
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The linkers in fluorene-labeled 2′-deoxyuridines affect fluorescence discriminating phenomena upon duplex formation†
So Young Leea,
Seung Woo Honga,
Hyeonuk Yeo *b and
Gil Tae Hwang*a
*b and
Gil Tae Hwang*a
aDepartment of Chemistry and Green-Nano Materials Research Center, Kyungpook National University, Daegu 41566, Republic of Korea. E-mail: giltae@knu.ac.kr
bDepartment of Chemistry Education, Kyungpook National University, Daegu 41566, Republic of Korea. E-mail: yeo@knu.ac.kr
First published on 19th May 2020
Abstract
Three fluorene-labeled 2′-deoxyuridines that differ in terms of their linkers—UF (without linker), UFL (with ethynyl linker), and UDF (with diethynyl linker)—have been introduced at the central positions of oligodeoxynucleotides to examine the effects that their linkers have on the fluorescence emission properties upon duplex formation with fully matched and single-base-mismatched targets. Here, we describe the influence of the linkers on the emission behavior, the intramolecular electron transfer between the fluorene moiety and the uracil base after photoexcitation, and the structural stability upon duplex formation. The probe containing the UFL residue (with an ethynyl linker) and cytosine residues as flanking bases exhibited the greatest fluorescence turn-on selective behavior toward the perfectly matched target.
Introduction
Detection of single nucleotide polymorphisms (SNPs) is an important aspect of the identification and diagnosis of disease-causing genes.1 Molecular beacons (MBs) featuring fluorophore and quencher units at the ends of their stems are used widely for the detection and analysis of SNPs.2 In the absence of a specific target, the emission of an MB is quenched as a result of the proximity of the fluorophore and quencher units; in contrast, duplex formation with the target nucleic acid results in a large increase in the fluorescence intensity. Interestingly, sequence-specific detection of nucleic acids is also possible when using fluorescently labeled oligonucleotide probes that feature no quencher unit.3 Recently, we developed a quencher-free MB system in which a 2′-deoxyuridine residue was linked to fluorene derivatives as labels through an ethynyl linker.4,5 That system could distinguish, through changes in fluorescence intensity, perfectly matched from single-base-mismatched sequences. Based on differences in the reducibility of nucleobases, intramolecular charge transfer (ICT) from fluorene derivatives to flanking pyrimidine residues (C or T) upon photoexcitation results in efficient quenching of single-stranded oligodeoxynucleotides (ssODNs) and single-base mismatched double-stranded ODNs (Fig. 1a).6 In addition, ODNs featuring C-flanking bases (C-FBs) can display additionally decreased fluorescence when hybridized with all-mismatched targets, when compared with that of ssODNs. This dramatic quenching arises because pairs of guanine bases (i.e., the complementary bases of C-FBs) act as internal quenchers when approached by the fluorene derivative.7 When the ODN encounters a perfectly matched target, however, this quenching is inhibited and the emission of the fluorene derivative is restored. Because an acetylene unit linking the fluorene derivatives to the 2′-deoxyuridine residue is efficient at mediating these electron transfer processes, fluorophore-labeled deoxyuridines featuring ethynyl linkers have been studied widely as fluorescent probes.8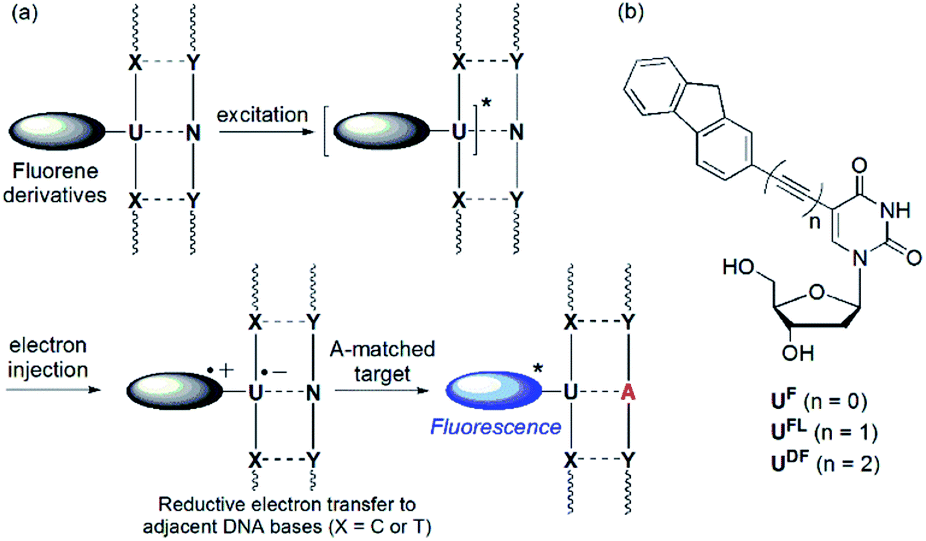 | ||
| Fig. 1 (a) Mechanism of operation of quencher-free MB systems. (b) Structures of fluorene-labeled 2′-deoxyuridines featuring various linkers. | ||
The ICT process is dependent on two features: (i) the energy difference between the donor's highest occupied molecular orbital (HOMO) and the acceptor's lowest unoccupied molecular orbital (LUMO) and (ii) the nature of the spacer mediating electron transfer between the two groups. In this study, we investigated how SNP probes would operate in the absence of a linker, and when a diethynyl unit was used as an alternative to an ethynyl moiety, linking the fluorene and 2′-deoxyuridine moieties (Fig. 1b). In previous studies, 2′-deoxyuridine residues labeled with chromophores through by a single C–C bond (i.e., linker-free systems) have been developed as fluorescence turn-on probes for the detection of an abasic site,9 triple-helix formation,10 a matched adenosine target11 and isomorphic thymine analogues.12 The only previously reported example of a DNA probe featuring a 2′-deoxyuridine residue labeled with a diethynyl linker was described by the Brown group,13 but their anthracene-labeled 2′-deoxyuridine residue featuring a diacetylenic unit could not discriminate effectively between matched and one-base-mismatched targets through changes in fluorescence intensity.
