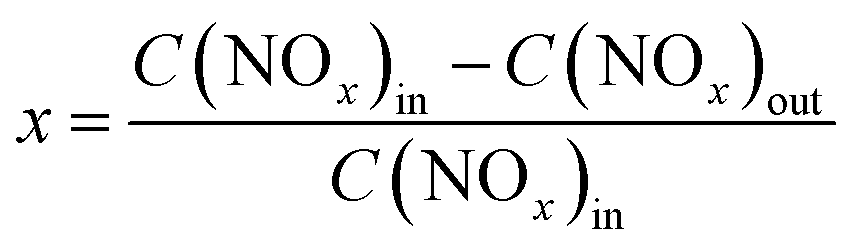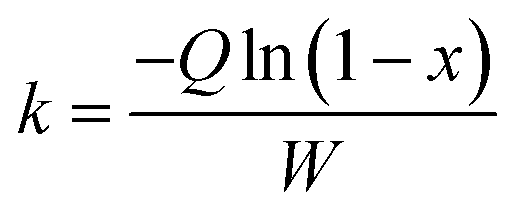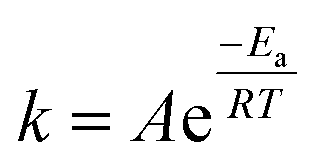DOI:
10.1039/D0RA01654C
(Paper)
RSC Adv., 2020,
10, 12908-12919
Insights into the promotion role of phosphorus doping on carbon as a metal-free catalyst for low-temperature selective catalytic reduction of NO with NH3†
Received
21st February 2020
, Accepted 25th March 2020
First published on 31st March 2020
1. Introduction
In recent years, the widespread use of fossil fuels has produced nitrogen oxides (NOx, mainly NO and NO2) that cause serious environmental problems, such as acid rain, photochemical smog, and particulate matter formation.1,2 Selective catalytic reduction of NO with NH3 (NH3-SCR) is a widely used technique for NO emission control during industrial production, with which an expensive and toxic V2O5–WO3/TiO2 catalyst is usually used. However, the optimum operating temperature of the V2O5–WO3/TiO2 catalyst is high (300–400 °C), so the SCR device must be installed upstream of the desulfurizer and dust collector to avoid reheating the flue gas, which shortened service life of the catalyst by the deactivation from the poisoners of high-concentration SO2 and dust.3–5 The difficulty in recovery of toxic vanadium species from the TiO2 support is another problem for this catalyst system. Therefore, the development of environment-friendly catalysts with high catalytic activity at relatively low temperature (100–200 °C) has become a research hotpot.
Carbon materials have been attracted great attention owning to their adjustable physical characteristics and surface chemistry properties. Except being used as the supports for the metal-based catalysts, carbon materials can also act as the metal-free catalysts by doping heteroatoms such as oxygen (O), nitrogen (N), boron (B), sulfur (S), and phosphorus (P), to tailor the electron donor–acceptor properties of carbon matrix. Oxygen, is the most common heteroatom on edges of carbon sheets, whose effects on the catalytic activity of carbon-based catalysts for NO reduction have been extensively studied. Many researchers have shown that the catalytic activity of carbon materials for NO conversion increases with the numbers and kinds of surface oxygen groups, as the adsorption sites for NO and NH3 molecules.6–9 Recently, Zhang et al. proposed that the carboxylic acid groups can directly participate in NO reduction.10 In addition, some studies have indicated that the incorporation of N into the carbon sheets can enhance the NH3-SCR activity.11–14 It is speculated that the nitrogen species can increase the carbon basicity and create the extra delocalized electrons for easy chemisorption of weakly acidic NO and O2 to facilitate NO2 formation.11,12 The NO conversion can be increased effectively by nitrogen-doping for the pre-oxidized carbon samples, suggesting the presence of acidic surface groups and N-containing functional groups is important for the activity and the selectivity of N2.11,12 Moreover, experimental and theoretical studies have revealed that N or S-doped graphene can be applied for direct catalytic decomposition of NO, the surface functional groups play a key role. The unique functional groups (pyridinic N and thiophene S) can transfer extra electrons to the π-antibond orbital of NO, which leads to lengthening the length of N-O bonds and reducing the order of N–O bonds, thereby weakening the NO stability. Eventually, the formation of Nads and Oads would further form N2 and O2. But the reaction temperature is pretty high (600–850 °C), which resulted in graphene oxidation that limit its long-term stability.15–18 Ji et al. systematically explored the possibility of boron-doped graphene (BG) for the NO electrochemical reduction (NOER) reaction through density functional theory (DFT). The results showed the introduction of B atom enhances the interaction of graphene with the HNO* intermediate, in which the preferable NO → HNO* → NH2O* → NH2OH* → NH2* → NH3 with a limiting potential of −0.35 V.19
P, situated in the same main group as N, possesses the same number of valence electrons as nitrogen. But it has a larger atomic radius and higher electron-donating ability than nitrogen, which make it an interesting dopant to alter the surface chemical properties of carbon sheets in specific applications.20–23 P modification can increase the specific capacitance of carbon electrode materials by the additional faradaic redox reactions, improve the cycle stability and widen the potential window as an oxidation protector in supercapacitors.23,24 The P-doped carbon materials as metal-free electrodes have exhibited higher electrocatalytic activity, lower resistance and much higher stability than commercial Pt/C catalysts for oxygen reduction reactions (ORR) in methanol oxidation.20,21,25–29 The P-doped carbon can be used as a metal-free catalyst to efficiently catalyze aerobic oxidation of benzyl alcohols to aldehydes or ketones.21 In addition, similar to nitrogen doping, the P-doping of carbon can increase the catalytic selectivity to alkene and decrease the consumption of hydrocarbons as a metal-free catalyst for selective hydrogenation of hydrocarbons.30,31 Furthermore, the catalytic role of phosphorus oxide clusters on carbon surface for alkane oxidative dehydrogenation was reported recently by Huang et al.32 They found that the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O configurations, exhibiting the structure and reactivity features similar to those of the traditional active metal oxide clusters, tended to abstract hydrogen atoms and favored the alkene selectivity. Despite multiple advantages of P-doping shown in above application fields, to the best of our knowledge, the effects of P-doping on the NH3-SCR performance of carbon-based catalysts is seldom concerned and the underlying principles remain unknown.
O configurations, exhibiting the structure and reactivity features similar to those of the traditional active metal oxide clusters, tended to abstract hydrogen atoms and favored the alkene selectivity. Despite multiple advantages of P-doping shown in above application fields, to the best of our knowledge, the effects of P-doping on the NH3-SCR performance of carbon-based catalysts is seldom concerned and the underlying principles remain unknown.
In the present work, P-doped carbon aerogels (P-CAs) were prepared by a one-pot sol–gel method using phosphoric acid as the phosphorus source, followed by carbonization at 600–900 °C. The contributions of P-containing functional groups to the NH3-SCR performance of the P-CAs at low temperatures (120–200 °C) were systematically investigated and discussed. We expect that these findings can broaden the insights into contributions of heteroatom-doping in carbon for NO reduction.
2. Experimental section
2.1 Catalyst preparation
In a typical synthesis, 8.15 g of phenol and 1.87 g of m-cresol were dissolved in 100 mL of 1-propanol to form a solution, then 2.98 g of phosphoric acid (85 wt%) was added to the solution under stirring and heated at 70 °C for 1 h. After the above solution was cooled to room temperature, 19.97 g of furfural was added to the solution under agitating for 15 min. The obtained mixed solution was poured into several ampules (30 mL), sealed, placed in a water bath at 80 °C for 5 days, and then at room temperature for 1 day, finally dried at 60 °C to obtain cylindrical organic xerogels. At last, the organic xerogels were carbonized in a vertical tube furnace at 600 °C, 700 °C, 800 °C and 900 °C for 3 h with a heating rate of 5 °C min−1 in nitrogen atmosphere, to obtain the P-CAs, which were denoted as P-CA-x, where x represents the carbonization temperature. Meanwhile, the P-CA-800 sample was subjected to heat treatment at 380 °C for 8 h in vacuum to obtain sample P-CA-800vac. For comparison, the P-free carbon aerogel was prepared following the same procedures and formulation without adding phosphoric acid under a carbonization temperature of 800 °C, which was denoted as CA-800.
2.2 Characterization of catalysts
The pore texture of the P-CAs and CA-800 was characterized by N2 adsorption at 77 K (Micromeritics, ASAP-2020). Samples were out-gassed at 150 °C for 12 h in vacuum prior to the measurements. The surface areas (SBET) was determined by the Brunauer–Emmett–Teller (BET) method using the N2 adsorption data. The mesopore volumes (Vmes) and micropore volumes (Vmic) were evaluated by the density functional theory (DFT) model. Surface compositions of samples were analyzed by X-ray photoelectron spectroscopy on a Thermo Scientific Escalab 250Xi system. The corresponding binding energies were calibrated by using C1s of 284.8 eV as a standard. The P content on the surface of samples was also examined by inductively coupled plasma atomic emission spectrometry (ICP-AES) on an Agilent 720 apparatus, in which the samples were digested by a microwave assisted pre-treatment. Fourier transform infrared (FTIR) spectra were recorded on a Nicolet Nexus 410 spectrometer at the range of 4000–400 cm−1 with a resolution of 4 cm−1 by collecting 256 scans. NH3-temperature programmed desorption (NH3-TPD) experiments were performed on a Micrometrics AutoChem II 2920 instrument. 100 mg of sample was pre-treated at 300 °C for 1 h in N2 and cooled to 50 °C, then was saturated with a 10 vol% NH3/Ar gas mixture at a flow rate of 30 mL min−1 for 1 h, followed by N2 purge for another 0.5 h, finally raised to 500 °C at a heating rate of 10 °C min−1 to obtain the NH3-TPD curves.
