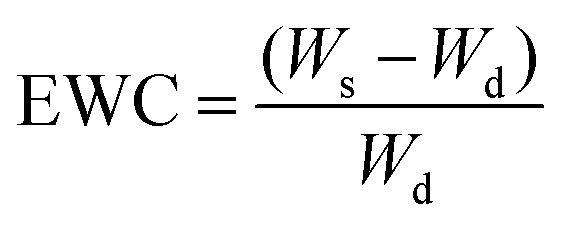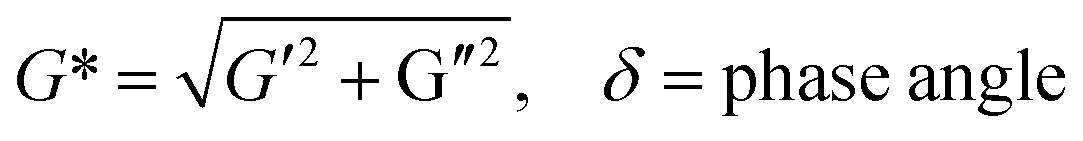DOI:
10.1039/D0RA02218G
(Paper)
RSC Adv., 2020,
10, 19587-19599
Dynamic crosslinked and injectable biohydrogels as extracellular matrix mimics for the delivery of antibiotics and 3D cell culture†
Received
10th March 2020
, Accepted 1st May 2020
First published on 22nd May 2020
Abstract
Antibiotics are widely used in clinical medicine. As an important member, vancomycin often plays an irreplaceable role in some serious infections but for its use, there is still a lack of suitable carriers and effective formulations. To find a vancomycin carrier with potential for clinical applications, a new class of poly(γ-glutamic acid)/dextran-based injectable hydrogels have been constructed through dynamic covalent hydrazone linkages. Adipic dihydrazide (ADH)-grafted poly(γ-glutamic acid) (PGAADH) and sodium periodate-oxidized dextran (OD) precursors were synthesized; then, the hydrogels were formed by blending PGAADH and OD buffer solutions without any additives under physiological conditions. The newly formed precursor structures, mechanical properties, morphologies, hydrogel degradation profiles, and the interaction between the drug and precursors were investigated with FTIR spectroscopy, 1H NMR spectroscopy, rheological experiments, compression tests, SEM, and isothermal titration calorimetric (ITC) measurements. The resulting hydrogels exhibited excellent antibacterial ability and ideal variable performances. Moreover, the hydrogels exhibited different drug release kinetics and mechanisms and were applied effectively towards the controlled release of vancomycin. Significantly, benefitting from the reversibly cross-linked systems and the excellent biocompatibility, the hydrogels can work as the ideal material for HeLa cell culture, leading to encapsulated cells with higher viability and capacity that is proliferative. Therefore, the injectable PGAADH/OD hydrogels demonstrated attractive properties for future applications in pharmaceutics and tissue engineering.
1 Introduction
Biomedical hydrogels can be composed of a range of natural and synthetic polymers1–5 or peptides.6–8 As it can soak up a significant amount of active liquids or biological liquids, this new carrier material has played a variety of roles in the biomedical field.9,10 Over the past decades, several R&D teams have developed a variety of crosslinking methods for the preparation of medical hydrogels. The physically crosslinked hydrogels usually realize a sol–gel transition though the action of various forces, such as hydrogen bonding,11 hydrophobic interaction12 and ionic force.13,14 What is quite different about the mechanism is that chemically crosslinked hydrogels usually form new chemical bonds through different chemical reactions to realize the transformation of sol–gels.
There are many mechanisms of reactions, such as oxidation–reduction reactions,15 light-initiated reactions,16 chemoselective reactions,17–19 Michael reactions,20 and Schiff base reactions.21 Chemical crosslinking improves the properties of biomaterials steadily, but the introduced crosslinking agent often causes severe cytotoxicity. On the other hand, physically crosslinked hydrogels become unstable under the influence of environmental changes. Therefore, the formation of biocompatible bio-hydrogels using different chemical reactions is always faced with various challenges.
Hydrazone crosslinking, which represents a type of aldehyde-hydrazide coupling reaction, is a facile protocol that creates non-toxic products or byproducts.22 It falls in the group of pseudo-click chemistry reactions that are reversible and versatile. Due to its high reactivity, this reaction enables the design of injectable hydrogels with excellent biocompatibility and biodegradability.23 The characteristics of hydrazone crosslinked hydrogels can be designed by altering the amount of precursors. The hydrogel injectability is particularly good for various drug delivery and clinical applications.
Dextran, one of the hydrogel components, was selected due to its biocompatibility, biodegradability, solubility in water and ease of chemical modification, making it an attractive biopolymer for designing hydrogels. Dextran is an extracellular bacterial polysaccharide bearing over 95% of linear α-1,6 glucopyranose units and includes some degree of 1,3-branching. This biopolymer is prepared in sucrose-containing media by Lactobacillus, Leuconostoc and Streptococcus and now is a large-scale commercial product with different molecular weights.
Poly(γ-glutamic acid) (PGA), which is the Bacillus natto's adhesive ingredient, could usually be considered a linear polymeric polypeptide. After degradation, the substance can form non-toxic short peptide molecules, so its biocompatibility is good. Because of these benefits, many research groups have developed several biomedical products such as drug carriers, tissue repair materials, coagulation sponges and so on with the help of PGA and its derivatives.10,24–26
Polysaccharide–polypeptide conjugates are anticipated to exert a synergistic influence on the tissue regeneration properties and biocompatibility. Therefore, the PGAADH/OD (PD) hydrogel crosslinked via the hydrazone reaction was investigated in this research. Two precursors (PGAADH and oxidized dextran) were first synthesized and then confirmed by 1H NMR or FTIR. Following this, these precursors' crosslinking reaction was triggered by just mixing their buffer solutions, resulting in the formation of the final hydrogels. The performance characteristics of the hydrogels including dynamic and static mechanical properties, degradation rate, SEM morphology and swelling ratio, and equilibrium water content were determined. Finally, the drug release mechanism, antimicrobial ability and in vitro 3D encapsulation of cells were also evaluated.
2 Materials and methods
2.1 Materials
Dextran (100 kDa) and PGA (1000 kDa) were supplied by Herbon International Polysaccharide Biotechnology and Shineking Biotechnology Co., Ltd., respectively. Shanghai Adamas-beta Chemical Company provided 1-ethyl-(3-3-dimethylaminopropyl)carbodiimide hydrochloride, sodium periodate (SP), methyl orange, adipic dihydrazide, hydroxylamine hydrochloride and N-hydroxysuccinimide (NHS). Vancomycin (900 μg mg−1) was purchased from Baomanbio Biotechnology Co., Ltd. (Shanghai, China). Most of the cell culture media and ingredients, including 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), DMEM, FBS, PBS buffer, etc., were purchased from HWB scientific Co. The HeLa cells, which were provided by Suer Biotech Co., were cultured in complete growth culture medium (DMEM supplemented with penicillin (100 U mL−1), streptomycin (100 μg mL−1), FBS (10%), and L-glutamine (1 mM)) in a 37 °C atmosphere with 5% CO2.
2.2 ADH grafted PGA precursor
Functional molecular adipic dihydrazide (ADH) was grafted to PGA side chains to form the PGAADH precursor using an EDC/NHS protocol in water. Three substances, 0.5 g PGA, 2.226 g EDC and 1.337 g NHS, were sequentially added to 50 mL H2O to activate the carboxyl groups on PGA at pH 5 for 2 h. Following this, the activated PGA was dropped into 60 mL ADH solution, which contained 6.74 grams ADH. The mixed liquids were stirred overnight with pH adjusted to 5.8. To obtain a more pure lyophilized product, the reaction system ended up being dialyzed using a dialyzed bag with MWCO = 1000 Da. 1H NMR was employed to verify the PGAADH structure27,28 (Fig. S1 in ESI†): 1H NMR (500 MHz, D2O) δ1.52 (s, 2H), 2.48–1.70 (m, 7H), 2.78 (d, J = 7.2 Hz, 1H), 3.28 (d, J = 79.4 Hz, 1H). The substitution ratio of PGAADH could be defined as the number of ADH molecules grafted per 100 PGA repeat units. The calculated value is about 39.2%.
2.3 Oxidized dextran precursor
Oxidized dextran was fabricated in an aqueous solution. First, the dextran solution (400 mL) with a concentration of 1.25% (w/v) was mixed with 3.28 g NaIO4 and allowed to react in the dark for 12 hours and then, a certain amount of diethylene glycol was dripped in to terminate the reaction. Before being lyophilized, the dialysis process of the reaction system was conducted using a dialysis bag with MWCO = 1000 Da.29 The oxidation degree of OD was determined by the hydroxylamine hydrochloride method30 and the calculated value was about 20%.
2.4 Hydrogel formation
Briefly, OD and PGAADH solutions with 5% concentration were mixed with each other in different proportions of PGAADH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OD (3:1, 2:1, 1:1, 1:2 and 1:3), resulting in PD31, PD21, PD11, PD12 and PD13 sample solutions, respectively. A 5 mm height cylindrical mold was used to hold the sample solution and to keep it solidified overnight. After demolding, the gel samples were sterilized in 75% ethanol, and then cleaned with pure water three times. The resulting samples were subsequently characterized with different techniques. The hydrogel gelation time was calculated by the vial tilting method,31 i.e., no obvious flow in 1 min after inverting the vial can be regarded as reaching the gel state.
:OD (3:1, 2:1, 1:1, 1:2 and 1:3), resulting in PD31, PD21, PD11, PD12 and PD13 sample solutions, respectively. A 5 mm height cylindrical mold was used to hold the sample solution and to keep it solidified overnight. After demolding, the gel samples were sterilized in 75% ethanol, and then cleaned with pure water three times. The resulting samples were subsequently characterized with different techniques. The hydrogel gelation time was calculated by the vial tilting method,31 i.e., no obvious flow in 1 min after inverting the vial can be regarded as reaching the gel state.