Results and discussion
UF and UDF were synthesized from 2′-deoxy-5-iodouridine (1) through a Suzuki reaction (Scheme 1a) with fluorene-2-boronic acid pinacol ester (2) and a Sonogashira reaction (Scheme 1b) with 2-(buta-1,3-diynyl)fluorene (6), respectively. UFL was synthesized using previously reported procedures.4f To obtain 2-(buta-1,3-diynyl)fluorene (6), we performed Cadiot–Chodkiewicz coupling14 of (bromoethynyl)triisopropylsilane (3)15 and 2-ethynylfluorene (4),16 followed by deprotection of the triisopropylsilyl group using tetrabutylammonium fluoride. After protecting the OH groups of UF and UDF with DMTr units, we converted the nucleosides to the phosphoramidites 9 and 10, respectively (Scheme 1c).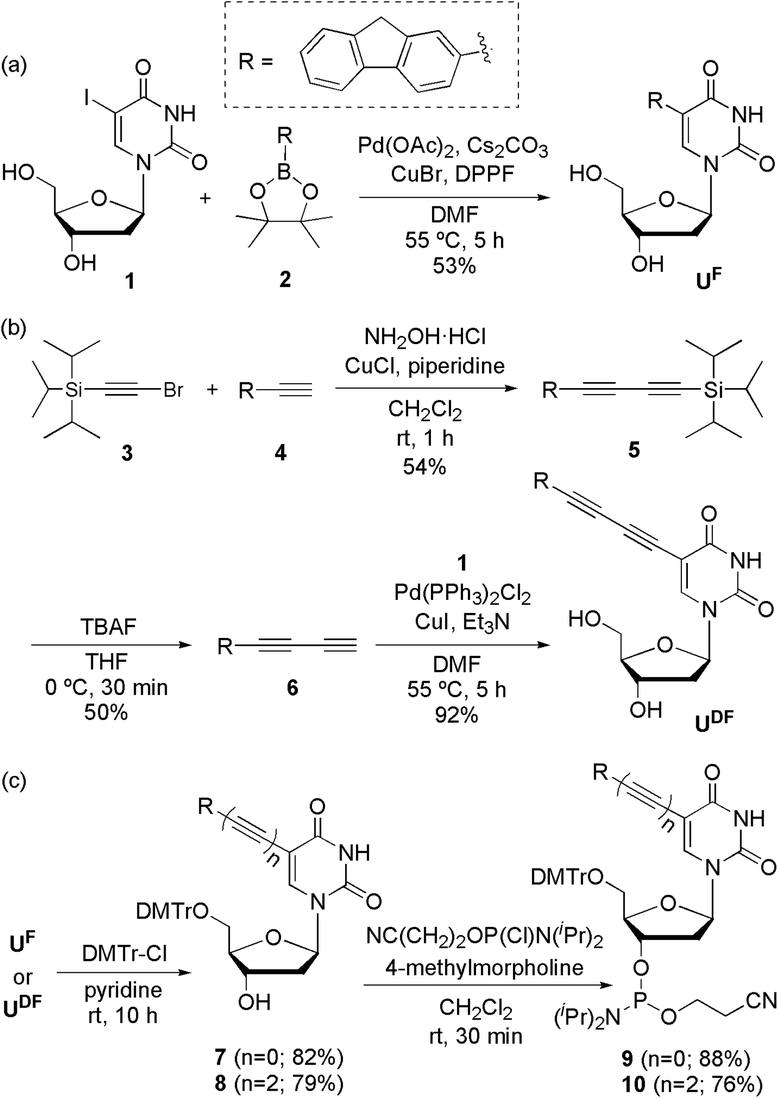 | ||
| Scheme 1 Synthesis of (a) UF, (b) UDF and (c) their phosphoramidites 9 and 10, respectively. | ||
Initially, we measured the absorption and emission spectra of the free nucleosides in MeOH (Table 1 and Fig. S1†). UF exhibited the highest absorbance among our three tested nucleosides; UFL displayed red-shifted absorption and emission signals relative to those of the more highly conjugated UDF. To determine the causes of these phenomena, we employed time-dependent density functional theory (TDDFT) to calculate the optimized structures of the ground and excited states of these nucleosides in vacuo (Fig. S2 and S3†). The dihedral angles between the fluorene and uracil moieties in the optimized structures of the ground states were 23.29° for UF, 0.77° for UFL and 10.21° for UDF; in their excited states they were 4.94, 3.95 and 26.87°, respectively. For both UF and UDF, the absorption maximum was blue-shifted relative to that of UFL because the dihedral angles between their fluorene and uracil moieties were twisted in the ground state (i.e., less effective conjugation). In addition, the emission maximum of UDF was similar to that of UF (which featured a planar dihedral angle in the excited state) and much more blue-shifted than that of UFL, because UDF remained relatively twisted in its excited state. This twist was presumably responsible for the low fluorescence yield of the UDF.