2.3 Catalytic activity measurement
The NH3-SCR activities were measured in a fixed-bed glass reactor with an internal diameter of 15 mm. The mass of the catalyst in the fixed-bed was 3.61 g. The simulated flue gas was composed of 500 ppm NO, 500 ppm NH3, 5.0 vol% O2, and N2 as the balance gas at a total flow rate of 88 mL min−1, corresponding to the gas hourly space velocity (GHSV) of 500 h−1. The catalytic reaction was tested from 100 to 200 °C. The outlet concentration of NO and NO2 were measured by a chemiluminescence method with an ECO PHYSICS nCLD62s NO/NOx analyzer and the concentration of N2O was detected by gas chromatography with a Techcomp GC 7900 equipment. The concentration of NH3 was measured by a laser method on a Mic-500S–NH3-Tdlas NH3 analyzer. The NO conversion was calculated using the following eqn (1),| |
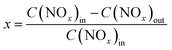 | (1) |
where the concentrations of NOx in the inlet and outlet are represented by C(NOx)in and C(NOx)out, respectively.
In this work, to better evaluate the catalytic activity of the P-CAs and CA-800, a steady state kinetics investigation of the catalysts was conducted. The calculation of the reaction rate constant k in the kinetics equation was based on the assumptions that the reaction is first order dependence of NO and zero order dependence of NH3.33,34 The calculation formula shown in the eqn (2):
| |
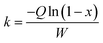 | (2) |
where
k is the reaction rate constant (cm
3 g
−1 s
−1),
Q means the total gas flow (mL min
−1),
W is the mass of the catalyst in the fixed reaction bed (g) and
x is the NO conversion. The apparent activation energy of the reaction is calculated according to the Arrhenius
eqn (3):
| |
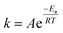 | (3) |
where
Ea (kJ mol
−1) represents the apparent activation energy of catalyst, which could be calculated from the slope of the curve ln(
k)
versus 1/
T.
3. Results and discussion
XPS measurements were used to characterize the types and contents of P-containing groups on the surface of as-obtained P-CAs. As shown in Fig. S1,† the peaks at 132.9 eV, 284.8 eV and 532.6 eV on the XPS full spectra of the P-CAs are assigned to P2p, C1s and O1s, respectively. The corresponding atomic contents of P, C and O are calculated and listed in Table 1. With increasing the carbonization temperature, the P content on the surface of carbon aerogels increases, reaching the highest value of 2.50% for the P-CA-800 sample, and then decreases to 1.37% for the sample P-CA-900. Meanwhile, the P content of the samples is also examined by ICP-AES and listed in Table 1. It is shown that the changing trend of the ICP results with the carbonization temperature is consistent with that of the XPS measurements. The ICP results represent the global atomic contents while the XPS measurements the surface atomic contents that depend on the penetration depth of XPS.
Table 1 Atomic compositions of the P-CAs
| Sample |
XPS analysis |
ICP analysis |
| C (%) |
PXPS (%) |
O (%) |
PICP (%) |
| P-CA-600 |
87.87 |
2.21 |
9.92 |
2.30 |
| P-CA-700 |
91.74 |
2.31 |
5.95 |
3.01 |
| P-CA-800 |
90.83 |
2.50 |
6.67 |
3.28 |
| P-CA-800vac |
90.38 |
2.22 |
7.40 |
2.34 |
| P-CA-900 |
90.94 |
1.37 |
7.69 |
1.64 |
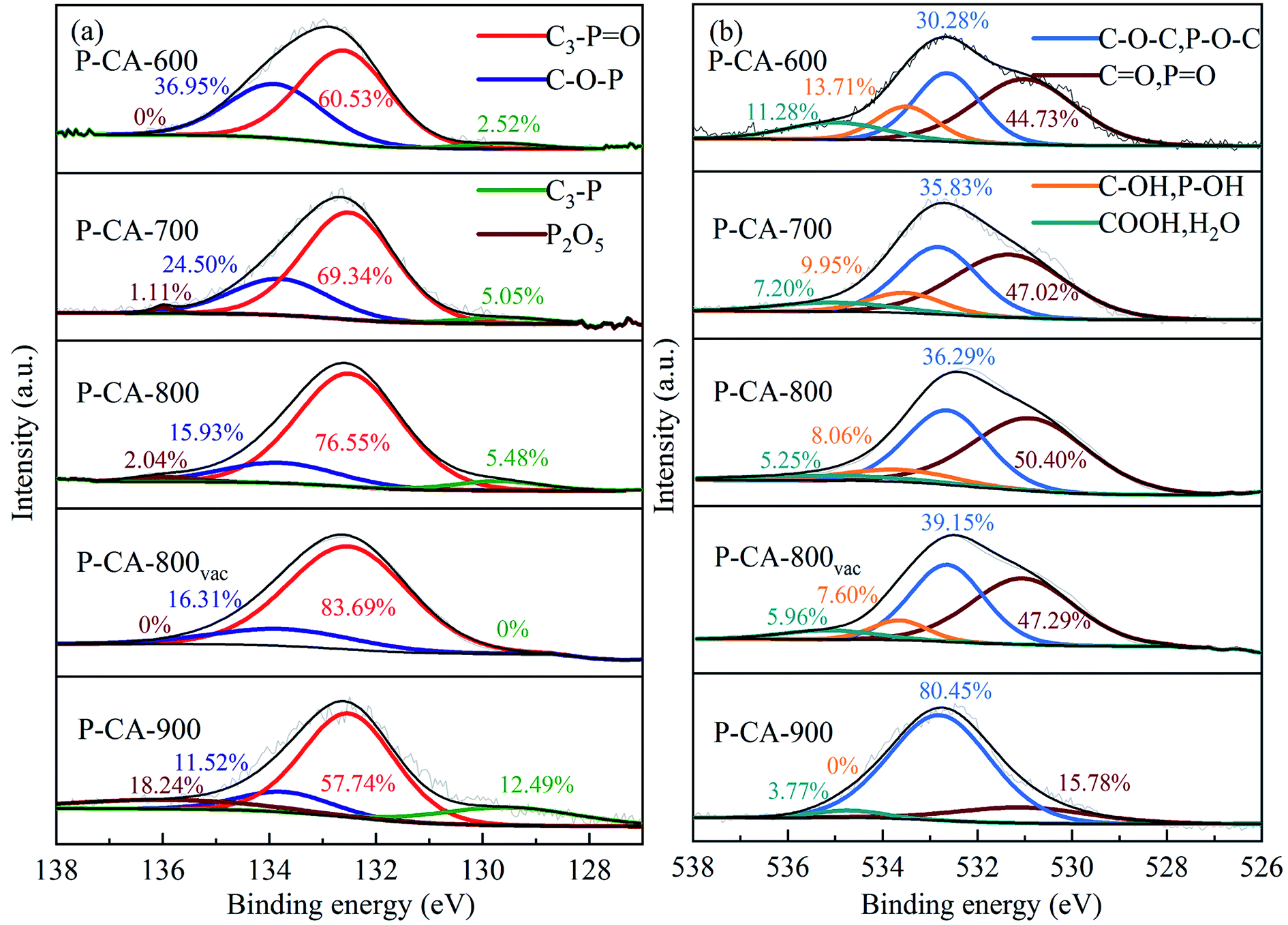
The high-resolution P2p spectra can be fitted to four peaks at 130.2 eV, 132.2 ± 0.4 eV, 134.0 ± 0.2 eV and 136 ± 0.5 eV, which corresponds to C3–P, C3–PO, C–O–P, and phosphorus pentoxide (P2O5),35–38 respectively, as shown in Fig. 1. It can be seen that the contents of C3–P, C3–PO and P2O5 increase, whereas that of C–O–P decrease with increasing the carbonization temperature before 800 °C. The formation of more C3–P and C3–PO (sum of the two) configurations at a higher carbonization temperature indicates that more P atoms are doped into the C–C lattice at a higher carbonization temperature.39 But thermal decomposition of the C3–PO group is found beyond 800 °C (the sample P-CA-900). It is reported that the presence of P2O5 may result in the pore blocking that affects the surface activity of carbon.40 Therefore, the P-CA-800 sample was subjected to heat treatment at 380 °C for 8 h under vacuum to obtain sample P-CA-800vac. It is shown that P2O5 can be successfully sublimed during the vacuum treatment. Meanwhile, the C3–P configuration also vanishes, which may be attributed to the fact that the transformation of C3–P into C3–PO during the vacuum treatment via interaction with a trace amount of oxygen because of the lower formation energy of C3–PO and its highly stable structure.24 The O1s spectra can be deconvoluted into four peaks located at 531.2 ± 0.2 eV, 532.5 ± 0.3 eV, 533.5 ± 0.2 eV and 534.9 ± 0.2 eV, which are assigned to CO and PO, C–O–C and C–O–P, C–OH and P–OH, and carboxylic groups COOH and/or H2O, respectively.22,23,35,40 The P–OH groups may be mainly linked on the C–O–P structure as the forms of CO–P(O)(OH)2 and (CO)2–P(O)(OH) groups located at the edge of graphene layer.41 It is shown that the relative proportion of O1 increases firstly and then decreases with increasing the carbonization temperature, in consistence with the evolution of C3–PO. The concentrations of O3 and O4 decrease gradually with the rise of the carbonization temperature due to their instability at high temperatures.
 |
| | Fig. 1 The deconvoluted and the fitted results of (a) P2p and (b) O1s spectra of the P-CAs. | |
The pore textures of the CA-800 and P-CAs were analyzed by N2 adsorption, as shown in Fig. S2a,† the adsorption isotherms of all samples exhibit a typical type II isotherm curve with a H3 hysteresis loop, indicating the existence of mesopores in the texture. Meanwhile, the high nitrogen uptakes at a low relative pressure and a high relative pressure above 0.95 can be observed due to the coexistence of micropores and macropores, in consistence with the corresponding pore size distributions displayed in Fig. S2b.† As displayed in Table 2, the BET surface area and total pore volume of the P-CAs decrease gradually with increasing the carbonization temperature from 600 to 900 °C, which are lower than that of the CA-800. It is worth noting that the specific surface area of the P-CA-800vac is significantly higher than that of the P-CA-800 due to the removal of P2O5 that is mainly filled in the micropores, resulting in the distinct increase of micropore volume.
