involving Human Subjects Act (WMO), approved by the Medical Ethics Review Committee (METC) of the University Medical Center Utrecht. Leftover blood samples from HIV-infected patients (who had given prior written informed consent for the use of their anonymized samples for research purposes) at the UMC Utrecht that had been drawn by venipuncture into EDTA blood collection tubes and been used for routine analysis (including a CD4 count), were used to evaluate the performance of our approach. One slide with two printed CD4 counting chambers on each, as depicted in Fig. 1 and S2a,† was used per patient sample. The slides were stored over silica gel at 4 °C (4sg). The CD4 counts obtained using our chambers were compared with the reference counts from flow cytometry, using MultiTest™ reagents and MultiSet™ four color automated immunophenotyping software (Becton Dickinson) on a FACS Canto II v3.0 (Becton Dickinson) flow cytometer at the UMC Utrecht.
Results and discussion
The concept of our CD4 counting assay is based on simple, complete on-chip sample preparation and robust image analysis using a portable, battery powered fluorescence image cytometer, resulting in accurate CD4 counts. In order to achieve this, the counting chambers have to meet the requirements outlined in the following.The first step of our complete on-chip sample preparation concept is capillary-driven filling, which relies on smooth and hydrophilic inner surfaces of the chamber. Polysaccharide matrices are very hydrophilic, due to their high polarity, and therefore well suited as coatings. In addition, hydrophilic polysaccharides are well compatible with glass substrates. Glass is preferable to polymer substrates, as the low thermal expansion coefficient of glass helps to reduce heat-induced sample movement during imaging. This allows us to extend the scope of our CD4 count to non-coagulated blood, i.e. blood collected by venipuncture in tubes containing an anticoagulant and blood from patients with coagulation disorders. Furthermore, as the blue excitation light is strongly absorbed by the hemoglobin contained in red blood cells, the height of the counting chamber should not considerably exceed 50 μm. Sufficient sample volume (>1 μL) is required for acceptable statistical errors in cell counting (for the clinically relevant range of CD4 counts of tens to hundreds per μL). Given the low chamber height, a large area (tens of mm2) has to be imaged to analyze volumes of >1 μL. In addition, we aim for an affordable and reliable imaging system with large focal depth and without any moving parts, as this allows us to construct a robust device without the need for readjusting the focus manually or including autofocus. These requirements necessitate an imaging system with a large field of view to avoid scanning and a small numerical aperture, which results in a rather limited lateral resolution, not allowing to resolve any features of the cells of interest other than their fluorescence intensities. Neither can light scattering be used as an additional parameter, due to the large number of erythrocytes in whole blood.
Therefore, uniform intensities of stained cells, as well as a homogeneous background intensity in the image, are required for the unambiguous identification of stained cells, which makes a homogeneous Ab distribution in the chamber essential. Clearly, incorporating fluorophore labeled Ab embedded in a polysaccharide matrix deposited inside our counting chambers is a good approach to meet this requirement. In our counting chambers, a well-designed and optimized polysaccharide matrix will facilitate capillary-driven sample filling due to its high hydrophilicity but also be used to safely store, and control the release of embedded Ab for homogeneous cell staining.
Screening polysaccharide materials