| Nucleoside | λmax/nma | ε/M−1 cm−1 | λem/nmb | ΦFc |
|---|---|---|---|---|
| a Only the largest absorption maxima are listed.b Wavelength of emission maximum when excited at the absorption maximum.c Quantum efficiencies determined using a cyclohexane solution of pyrene (λex = 313 nm) for UF, a solution of quinine sulfate in 0.1 N H2SO4 (λex = 350 nm) for UFL and a solution of fluorescein in 0.1 N NaOH (λex = 366 nm) for UDF as standards.18 Data are presented as mean values from three independent experiments.d Taken from ref. 17. | ||||
| UF | 310 | 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
419 | 0.16 |
| UFLd | 369 | 25000 |
453 | 0.26 |
| UDF | 363 | 26000 |
418 | 0.022 |
We used a DNA synthesizer and the phosphoramidites 9 and 10 to incorporate the UF and UDF residues, respectively, at the central positions of ODNs (Table 2). We characterized these ODNs using matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (Table S1†). To determine the suitability of these ODNs as SNP probes, we positioned pyrimidine bases (C or T) as FBs for the UF and UDF residues and compared their effects with those previously reported for UFL residues.
First, we investigated the discrimination against single-nucleotide variants exhibited by the fluorescent ODN1 featuring T-FBs (Fig. 2). When the ODN1s containing UF and UFL residues formed their matched duplexes, their emissions were enhanced 37 and 4.6 times, respectively, relative to those of the ssODN1s (Table 3). Notably, the fluorescence of ODN1(UF) itself was almost completely quenched through effective ICT; it recovered completely upon formation of its matched duplex. ODN1(UFL), featuring an acetylenic linker, did not experience ICT as effective as that of UF. Furthermore, when forming mismatched duplexes, the ODN1s containing UF and UFL residues provided fluorescence increases greater than those of the ssODN1s, in most cases, making it difficult to use them as A-selective SNP probes. All of the matched and mismatched duplexes of ODN1(UDF) displayed emissions slightly lower than those of ssODN1(UDF). These results indicate that ODN1(UDF) featuring T-FBs and diacetylenic linker did not produce effective ICT or its formation of duplex with A-matched target did not inhibit fluorescence quenching caused by the ICT process.
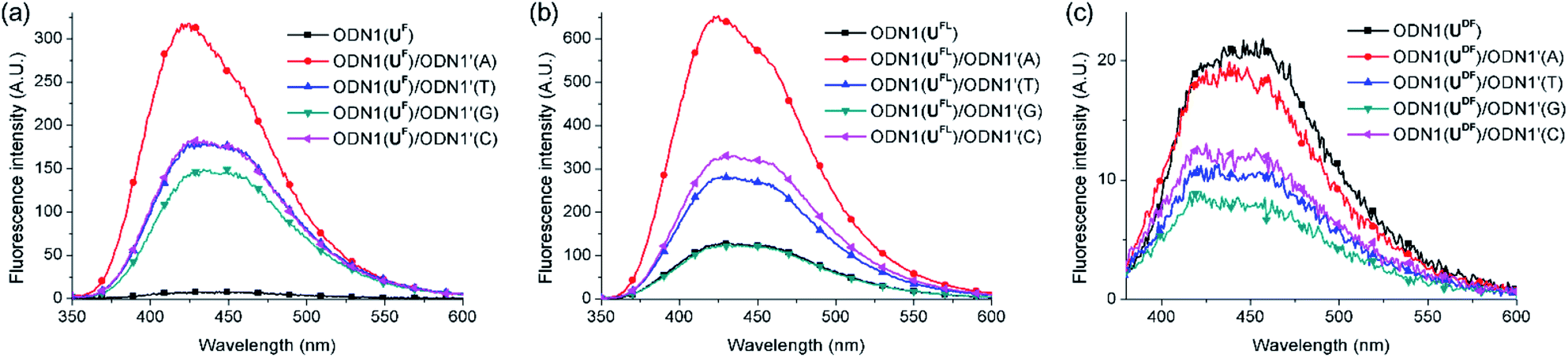 | ||
| Fig. 2 Fluorescence spectra of probes featuring T-FBs, recorded in 10 mM Tris–HCl buffer (pH 7.2; 100 mM NaCl, 20 mM MgCl2): ssODN1s and their duplexes (each concentration: 1.5 μM) formed between (a) ODN1(UF)/ODN1′(N), (b) ODN1(UFL)/ODN1′(N) and (c) ODN1(UDF)/ODN1′(N); N = A, T, G or C. Excitation wavelength: absorption maximum. | ||
| Duplexb | UF | UFLc | UDF |
|---|---|---|---|
| a Area ratio of fluorescence intensity relative to those of corresponding ssODNs.b X: UF, UFL or UDF.c Taken from ref. 4a. | |||
| ODN1(X)/ODN1′(A) | 37 | 4.6 | 0.91 |
| ODN1(X)/ODN1′(T) | 23 | 2.1 | 0.54 |
| ODN1(X)/ODN1′(G) | 19 | 0.96 | 0.43 |
| ODN1(X)/ODN1′(C) | 23 | 2.5 | 0.63 |
| ODN2(X)/ODN2′(A) | 5.7 | 4.0 | 2.6 |
| ODN2(X)/ODN2′(T) | 1.6 | 0.39 | 0.72 |
| ODN2(X)/ODN2′(G) | 1.1 | 0.35 | 0.67 |
| ODN2(X)/ODN2′(C) | 2.9 | 0.27 | 0.91 |
Next, we investigated the fluorescence behavior of the ODN2 featuring C-FBs (Fig. 3). The fluorescence of the matched duplex of ODN2(UFL) was four-fold greater than that of ssODN2(UFL); the mismatched duplexes exhibited decreases in emission that were more than twice that of ssODN, resulting from the quinine-quenching effect (G-effect). Similar to the situation for ODN1(UDF) having T-FBs, ODN2(UDF) featuring C-FBs displayed decreased fluorescence relative to that of ssODN when forming duplexes with mismatched targets. However, the formation of duplex with A-matched target showed only a 2.6-fold increase in fluorescence relative to that of ssODN2(UDF). In other words, the diacetylenic