Table 2 Textual parameters of the CA-800 and P-CAs
| Samples |
SBET (m2 g−1) |
Vtotal (m3 g−1) |
Vmes (m3 g−1) |
Vmic (m3 g−1) |
| P-CA-600 |
367 |
0.763 |
0.244 |
0.097 |
| P-CA-700 |
323 |
0.695 |
0.234 |
0.077 |
| P-CA-800 |
217 |
0.625 |
0.217 |
0.055 |
| P-CA-800vac |
315 |
0.673 |
0.221 |
0.080 |
| P-CA-900 |
202 |
0.597 |
0.222 |
0.059 |
| CA-800 |
447 |
0.768 |
0.597 |
0.118 |
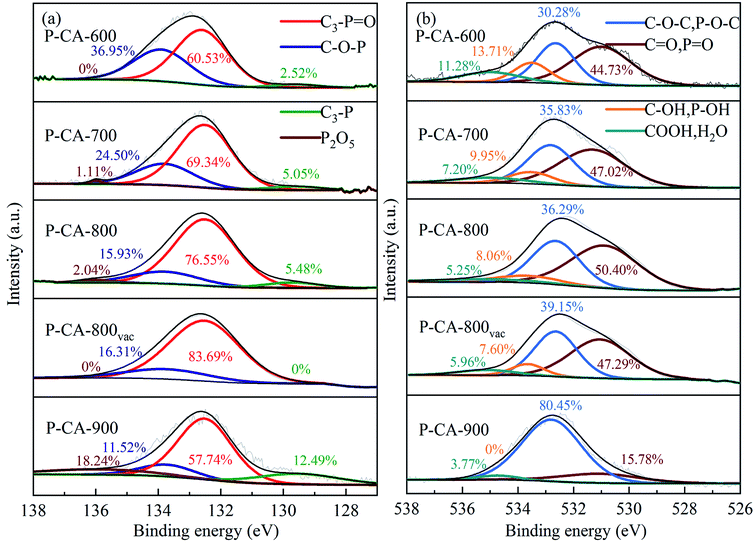
NH3-TPD tests were used to explore the number and intensity of acid sites on the obtained samples. As shown in Fig. 2, a distinct NH3 desorption peak at 100–250 °C is observed from the TPD profiles of the P-CA-800 and P-CA-800vac samples, whereas no obvious peak is detected for the CA-800 sample. The NH3 adsorption amounts for the P-CA-800 and P-CA-800vac are higher than that of the CA-800. It is generally believed that the peaks below 400 °C on the NH3-TPD profiles is attributed to NH4+ ions coordinated on the Brønsted acid sites and the NH3 molecule is coordinated to the Lewis acid sites above 400 °C.42–44 The Brønsted acidity may be originated mainly from the surface C–OH (CA-800, P-CA-800 and P-CA-800vac) and P–OH (P-CA-800 and P-CA-800vac) groups revealed by above XPS analysis, which are favorable for coordination with NH3 to form NH4+ and facilitate the NH3-SCR reaction.9,45–49 It is worth noting that the Lewis acid sites of P-CA-800 and P-CA-800vac samples disappeared and the number of Brønsted acid sites increased significantly. This is because phosphoric acid can inhibit the generation of strong acid sites and generate weak acid sites.50 The COOH group is unstable at a high temperature, therefore the contribution of COOH group to the Brønsted acid sites is less obvious than that of C–OH and P–OH groups. After the vacuum treatment at 380 °C for 8 h, more Brønsted acid sites are exposed as a result of removal of P2O5 by sublimation as shown in Fig. 2. So the amount of adsorbed NH3 follows the order, CA-800 < P-CA-800 < P-CA-800vac.
 |
| | Fig. 2 NH3-TPD profiles and the corresponding amounts of adsorbed NH3 over the CA-800, P-CA-800 and P-CA-800vac. | |
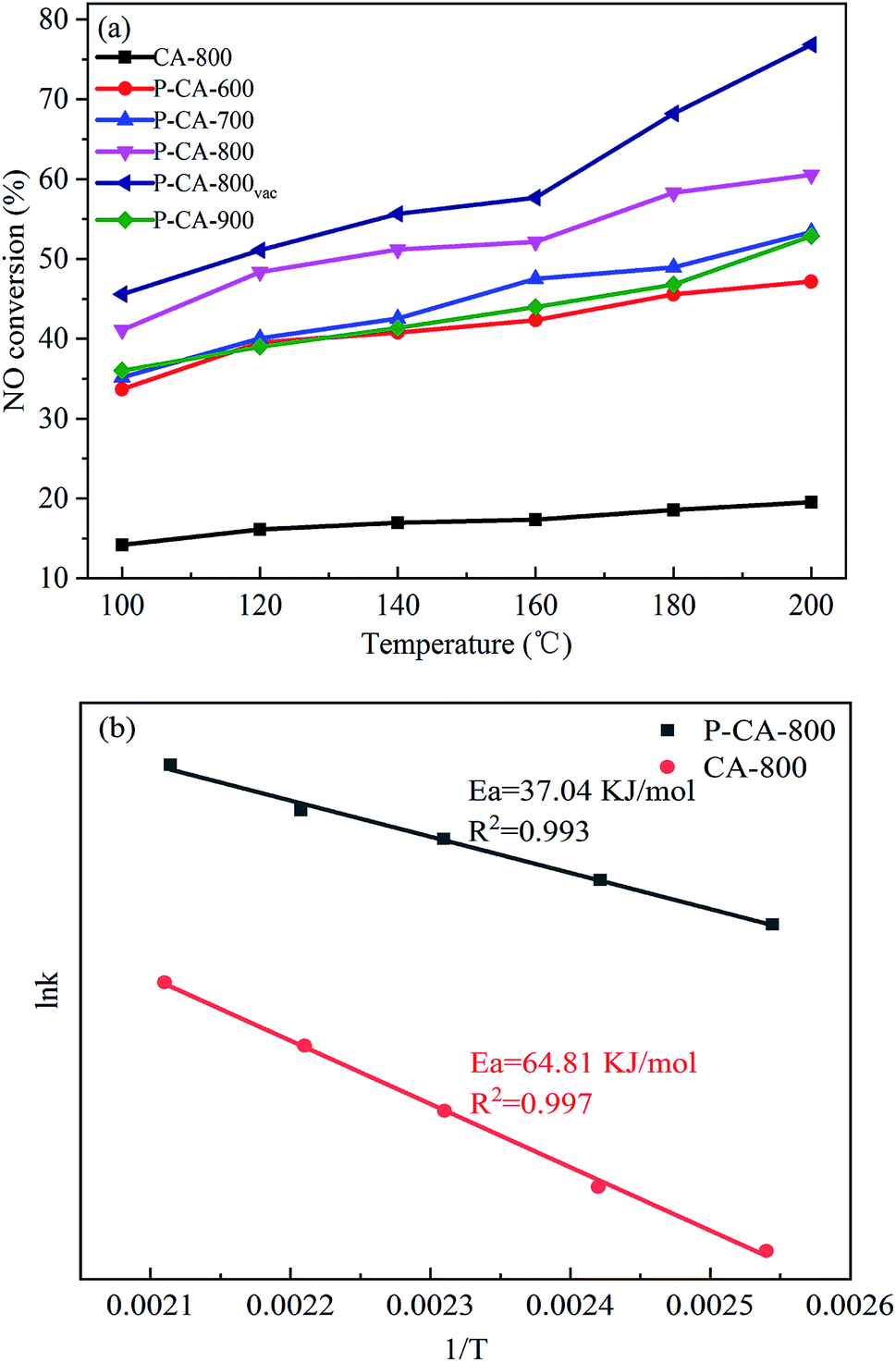
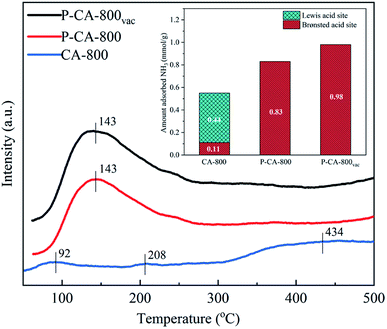
The NH3-SCR performance of the P-CAs were examined and shown in Fig. 3a. It is interesting to note that all the P-CAs exhibit higher catalytic activity for NO reduction compared with that of the CA-800. It is noted that the catalytic activity of the P-CAs is related to their surface P contents. The NO conversion over the P-CA-800 sample is 41.1–60.6% in the temperature range of 100–200 °C, whereas it is only 14.2–19.5% for the CA-800. To further compare the catalytic performance of the CA-800 and P-CA-800, their Arrhenius plots are tested and the corresponding apparent activation energies (Ea) are calculated from kinetics study, as displayed in Fig. 3b. The apparent activation energy of the P-CA-800 (37.04 kJ mol−1) is significantly lower than that of CA-800 (64.81 kJ mol−1), in accordance with their catalytic activity. In addition, it is shown that the catalytic activity of the P-CA-800vac is improved significantly compared with that of the P-CA-800, which is 45.6–76.8% at 100–200 °C, suggesting more active sites are exposed after the vacuum treatment.
 |
| | Fig. 3 (a) NO conversions over the CA-800 and P-CAs (reaction conditions: [NO] = 500 ppm, [NH3] = 500 ppm, [O2] = 5%, N2 balance, GHSV = 500 h−1) and (b) Arrhenius plots of the P-CA-800 and CA-800 samples. | |
In order to explore the promotion effects of P-doping on the NH3-SCR performance, the fresh P-CA-800vac sample was subjected to NH3-SCR reaction at 120, 140, 160, 180, 200 °C for 1 h to obtain P-CA-800vac-120, P-CA-800vac-140, P-CA-800vac-160, P-CA-800vac-180, P-CA-800vac-200, respectively. The XPS analysis results of above used samples are shown in Fig. S3† and Table 3. It can be clearly seen that the content of C decreases, the content of O increases, whereas the content of P is almost unchanged for the used samples compared with that of the fresh P-CA-800vac sample, indicating that the increase of oxygen can be regarded as the adsorption of NO on the catalyst surface during the NH3-SCR reaction. It is worth noting that oxygen content in XPS results have a linear relationship with the removal efficiency of NO.