2.5 Test of equilibrium water content
The obtained hydrogels (PD31, PD21, PD11, PD12 and PD13), which were mixed in different volume ratios of PGAADH:OD (3:1, 2:1, 1:1, 1:2 and 1:3, respectively), were incubated in PBS buffer solutions (pH 6.0 or 7.4) to reach a swelling state. After removing free water from the surface, each sample's swollen weight (Ws) was recorded. The hydrogels were then frozen at −20 °C and lyophilized to dry the samples.
The dry weight (Wd) of each sample was used to calculate the samples' equilibrium water content (EWC) value:
All the samples were tested in triplicate for each group.
2.6 Observation of morphology
PD11 was lyophilized in a lyophilizer (Benchtop Pro 31, Virtis) and observed by SEM (GeminiSEM 300, Zeiss). After the sample was placed on the platform and sputter-coated with gold, the morphologies of the samples were viewed at a 5 kV accelerating voltage. The value was evaluated by randomly determining the pore diameters in three different visual points.
2.7 Mechanical properties
Different samples (PD31, PD21, PD11, PD12 and PD13) with a fixed size (10.7 mm diameter and 5 mm height) were tested using a universal system (WDW-50, Hengle Xingke Instrument Co., Ltd, Jinan, China). The compression rate was set to 20 mm min−1. The load–displacement curve was used to calculate the compressive strength three times for each sample.
2.8 Dynamic rheological test
Dynamic rheological experiments were performed using an Anton Paar MCR 302 Rheometer equipped with a Peltier temperature control system having an accuracy of ±0.01 °C. The rheological study of an injectable hydrogel is very important for its practical application. The storage modulus (G′) and loss modulus (G′′) of hydrogel samples, PD12, PD11, and PD21, in different PGAADH:OD volume ratios (1:2, 1:1 and 2:1, respectively) were measured using parallel plates at room temperature in an oscillatory mode for 10 min (gap size = 1 mm, strain = 1.0%). Moreover, the stress sweep was performed at a constant frequency of 10 Hz.32
2.9 Investigation of swelling and degradation behavior
In order to test the degradation rate and swelling ratio, the PD11 hydrogels were immersed in PBS (0.01 M) containing papain (0.05 mg mL−1) under constant shaking at 100 rpm for 2, 4, 8 and 24 h at 37 °C to accelerate degradation. At each pre-determined time interval, the samples were washed with distilled water and then lyophilized. The in vitro degradation rate was calculated by dividing the dry weight after degradation (Wt) with the initial dry weight of the gel (W0) as follows:
| Fractional mass remaining = (Wt/W0) × 100% |
The swelling ratio (Q) was calculated by dividing the swollen weight after degradation (Ws) with the initial weight of the gel (W0) as follows:
All the samples were tested in triplicate for each group.
2.10 In vitro drug release study of the hydrogels
2.10.1 Drug loaded hydrogel formation and drug release experiments. To form the drug-loaded hydrogels, an improved strategy was adopted to encapsulate vancomycin into the hydrogels, which is slightly different from the previous protocol to create drug-loaded hydrogels.33 Specifically, 9 mg vancomycin was added to OD solution (2.5 mL, 3.4 mL, 5 mL, 6.6 mL and 7.5 mL, respectively), which was further mixed with PGAADH buffer solution (7.5 mL, 6.6 mL, 5 mL, 3.4 mL and 2.5 mL, respectively) to form the mixture (10 mL). The mixture was incubated at room temperature by vortexing to obtain drug-loaded hydrogels. In this case, the drug loading ability could be considered 100%.In the drug release experiment, typically, vancomycin-loaded hydrogels (10 mg) were soaked in 2 mL PBS (pH 7.4) with continuous shaking at 37 °C. At predetermined time points, 100 μL aliquots of the supernatant were withdrawn and replaced by an equal volume of fresh PBS solution. The cumulative release of vancomycin was measured using a UV spectrophotometer (S-3100, SCINCO) at a wavelength of 280 nm. Before characterizing the drug release quantitatively, the calibration curves of vancomycin in PBS (pH 7.4) solution were established.34
2.10.2 Study of drug release kinetics and mechanism. Drug release behavior of the PD hydrogels was analyzed employing several kinetic models (Zero-order, First-order, Higuchi and Ritger–Peppas). The optimal model by the best fit correlation was determined. The equations for each model are shown below:
| Zero-order model: Mt = K0t |
| First-order model: Mt = 1 − e−K1t |
| Higuchi model: Mt = Kht1/2 |
| Ritger–Peppas model: Mt/M∞ = krtn |
where Mt: the fraction of released drug at each time point (t), K0: the zero-order release kinetic constant, K1: the first-order release kinetic constant, Kh: the Higuchi release kinetic constant, and Kr: the Ritger–Peppas release kinetic constant.Drug release mechanisms were analyzed using the Ritger–Peppas model, which is commonly used when the mechanism is unclear or when more than one mechanism is involved.35 The release exponent (n) indicates the mechanism of drug release. When n is <0.45, drug release follows a Fickian diffusion mechanism that indicates drug release occurs by diffusion. At 0.45 ≤ n < 0.89, it follows non-Fickian transport. At n = 0.89, it follows case II transport that indicates drug release occurs by erosion. At 0.89 < n, it follows a super case II transport mechanism.36,37
2.11 Isothermal titration calorimetric measurement
The heat flow resulting from the binding interaction of vancomycin with PGAADH or OD was measured by an isothermal titration calorimeter (ITC200, MicroCal, USA). The reference cell (effective volume of 205 μL) of the calorimeter was loaded with Milli-Q water. For the PGAADH + vancomycin system, the sample cell was loaded with vancomycin (2 mM) and the titrant syringe was filled with PGAADH (30 mM). In addition, the sample cell and titrant syringe were loaded with OD (370 mM) and vancomycin (5 mM) for the system of OD + vancomycin, respectively. The drug was loaded into different positions in order to better understand their interactions. In each titration experiment, 20 injections of 2 μL syringe sample solution (with an initial injection of 0.4 μL) were titrated into the cell. In order to ensure proper mixing after each injection, a constant stirring speed of 750 rpm was maintained during the experiment. Control experiments were performed by injecting the syringe solution into the PBS (solvent) and the PBS into the cell solution, respectively, in which the concentrations of sample solutions were consistent with those in the above titration experiments. The resulting corrected injection heat was plotted as a function of titrant/titrand molar ratio and fitted to different models presented in the Windows-based Origin 7.0 software package provided by the ITC200 calorimeter with the first injection peak discarded. In the non-linear least squares fitting of the experimental data, the best-fit values of the stoichiometry (n), complexation constants (KC) and standard enthalpy change (ΔHo) were obtained. Based on the above thermodynamic parameters, the standard Gibbs free energy change (ΔGo) and standard entropy change (ΔSo) were calculated as per the equation:38
| ΔGo = −RTlnKC = ΔHo − TΔSo |
2.12 Antibacterial activity assay
The morphological changes of the bacteria were viewed by scanning electron microscopy (GeminiSEM 300, Zeiss) before and after they were co-cultured with different hydrogels and reference drug solutions. E. coli and S. aureus were selected as two kinds of strains, which were cultured to an exponential phase and then harvested by centrifugation. Subsequently, the strains were washed once with Luria Bertani medium and twice with PBS and re-suspended in PBS. Bacteria were incubated at 37 °C for up to 60 min with 100 mg of hydrogels (blank and vancomycin-loaded hydrogels) and reference drug solution (configured with the dissolution rate of 7%). Controls were performed in the absence of hydrogels. Cells were fixed with 2.5% (w/v) glutaraldehyde in PBS, washed extensively with PBS, and then dehydrated with a graded ethanol solution (30%, 50%, 70%, 95% and 100%, 15 min each). The dehydrated samples were lyophilized before spraying with gold twice. The acceleration voltage was set to 3 kV.