linker did not facilitate effective ICT. Interestingly, when ODN2(UF) formed its matched duplex, there was a 5.7-fold increase in fluorescence relative to that of ssODN2(UF)—slightly higher than the 4.0-fold increase of ssODN2(UFL). Thus, it appeared that the linker-free UF residue was slightly more suitable for effective electronic coupling between the fluorene and uracil moieties for ICT than was the UFL residue featuring an acetylenic linker. Nevertheless, ODN2(UF) exhibited slightly increased emissions upon hybridization with all of its mismatched targets, resulting in 1.6 (T)-, 1.1 (G)- and 2.9 (C)-fold enhancements in its emission intensities relative to that of ssODN2(UF). That is, the G-effect observed for ODN2(UFL) did not operate for ODN2(UF). We suspect that the fluorene unit was located too close to the duplexes of ODN2(UF), such that partial distortion of the duplex structure (or of the dihedral angle between the fluorene and uracil moieties) occurred when forming mismatched duplexes; thus, it was less likely that the G-effect or ICT through photoexcitation could be generated. We measured melting temperatures to compare the thermal stabilities of the duplexes formed using the ODN2s (Table 4). The A-selective thermal stability observed for the duplexes of ODN2(UFL) was not evident for ODN2(UF) and ODN2(UDF). That is, the values of Tm for the mismatched duplexes of ODN2(UF) and ODN2(UDF) were higher than those of ODN2(UFL). This phenomenon was presumably caused by the structural modification of the UF and UDF residues, and additionally weaker G-effects, making them ineffective DNA probes.
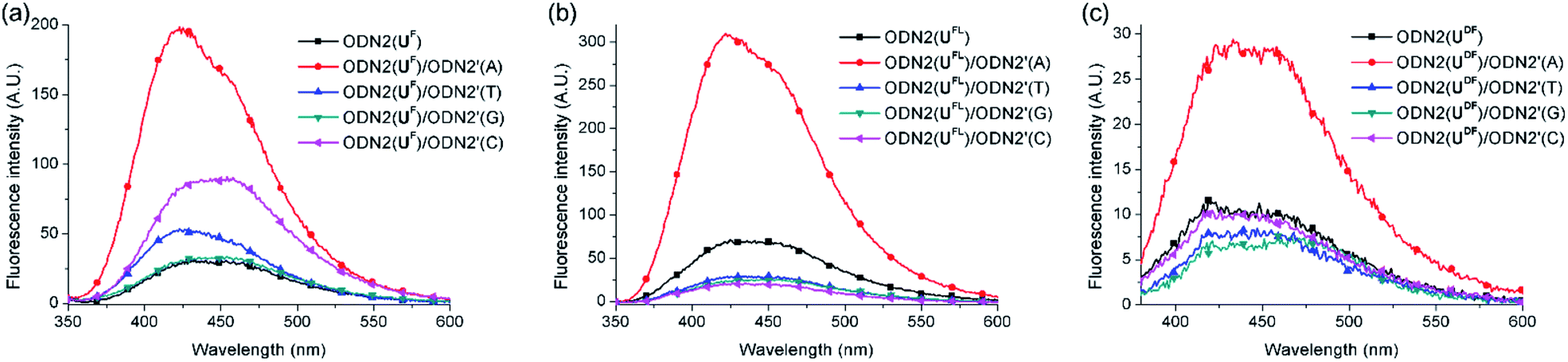 | ||
| Fig. 3 Fluorescence spectra of probes featuring C-FBs, recorded in 10 mM Tris–HCl buffer (pH 7.2; 100 mM NaCl, 20 mM MgCl2): ssODN2s and their duplexes (each concentration: 1.5 μM) formed between (a) ODN2(UF)/ODN2′(N), (b) ODN2(UFL)/ODN2′(N) and (c) ODN2(UDF)/ODN2′(N); N = A, T, G or C. Excitation wavelength: absorption maximum. | ||
| Duplex | X = T | X = UF | X = UFL | X = UDF |
|---|---|---|---|---|
| a All values of Tm (1.5 μM) were measured in 10 mM Tris–HCl buffer (100 mM NaCl, 20 mM MgCl2; pH 7.2) and are provided as averages from three independent measurements. | ||||
| ODN2(X)/ODN2′(A) | 57.3 | 48.3 | 48.7 | 49.5 |
| ODN2(X)/ODN2′(T) | 49.4 | 46.9 | 42.8 | 46.5 |
| ODN2(X)/ODN2′(G) | 51.8 | 45.6 | 43.6 | 47.8 |
| ODN2(X)/ODN2′(C) | 47.1 | 47.8 | 45.8 | 47.9 |
In summary, effective electron transfer between the two moieties occurred when the fluorene and uracil units were connected directly without a linker. This tendency was more pronounced when T-FBs were involved, resulting in a 37-fold increase in fluorescence when forming duplexes with A-matched targets. The application of a UF residue, however, was not suitable for discriminating mismatched duplexes. In addition, ODNs featuring a UDF residue, bearing a diacetylenic linker, were not useful as SNP probes because of low quantum yields and no electron transfer between the fluorene and uracil moieties, due to their twisted dihedral angle in the excited state. The UFL residue, bearing an ethynyl linker, did, however, feature an appropriate distance between the fluorene and uracil moieties to ensure effective electron transfer and structural stability of its duplexes. Nevertheless, the electron transfer of UFL was less effective than that of UF; furthermore, ODN2(UFL) featuring C-FBs also displayed an effective G-effect and could, therefore, act as an A-selective SNP probe.19
Conclusions