There are a plethora of naturally occurring, modified and synthetic polysaccharides available with very diverse properties. To identify an appropriate polysaccharide material which is capable of fulfilling the relevant functions as a storage and release matrix in our counting chambers, we defined 4 screening parameters (topography of dry layers, Ab distribution after inflow, intensity of Ab stained cells and matrix stability) in the step-wise selection procedure (Fig. S3†). We started with 11 materials, selected from literature as promising candidates with regard to the above criteria, which were categorized into 3 groups (pure polysaccharides: chitosan (chi), gellan (gell), xanthan (xan) and carrageenan (carr); polysaccharide blends: alginate (alg)/chi, pectin (pec)/chi, carr/chi, gell/chi and xan/chi; ionically crosslinked polysaccharides: alg/Ca2+ and pec/Ca2+). During the screening, the materials were tested for their performance with regard to each parameter, sequentially, so that at each stage a number of materials were eliminated from further testing. First, the roughness of cast polysaccharide materials was assessed. A smooth surface of the dry polysaccharide layer cast in the counting chambers is critical to ensure bubble-free sample filling for correct identification of cells and precise measurement of sample volume and uniform cell distribution, which are required for accurate cell counts. The roughness of all prepared layers of polysaccharide materials is shown in Fig. 2a. Fully dissolved chi, gell and carr solutions resulted in smooth layers with a coefficient of variance (CV) of the layer thickness of less than 0.11 (Fig. 2a, green labels). In contrast, xan, all polysaccharide blends, and ionically crosslinked polysaccharides were poorly soluble, partially aggregating and therefore, resulted in rough layers (Fig. 2a, red labels), which prevented homogeneous filling of the counting chambers. It should be mentioned, that when casting polysaccharide/Ab mixtures, the CV of dry layer fluorescence intensity, representing the Ab distribution in the dry matrix, correlates well with the CV of dry layer thickness (Fig. 2a), indicating that there is no significant phase separation between Ab and polysaccharide materials. Therefore, layer homogeneity is a good measure of Ab distribution. Only chi, gell and carr matrices were deemed sufficiently smooth and therefore the only materials used in further screening.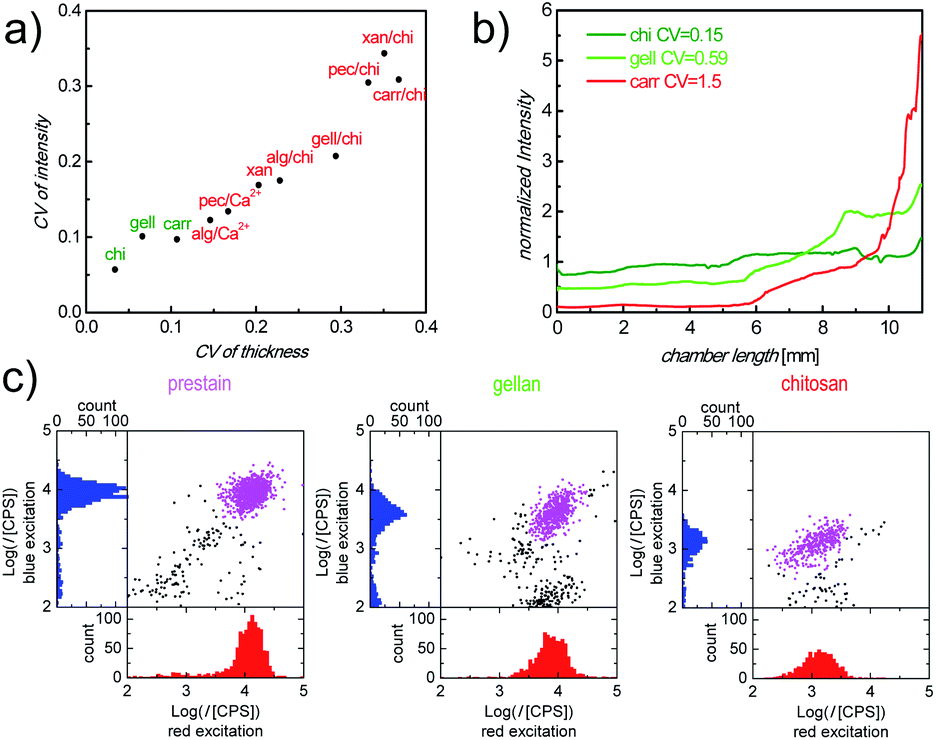 | ||
| Fig. 2 (a) Correlation of CV of roughness and CV of fluorescence intensity (APC-αCD3 readout) in polysaccharide/Ab layers. (b) Normalized and background corrected intensity ratios, along inflow direction, indicating the distribution of APC-αCD3 in the counting chambers with different cast layers after/before sample inflow. (c) Representative scatter plots and histograms obtained using our image cytometry method. Plotted are the fluorescence intensity (I) of APC-αCD3 (red excitation) and PerCP-αCD4 (blue excitation) stained cells in counting chambers with: prestained cells (left), cells stained in chambers with cast gell/Ab layers (center) and cells stained in chambers with cast chi/Ab layers (right), all after 30 min incubation. Purple dots are cells identified as the double positive cell population. | ||
Ab distribution after inflow is a good indication of the (delayed) release of Ab from the polysaccharide matrix during sample inflow. As a second step during the screening process, this parameter was evaluated by quantifying the Ab concentration gradient after inflow using fluorescence imaging.
In Fig. 2b, normalized fluorescence intensities, as ratios of Ab concentrations before and after sample inflow, are plotted against the distance from the sample filling port of the chamber for the remaining 3 materials, carr, chi, and gell. Only counting chambers with cast carr/Ab layers show a steep Ab concentration gradient (Fig. 2b, red line) after sample inflow. We determined in dipping experiments that carr rapidly dissolves in aqueous media (data not shown). The loss of integrity of the matrix during inflow can obviously cause strong Ab wash-off. In contrast to the results obtained for carr, the Ab wash-off from gell/Ab layers is largely prevented, while chambers with cast chi/Ab layers show no significant Ab concentration gradient at all. The degree of the homogeneity of the Ab distribution was also quantified as CV of the normalized fluorescence intensity across the chamber. The CVs from the chambers with cast gell/Ab and chi/Ab layers were below 1 (Fig. 2b, green lines) and were deemed promising enough for both materials to be tested regarding cell staining performance (screening step 3), while carr was excluded from further testing.