2.13 Cytotoxicity assay in vitro
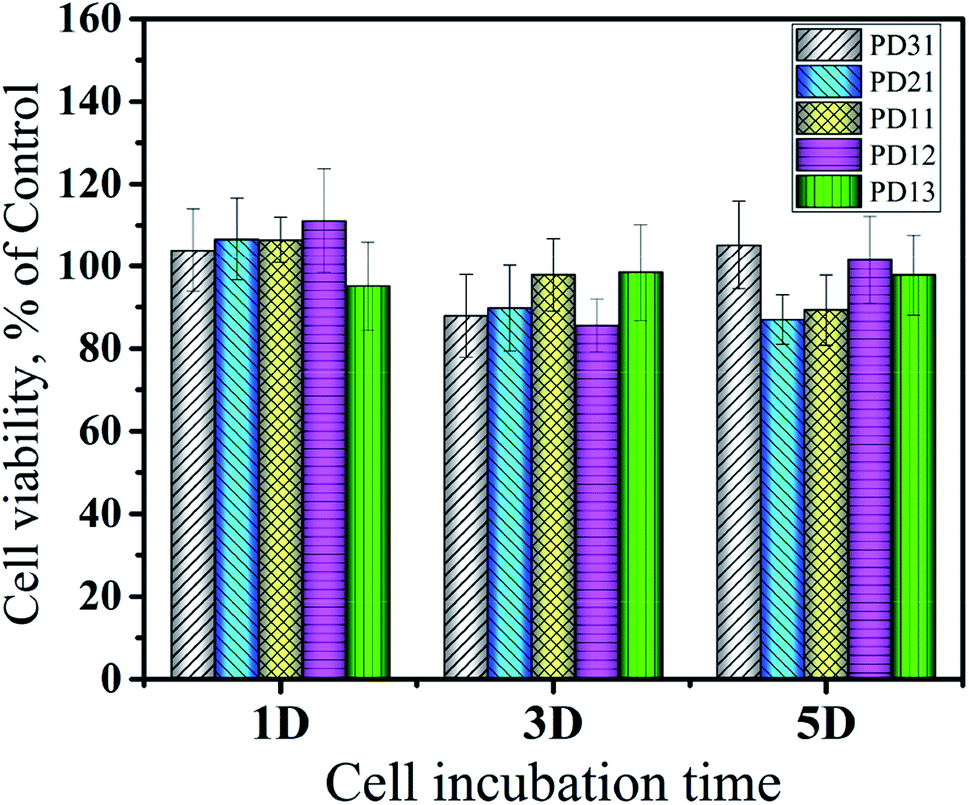
The evaluation of the cytotoxicity of PD hydrogels (PD31, PD21, PD11, PD12 and PD13) was accomplished on an extracted solution of the hydrogel via MTT assay as per the ISO 10993-5 standard. Sterilized hydrogels were extracted using DMEM at an extraction ratio of 1 cm2 mL−1 at 37 °C for 24 h. A 100 μL media suspension containing a total of 104 cells and 100 μL extract solution were plated into each well of the 96-well plate. As a blank group, 200 μL of complete medium was used. Both the sample and blank group were then incubated at 37 °C in 5% CO2 atmosphere. On the 1st, 3rd and 5th day, 20 μL of MTT (5 mg mL−1 in PBS) was added and cultured for 4 h to allow for the formation of formazan crystal. After removal of the supernatant, 150 μL DMSO was added to each well and the absorbance was measured at 490 nm using an ELISA reader (Elx808, BioTek Instrument Inc., VT). The results were expressed as percentages relative to the data obtained with the blank control. Six samples were tested for each group.
2.14 Cells 3D encapsulation
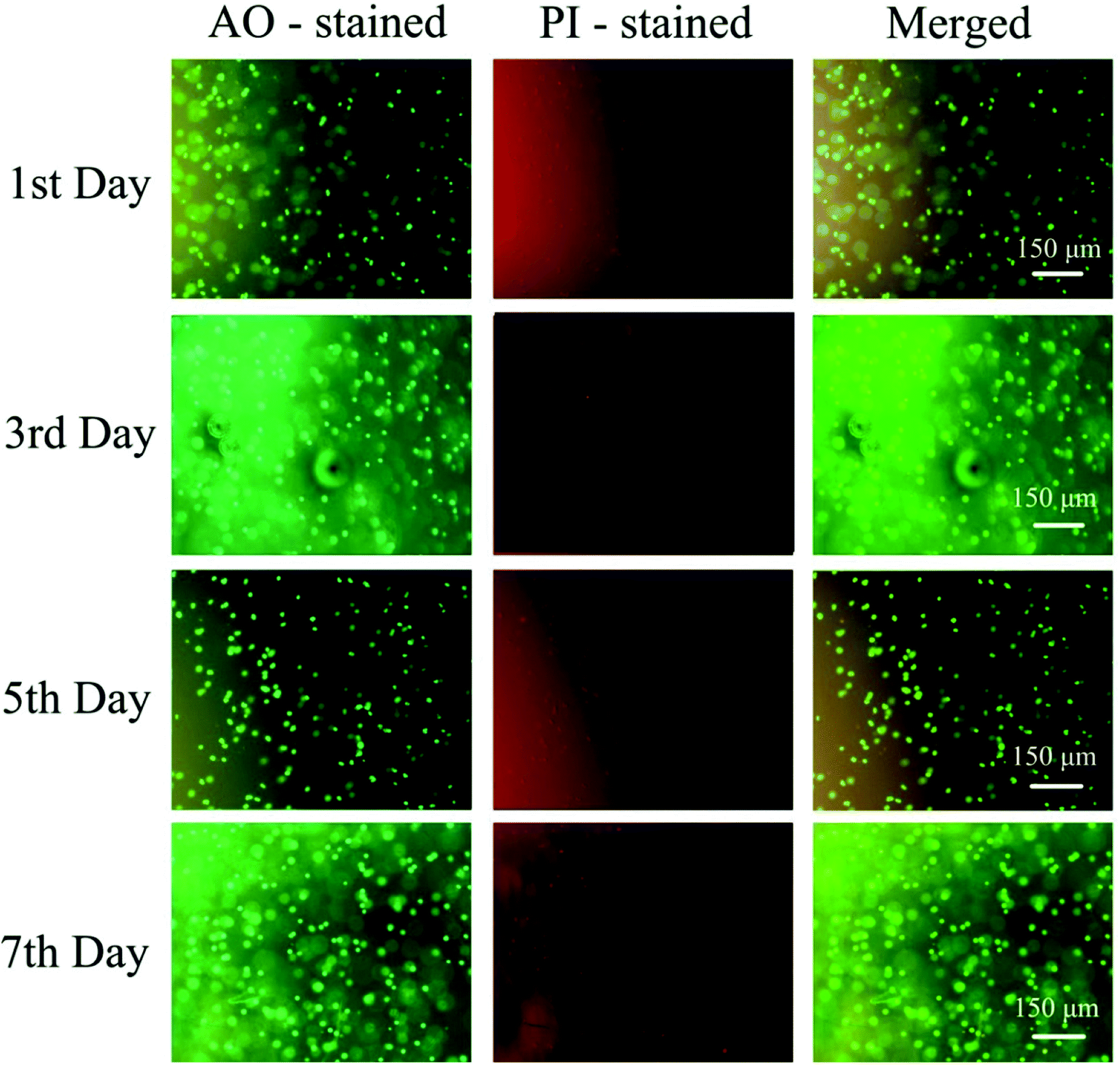
To further verify the biocompatibility of hydrogels, a HeLa cell encapsulation experiment was carried out. First, 5% (w/v) PGAADH solution and 5% (w/v) OD solution containing HeLa cells were mixed (1:1 v/v) homogeneously, followed by immediate placing into a 24-well culture plate (2.0 × 105 cells per well). The HeLa cell encapsulated hydrogels were rapidly formed at 37 °C via the hydrazone crosslinking reaction. DMEM medium (1 mL) was then added onto the surface of the hydrogels, and the medium was refreshed every 2 days. At a predetermined time, the hydrogel sheets encapsulated with HeLa cells were immersed in PBS solution containing acridine orange (AO, 2 μM) and propidium iodide (PI, 2 μM) for 30 minutes at 37 °C. After cleaning the dyes with PBS solution, the images of stained cells were photographed under an inverted fluorescence microscope.
2.15 Statistical analysis
Experiments were performed in triplicate and results are reported as mean ± SD. Comparisons between groups were computed using Origin Pro8 software for t tests and the significant difference between means was asserted at 95% confidence intervals (p < 0.05).
3 Results and discussion
3.1 Synthesis of two precursors
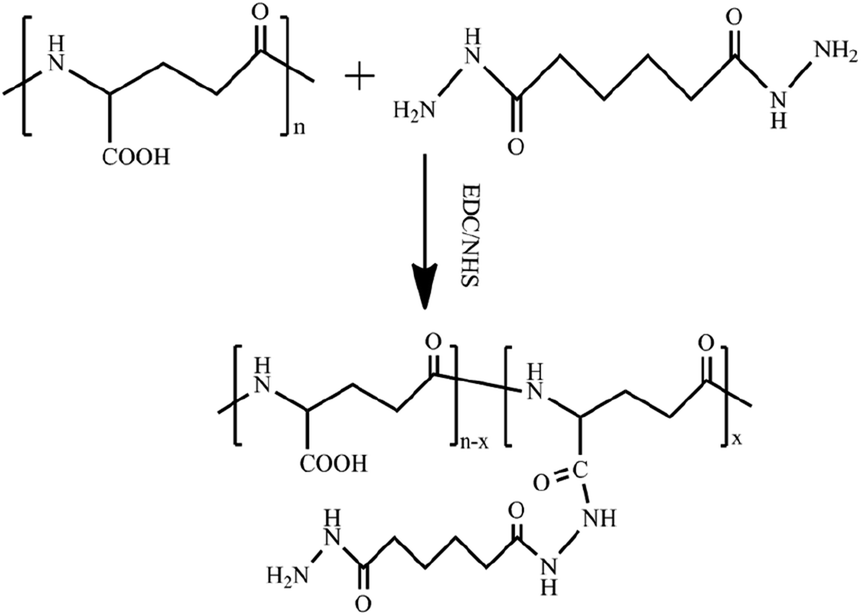
The PGAADH precursor was synthesized using the EDC/NHS protocol as described in our previous article and shown in Fig. 1.4 Comparing the 1H NMR spectra of PGAADH and PGA (ESI, Fig. S0 and S1†), the additional signals of the latter at 1.40–1.60 can be respectively attributed to the hydrogens on the methylene of ADH, indicating that the amidation was successful. The degree of substitution (the number of ADH molecules per 100 repeating units of PGA) in the PGAADH precursor is about 39.2%.