A previously reported quencher-free MB system featuring a 2′-deoxyuridine residue (UFL) labeled with a fluorene moiety (separated by an ethynyl linker) and C-FBs could discriminate a perfectly matched DNA target through an enhancement in fluorescence intensity. This discrimination originated from (i) effective ICT between the fluorene and uracil moieties after photoexcitation and (ii) the quenching effect of guanine residues that were complementary to the cytosine bases positioned as the FBs of the UFL residue. As potential alternatives to the ethynyl linker, in this present study we prepared the linker-free UF and the UDF featuring a diacetylenic linker, as well as their corresponding DNA probes. The emission of the isolated probe containing the UF residue was quenched through prominent ICT, but a large increase in fluorescence occurred when it met its matched target. Nevertheless, the fluorene moiety (in the absence of a linker) affected the three-dimensional structure of the mismatched duplexes, such that an effective G-effect was rare, resulting in low discrimination of mismatched targets. On the other hand, the presence of a diethynyl linker twisted the dihedral angle between the fluorene moiety and the uracil base, inhibiting electron transfer between the two moieties, such that its ODNs did not function as probes at all. Overall, the ethynyl linker was most suitable for positioning between the fluorene and uracil moieties. Because the π-conjugated bridge in UFL was quite planar, an electron could be transferred smoothly from the fluorene donor to the uracil acceptor. On the other hand, none of the probes featuring T-FBs exhibited selective fluorescence increases for their matched targets, due to the lack of a G-effect. Thus, the nature of the linker had a great influence on the dihedral angle and fluorescence properties of the nucleosides, as well as their propensity for electron transfer. Therefore, the selection of a suitable linker is one of the most important factors when designing a probe.Experimental
General
All reactions were performed in dry glassware under Ar atmospheres. Analytical thin layer chromatography (TLC) was performed using Merck 60 F254 silica gel plates; column chromatography was performed using Merck 60 silica gel (230–400 mesh). Melting points were determined using an Electrothermal IA 9000 series melting point apparatus and are uncorrected. Optical rotations [α]D were measured on a Rudolph Research Analytical AUTOPOL I polarimeter. Infrared (IR) spectra were recorded using a JASCO FT/IR-4100 spectrometer. 1H, 13C and 31P NMR spectra were recorded using a Bruker NMR spectrometer (AVANCE III 500 MHz). High-resolution fast atom bombardment (FAB) mass spectra were recorded using a JEOL JMS-700 mass spectrometer, at the Daegu Center of KBSI, Korea. All commercially available chemicals were used without further purification; solvents were carefully dried and distilled prior to use. The synthesis of UFL has been reported previously.4fSynthetic procedures
:1) to yield UF (159 mg, 53%): mp > 150 °C dec; [α]13D = −10.0° (c = 1.00 in MeOH); IR (film): ν 3364, 3039, 2933, 1659, 1607, 1591, 1461, 1422, 1351, 1275, 1231, 1089, 1017, 833, 734 cm−1; 1H NMR (500 MHz, CD3OD): δ 8.23 (s, 1H; H-6), 7.70 (d, J = 7.9 Hz, 1H; fluorene-H), 7.65 (s, 1H; fluorene-H), 7.47–7.42 (m, 2H; fluorene-H), 7.25 (t, J = 7.4 Hz, 1H; fluorene-H), 7.19 (td, J = 7.4, 0.96 Hz, 2H; fluorene-H), 6.27 (t, J = 6.6 Hz, 1H; H-1′), 4.37 (q, J = 3.6 Hz, 1H; H-3′), 3.80 (q, J = 3.1 Hz, 1H; H-4′), 3.81 (s, 2H; fluorene-CH2), 3.75–3.64 (m, 2H; H-5′), 2.27–2.23 (m, 2H; H-2′); 13C NMR (125 MHz, CD3OD): δ 167.0, 164.9, 152.0, 145.0, 144.7, 142.7, 142.6, 139.7, 132.9, 128.3, 128.1, 127.9, 126.2, 121.0, 120.7, 116.6, 89.2, 87.0, 72.2, 62.7, 41.8, 37.8; HRMS-FAB (m/z): [M + H]+ calcd for C22H21N2O5, 393.1450; found, 393.1447.:1) to yield UDF (87 mg, 92%): mp > 220 °C dec; [α]13D = −37.7° (c = 1.00 in MeOH); IR (film): ν 3404, 3187, 3122, 2951, 2813, 2144, 1721, 1660, 1612, 1465, 1316, 1271, 1099, 1049, 829 cm−1; 1H NMR (500 MHz, DMSO-d6): δ 11.77 (s, 1H; NH), 8.50 (s, 1H; H-6), 7.96 (d, J = 7.8 Hz, 2H; fluorene-H), 7.79 (s, 1H; fluorene-H), 7.63 (dd, J = 7.2, 2.5 Hz, 2H; fluorene-H), 7.42 (t, J = 7.3 Hz, 1H; fluorene-H), 7.37 (td, J = 7.4 Hz, 1.1 Hz, 1H; fluorene-H), 6.10 (t, J = 6.4 Hz, 1H; H-1′), 5.27 (d, J = 4.3 Hz, 1H; OH-3′), 5.19 (t, J = 4.9 Hz, 1H; OH-5′), 4.29–4.25 (m, 1H; H-3′), 3.96 (s, 2H; fluorene-CH2), 3.82 (q, J = 3.3 Hz, 1H; H-4′), 3.68–3.57 (m, 2H; H-5′), 2.18–2.16 (m, 2H; H-2′); 13C NMR (125 MHz, DMSO-d6): δ 161.6, 149.2, 146.2, 143.7, 143.4, 142.7, 140.1, 131.4, 128.9, 127.7, 127.0, 125.3, 120.8, 120.5, 118.3, 96.7, 87.7, 85.2, 82.6, 76.6, 75.7, 78.8, 69.8, 60.7, 40.3, 36.3; HRMS-FAB (m/z): [M]+ calcd for C26H20N2O5, 440.1372; found, 440.1373.:1) to yield 7 (290 mg, 82%): mp > 137 °C dec; [α]13D = −6.7° (c = 1.40 in CHCl3); IR (film): ν 3476, 3056, 2926, 1691, 1607, 1508, 1461, 1421, 1274, 1249, 1091, 1032, 862, 736 cm−1; 1H NMR (500 MHz, CDCl3): δ 8.45 (s, 1H; NH), 7.87 (s, 1H; H-6), 7.71 (d, J = 7.5 Hz, 1H; fluorene-H), 7.52 (d, J = 7.9 Hz, 1H; fluorene-H), 7.45–7.30 (m, 7H; fluorene-H + DMTr-H), 7.22–7.12 (m, 7H; fluorene-H + DMTr-H), 6.64 (dt, J = 8.7, 1.9 Hz, 4H; DMTr-H), 6.44 (t, J = 6.0 Hz, 1H; H-1′), 4.48–4.42 (m, 1H; H-3′), 4.10 (q, J = 3.7 Hz, 1H; H-4′), 3.63 (s, 6H; OCH3), 3.50–3.27 (m, 4H; fluorene-CH2 + H-5′), 2.56–2.34 (m, 2H; H-2′); 13C NMR (125 MHz, CDCl3): δ 162.1, 158.5, 149.9, 144.3, 143.5, 143.3, 141.4, 141.2, 136.5, 135.3, 135.4, 130.3, 130.0, 129.8, 129.7, 127.8, 127.0, 126.7, 126.6, 124.9, 119.9, 119.6, 116.3, 113.1, 86.6, 86.3, 85.3, 72.3, 63.3, 55.0, 41.4, 36.5; HRMS-FAB (m/z): [M]+ calcd for C43H38N2O7, 694.2679; found, 694.2683.:1) yielded 9 (227 mg, 88%): mp > 80 °C dec; IR (film): ν 2960, 2923, 2851, 2358, 1685, 1607, 1509, 1459, 1249, 1177, 1031, 977, 878, 827, 756, 736, 701, 641, 603 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.85 and 7.80 (2s, 1H; NH), 7.63 and 7.61 (2s, 1H; H-6), 7.41–7.31 (m, 4H; fluorene-H), 7.30–7.40 (m, 12H; fluorene-H + DMTr-H), 6.60–6.50 (m, 4H; DMTr-H), 6.39–6.32 (m, 1H; H-1′), 4.58–4.52 (m, 1H; H-3′), 4.19–4.23 (m, 1H; H-4′), 3.85–3.74 (m, 1H; OCH2), 3.72 (s, 2H; fluorene-CH2), 3.70–3.59 (m, 1H; OCH2), 3.54 and 3.53 (2s, 6H; OCH3), 3.43–3.12 (m, 4H; NCH + H-5′), 2.62–2.49 (m, 2H; CH2CN + H-2′), 2.39–2.21 (m, 2H; CH2CN + H-2′), 1.20–1.18 (m, 12H, NCHCH3); 31P NMR (202 MHz, CDCl3): δ 149.1, 148.6.Synthesis of oligonucleotides
ODNs were prepared using the β-cyanoethylphosphoramidite method on controlled pore glass supports (1 μmol) with a POLYGEN Professional 12-Column DNA synthesizer and standard methods.20 After automated synthesis, the oligonucleotides were cleaved from the solid support and deprotected through treatment with 30% aqueous NH4OH (1.0 mL) for 10 h at 55 °C. The crude products from the automated ODN synthesis were lyophilized and diluted with distilled water (1 mL); they were then purified using high-performance liquid chromatography (HPLC; Grace VyDAC™ C18 column, 250 × 10 mm; pore size: 120 Å). The HPLC mobile phase was held isocratically for 10 min using 5% MeCN/0.1 M triethylammonium acetate (TEAA; pH 7.0) at a flow rate of 2.5 mL min−1. The gradient was then increased linearly over 10 min from 5% MeCN/0.1 M TEAA to 50% MeCN/0.1 M TEAA at the same flow rate. The fractions containing the purified ODN were pooled and lyophilized. Aqueous AcOH (80%) was added to the ODN; after standing for 30 min at ambient temperature, the AcOH was evaporated under reduced pressure. The residue was diluted with water (1 mL) and then the solution was purified through HPLC under the same conditions described above. The concentrations of the ODNs were determined through measurement of UV-Vis absorptions. MALDI-TOF mass spectra of the ODNs were recorded using a Kratos Analytical AXIMA LNR MALDI TOF mass spectrometer operated in the linear mode with an 8:1 mixture of 3-hydroxypicolinic acid (0.35 M) and ammonium citrate (0.1 M) as the matrix; the accelerating voltage was 20 kV.