When carrying out full CD4 counting assays, the intensity of stained cells, after the incubation period, reflects the availability of Ab in the sample fluid. Sufficient Ab release from the polysaccharide matrices within the incubation period is necessary to ensure sufficient cell staining, ideally with staining intensities close to saturation levels. Generally, rapid diagnostic POC tests require on-chip immunostaining to approach saturation within 30 min. Therefore, staining efficiency was assessed by comparing the fluorescence intensities of CD4+ T-cells in whole blood samples stained in counting chambers with cast chi/Ab and gell/Ab layers, containing equal amounts of Ab, after 30 min of incubation with the corresponding saturated intensities of prestained cells in chambers without Ab release layer. The scatter plots of the fluorescence intensities of cells stained with allophycocyanin labeled antiCD3 IgG (APC-αCD3, red excitation) vs. peridinin chlorophyll labeled antiCD4 IgG (PerCP-αCD4, blue excitation) were analyzed. Fig. 2c shows that in each of the counting chambers a clearly defined double-positive population can be identified by our image cytometry method, when plotting the intensities of each object in the image under blue excitation against their intensities under red excitation. Fluorescence intensities of cells stained in the chambers with cast chi/Ab layers were much lower than those in the reference chambers filled with prestained cells (intensities of cells in the chambers with cast chi/Ab layers compared with prestained cells: 10 ± 4% for APC, 14 ± 2% for PerCP). In contrast, intensities of stained cells in the chambers with cast gell/Ab layers were much closer to the intensities of prestained cells, used as reference (70 ± 4% for APC, 53 ± 5% for PerCP). The discrepancy of the resulting cell staining intensities between chi/Ab layers and gell/Ab layers can be explained by their difference in the mass fraction of the released Ab within the relevant 30 min incubation period (M30min/Mtotal). As dipping experiments have shown, the majority of Ab is released from the gell matrix (95 ± 2%, n = 3) after 30 min incubation and is therefore available to stain cells. In contrast, a much smaller portion (24 ± 3%, n = 3) of Ab is released from the chi matrix during the same period of time. Additionally, the substantial amount of Ab retained in chi matrices contributes to the background intensity. The increased background intensity together with the reduced cell staining intensity makes the identification of stained cells in the chambers with cast chi difficult. Therefore, gell was selected as the most promising polysaccharide material for the optimization of storage and release of fluorophore labeled Ab in our counting chambers.
Clearly, the release kinetics of Ab from the matrix is vital for both uniform and strong cell staining. In order to evaluate the long-term assay performance of the counting chambers with cast gell/Ab layers, the stability of gell as a matrix was assessed. To this end, the fractional release of APC-αCD3 from cast gell/Ab layers stored at three different temperatures was measured for different storage times up to 3 months. Two characteristic release fractions, after 10 s (inflow period) and 30 min (incubation period), were recorded. Fig. S4† shows that the resulting M10s/Mtotal and M30min/Mtotal are storage temperature and time independent, indicating the excellent stability of gell as Ab release matrix.
Preparation and optimization of the CD4 counting chambers with printed gellan/Ab layers
For the fabrication of affordable counting chambers, we used inkjet printing to deposit gell/Ab layers on glass substrates. Inkjet printing, as demonstrated earlier,26 is a potent tool to reduce labor and can facilitate future up-scaling to large volume production. We first assessed the performance of counting chambers with printed gell/Ab layers. To this end, chambers containing printed gell/Ab layers were compared with chambers with cast gell/Ab layers in CD4 counting assays, by comparing the respective fluorescence intensities of stained cells after 30 min incubation. In comparison with the chambers with cast layers (Fig. 2c middle), APC-αCD3 and PerCP-αCD4 stained cells in the chambers with printed layers (Fig. 3e) were 14% and 56% dimmer, respectively. This intensity drop could be due to reagent degradation during the