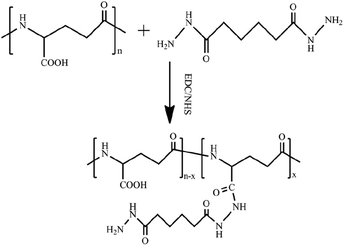 |
| | Fig. 1 Synthesis of adipic acid dihydrazide (ADH)-grafted PGA precursor PGAADH using EDC/NHS protocol in the aqueous phase at room temperature. | |
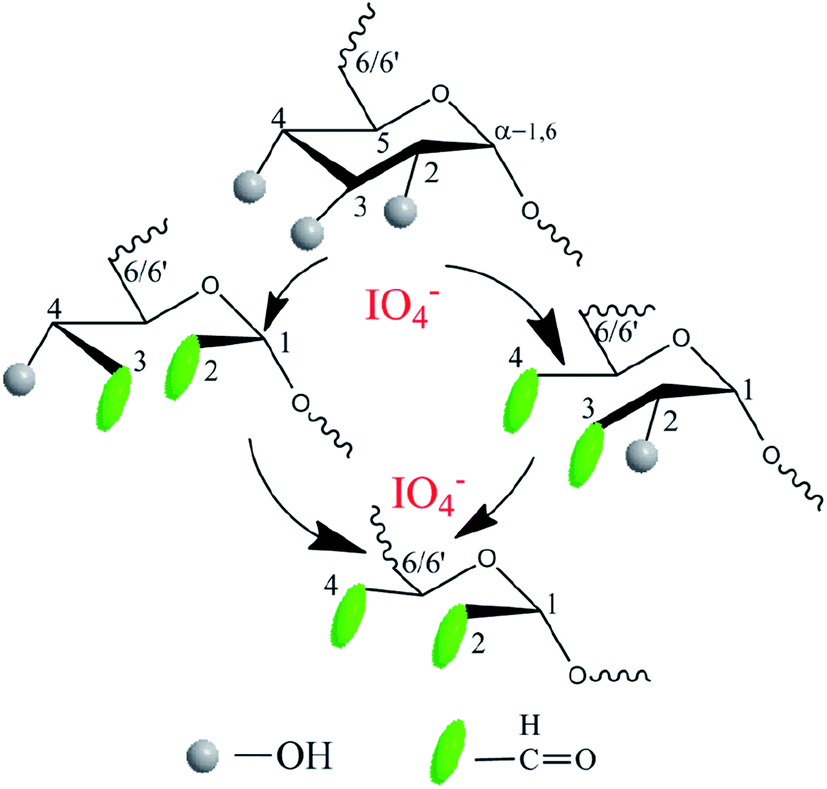
Another precursor OD was also synthesized by aqueous phase synthesis as illustrated in Fig. 2. Comparing the 1H NMR spectra of dextran and OD, the additional signals of the latter at 5.40 and 5.60 ppm can be respectively attributed to the hemiacetal protons formed from the aldehyde and neighboring hydroxyl groups39 (ESI, Fig. S2 and S3†). In addition, the comparison of infrared spectra can also verify that a successful oxidation reaction took place. The peak at 1725 cm−1 was assigned to the aldehyde groups, suggesting the formation of OD (ESI, Fig. S4 and S5†). The degree of oxidation of OD, defined as the number of aldehyde groups per 100 saccharide units, was determined to be about 18%, based on the hydroxylamine hydrochloride method.
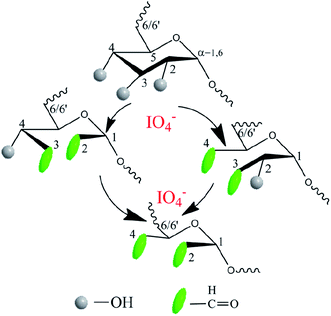 |
| | Fig. 2 Synthesis of oxidized dextran precursor OD using sodium periodate as the oxidizing agent in an aqueous solution under dark conditions. | |
3.2 Hydrogels formation
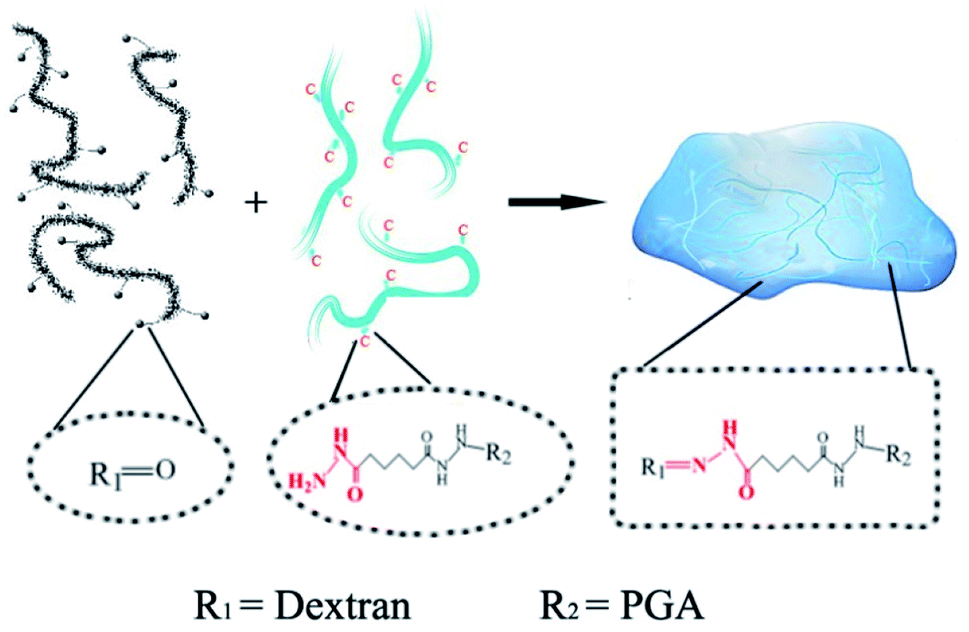
The PD hydrogels were prepared as illustrated in Scheme 1 with different precursor compositions. Among all the samples, PD11 hydrogels provided the fastest gelation time (≈15 s) in PBS 7.4 under physiological conditions. The gelation time of other hydrogel samples ranging from 15 s to approx. 1 min was also established by the tilting method.40 Compared with our previous native chemical ligation (NCL) system hydrogels,4 the PD hydrogels in this paper utilized a completely different crosslinking method. The design of the hydrazone crosslinking reaction greatly reduced the gelation time of this system hydrogel from 3.5 hours of the NCL hydrogel to less than 1 minute. In terms of materials, NCL system hydrogels only choose polyglutamic acid as the base material, while PD hydrogels combined the polyglutamic acid with dextran, which is more similar to the composition of the extracellular matrix and is expected to have better clinical application prospects.
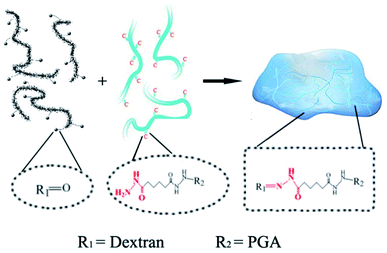 |
| | Scheme 1 The mechanism of hydrazone hydrogel formation under physiological conditions. The dotted boxes indicate the functional groups before and after the crosslinking reaction. | |
3.3 Test of equilibrium water content
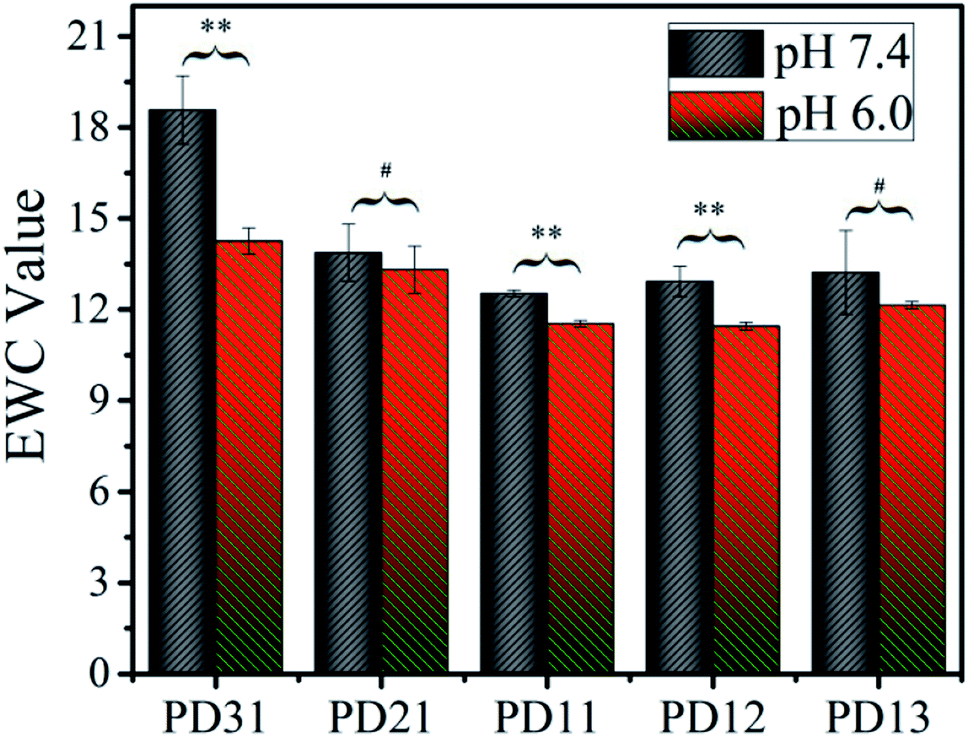
To explore the applicability of PD hydrogels in different human tissues and lesions, which have different acid–base environments, the EWC values of the hydrogels were measured in two buffer solutions with different pH values (S210, METLER TOLEDO), as illustrated in Fig. 3. Among all the samples, EWC values decreased as the OD precursor content ratio increased. This suggests that the hydrogel network formed more efficiently as the OD content ratio increased. However, when the ratio of PGAADH:OD exceeded 1:1, the EWC value of PD12 and PD13 increased to 12.9 and 13.2 in pH 7.4 buffer solution and 11.5 and 12.1 in pH 6.0 buffer solution, respectively. This phenomenon could be attributed to the low content of PGAADH leading to a decrease of crosslinking points. Under certain hydrogel composition ratios, the EWC value of hydrogels in pH 7.4 medium is generally higher than that in pH 6.0 medium, which can be attributed to the sensitivity of polymer materials to a different acidic environment. Because of the different composition ratio and different crosslinking conditions, the equilibrium water content of the three groups of PD31, PD11 and PD12 hydrogels showed significant differences (p < 0.05) in the two buffer solutions (pH 7.4 and 6.0).