Table 3 Atomic compositions of the P-CA-800vac after the NH3-SCR reaction for 1 h at different temperatures as examined by XPS
| Sample |
XPS analysis |
| C |
O |
P |
| P-CA-800vac-120 |
81.23 |
16.59 |
2.18 |
| P-CA-800vac-140 |
77.26 |
20.55 |
2.19 |
| P-CA-800vac-160 |
75.42 |
22.43 |
2.15 |
| P-CA-800vac-180 |
70.14 |
27.68 |
2.18 |
| P-CA-800vac-200 |
65.77 |
32.06 |
2.17 |
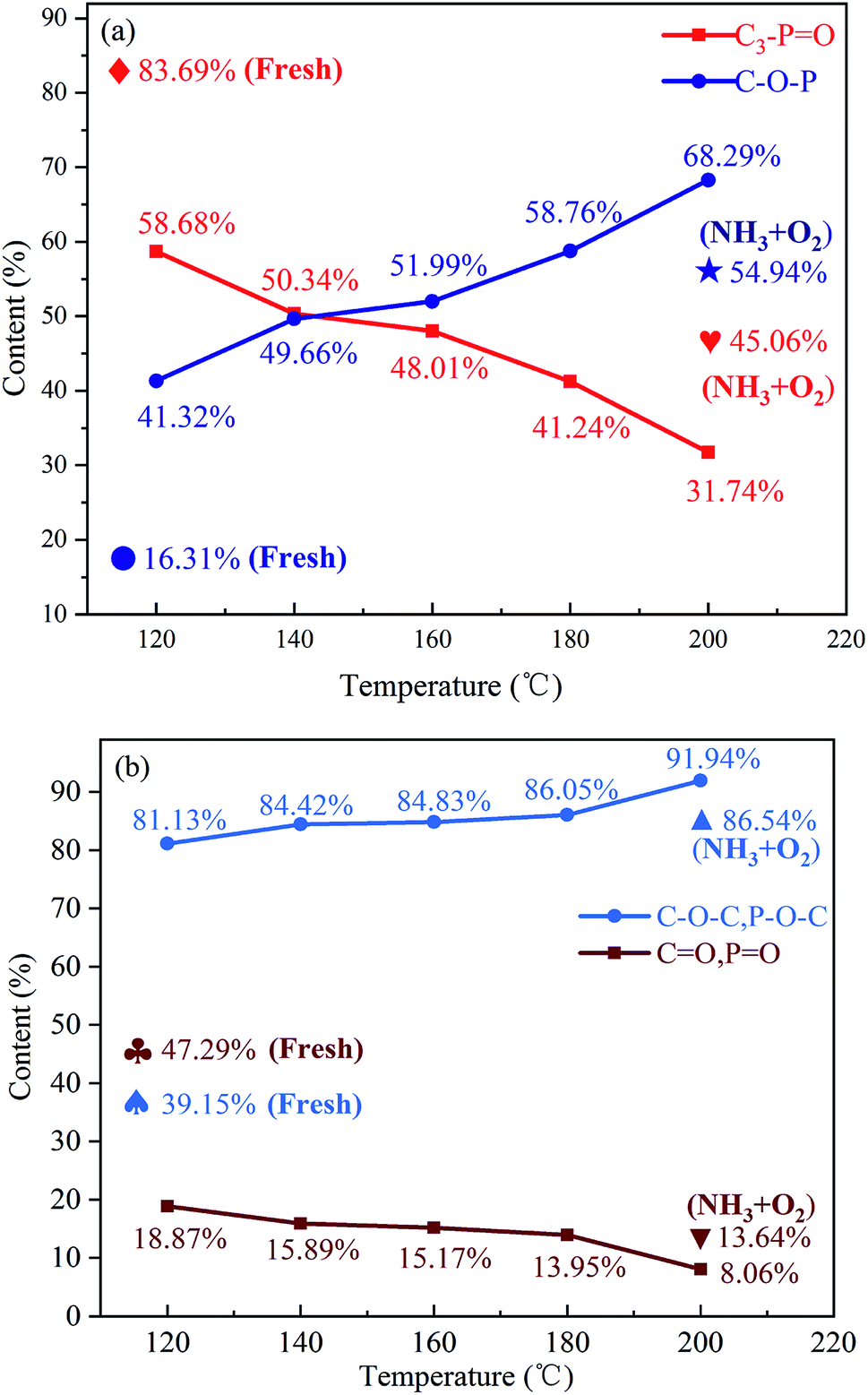
The P2p and O1s spectra and the corresponding deconvoluted results are shown in Fig. S4† and 4. It can be observed from the P2p spectra that the content of C3–PO decreases rapidly, while the content of C–O–P gradually increases with increasing the reaction temperature. The O1s spectra indicate that the –OH and COOH groups all disappear after reaction, which may be due to the oxidation of –OH to ketones and the consumption of COOH during reaction.10 It should be mentioned that the contribution of carboxylic acid groups to NO reduction is minor due to their low concentration, although the carboxylic acid groups can directly react with NO.10 The decrease in the contents of CO and PO, and the increase in the contents of C–O–C and P–O–C groups can be observed from the O1s spectra, indicating the oxygen have entered the C–P bond and/or the C–C bond during reaction.
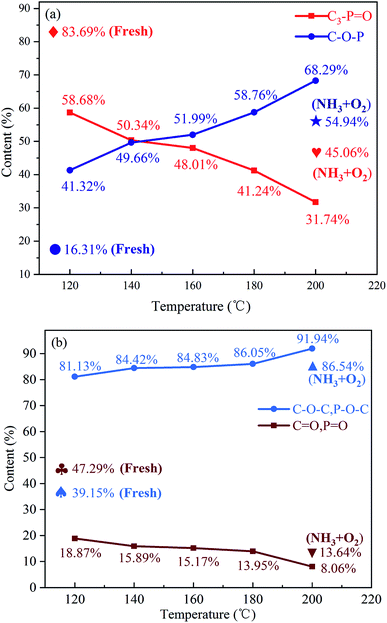 |
| | Fig. 4 Relative surface concentrations of phosphorous species for the P-CA-800vac sample after the NH3-SCR at different reaction temperatures obtained from (a) P2p XPS spectra and (b) O1s XPS spectra. Reaction conditions: [NO] = 500 ppm, [NH3] = 500 ppm, [O2] = 5%, N2 balance, GHSV = 500 h−1. | |
The P-CA-800vac sample was also treated in a mixed gas of NH3 and O2 at 200 °C for 1 h and then subjected to XPS test. It is shown that a part of C3–PO can also be transferred into C–O–P functional group without NO atmosphere, although the conversion of C3–PO is less than that of the used P-CA-800vac sample after reaction in NO + NH3 + O2 atmosphere, indicating that the presence of NO accelerates the oxidation of the C3–PO group.
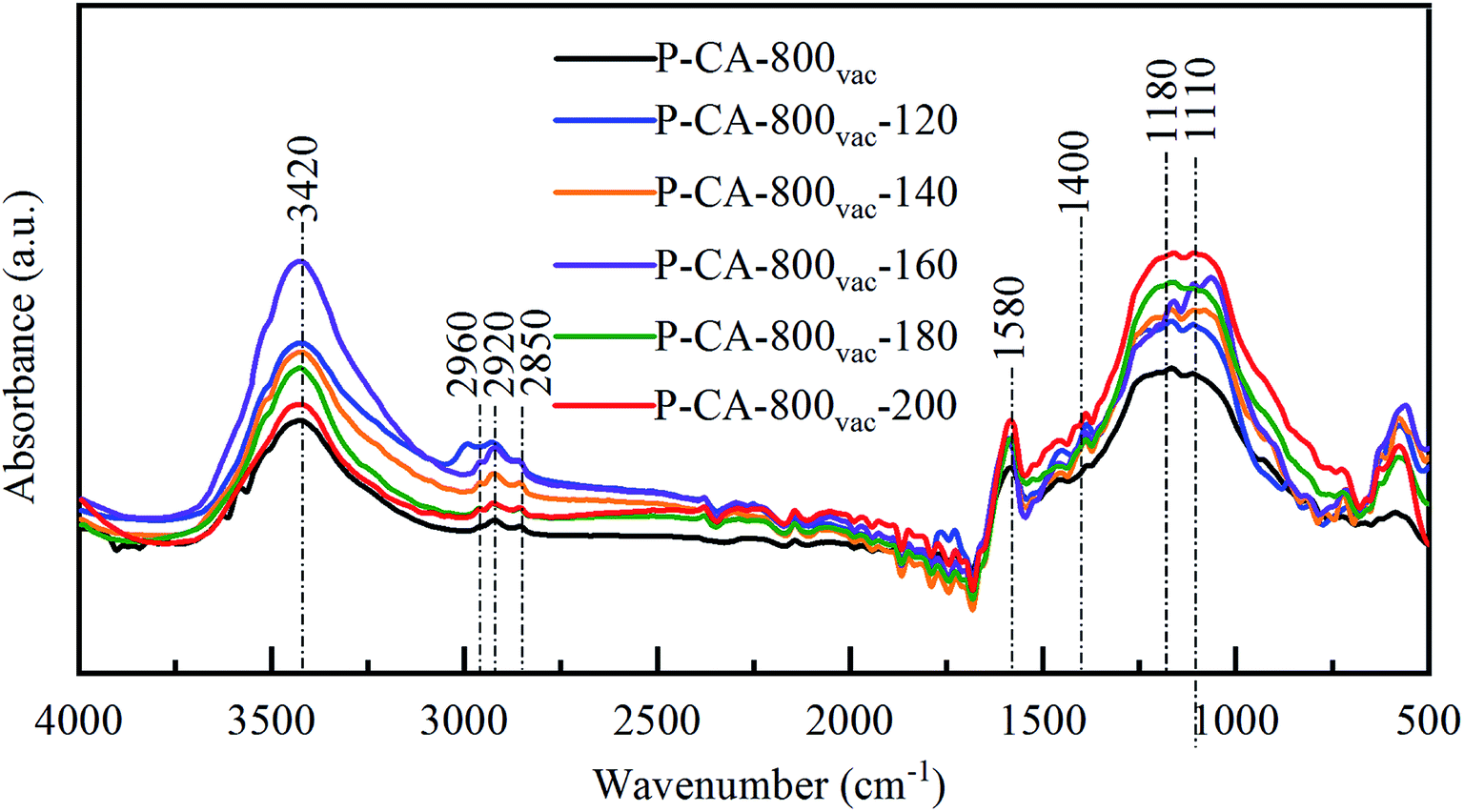
FTIR spectra were also used to detect the change of surface composition for the P-CA-800vac samples before and after the NH3-SCR reaction at different temperatures, as shown in Fig. 5. A strong broad absorption band at 3450 cm−1 (hydroxyl groups and/or the adsorbed water), a minor peak at 2960, 2920 and 2850 cm−1 (C–H stretching in CH2), an absorption band at 1580 cm−1 (CC stretching in aromatic ring), and an absorption band at 1480–1400 cm−1 (bending modes from CH2, CH3, and OH) are observed for all the samples.51–53 In addition, a broad absorption band at 1300–900 cm−1 is observed, which should be attributed to the overlapping signals for vibration of oxygen and phosphorus functional groups. The peak at 1180 cm−1 is generally assigned to stretching vibration from PO, C–O in C–O–P, and OP–OH,54 and the band at 1115 cm−1 to bending vibration of C–O–C group.55 The absorption signals in this region become stronger after the NH3-SCR reaction, indicating that more C–O–C and/or C–O–P functional groups are generated during reaction, which is consistent with above XPS analysis results.