jetting process. We therefore determined the emission intensity of the fluorophores in the ink after jetting, compared with fluorophore intensity from the ink in the reservoir: the intensities of APC and PerCP decrease only by 3% and 24% during jetting, respectively. The fact that the decrease of cell staining intensity, when comparing chambers with printed layers with chambers with cast layers, is larger than the reduction in fluorophore intensity upon jetting, implies that the jetting process not only damages the fluorophores, but also compromises the viability of the Abs. To maintain the viability of fluorophore labeled Ab and optimize cell staining in the counting chamber, trehalose, well-known for its protein-stabilizing abilities,43–45 was tested as protein preservative and added to the gell matrix. The new composite gell/trehalose/Ab layers, deposited by inkjet printing, were then used as storage and release matrices in our counting chambers.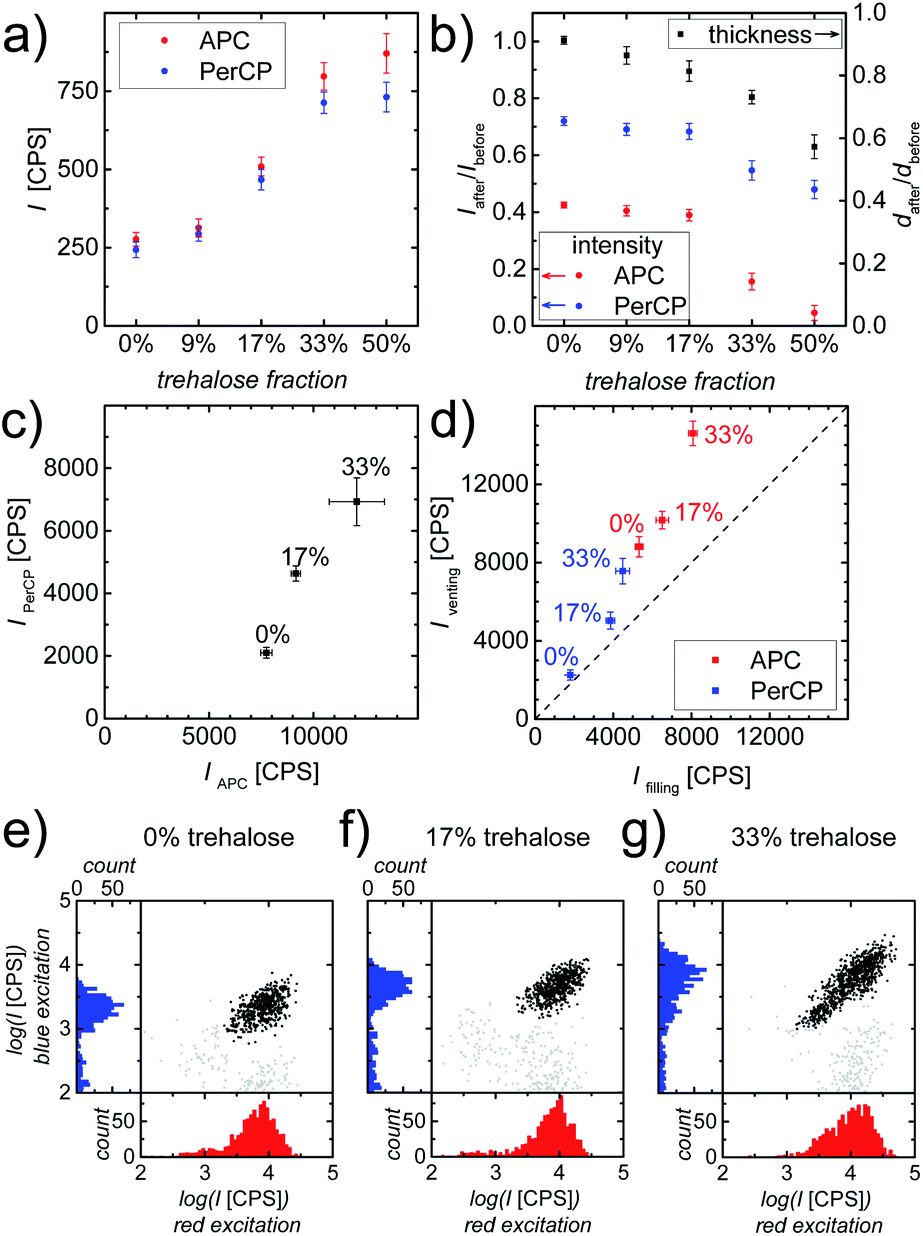 | ||
| Fig. 3 (a) Fluorescence intensities (I) of dry printed layers with varying fractions of trehalose but equal amounts of gell and fluorophore labeled Ab. (b) The intensity and thickness (d) ratios of the layer after and before PBS immersion. Data points represent mean ± standard deviation (n = 4). (c) Average intensities (PerCP vs. APC readout) obtained using our image cytometry method of CD4+ T-cells stained in counting chambers containing printed gell/Ab layers with varying trehalose contents. Data points represent mean ± standard deviation (n = 3). (d) Summary of average intensities (PerCP vs. APC readout) obtained using our image cytometry method of CD4+ T-cells at “filling” vs. “venting regions” stained in counting chambers containing printed gell/Ab layers with varying trehalose contents. Data points represent mean ± standard deviation (n = 4). The dashed diagonal line represents the ideal case, where the average intensities of stained cells located at the “filling region” and the “venting region” are identical, which implies the absence of Ab wash-off. (e–g) Scatter plots and histograms of fluorescence intensities obtained using our image cytometry method of APC-αCD3 (red excitation) and PerCP-αCD4 (blue excitation) stained cells in counting chambers containing: (e) pure gell (no trehalose), (f) gell/trehalose (17%) and (g) gell/trehalose (33%) matrices. Black solid circles represent all double (CD3 and CD4) positive cells (i.e. CD4+ T-cells) found in the entire image area. | ||
Prior to evaluating the cell staining process in chambers with printed composite layers, the influence of additional trehalose on the properties of these printed layers was examined. It is widely acknowledged that the viability of most proteins relies on their structural integrity. Even minute changes in the tertiary structure of proteins, i.e. denaturation, are associated with changes in their functionality, e.g. the fluorescence characteristics of fluorescent proteins, like GFP.46–48 Thus, the fluorescence intensity of dry printed layers was used to assess the effect of trehalose on the preservation of the structural integrity of the fluorophores, APC and PerCP, which are large proteins with multiple phycobilin, or carotene and porphyrin derived chromophores, respectively.49,50 As can be clearly seen in Fig. 3a, the addition of trehalose to gell/Ab layers helps to maintain the fluorescence intensities of both APC and PerCP, probably linked to the retention of their structural integrity. The fluorophores, in composite matrices containing ≥33% trehalose are well preserved, resulting in APC and PerCP being ∼3 times as bright as those in pure gell matrix. However, trehalose readily dissolves upon coming into contact with the aqueous sample fluid, thus causing partial disintegration of the composite matrix. To determine the impact of trehalose on matrix integrity and Ab release kinetics during sample inflow, printed composite layers were immersed in PBS for 10 s. The resulting reductions in thickness and fluorescence intensity of the printed composite layers are shown in Fig. 3b. These results clearly indicate that both the dissolution of the release layer as well as the Ab wash-off increase with trehalose content. While the addition of trehalose to gell matrices improved the fluorophore preservation, it negatively affected the layer integrity and increased Ab wash-off.