Melting temperatures (Tm)
All values of Tm of the ODNs (1.5 μM) were recorded in 10 mM Tris–HCl buffer (pH 7.2) containing 100 mM NaCl and 20 mM MgCl2. Absorbance–temperature profiles were measured at 260 nm using a Cary 100 Conc UV-Vis spectrophotometer equipped with a temperature controller (cell path length: 1 cm). The absorbance of the samples was monitored at 260 nm upon varying the temperature from 5 to 90 °C at a heating rate of 1 °C min−1. Melting temperatures were determined using a derivative method and Cary Win UV thermal application software. Each measurement was run in triplicate.UV and fluorescence spectroscopy
ODN solutions were prepared as described above for the measurement of the melting temperatures. Absorption spectra were recorded using a Cary 100 Conc UV-Vis spectrophotometer (cell path length: 1 cm). Fluorescence spectra were recorded using a Cary Eclipse fluorescence spectrophotometer (cell path length: 1 cm; excitation at absorption maximum).Optimized structure calculations
To understand absorption and emission phenomena of the nucleosides, the theoretical calculations were carried out by using the software package Gaussian 09 D.21 The calculation using B3LYP level with 6-31G basis set provided the optimized geometries at ground states (S0) and excited states (S1) and the images of the optimized structures were described in ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2017R1D1A1B03034920).Notes and references
- (a) M. Sharifi, M. Futema, D. Nair and S. E. Humphries, Curr. Cardiol. Rep., 2019, 21, 43 CrossRef PubMed; (b) X. M. Wei, X. C. Ju, X. Yi, Q. Zhu, N. Qu, T. F. Liu, Y. Chen, H. Jiang, G. H. Yang, R. Zhen, Z. Z. Lan, M. Qi, J. M. Wang, Y. Yang, Y. X. Chu, X. Y. Li, Y. F. Guang and J. Huang, PLoS One, 2011, 6, e29500 CrossRef CAS PubMed; (c) G. H. Luo, L. Zheng, X. Y. Zhang, J. Zhang, P. Nilsson-Ehle and N. Xu, Anal. Biochem., 2009, 386, 161–166 CrossRef CAS PubMed.
- (a) S. Tyagi, D. P. Bratu and F. R. Kramer, Nat. Biotechnol., 1998, 16, 49–53 CrossRef CAS PubMed; (b) A. S. Piatek, S. Tyagi, A. C. Pol, A. Telenti, L. P. Miller, F. R. Kramer and D. Alland, Nat. Biotechnol., 1998, 16, 359–363 CrossRef CAS PubMed; (c) S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS PubMed.
- G. T. Hwang, Molecules, 2018, 23, 124 CrossRef PubMed.
- (a) J. W. Lee, Y.-S. Son, J. Y. Hwang, Y. Park and G. T. Hwang, Org. Biomol. Chem., 2017, 15, 7165–7172 RSC; (b) J. Lee, H. Y. Cho and G. T. Hwang, ChemBioChem, 2013, 14, 1353–1362 CrossRef CAS PubMed; (c) J. H. Ryu, J. Y. Heo, E. K. Bang, G. T. Hwang and B. H. Kim, Tetrahedron, 2012, 68, 72–78 CrossRef CAS; (d) Y. A. Lee and G. T. Hwang, Bull. Korean Chem. Soc., 2010, 31, 2011–2014 CrossRef CAS; (e) J. H. Ryu, Y. J. Seo, G. T. Hwang, J. Y. Lee and B. H. Kim, Tetrahedron, 2007, 63, 3538–3547 CrossRef CAS; (f) G. T. Hwang, Y. J. Seo and B. H. Kim, J. Am. Chem. Soc., 2004, 126, 6528–6529 CrossRef CAS PubMed.
- (a) K. Murayama and H. Asanuma, ChemBioChem, 2020, 21, 120–128 CrossRef CAS PubMed; (b) K. T. Kim, R. N. Veedu, Y. J. Seo and B. H. Kim, Chem. Commun., 2014, 50, 1561–1563 RSC; (c) C. Boonlua, B. Ditmangklo, N. Reenabthue, C. Suparpprom, N. Poomsuk, K. Siriwong and T. Vilaivan, RSC Adv., 2014, 4, 8817–8827 RSC; (d) K. K. Karlsen, A. Okholm, J. Kjems and J. Wengel, Bioorg. Med. Chem., 2013, 21, 6186–6190 CrossRef CAS PubMed; (e) A. Okamoto, Y. Saito and I. Saito, J. Photochem. Photobiol., C, 2005, 6, 108–122 CrossRef CAS.