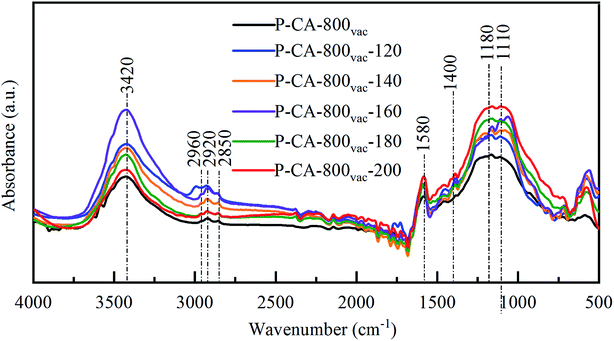 |
| | Fig. 5 FTIR spectra of the P-CA-800vac samples before and after the NH3-SCR reaction for 1 h at different temperatures. | |
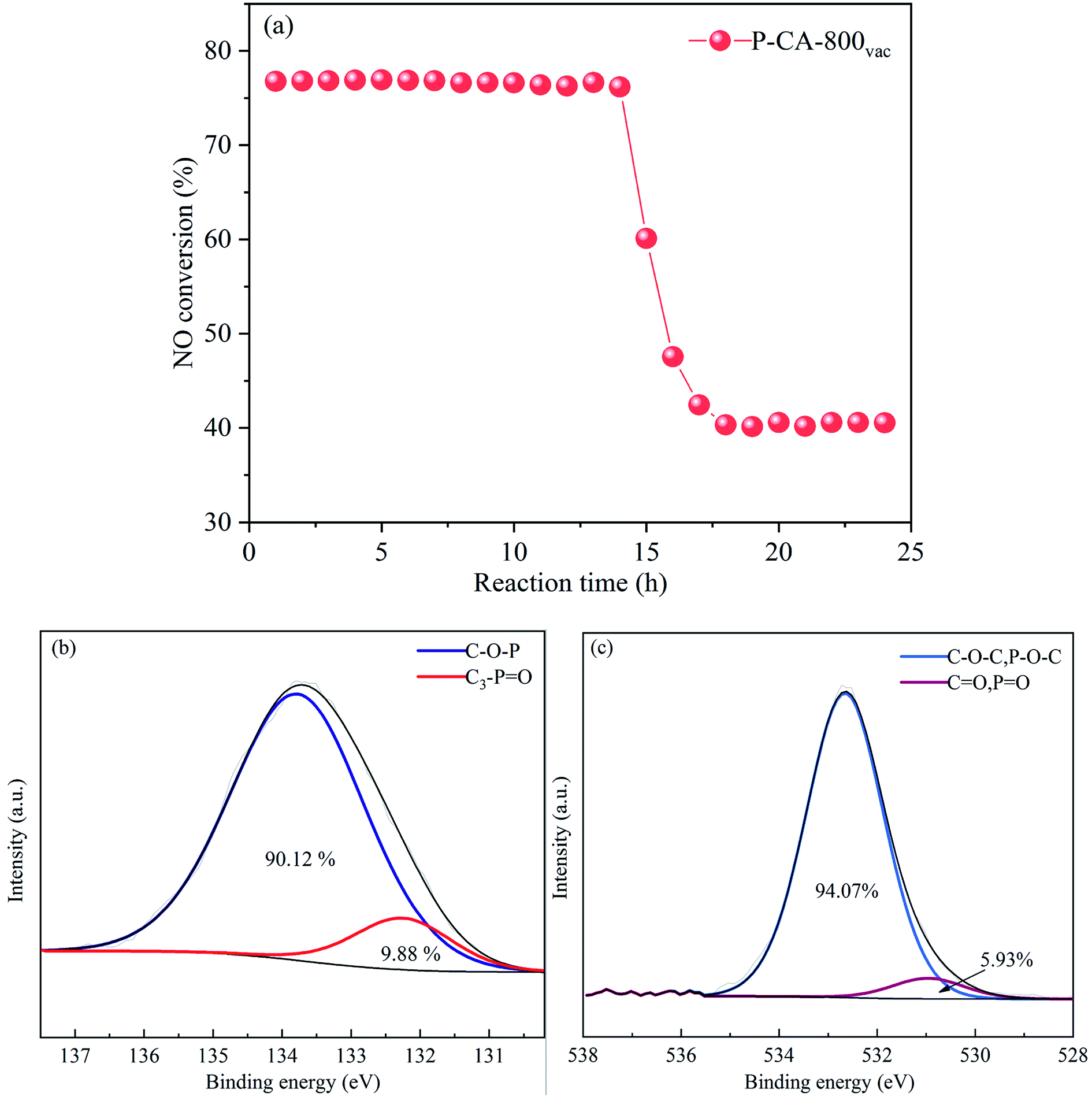
The long-term stability of the P-CA-800vac sample for the NH3-SCR reaction was further evaluated, as shown in Fig. 6a. The NO conversion maintains at ca. 77% during the first 14 h, which then decreases sharply to ca. 40% for the rest of test time. The P2p and O1s XPS results of the used sample after stability test show that most of the C3–PO configurations on the catalyst surface have been converted into C–O–P groups (Fig. 6b and c), suggesting that the C3–PO groups may be the active sites for the high NH3-SCR activity of the P–CAs. The sudden deactivation of the catalyst at the fourteenth hour may be due to the fact that the C3–PO groups gradually converted to C–O–P groups that are less active after NH3-SCR reactions for 14 h. At the 14 h, the number of C3–PO groups as active sites became too small to maintain the high NH3-SCR activity of the P-CA-800vac, resulting in a partial deactivation of the catalyst. The C3–PO group decreases by 88% after reaction for 14 h while the NH3-SCR activity decreases by 63% of the original level. The remaining activity may be due to the Brønsted acid sites contributed by the C–O–P groups (CO–P(O)(OH)2 and (CO)2–P(O)(OH) groups) that are active in NH3-SCR reaction.