These two effects need to be balanced and the optimal trehalose content for best assay performance needs to be determined. To this end, the most promising composite matrices with 17% and 33% trehalose were compared to trehalose-free layers in cell staining experiments. The evaluation of on-chip immunostaining was carried out by quantifying the intensity of stained cells in the counting chambers after 30 min incubation at room temperature (RT) using whole blood as sample fluid. The fluorescence intensities of APC-αCD3 (red excitation) and PerCP-αCD4 (blue excitation) stained cells were analyzed and are shown in Fig. 3c. As expected, double positive cells (CD4+ T-cells) stained in printed chambers with gell/trehalose/Ab layers were much brighter than their counterparts in printed chambers with trehalose-free layers, showing that trehalose preserves the fluorophores (APC and PerCP) in printed layers contributing to improved immunostaining. Fig. 3e and f show that double positive objects form defined and compact (cell) populations in chambers with printed trehalose-free layers and 17% trehalose layers. A much more smeared-out population of double positive objects (Fig. 3g) is observed in chambers with printed 33% trehalose layers. This large range in intensities is indicative of Ab wash-off, which leads to inhomogeneous Ab distribution and hence inhomogeneous cell staining resulting in low intensities close to the “filling port” (Fig. 1a) and high intensities close to the “venting slit” (Fig. 1a). In principle, this is unproblematic as long as the cells can still be distinguished from the background easily. However, if the effect is too strong, poorly stained cells can hardly be discriminated from the noise, whereas high Ab concentration leads to large background inhomogeneity due to the abundance of erythrocytes in the sample, thus making a clear identification of stained cells difficult. Fig. 3d summarizes the intensities (APC and PerCP readout) of double positive cells at “filling” versus “venting” regions of counting chambers containing printed gell/Ab layers containing different amounts of trehalose. As can be seen, the addition of 33% trehalose to the gell layer helps to achieve intense cell staining. However, the resulting inhomogeneity of cell intensities causes severe problems in cell identification, which outweighs the benefits. A trehalose content of 17% proved a good compromise between improved staining intensity and increased Ab wash-off. Therefore, all further experiments were carried out with chambers containing printed composite gell/trehalose (17%)/Ab layers.
Shelf life of printed gellan CD4 counting chambers
In addition to the performance of the counting chambers shortly after their fabrication, we determined the dependence of the immunostaining on storage conditions and storage time. To this end, a batch of optimized counting chambers (with printed gell/trehalose (17%)/Ab layers), subject to different storage conditions (4 °C, 20 °C and 40 °C; stored in vacuum, over silica gel, or at high relative humidity, RH = 85%), were repeatedly tested in CD4 counting assays using whole blood from healthy anonymous donors over a period of ∼3 months. Intensities of the dry layers, as well as cell staining intensities and counting accuracy were analyzed, to evaluate the shelf life. Storage over silica gel at 4 °C (4sg) was found to be the optimal condition for long-term preservation of our printed counting chambers. As can be clearly seen in Fig. 4a, more than 70% of the fluorescence intensities of the dry printed layers were retained over a period of ∼3 months, indicating good stability of both fluorophores. Fig. S5† shows the intensities of APC-αCD3 and PerCP-αCD4 stained cells in the counting chambers stored under 4sg conditions up to ∼3 months. In spite of a moderate decrease of cell staining intensities (65% of the initial value in both APC and PerCP readouts) in ∼3 month old chambers, the double positive population can still be identified without any difficulty.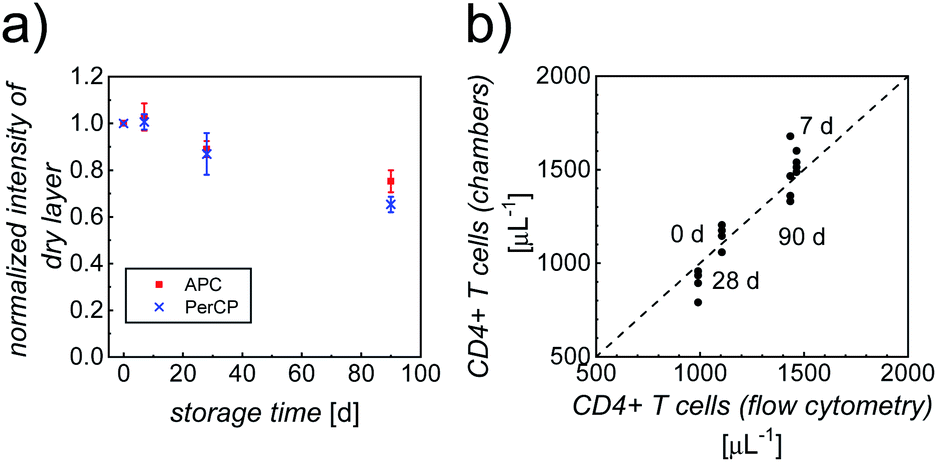 | ||
| Fig. 4 (a) The ratio of fluorescence intensity (I) in both APC (red squares) and PerCP (blue crosses) readouts of dry printed layers after storage over silica gel at 4 °C (4sg) and the intensity of comparable fresh layers. Data points represent mean ± standard deviation (n = 4). (b) Comparison between CD4 counts (#cells per μL) of whole blood from healthy donors, measured using our image cytometry approach and standard flow cytometry. For each sample with a given leukocyte concentration, 4 separate counting chambers were used and 3 separate flow cytometry analyses were performed. The averages of the flow cytometry results were plotted against the individual counts using our image cytometry approach. | ||
Fig. 4b shows the very good correlation of cell counts obtained from chambers stored under 4sg conditions for different periods of time, when compared to standard flow cytometry results, confirming excellent assay performance of chambers stored up to ∼3 months. The parameters of accuracy (average counting error) obtained from the counting chambers subject to 4sg conditions are 4.8%, 5.7% and 7.9% for 1 week, 1 month and 3 month storage, respectively.