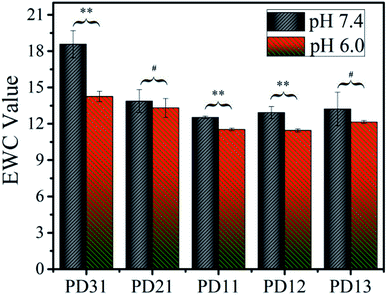 |
| | Fig. 3 The equilibrium water content of five PD hydrogels in two PBS buffer solutions (pH 6.0 or 7.4) at room temperature (**p < 0.05: statistically significant difference under certain hydrogel composition ratio, #p > 0.05: No statistically significant difference under certain hydrogel composition ratio). | |
Among all the samples, PD11 hydrogels in pH 6.0 buffer solution showed the lowest EWC value of 11.4, which may be ascribed to its highest crosslinking density. The PD31 hydrogel was formed at a low OD content ratio, resulting in a relatively loose structure with the highest EWC value of 18.6.
3.4 Observation of morphology
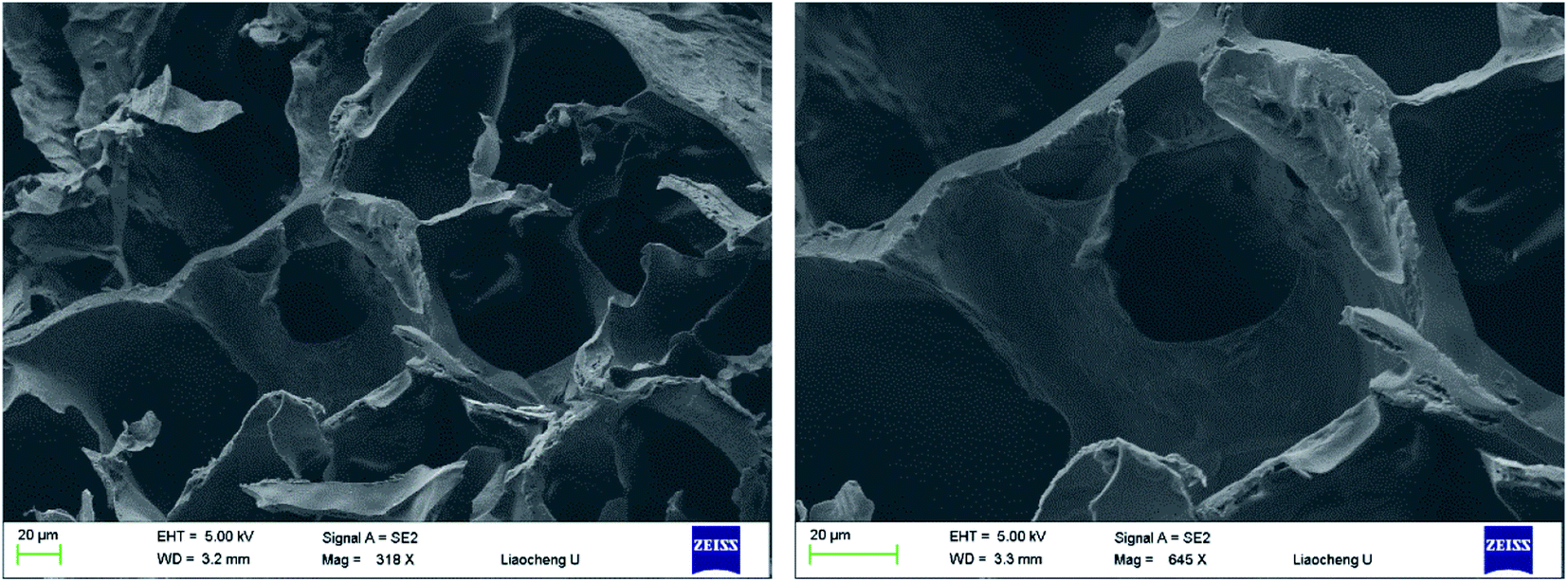
Pore size and pore interconnectivity are critical parameters determining the performance of hydrogels in tissue engineering. Pore size affects the cell attachment, migration, morphology and proliferation; pore structures also have a strong influence on the mechanical properties of the matrix, the supply of nutrients and the removal of waste products. SEM was used to characterize the microstructures and morphologies of lyophilized PD hydrogels. As shown in Fig. 4, the PD11 hydrogel used as an example was highly porous with a well-interconnected pore structure. It was characterized by a wide pore size distribution; the estimated pore size, which was randomly selected in the images, was in the range of 10–100 μm. It is assumed that these properties are satisfactory for cell encapsulation and other biomedical applications as reported in the literature.
 |
| | Fig. 4 SEM images of lyophilized PD11 hydrogel samples, which were sputter-coated with gold and viewed under a 5 kV accelerating voltage (left: 318×, right: 645×). | |
3.5 Mechanical properties
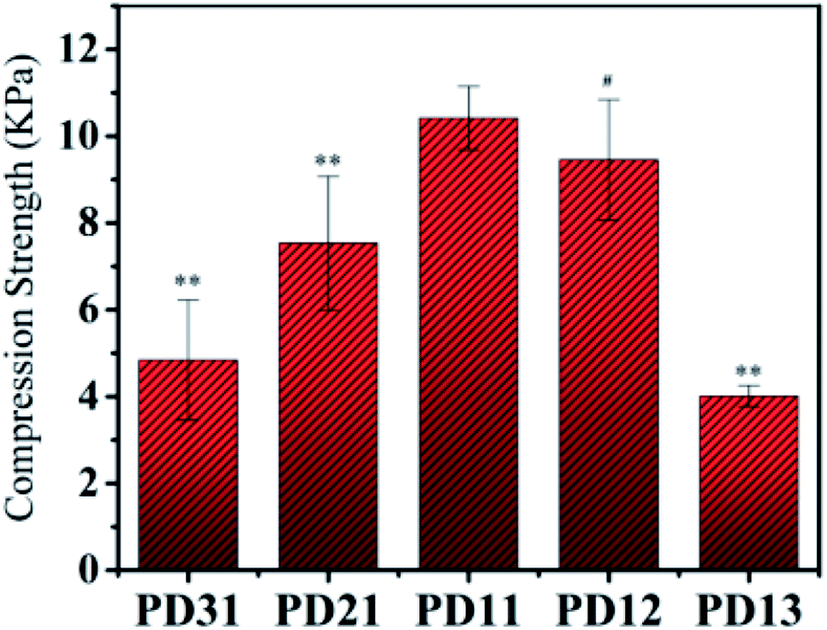
The compressive strength of various PD hydrogels is shown in Fig. 5. In the PD31 group, the structure of the hydrogel was relatively unstable due to a lower number of crosslinking points and low crosslinking density, resulting in a significantly lower (p < 0.05) compressive strength than that of PD11; the reduction was 53.6% (4.84 kPa vs. 10.42 kPa). The compressive strength of PD21 and PD11 groups increased gradually with increasing OD content ratio. It indicated that the crosslinking density was enhanced, resulting in a more compact hydrogel structure. However, the excess content ratio of OD may also cause a relatively lower concentration of PGAADH in the hydrogel preparation, leading to a decrease of crosslinking points and resulting in the lower compressive strength of PD12 and PD13 hydrogels. Among all these groups, PD11 showed the highest compressive strength of 10.42 kPa, which is significantly higher (p < 0.05) than that of the PD31 and PD21 samples. The PD13 group also showed a significantly lower (p < 0.05) compressive strength than that of PD11 by 61.5% (4.01 kPa vs. 10.42 kPa). This phenomenon was similar to those found in the EWC study in section 3.3. The mechanical strength of PD hydrogels is in the range of 4.01–10.42 kPa. With its rapid crosslinking rate, it can be used as an injectable material. However, as a clinical implant, the optimization and improvement of mechanical strength should also be considered for specific applications.