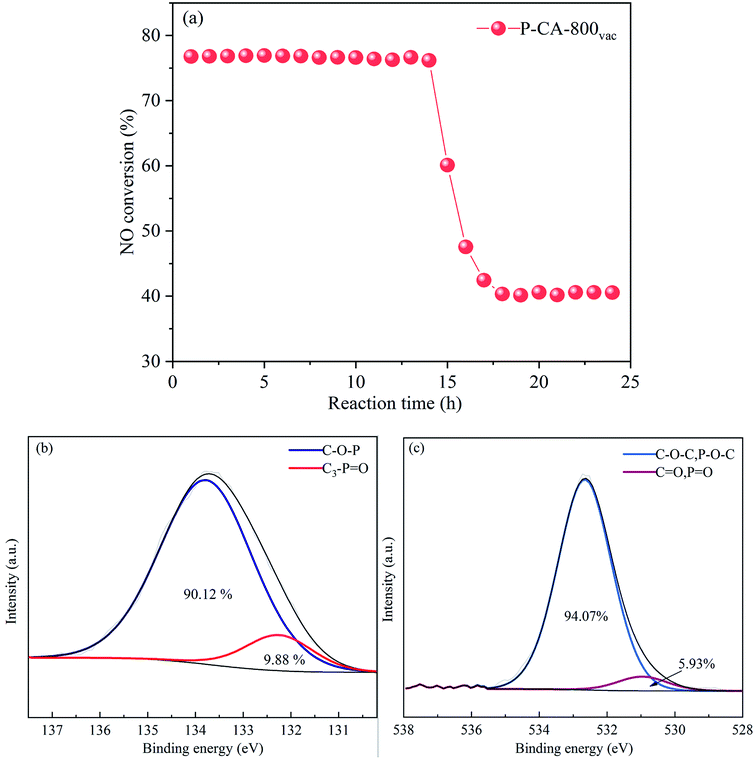 |
| | Fig. 6 (a) Stability test of NH3-SCR for the P-CA-800vac sample, (b) P2p and (c) O1s XPS spectra of the P-CA-800vac after NH3-SCR reaction for 24 h. Reaction conditions: [NO] = 500 ppm, [NH3] = 500 ppm, [O2] = 5%, N2 balance, GHSV = 500 h−1, 200 °C. | |
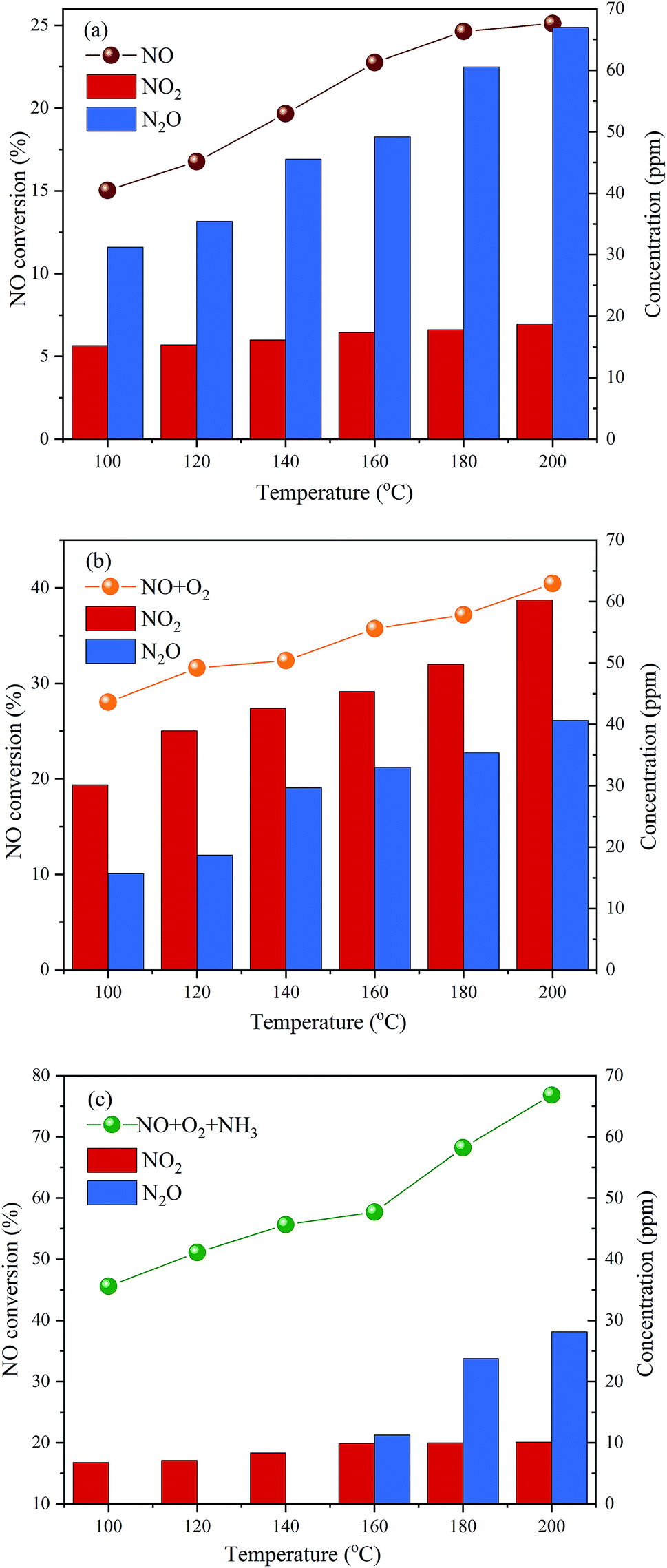
The above results show that P-doping significantly enhances the NH3-SCR activity of carbon aerogels as a metal-free catalyst, and the C3–PO structure seems to be the most active sites for the reaction. To explore the possible mechanism for NO reduction on P-CA surface, the NO conversions, NO2 and N2O concentrations in the outlet gas over the P-CA-800vac sample under NO, NO + O2 and NO + NH3 + O2 atmosphere at 100–200 °C were also tested, as shown in Fig. 7. It should be mentioned that NH3 can not be detected in the tail gas under the atmosphere of NO + NH3 + O2, which may be due to the adsorption of NH3 on the acid sites and the subsequent consumption via reaction. It is shown that the NO removal efficiency is in the order of NO + NH3 + O2 > NO + O2 > NO, demonstrating that the coexistence of NH3 and O2 is essential for the high conversion of NO on the P-CAs.
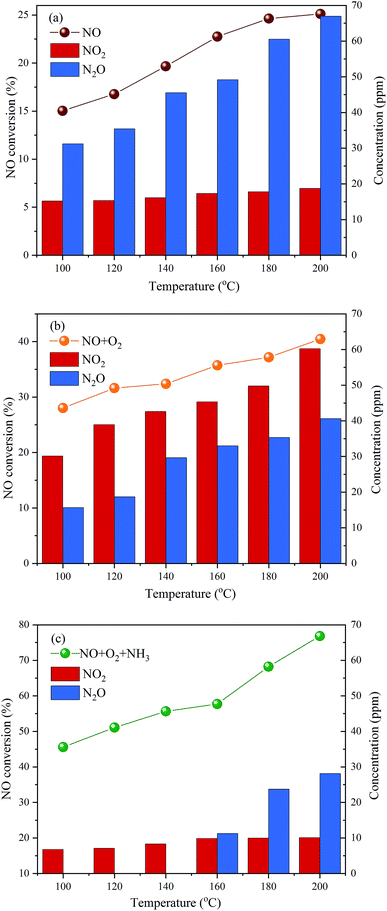 |
| | Fig. 7 NO conversions, NO2 and N2O concentration in the outlet gas over the P-CA-800vac sample for the different inlet gases of NO, NO + O2 and NO + NH3 + O2. Reaction conditions: [NO] = 500 ppm, [NH3] = 500 ppm (when used), [O2] = 5% (when used), N2 balance, GHSV = 500 h−1. | |
Molecule NO contains an unpaired π-antibonding orbit and prefers to accept electron from substrates.17 The density functional theory (DFT) calculations have shown that NO molecule can be adsorbed moderately on the P-doped carbon surface due to the electron-donating nature of P atom that can create localized electronic state into the carbon skeleton.56 The electron transfer from P-doped carbon to NO molecule could elongate N–O bond length and decrease N–O bond order.56 The mechanisms for the adsorption and decomposition of NO on the N-doped graphene (NG) have been studied by the DFT calculations.15,16 It is reported that the dimer mechanism is more facile than the direct dissociation NO to N* and O*, which involves the adsorption of two NO molecules on the surface of NG, followed by the decomposition of (NO)2 dimer into N2O and adsorbed O*, then N2O can dissociate to N2 and adsorbed O* and NO can react with adsorbed O* to form NO2, as depicted in the eqn (4)–(7). The catalytic reduction of NO on the P-CAs is proposed to follow the above dimer mechanism, because NO2 and N2O are detected in the outlet gas when the P-CA-800vac is exposed to only NO diluted in N2, as shown in Fig. 7a.