- (a) M. Tanaka, B. Elias and J. K. Barton, J. Org. Chem., 2010, 75, 2423–2428 CrossRef CAS PubMed; (b) K. Tainaka, M. Fujitsuka, T. Takada, K. Kawai and T. Majima, J. Phys. Chem. B, 2010, 114, 14657–14663 CrossRef CAS PubMed; (c) P. Daublain, A. K. Thazhathveetil, Q. Wang, A. Trifonov, T. Fiebig and F. D. Lewis, J. Am. Chem. Soc., 2009, 131, 16790–16797 CrossRef CAS; (d) S. T. Gaballah, J. D. Vaught, B. E. Eaton and T. L. Netzel, J. Phys. Chem. B, 2005, 109, 5927–5934 CrossRef CAS PubMed; (e) N. Amann, E. Pandurski, T. Fiebig and H. A. Wagenknecht, Chem.–Eur. J., 2002, 8, 4877–4883 CrossRef CAS PubMed.
- (a) G. H. Luo, L. Zheng, X. Y. Zhang, J. Zhang, P. Nilsson-Ehle and N. Xu, Anal. Biochem., 2009, 386, 161–166 CrossRef CAS PubMed; (b) C. A. M. Seidel, A. Schulz and M. H. M. Sauer, J. Phys. Chem., 1996, 100, 5541–5553 CrossRef CAS.
- (a) N. P. F. Barthes, I. A. Karpenko, D. Dziuba, M. Spadafora, J. Auffret, A. P. Demchenko, Y. Mely, R. Benhida, B. Y. Michel and A. Burger, RSC Adv., 2015, 5, 33536–33545 RSC; (b) A. A. Tanpure and S. G. Srivatsan, ChemBioChem, 2014, 15, 1309–1316 CrossRef CAS PubMed; (c) D. G. Lee, I. S. Kim, J. W. Park and Y. J. Seo, Chem. Commun., 2014, 50, 7273–7276 RSC; (d) M. Tanaka, K. Oguma, Y. Saito and I. Saito, Bioorg. Med. Chem. Lett., 2012, 22, 4103–4105 CrossRef CAS PubMed; (e) S. S. Bag, R. Kundu, K. Matsumoto, Y. Saito and I. Saito, Bioorg. Med. Chem. Lett., 2010, 20, 3227–3230 CrossRef CAS PubMed; (f) H. S. Jeong, S. Kang, J. Y. Lee and B. H. Kim, Org. Biomol. Chem., 2009, 7, 921–925 RSC; (g) Q. Xiao, R. T. Ranasinghe, A. M. P. Tang and T. Brown, Tetrahedron, 2007, 63, 3483–3490 CrossRef CAS; (h) Y. Saito, Y. Miyauchi, A. Okamoto and I. Saito, Tetrahedron Lett., 2004, 45, 7827–7831 CrossRef CAS; (i) A. Okamoto, K. Kanatani and I. Saito, J. Am. Chem. Soc., 2004, 126, 4820–4827 CrossRef CAS PubMed.
- A. A. Tanpure and S. G. Srivatsan, ChemBioChem, 2012, 13, 2392–2399 CrossRef CAS PubMed.
- T. Kanamori, H. Ohzeki, Y. Masaki, A. Ohkubo, M. Takahashi, K. Tsuda, T. Ito, M. Shirouzu, K. Kuwasako, Y. Muto, M. Sekine and K. Seio, ChemBioChem, 2015, 16, 167–176 CrossRef CAS PubMed.
- S. S. Bag, M. K. Pradhan, S. K. Das, S. Jana and R. Bag, Bioorg. Med. Chem. Lett., 2014, 24, 4678–4681 CrossRef CAS PubMed.
- (a) M. S. Noe, R. W. Sinkeldam and Y. Tor, J. Org. Chem., 2013, 78, 8123–8128 CrossRef CAS PubMed; (b) R. W. Sinkeldam, P. Marcus, D. Uchenik and Y. Tor, ChemPhysChem, 2011, 12, 2260–2265 CrossRef CAS PubMed; (c) R. W. Sinkeldam, N. J. Greco and Y. Tor, ChemBioChem, 2008, 9, 706–709 CrossRef CAS PubMed.
- Q. Xiao, R. T. Ranasinghe, A. M. P. Tang and T. Brown, Tetrahedron, 2007, 63, 3483–3490 CrossRef CAS.
- K. S. Sindhu, A. P. Thankachan, P. S. Sajitha and G. Anilkumar, Org. Biomol. Chem., 2015, 13, 6891–6905 RSC.
- D. Listunov, N. Saffon-Merceron, E. Joly, I. Fabing, Y. Genisson, V. Maraval and R. Chauvin, Tetrahedron, 2016, 72, 6697–6704 CrossRef CAS.
- T. F. Tani Hisaya and M. Koreyasu, Bull. Chem. Soc. Jpn., 1963, 36, 391–396 CrossRef.
- H. Y. Cho, S. K. Woo and G. T. Hwang, Molecules, 2012, 17, 12061–12071 CrossRef CAS PubMed.
- C. A. Parker, Photoluminescence of solutions. With applications to photochemistry and analytical chemistry, Elsevier Pub. Co., Amsterdam, New York etc., 1968 Search PubMed.
- (a) S. Kamoto, M. Hyuga and T. Kato, Analyst, 2016, 141, 6087–6092 RSC; (b) A. Okamoto, K. Tainaka, Y. Ochi, K. Kanatani and I. Saito, Mol. BioSyst., 2006, 2, 122–126 RSC.
- M. J. Gait, Oligonucleotide synthesis: a practical approach, IRL Press, Oxford Oxfordshire, Washington, DC, 1984 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D. 01, Gaussian. Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01651a |
| This journal is © The Royal Society of Chemistry 2020 |