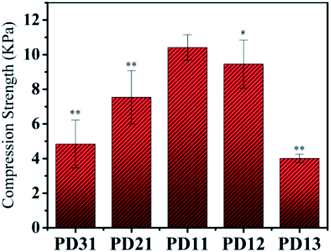 |
| | Fig. 5 The compressive properties of five PD hydrogels with the compression rate of 20 mm min−1 at room temperature (**p < 0.05: statistically significant difference compared with PD11 hydrogel, #p > 0.05: no statistically significant difference compared with PD11 hydrogel). | |
By analogy with other reports in the literature, PD hydrogels could be constructed as a novel drug delivery system in pharmaceutical fields.41 Furthermore, the mechanical properties (≈10 kPa) obtained in this study for PD hydrogels also suggest potential application in cell 3D encapsulation and cell regulation.42,43
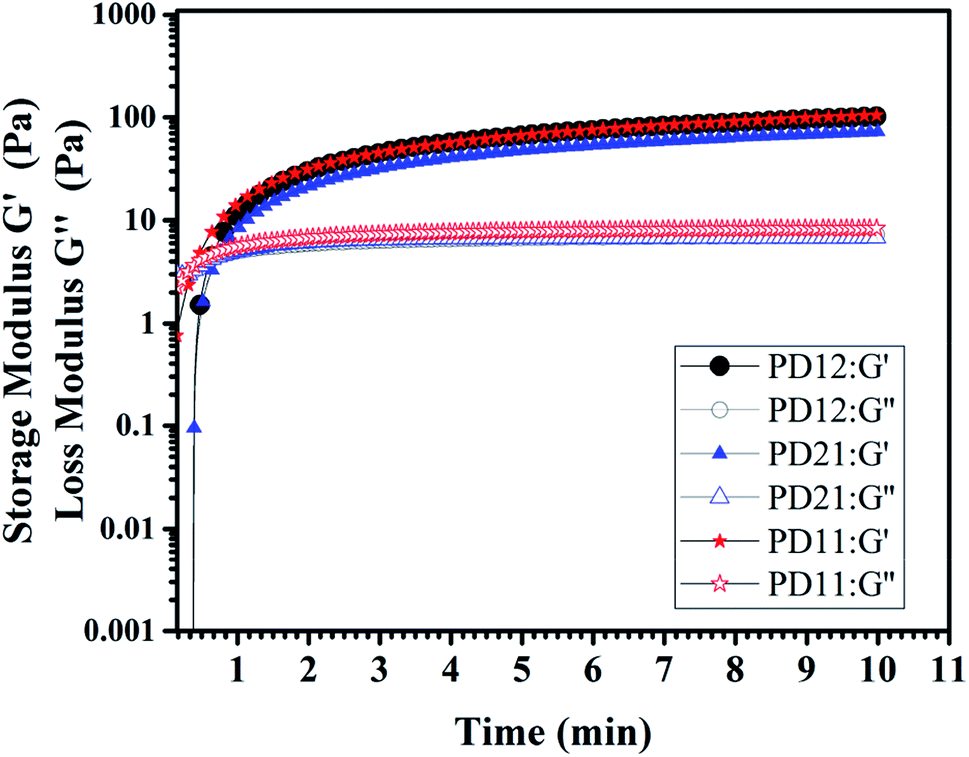
3.6 Dynamic rheological test
The viscoelastic behaviors of the PD hydrogels obtained with a rheometer are presented in Fig. 6. The storage modulus, G′, describes the energy stored in the hydrogel by the applied force and represents the elastic nature of the hydrogel. The loss modulus, G′′ indicates the viscous nature of the hydrogel. These factors can be calculated by the following eq.:
| G′ = G*cosδ, G′′ = G*sinδ |
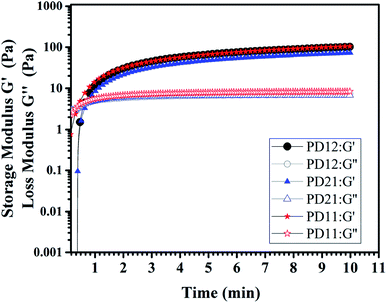 |
| | Fig. 6 The storage modulus G′ and loss modulus G′′ of three PD hydrogels (PD12, PD11, PD21) with a concentration of 5% (w/v) at room temperature in oscillatory mode and a frequency of 10 Hz. | |
The tanδ represents G′′/G′, which is a value that represents the internal bonding force. If the property of elasticity is larger (G′ > G′′), tanδ < 1, it means that the material tends to be in a solid state and the internal bonding force is strong. In contrast, if the viscosity property is larger (G′ < G′′), tanδ > 1, this means that the material is close to being a fluid state and the internal bonding force is weak. In this experiment, an oscillatory test was performed at a frequency of 10 Hz. As shown in Fig. 6, there is an obvious crossover of G′ and G′′ in the early stage of hydrogel formation, indicating the hydrogel undergoes a rapid sol–gel transition. The time corresponding to the crossover point is usually regarded as gelation time (Tgel), and the gelation time of three PD hydrogels are all within 1 min, which is in good agreement with what we have observed in the actual experiment (ESI, Movie S1†). All the G′ and G′′ moduli of the three samples reached the plateau stage after 5 minutes, indicating that the crosslinking process was completed. Among the three samples, the PD11 hydrogel has the highest storage modulus (105.1 Pa), and the storage modulus G′ of PD12 is about 29% higher than that of PD21 (101.8 Pa vs. 72.7 Pa). This phenomenon was also very consistent with the static mechanical test data. These results revealed that the storage modulus of the hydrogels could be readily manipulated by varying the precursor ratios. It has been widely recognized that the storage modulus of the extracellular matrix plays a key role in the migration, proliferation and differentiation of cells. For example, neural stem cells are prone to neuronal differentiation in reasonably soft materials (storage modulus ≈ 0.1–1 kPa).44 Therefore, the PD hydrogels with a tunable storage modulus have great potential for applications in the extracellular matrix.
3.7 Investigation of swelling and degradation behavior
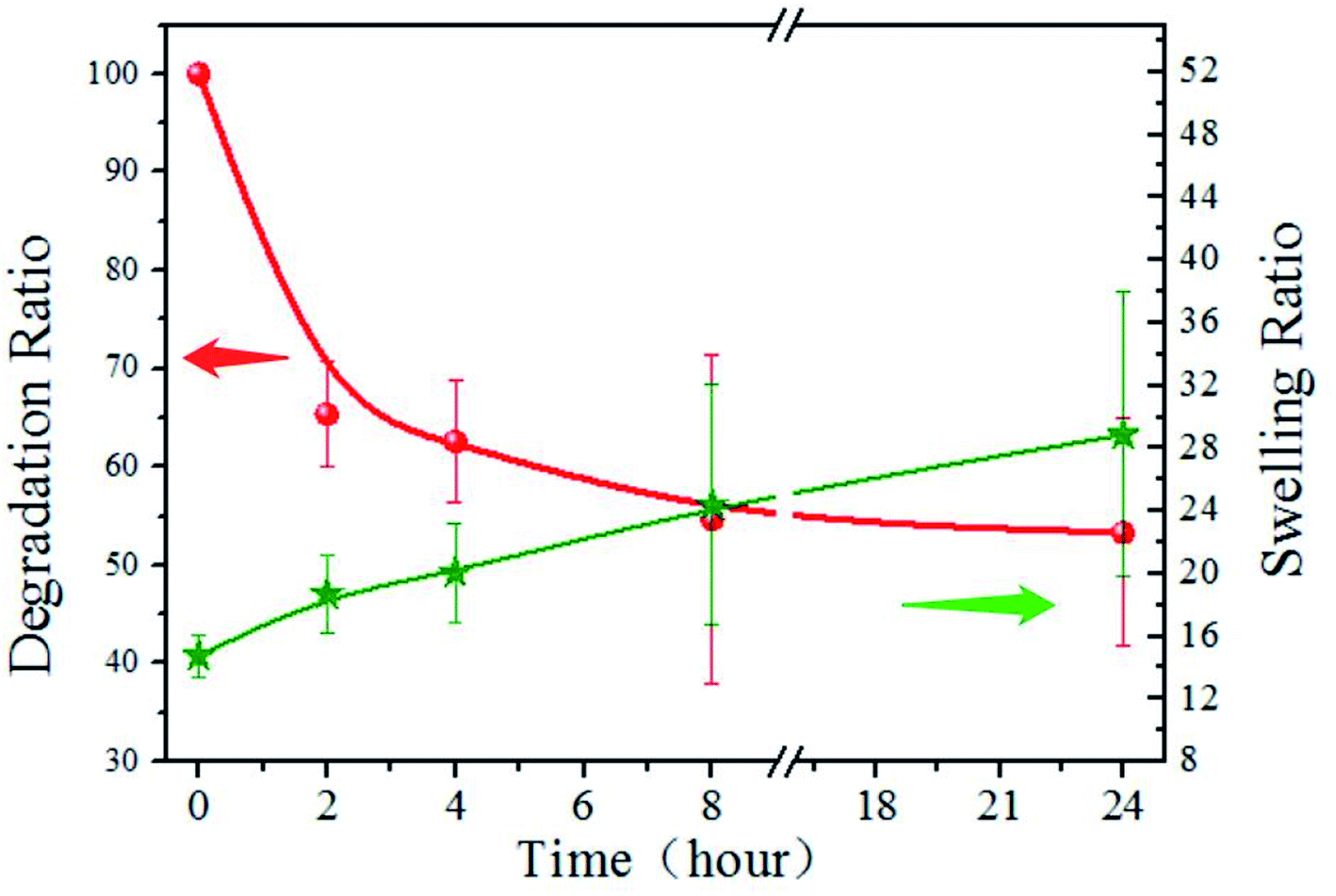
The degradation and swelling properties of the PD11 hydrogels were characterized by measuring the hydrogel weight change in PBS at 37 °C, as shown in Fig. 7. As can be seen, the mass of PD11 decreased by 34.6% within 2 h, and 53.3% remained after 24 hours. The swelling ratio increased from 14.8 to 18.7 in the first 2 hours and reached 28.9 after 24 hours of degradation. This indicates that the hydrogel internal structure undergoes dynamic changes during the degradation. The degradation curve illustrated the rapid decline in the initial stage, which should be attributed to the faster enzymatic hydrolysis rate of hydrogel materials in the initial stage. During the next hours, the degradation profile of PD11 tended to be more regular and controllable. The increase of swelling ratio in the degradation process showed that although the mass of hydrogel polymers decreased during the hydrogel degradation, the overall water holding capacity of the hydrogel could remain ideal for a long time. The combination of the biodegradability of naturally derived polymers and good water holding capacity of hydrogels establishes PD hydrogels as an ideal material of choice for drug release and tissue engineering.