The depletion efficiency of NO is enhanced when O2 is introduced into the system compared with the situation when NO is solely present in the inlet gas, as shown in Fig. 7b. It is shown that the NO2 concentration in the tail gas is also higher under the atmosphere of NO + O2 than the NO atmosphere, indicating that the presence of O2 may facilitate the oxidation of NO to NO2. The catalytic oxidation mechanisms of NO (2NO + O2 → 2NO2) over carbon have been studied extensively in the last decades,57–59 namely, Eley–Rideal (E–R) and Langmuir–Hinshelwood (L–H) mechanisms are proposed. For the L–H mechanism, adsorbed NO reacts with dissociated O* activated by carbon, and for the E–R mechanism, gaseous NO and O2 directly reacts in narrow micropores.60–62 By comparing P-CA-800 and P-CA-800vac catalysts, we can find that there are more phosphorus-containing functional groups on the exposed surface of P-CA-800vac catalysts, and at the same time, partially blocked micropores by P2O5 are released, which may provide another routine for denitration by reactions of gaseous NO and O2 in the micropores,61 thereby improving the denitration efficiency of the catalyst. However, very few micropores are present in the P-CAs as demonstrated from the N2 adsorption results, hence the existence of micropores can only promote the oxidation of NO and is not a key factor affecting the denitrification mechanism of catalyst. So, we considered that the enhanced NO oxidation activity should be attributed to the P-doping on carbon surface, which may facilitate the adsorption and dissociation of O2 molecule that have been revealed in ORR process.21,25–29 Due to the difference of electron negativity between heteroatoms and carbon, the incorporation of heteroatoms can cause charge redistribution of carbon skeletons to create charged sites favorable for O2 adsorption. The catalytic active sites in the P-doped carbon are believed to be the positively charged P atoms because the electronegativity of P (2.19) is less than that of carbon (2.55) on the basis of three-coordinated P-doped graphene (PC3G) structure.21,28 In present work, the dominant active sites should be C3–PO, in which the dangling-bond of P atom is saturated by O atom, making the adjacent C atoms become the sole active sites for O2 adsorption.25–27,29 The O atom with the highest electronegativity (3.44) will redistribute atomic charge of (PC3G) structure, namely, O and P atom can coordinate to adjust the electrons of C atom by using P atom as a connector. As a result, although the C atoms exhibit negative charge, they can also be the optimal active sites for favorable adsorption and dissociation of O2 according to the DFT calculation results,29 as described in the eqn (8). The presence of O2 can promote the generation of NO2 and reduce the spillover of N2O, which is also consistent with the results of DFT calculations based on the N-doped graphene.15
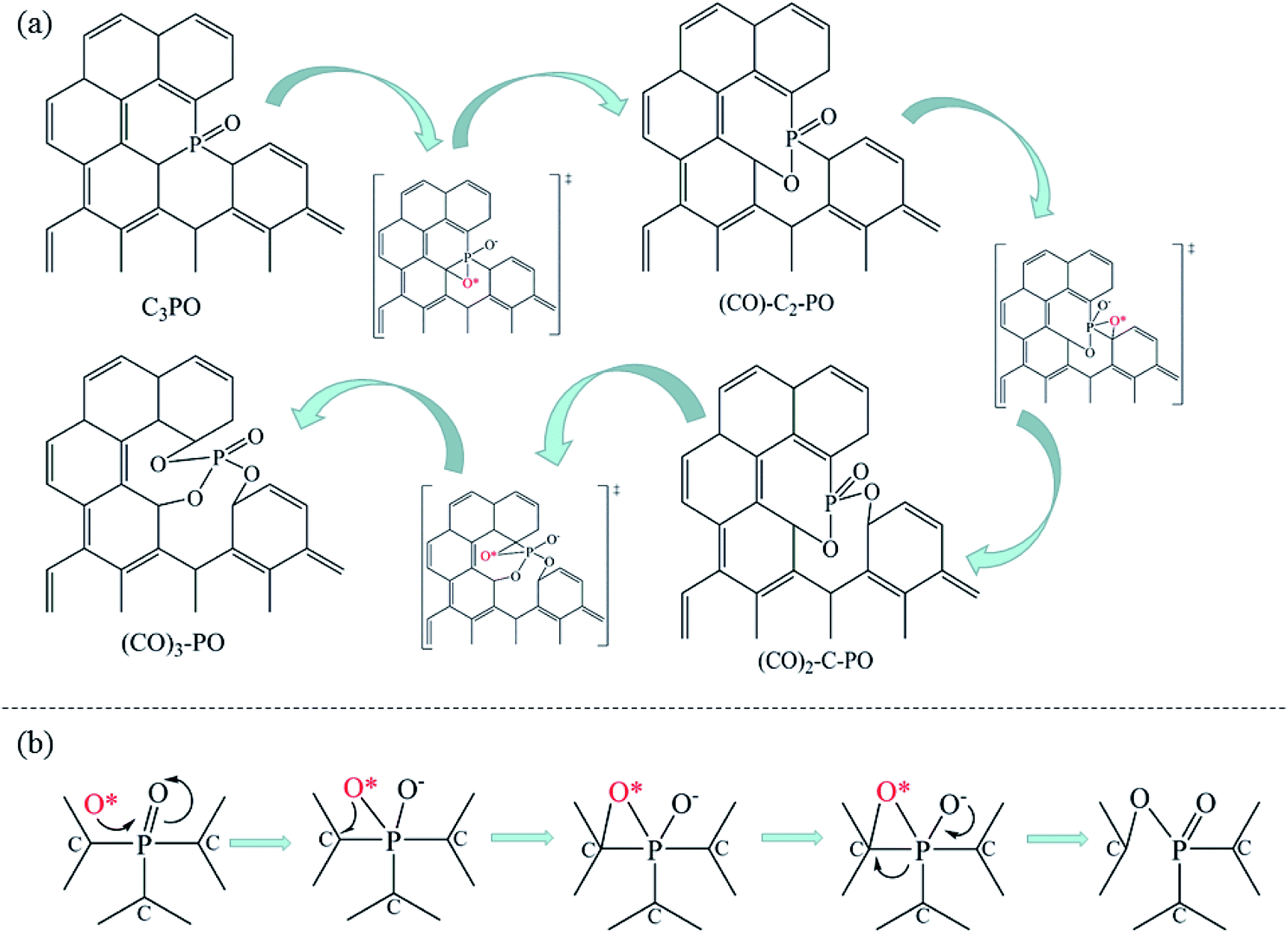
The introduction of NH3 can further improve the NO conversion and decrease the spillover of NO2 and N2O, as shown in Fig. 7c. Based on the experimental results, a mechanism for NH3-SCR of NO over P-CAs is proposed, as depicted in the eqn (7)–(10). Firstly, the O2 molecular is adsorbed on the carbon regions activated by P-doping and dissociated to O* that can readily interact with NO to generate NO2, meanwhile NH3 can be coordinated on the Brønsted acid sites to form NH4+, then NO2 can react with NH4+ to generate an ammonium nitrite intermediate,23,24 which can decompose directly into N2 and H2O. Therefore, the simultaneous presence of acidic groups for NH3 adsorption and the active sites for NO2 generation due to activation of O2 molecular should be responsible for the significant increase in SCR activity over the P–CAs as compared with the undoped one. In addition, the C3–PO groups transfer into C–O–P bonds after the stability test as revealed by the XPS results, which could be assigned to the oxidation of C3–PO by the dissociated O*, as described by the eqn (11)–(13). We propose a mechanism for structure evolution from C3–PO to C–O–P, as illustrated in Fig. 8. The dissociated O* atom could be easily adsorbed at the bridge site of C–P bonds of C3–PO configuration due to the accumulated electrons on the C–P bonds.29 The highly reactive O* would attack the C–P bonds and insert into C–P bonds to form C–O–P bonds,23,24 resulting in the deactivation of catalytic sites for SCR reaction. Besides, the oxidation of carbon may also occur as in other carbon materials that can be described by the eqn (14).10,17 Certainly, much more work is undergoing to elucidate the exact mechanism via a theoretical DFT calculation.
| | |
(NO)2(ad) → N2O + O*(ad)
| (5) |
| | |
NH3 + H+(Brønsted acid sites) → NH4+(ad)
| (9) |
| | |
NH4+(ad) + NO2 → NH4NO2 → N2 + 2H2O
| (10) |
| | |
–C3PO + O*(ad) → –CO–C2–PO
| (11) |
| | |
–CO–C2–PO + O*(ad) → –(CO)2–C–PO
| (12) |
| | |
–(CO)2–C–PO + O*(ad) → –(CO)3–PO
| (13) |
| | |
–C–C– + O*(ad) → CO(CO2)
| (14) |
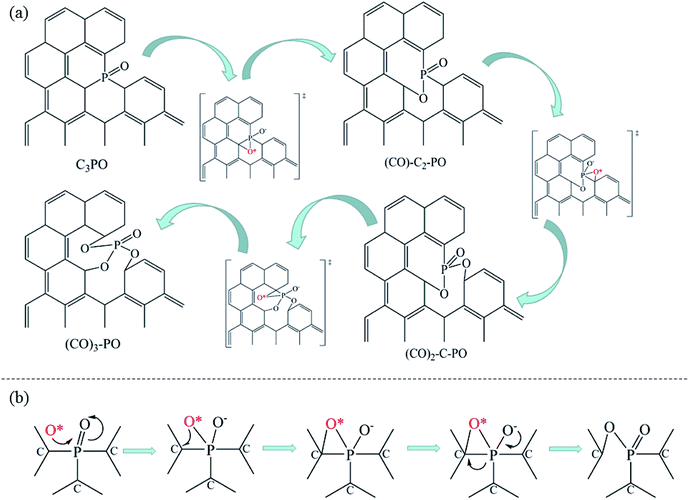 |
| | Fig. 8 Proposed mechanism for structure evolution from C3–PO to C–O–P. | |
It should be noted that although the P-CA-800 has a low apparent activation energy and a high activity at low temperatures, its GHSV is only 500 h−1, significantly lower than other inorganic oxide based catalysts. This could be caused by a small number of active species. Therefore, a further work is necessary to increase the number of active sites for this catalyst system.
4. Conclusion
This work studied the influence of P doping on the NH3-SCR activity over carbon aerogels as a metal-free catalyst at low temperatures. The results show that the incorporated P species on the carbon sheets can effectively enhance the catalytic activity of carbon aerogels for NO reduction. The NH3-TPD results show that Brønsted acid sites are introduced to the carbon surface due to mainly the presence of a large portion of C–OH and P–OH species, and a small portion of COOH group. Moreover, the C3–PO structure may act as the most active sites for easy adsorption and dissociation of O2 to facilitate NO2 generation. Therefore, the improvement of SCR activity over the P-CAs is assigned to simultaneous presence of acidic groups for NH3 adsorption and the active sites for NO2 generation due to activation of O2 molecular. Based on the experimental results, a mechanism for NH3-SCR of NO over the P-CAs is proposed. Firstly, the O2 molecular is adsorbed on the carbon regions activated by P-doping and dissociated to O* that can readily interact with NO to generate NO2, meanwhile NH3 can be coordinated on the Brønsted acid sites to form NH4+, then NO2 can react with NH4+ to generate an ammonium nitrite intermediate, which decomposes directly into N2 and H2O. The C3–PO configuration is unstable and oxidized to C–O–P groups probably by O2 during reaction, leading to an apparent decrease of catalytic activity for P-CAs for a reaction time beyond 14 h. These results enrich the understanding on contributions of heteroatoms to the catalytic activity of carbon for the low-temperature NH3-SCR reaction. More experimental and theoretical work will be done to demonstrate and improve the proposed mechanisms. Moreover, some efforts can be made to develop the oxidation resistance of the C–P bonds to enhance the durability of P-CA catalysts and to increase the number of active sites.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. U1710252).
References
- Q. Zhao, J. Xiang, L. Sun, S. Su and S. Hu, Energy Fuels, 2009, 23, 539–1544 CrossRef.
- W. Shan and H. Song, Catal. Sci. Technol., 2015, 5, 4280–4288 RSC.
- R. Jin, Y. Liu, Z. Wu, H. Wang and T. Gu, Chemosphere, 2010, 78, 1160–1166 CrossRef CAS PubMed.
- Z. Liu, Y. Li, T. Zhu, H. Su and J. Zhu, Ind. Eng. Chem. Res., 2014, 53, 12964–12970 CrossRef CAS.