It should be pointed out that counting chambers stored at 4 °C, sealed in vacuum packs perform equally well as those stored under 4sg conditions with accuracies of 2.5%, 5.6% and 7.0% for 1 week, 1 month, and 3 month storage, respectively. However, as vacuum packaging adds extra labor and cost to chamber production, storage over silica gel at 4 °C was chosen as the default approach to preserve counting chambers.
The other tested storage conditions: high temperature (20, 40 °C) and high humidity condition (∼85% RH) do not allow for long-term storage of the chambers with printed gell/trehalose (17%)/Ab layers. However, even under these, less favorable conditions, our counting chambers will retain excellent quality over a period of 1 week. Fig. S6† demonstrates a slight decrease (<20%) of APC and PerCP intensities in the dry layers and still acceptable counting accuracy (<10% average counting error) for chambers stored for 1 week under less favorable conditions. Even though counting chambers will not perform well after extended storage at higher temperatures and humidity, chambers stored under milder conditions perform very well for storage times of at least 3 months.
Performance of CD4 counting chambers with printed, optimized composite layers using whole blood samples from healthy donors and HIV-infected patients
After the successful fabrication of optimized counting chambers containing our new composite gellan/trehalose/Ab layer and demonstrating the reliable performance of our assay over a period of 3 months, we proceeded to validate our CD4 counting assay. Flow cytometry is considered the gold standard in CD4 counting. Thus, to validate our CD4 counting assay using image cytometry, the correlation of CD4 counts obtained using our optimized counting chambers containing printed gell/trehalose/Ab layers with CD4 counts obtained using flow cytometry, was assessed. First, a validation study was performed using whole blood from healthy, anonymous donors. Blood samples with 7 different concentrations of leukocytes were prepared. As can be seen in Fig. S7,† the results obtained from the two approaches correlate very well over a large range of cell concentrations (∼5–1000 μL−1).To substantiate this first validation with samples from healthy donors, we used leftover blood samples from HIV-infected patients (for details see methods section) for which reference CD4 counts were obtained by routine flow cytometry. One slide with two counting chambers each was used per patient sample. A linear regression of the results per chamber, also as a Bland–Altman plot,51–53 shown in Fig. 5, demonstrates the excellent agreement (coefficient of determination R2 = 0.950, Lin's concordance correlation:54 0.974 (95% CI: 0.962–0.982)) between the two methods.
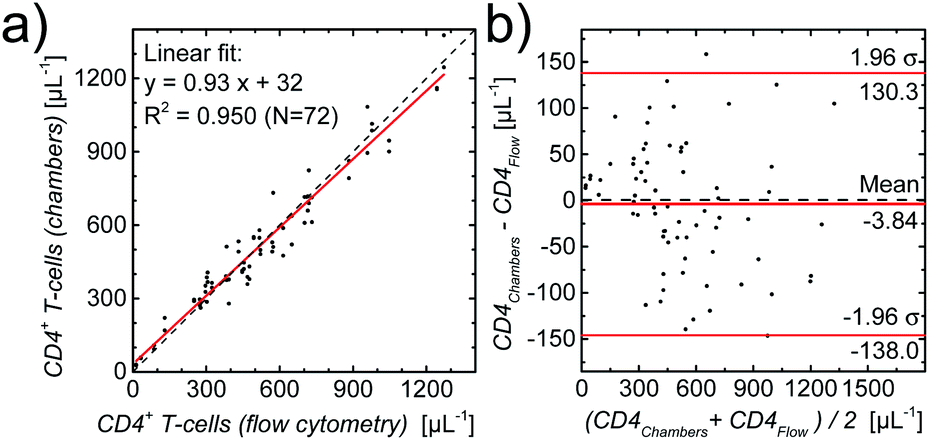 | ||
| Fig. 5 (a) Comparison between CD4 counts (number of CD4+ T-cells per μL) of HIV-infected patients obtained with standard flow cytometry and with image cytometry using fully printed CD4 counting chambers, with the linear regression demonstrating excellent agreement. (b) Bland–Altman plot of the same data. | ||
Conclusions
Here, we show the development of a new polysaccharide matrix, tailor-made for the storage and controlled release of fluorophore labeled antibody (Ab) in our simply designed and fully printed microfluidic chambers with complete on-chip sample preparation.By carefully tailoring the matrix material, taking into account suitability for inkjet printing, layer properties, antibody release kinetics, and shelf life, we managed to further simplify our point-of-care CD4 count and to extend the shelf life of the disposables to more than 3 months. A composite of gellan and trehalose emerges as the optimal release matrix out of a number of widely differing polysaccharide materials due to its superior performance in our counting chambers. This matrix is not only biocompatible and, thus, very well suited for the use with fluorophore labeled Ab, but its Ab release kinetics enables intense and homogenous immunostaining of cells in our chambers, without any preparative, bench-top treatment of whole blood samples. Our counting chambers are fabricated via automated printing techniques to minimize the labor and cost needed for their production, as well as making up-scaling to large volume production feasible. Furthermore, the gellan/trehalose matrix is compatible with the use of glass slides as chamber material. The use of glass substrates leads to minimized sample movement during measurements and thereby extends the scope of the assay to include non-coagulated blood samples.