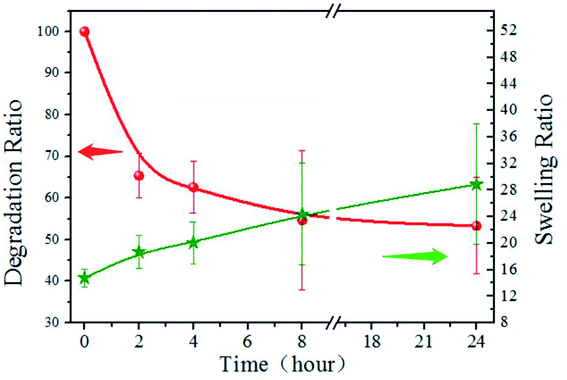 |
| | Fig. 7 Degradation and swelling property of PD11 hydrogels in pH 7.4 PBS (0.01 M) containing papain (0.05 mg mL−1) under constant shaking at 100 rpm at 37 °C. | |
3.8 Drug release behavior and mechanism studies
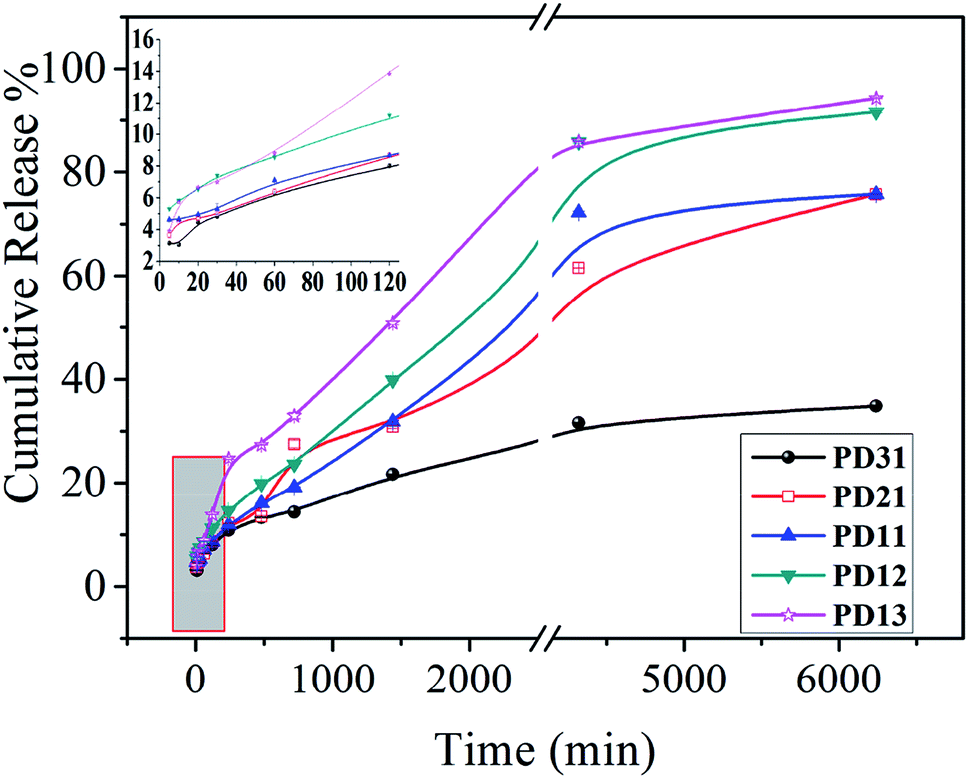
The UV spectrophotometer was utilized to monitor the vancomycin release profile. As demonstrated in Fig. 8, vancomycin released from the PD hydrogels in a sustained manner. Moreover, the release behavior of drug-loaded PD hydrogels showed a similar trend. However, the amount of released drug decreased as the PGAADH precursor content in hydrogels decreased, indicating that the interaction between drug and polymers has a great influence on the drug release behavior. Two hydrogel samples, PD12 and PD13, showed an obvious burst release, where the release amount was up to 11.21% and 13.86% during the initial 120 min, respectively. However, for PD31 (8.01%), PD21 (8.66%) and PD11 (8.68%) hydrogels, the release amount was only around 8% over the same time. After nearly 5 days of release, the cumulative release percent of PD31 hydrogels increased gradually and reached 34.87%, demonstrating its good sustained release behavior.
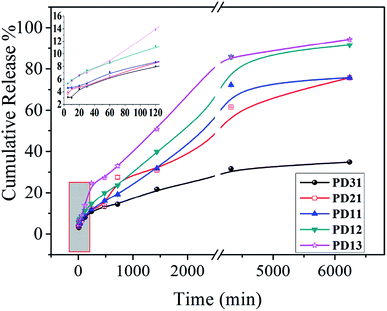 |
| | Fig. 8 In vitro drug release profiles of five hydrogels (PD31, PD21, PD11, PD12 and PD13) in pH 7.4 PBS at 37 °C. The inset shows an enlarged picture of five hydrogels' drug release behavior within the initial 120 minutes. | |
The reasons for the differences in the PD hydrogel release behaviors may be complicated. Many research groups have also verified the different influencing factors for different porous material drug release behaviors from the interaction between the drug and the matrix,45–47 or from the internal structural factors of the porous material.48–50 For ordinary polymer hydrogels, the internal structure affects the EWC, which could generally affect the drug release behavior. However, unlike some reported hydrogels,33,37 the drug release behavior of PD hydrogels is not proportional to its EWC value. Therefore, it is inferred that there may be other factors that play a decisive role in the drug release behavior. Moreover, by analyzing the precursor ratio of different PD hydrogels, it can be seen that PGAADH has a significant effect on drug release. This phenomenon may be because the vancomycin can strongly interact with PGAADH, so with more PGAADH in the hydrogel, the release of vancomycin is lower, which is also confirmed in the ITC study section.
To investigate the drug release mechanism,51 Zero-order, First-order, Higuchi, and Ritger–Peppas models were employed to fit the accumulative drug release curves presented in Fig. 8 and the fitted parameters are given in Table 1. According to published reports,34,37,52 the determination of different hydrogel release models is usually based on the release data of hydrogels, which were generally affected by the combination of composition and structure factors. In this experiment, Origin Pro 8.0 software was used for automatic fitting to obtain correlation coefficients corresponding to different models. The model with the highest correlation coefficient value is generally regarded as the model that best matches the drug release behavior. For the typical PD31 and PD13, their drug release behavior in 7.4 buffer solutions can be described using Ritger–Peppas (R2 = 0.987) and Higuchi (R2 = 0.991), respectively, which may be determined by the interaction between the matrix and the drug and its internal structure and related factors. The Ritger–Peppas model is a semi-experimental exponential function equation considering the diffusion of drug and the erosion of the polymer matrix,53 wherein n is the diffusional exponent and indicative of the release mechanism. When n < 0.45, Fickian diffusional release takes the dominant role; when 0.45 ≤ n < 0.89, non-Fickian diffusional release prevails and the drug releases by the dissolution, diffusion and erosion of the matrix; when n ≥ 0.89, the drug mainly releases through the erosion of the polymer matrix.54,55 At pH 7.4 buffer solution, the n of PD31 is 0.39, indicating that the diffusion release mechanism is Fickian. For PD21, PD11, PD12 and PD13, the value of n ranges from 0.45 to 0.56, suggesting that the combined effect of diffusion and erosion is largely responsible for the drug release. The difference of drug release mechanism between PD hydrogels was probably influenced by the crosslinking degree56–58 and hydrogen bonds between precursors and vancomycin.