- Y. Yang, M. Wang, Z. Tao, Q. Liu, Z. Fei, X. Chen, Z. Zhang, J. Tang, M. Cui and X. Qiao, Catal. Sci. Technol., 2018, 8, 6396–6406 RSC.
- S. Ahmed, R. Baldwin, F. Derbyshire, B. Mcenaney and J. Stencel, Fuel, 1993, 72, 287–292 CrossRef CAS.
- B. Ku, J. Lee, D. Park and H. Rhee, Ind. Eng. Chem. Res., 1994, 33, 2868–2874 CrossRef CAS.
- J. Muniz, G. Marban and A. Fuertes, Appl. Catal., B, 1999, 23, 25–35 CrossRef CAS.
- H. Teng, Y. Tu, Y. Lai and C. Lin, Carbon, 2001, 39, 575–582 CrossRef CAS.
- J. Zhang, Q. Gao, X. Li, J. Zhou, X. Ruan, Q. Liu, G. Qian and Z. Xu, Phys. Chem. Chem. Phys., 2017, 19, 22462–22471 RSC.
- J. Muñiz, G. Marbán and A. Fuertes, Appl. Catal., B, 2000, 27, 27–36 CrossRef.
- M. Huang and H. Teng, Carbon, 2003, 41, 951–957 CrossRef CAS.
- G. Szymanski, T. Grzybek and H. Papp, Catal. Today, 2004, 90, 51–59 CrossRef CAS.
- T. Grzybek, J. Klinik, B. Samojeden, V. Suprun and H. Papp, Catal. Today, 2008, 137, 228–234 CrossRef CAS.
- X. Zhang, Z. Lu, Y. Tang, D. Ma and Z. Yang, Catal. Lett., 2014, 144, 1016–1022 CrossRef CAS.
- X. Zhang, Z. Lu, Y. Tang, Z. Fu, D. Ma and Z. Yang, Phys. Chem. Chem. Phys., 2014, 16, 20561–20569 RSC.
- Y. Wang, Y. Shen and S. Zhu, Catal. Commun., 2017, 9, 29–32 CrossRef.
- Y. Wang, Y. Shen, Y. Zhou, Z. Xue, Z. Xi and S. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 36202–36210 CrossRef CAS PubMed.
- S. Ji and J. Zhao, New J. Chem., 2018, 42, 16346–16353 RSC.
- D. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed.
- W. He, Y. Wang, C. Jiang and L. Lu, Chem. Soc. Rev., 2016, 45, 2396–2409 RSC.
- M. Patel, F. Luo, M. Khoshi, E. Rabie, Q. Zhang and C. Flach, ACS Nano, 2016, 10, 2305–2315 CrossRef CAS PubMed.
- Z. Bi, L. Huo, Q. Kong, F. Li, J. Chen, A. Ahmad, X. Wei, L. Xie and C. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 11421–11430 CrossRef CAS PubMed.
- R. Berenguer, R. Ruiz-Rosas, A. Gallardo, D. Cazorla-Amorós, E. Morallón, H. Nishihara, T. Kyotani, J. Rodríguez-Mirasol and T. Cordero, Carbon, 2015, 95, 681–689 CrossRef CAS.
- R. Li, Z. Wei, X. Gou and W. Xu, RSC Adv., 2013, 3, 9978–9984 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Qiao, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef CAS PubMed.
- Y. Liu, K. Li, Y. Liu, L. Pu, Z. Chen and S. Deng, J. Mater. Chem. A, 2015, 3, 21149–21158 RSC.
- X. Zhang, Z. Lu, Z. Fu, Y. Tang, D. Ma and Z. Yang, J. Power Sources, 2015, 276, 222–229 CrossRef CAS.
- N. Yang, X. Zheng, L. Li, J. Li and Z. Wei, J. Phys. Chem. C, 2017, 121, 19321–19328 CrossRef CAS.
- R. Gao, L. Pan, J. Lu, J. Xu, X. Zhang, L. Wang and J. Zou, ChemCatChem, 2017, 9, 4287–4294 CrossRef CAS.
- R. Gao, L. Pan, Z. Li, X. Zhang, L. Wang and J. Zou, Chin. J. Catal., 2018, 39, 664–672 CrossRef CAS.
- R. Huang, J. Wang, B. Zhang, K. Wu, Y. Zhang and D. Su, Catal. Sci. Technol., 2018, 8, 1522–1527 RSC.
- J. Liu, X. Li, Q. Zhao, J. Ke, H. Xiao, X. Lv, S. Liu, M. Tadé and S. Wang, Appl. Catal., B, 2017, 200, 297–308 CrossRef CAS.
- Y. Peng, K. Li and J. Li, Appl. Catal., B, 2013, 140, 483–492 CrossRef.
- Y. Wang, S. Zuo, J. Yang and J. Yoon, Langmuir, 2017, 33, 3112–3122 CrossRef CAS PubMed.
- X. Wu and L. Radovic, Carbon, 2006, 44, 141–151 CrossRef CAS.
- J. Rosas, J. Bedia, J. Rodríguez-Mirasol and T. Cordero, Fuel, 2009, 88, 19–26 CrossRef CAS.
- A. Puziy, O. Poddubnaya, R. Socha, J. Gurgul and M. Wisniewski, Carbon, 2008, 46, 2113–2123 CrossRef CAS.
- M. Patel, F. Luo, M. Khoshi, E. Rabie, Q. Zhang, C. Flach, R. Mendelsohn, E. Garfunkel, M. Szostak and H. He, ACS Nano, 2016, 10, 2305–2315 CrossRef CAS PubMed.
- W. Ma, L. Xie, L. Dai, G. Sun, J. Chen, F. Su, Y. Cao, H. Lei, Q. Kong and C. Chen, Electrochim. Acta, 2018, 266, 420–430 CrossRef CAS.
- M. Valero-Romero, F. García-Mateos, J. Rodríguez-Mirasol and T. Cordero, Fuel Process. Technol., 2017, 157, 116–126 CrossRef CAS.
- G. Dong, Y. Zhang, Y. Zhao and Y. Bai, J. Fuel Chem. Technol., 2014, 42, 1455–1463 CrossRef CAS.
- L. Gan, F. Guo, J. Yu and G. Xu, Catalysts, 2016, 6, 25 CrossRef.
- I. Mejri, F. Ayari, M. Mhamdi, G. Ksibi and A. Ghorbel, Microporous Mesoporous Mater., 2016, 220, 239–246 CrossRef CAS.
- A. Chughtai, M. Atteya, J. Kim, B. Konowalchuk and D. Smith, Carbon, 1998, 36, 1573–1589 CrossRef CAS.
- G. Marbán, T. Valdés-Solís and A. Fuertes, J. Catal., 2004, 226, 138–155 CrossRef.
- C. Liu, J. Shi, C. Gao and C. Niu, Appl. Catal., A, 2016, 522, 54–69 CrossRef CAS.
- L. Kang, L. Han, J. He, H. Li, T. Yan, G. Chen, J. Zhang, L. Shi and D. Zhang, Environ. Sci. Technol., 2019, 53, 938–945 CrossRef CAS PubMed.
- Z. Hao, Z. Shen, Y. Li, H. Wang, L. Zheng, R. Wang, G. Liu and S. Zhan, Angew. Chem., 2019, 58, 6351–6356 CrossRef CAS PubMed.
- K. Ramesh, C. Jie, Y. Han and A. Borgna, Ind. Eng. Chem. Res., 2010, 49, 4080–4090 CrossRef CAS.
- J. Bedia, J. Rosas, D. Vera, J. Rodríguez-Mirasol and T. Cordero, Catal. Today, 2010, 158, 89–96 CrossRef CAS.
- J. Bedia, R. Ruiz-Rosas, J. Rodríguez-Mirasol and T. Cordero, J. Catal., 2010, 271, 33–42 CrossRef CAS.
- B. Maia, J. Tjong and M. Sain, Materials Today Sustainability, 2019, 5, 100011 CrossRef.
- M. Guerrero-Pérez, M. Valero-Romero, S. Hernández, J. Nieto, J. Rodríguez-Mirasol and T. Cordero, Catal. Today, 2012, 195, 155–161 CrossRef.
- A. Puziy, O. Poddubnaya, A. Martínez-Alonso, A. Castro-Muñiz, F. Suárez-García and J. Tascón, Carbon, 2007, 45, 1941–1950 CrossRef CAS.
- H. Wang, H. Wang, Y. Chen, Y. Liu, J. Zhao, Q. Cai and X. Wang, Appl. Surf. Sci., 2013, 273, 302–309 CrossRef CAS.
- I. Mochida, N. Shirahama, S. Kawano, Y. Korai, A. Yasutake, M. Tanoura, S. Fujii and M. Yoshikawa, Fuel, 2000, 79, 1713–1723 CrossRef CAS.
- Z. Guo, Y. Xie, I. Hong and J. Kim, Energy Convers. Manage., 2001, 42, 2005–2018 CrossRef CAS.
- J. Sousa, M. Pereira and J. Figueiredo, Catal. Today, 2011, 176, 383–387 CrossRef CAS.
- S. Adapa, V. Gaur and N. Verma, Chem. Eng. J., 2006, 116, 25–37 CrossRef CAS.
- W. Zhang, S. Rabiei, A. Bagreev, M. Zhuang and F. Rasouli, Appl. Catal., B, 2008, 83, 63–71 CrossRef CAS.
- Z. Zhang, J. Atkinson, B. Jiang, M. Rood and Z. Yan, Appl. Catal., B, 2014, 148, 573–581 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01654c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yabin Weia,
Jiangcan Wanga,
Shuo Yanga,
He Wanga,
Minghe Yanga,
Yan Liua,
Wenming Qiaob,
Licheng Ling
*a,
Yabin Weia,
Jiangcan Wanga,
Shuo Yanga,
He Wanga,
Minghe Yanga,
Yan Liua,
Wenming Qiaob,
Licheng Ling