We tested this approach of our point-of-care (POC) CD4 counting assay using blood samples from healthy donors and HIV-infected patients and found excellent agreement with flow cytometry results.
Preliminary data suggests that the low-cost printing technique used for the fabrication of our CD4 counting chambers, in combination with the simple concept of on-chip sample preparation will be applicable to a large variety of other (immuno-)staining cytometry assays at the POC.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
XZ, DW, CB, JvD and MB acknowledge financial aid under European Research Council Starting Grant FP7-IDEAS-ERC-282276. MB and JvD acknowledge financial aid under European Research Council under Proof-of-Concept grant H2020-ERC-PoC-694045. BJC acknowledges financial aid under Marie Curie Intra-European Fellowship FP7-PEOPLE-2012-IEF-331131.Notes and references
- B. Weigl, G. Domingo, P. Labarre and J. Gerlach, Lab Chip, 2008, 8, 1999 RSC.
- P. Yager, T. Edwards, E. Fu, K. Helton, K. Nelson, M. R. Tam and B. H. Weigl, Nature, 2006, 442, 412 CrossRef CAS PubMed.
- S. Luchters, K. Technau, Y. Mohamed, M. F. Chersich, P. A. Agius, M. D. Pham, M. L. Garcia, J. Forbes, A. Shepherd, A. Coovadia, S. M. Crowe and D. A. Anderson, J. Clin. Microbiol., 2019, 57, e01277 CAS.
- T. R. Sterling, R. E. Chaisson and R. D. Moore, AIDS, 2001, 15, 2251 CrossRef CAS PubMed.
- D. S. Boyle, K. R. Hawkins, M. S. Steele, M. Singhal and X. Cheng, Trends Biotechnol., 2012, 30, 45 CrossRef CAS PubMed.
- M. T. Glynn, D. J. Kinahan and J. Ducree, Lab Chip, 2013, 13, 2731 RSC.
- X. Li, Q. Deng, H. Liu, Y. Lei, P. Fan, B. Wang, Y. Chen, Z. J. Smith, Y. Tang and T. Gao, Anal. Bioanal. Chem., 2019, 411, 2767 CrossRef CAS PubMed.
- S. Bystryak, R. P. Bandwar and R. Santockyte, J. Virol. Methods, 2019, 271, 113672 CrossRef CAS PubMed.
- M. Sher and W. Asghar, Biosens. Bioelectron., 2019, 142, 111490 CrossRef CAS PubMed.
- S. Sharma, J. Zapatero-Rodriguez, P. Estrela and R. O'Kennedy, Biosensors, 2015, 5, 577 CrossRef PubMed.
- S. Kumar, S. Kumar, M. A. Ali, P. Anand, V. V. Agrawal, R. John, S. Maji and B. D. Malhotra, Biotechnol. J., 2013, 8, 1267 CrossRef CAS PubMed.
- J. Park, D. H. Han and J. K. Park, Lab Chip, 2020, 20, 1191 RSC.
- M. Hitzbleck and E. Delamarche, Chem. Soc. Rev., 2013, 42, 8494 RSC.
- M. Hitzbleck, L. Gervais and E. Delamarche, Lab Chip, 2011, 11, 2680 RSC.
- E. Garcia, J. R. Kirkham, A. V. Hatch, K. R. Hawkins and P. Yager, Lab Chip, 2004, 4, 78 RSC.
- D. Y. Stevens, C. R. Petri, J. L. Osborn, P. Spicar-Mihalic, K. G. McKenzie and P. Yager, Lab Chip, 2008, 8, 2038 RSC.
- E. Fu, B. Lutz, P. Kauffman and P. Yager, Lab Chip, 2010, 10, 918 RSC.
- M. Beck, S. Brockhuis, N. van der Velde, C. Breukers, J. Greve and L. W. Terstappen, Lab Chip, 2012, 12, 167 RSC.
- M. Mortato, L. Blasi, G. Barbarella, S. Argentiere and G. Gigli, Biomicrofluidics, 2012, 6, 044107 CrossRef PubMed.
- M. Tijero, R. Diez-Ahedo, F. Benito-Lopez, L. Basabe-Desmonts, V. Castro-Lopez and A. Valero, Biomicrofluidics, 2015, 9, 044124 CrossRef CAS PubMed.
- H. T. Mitchell, S. A. Schultz, P. J. Costanzo and A. W. Martinez, Chromatography, 2015, 2, 436 CrossRef CAS.
- R. R. Niedl and C. Beta, Lab Chip, 2015, 15, 2452 RSC.
- X. Zhang, D. Wasserberg, C. Breukers, L. W. M. M. Terstappen and M. Beck, Analyst, 2016, 141, 3068 RSC.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181 CrossRef CAS PubMed.
- X. Zhang, D. Wasserberg, C. Breukers, L. W. M. M. Terstappen and M. Beck, ACS Appl. Mater. Interfaces, 2016, 8, 27539 CrossRef CAS PubMed.
- D. Wasserberg, X. Zhang, C. Breukers, B. J. Connell, E. Baeten, D. van den Blink, È. Solà Benet, A. C. Bloem, M. Nijhuis, A. M. J. Wensing, L. W. M. M. Terstappen and M. Beck, Biosens. Bioelectron., 2018, 117, 659 CrossRef CAS PubMed.
- P. Matricardi, F. Alhaique and T. Coviello, Polysaccharide Hydrogels: Characterization and Biomedical Applications, CRC Press, Baco Raton, FL, USA, 2016 Search PubMed.
- K. Imamura, T. Ogawa, T. Sakiyama and K. Nakanishi, J. Pharm. Sci., 2003, 92, 266 CrossRef CAS PubMed.
- N. B. Shelke, R. James, C. T. Laurencin and S. G. Kumbar, Polym. Adv. Technol., 2014, 25, 448 CrossRef CAS.
- V. Jokinen and S. Franssila, Microfluid. Nanofluid., 2008, 5, 443 CrossRef.
- A. Olanrewaju, M. Beaugrand, M. Yafia and D. Juncker, Lab Chip, 2018, 18, 2323 RSC.
- T. Coviello, P. Matricardi, C. Marianecci and F. Alhaique, J. Controlled Release, 2007, 119, 5 CrossRef CAS PubMed.
- G. Franz and U. Feuerstein, Macromol. Symp., 1997, 120, 169 CrossRef CAS.
- N. Ruocco, S. Costantini, S. Guariniello and M. Costantini, Molecules, 2016, 21, 551 CrossRef PubMed.
- Polysaccharides: Structural Diversity and Functional Versatility, ed. S. Dumitriu, CRC Press, Baco Raton, FL, USA, 2004 Search PubMed.
- UNAIDS, FACT SHEET – World AIDS Day, 2019, http://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf, accessed April, 2020 Search PubMed.
- M. Helbert and J. Breuer, J. Clin. Pathol., 2000, 53, 266 CrossRef CAS PubMed.
- WHO, Update on recommendations on antiretroviral regimens for treating and preventing HIV infection, https://apps.who.int/iris/bitstream/handle/10665/277395/WHO-CDS-HIV-18.51-eng.pdf, accessed April, 2020 Search PubMed.
- I. Braccini and S. Perez, Biomacromolecules, 2001, 2, 1089 CrossRef CAS PubMed.
- C. K. Kuo and P. X. Ma, Biomaterials, 2001, 22, 511 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671 CrossRef CAS PubMed.
- M. Beck, S. Brockhuis, N. van der Velde, C. Breukers, J. Greve and L. W. M. M. Terstappen, Lab Chip, 2012, 12, 167 RSC.
- J. L. Cleland, X. Lam, B. Kendrick, J. Yang, T. H. Yang, D. Overcashier, D. Brooks, C. Hsu and J. F. Carpenter, J. Pharm. Sci., 2001, 90, 310 CrossRef CAS PubMed.
- N. K. Jain and I. Roy, Protein Sci., 2009, 18, 24 CAS.
- J. K. Kaushik and R. Bhat, J. Biol. Chem., 2003, 278, 26458 CrossRef CAS PubMed.
- S. H. Bokman and W. W. Ward, Biochem. Biophys. Res. Commun., 1981, 101, 1372 CrossRef CAS PubMed.
- Z. Ganim and M. Rief, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11052 CrossRef CAS PubMed.
- B. D. N. Rao, M. D. Kemple and F. G. Prendergast, Biophys. J., 1980, 32, 630 CrossRef CAS PubMed.
- R. R. Sonani, G. D. Gupta, D. Madamwar and V. Kumar, PLoS One, 2015, 10, e0124580 CrossRef PubMed.
- E. Hofmann, P. M. Wrench, F. P. Sharples, R. G. Hiller, W. Welte and K. Diederichs, Science, 1996, 272, 1788 CrossRef CAS PubMed.
- D. G. Altman and J. M. Bland, Statistician, 1983, 32, 307 CrossRef.
- J. M. Bland and D. G. Altman, Lancet, 1986, 1, 307 CrossRef CAS.
- D. Giavarina, Biochem. Med., 2015, 25, 141 CrossRef PubMed.
- L. I.-K. Lin, Biometrics, 1989, 45, 255 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The fabrication process of chambers by casting (S1), the schematic representations of printed counting chambers (S2), the step-wise polysaccharide screening process (S3), the fractional release of reagent from cast layers stored for different periods of time (S4), the intensities of cells stained in counting chambers stored up to 3 months (S5), the stability of reagents and the cell count accuracy from counting chambers stored under different conditions (S6), and the comparison of CD4 counts between image cytometry and flow cytometry (S7). See DOI: 10.1039/d0ra01645d |
| This journal is © The Royal Society of Chemistry 2020 |