Table 1 The fitted parameters of different models for vancomycin release at pH 7.4
| Sample |
Model type |
Equations |
k |
b |
n |
R2 |
| PD31 |
Zero-order |
Mt = kt + b |
0.007 |
7.304 |
— |
0.805 |
| First-order |
Mt = k(1 − exp(−b × t)) |
31.228 |
0.0009 |
— |
0.870 |
| Higuchi |
Mt = kt1/2 + b |
0.471 |
2.442 |
— |
0.986 |
| Ritger–Peppas |
Mt/M∞ = ktn |
1.207 |
— |
0.39 |
0.987 |
| PD21 |
Zero-order |
Mt = kt + b |
0.012 |
6.953 |
— |
0.991 |
| First-order |
Mt = k (1 − exp(−b × t)) |
64.536 |
0.0008 |
— |
0.985 |
| Higuchi |
Mt = kt1/2 + b |
0.950 |
−1.015 |
— |
0.998 |
| Ritger–Peppas |
Mt/M∞ = ktn |
0.634 |
— |
0.55 |
0.999 |
| PD11 |
Zero-order |
Mt = kt + b |
0.012 |
7.654 |
— |
0.910 |
| First-order |
Mt = k (1 − exp(−b × t)) |
73.684 |
0.0006 |
— |
0.848 |
| Higuchi |
Mt = kt1/2 + b |
0.823 |
0.084 |
— |
0.929 |
| Ritger–Peppas |
Mt/M∞ = ktn |
0.552 |
— |
0.56 |
0.951 |
| PD12 |
Zero-order |
Mt = kt + b |
0.019 |
7.809 |
— |
0.838 |
| First-order |
Mt = k (1 − exp(−b × t)) |
66.624 |
0.0009 |
— |
0.557 |
| Higuchi |
Mt = kt1/2 + b |
0.812 |
2.713 |
— |
0.943 |
| Ritger–Peppas |
Mt/M∞ = ktn |
1.330 |
— |
0.45 |
0.884 |
| PD13 |
Zero-order |
Mt = kt + b |
0.017 |
6.221 |
— |
0.933 |
| First-order |
Mt = k (1 − exp(−b × t)) |
87.295 |
0.0012 |
— |
0.891 |
| Higuchi |
Mt = kt1/2 + b |
1.248 |
1.161 |
— |
0.991 |
| Ritger–Peppas |
Mt/M∞ = ktn |
1.584 |
— |
0.47 |
0.989 |
3.9 Isothermal titration calorimetric measurement
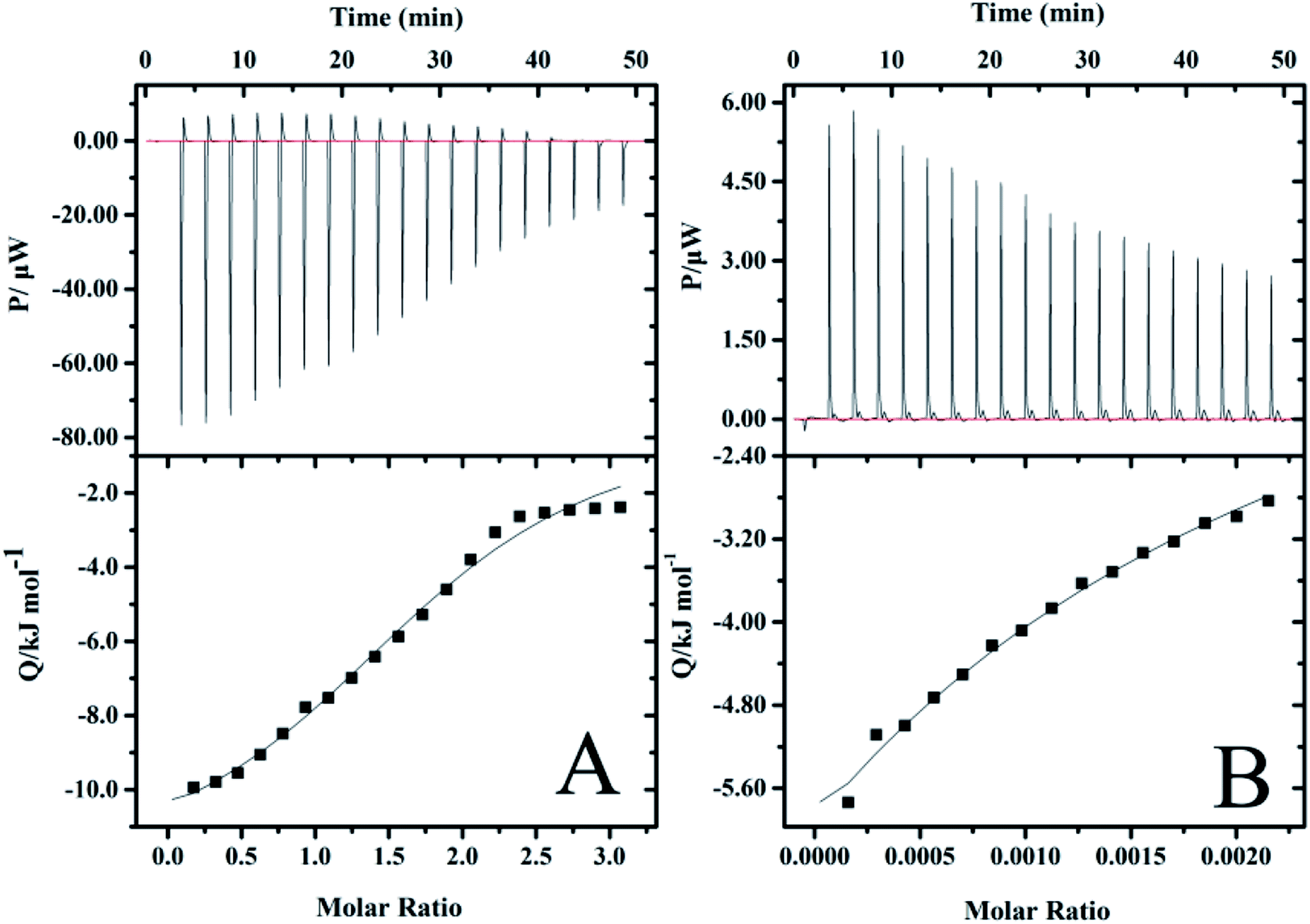
In order to further understand the nature of the interaction between vancomycin and precursors, and to explore the decisive factors affecting the release of vancomycin, ITC experiments were carried out, which allow for the simultaneous determination of the binding enthalpy, the binding constant and the reaction stoichiometry in a single experiment.59 Calorimetric titration profiles for the binding of 2 mM vancomycin to 30 mM PGAADH and 5 mM vancomycin to 370 mM OD at 298.2 K and pH 7.4 are shown in Fig. 9A and B, respectively. The upper panels represent the heat flow of each titration (μW) between the drug molecules and precursors over time (min). The lower panels show the integrated heat after correction for dilution effects as a function of ligand molar ratio. The results indicate that the PGAADH + vancomycin system exhibits the exothermic nature of the binding, while the OD + vancomycin presents the endothermic binding behavior. The binding isotherms were fit to the one-set of site model to determine the various thermodynamic parameters, including binding stoichiometry (n), ΔH° and binding constant (Kc), that were used to calculate the values of Gibbs free energy change (ΔGo) and entropy change (ΔSo),60,61 which are useful for the evaluation of driving forces of different systems. The agreement between the fitted curve and the experimental data in Fig. 9 confirms the rationality of the selected model.
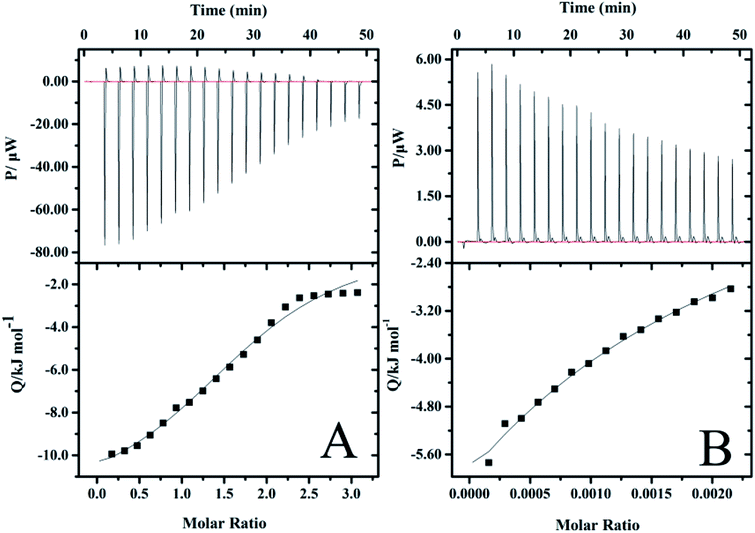 |
| | Fig. 9 ITC profiles for the binding of (A) vancomycin to PGAADH and (B) vancomycin to OD at 298.2 K in PBS buffer solution (0.01 mM, pH 7.4). | |
A negative value of Gibbs free energy change indicates that the binding of PGAADH or OD to vancomycin (−17.9 and −14.6 kJ mol−1, respectively) is a spontaneous process. The negative ΔHo (−895 kJ mol−1) and ΔSo (−2950 J mol−1 K−1) values for the interaction between OD and vancomycin reveal that hydrogen bonds and van der Waals forces might play a major role (enthalpy driven process). While according to the ΔHo (−12.4 kJ mol−1) and ΔSo (18.4 J mol−1 K−1) values obtained from the PGAADH + vancomycin system, the main force between PGAADH and vancomycin may be the electrostatic force (enthalpy and entropy driven process).
By exploring the interaction between vancomycin and two precursors, it is found that the binding constant of PGAADH + vancomycin is 1350 M−1, which is almost 3.7 times the binding constant of OD + vancomycin (364 M−1). Among all PD hydrogels, PD31 has the highest PGAADH content and is inferred to have the strongest binding strength with the drug, so its release period is the longest and the sustained release effect is the most obvious, which is highly consistent with the results of the drug release experiment. In addition, the existing literature also shows that vancomycin can tightly bind to the short peptide at the end of peptidoglycan, thereby hindering the formation of bacterial cell membranes to achieve antibacterial effects.62 This also further confirms the possibility of close binding of vancomycin and PGA (a peptide analog). Therefore, based on these studies and ITC data, it can be concluded that factors affecting the release of drugs in PD hydrogels may include structural factors and the interaction between the drug and the precursors, but the most important is the latter.
3.10 Antibacterial activity assay
The change of micromorphology can intuitively show the bacterial survival state. In this study, the morphology of bacteria was observed using SEM before and after co-culture with different samples and are presented in Fig. 10. For the control group, the bacteria surface was very smooth, indicating that both kinds of bacteria grew well. Similar to the control group, the cell morphology of both bacteria were still smooth after treatment with blank hydrogels, which could be attributed to the good biocompatibility of the blank hydrogels. This advantage could be conducive for expanding the types of delivery drugs, especially some unstable drugs which tend to react with large numbers of amino groups on traditional antimicrobial materials (such as chitosan). The c, c1, d and d1 in Fig. 10 are the SEM images of E. coli and S. aureus co-cultured with drug solution and drug-loaded hydrogels, respectively. It can be seen that the bacteria after treating with drug solution and vancomycin-loaded hydrogels were all shriveled or ruptured, and intracellular substances even spilled from the bacteria when dead. This indicates that the PD hydrogel has a good encapsulation and protective effect on the drug, while the released vancomycin still maintains good drug activity compared with the reference drug solution.
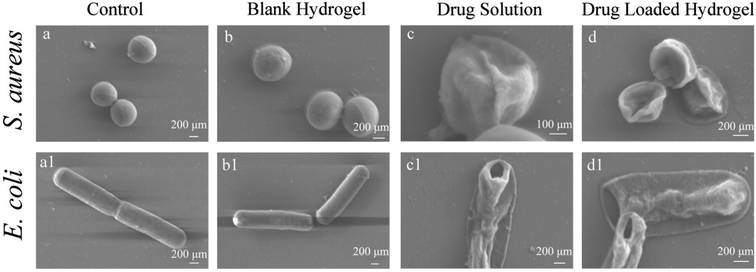 |
| | Fig. 10 SEM images of E. coli and S. aureus after being co-cultured with control (a and a1); blank hydrogels (b and b1); drug solution (c and c1); drug-loaded hydrogels (d and d1). All the samples were sputter-coated with gold and viewed under a 2 kV accelerating voltage. | |
3.11 Cytotoxicity assay in vitro
The in vitro cytotoxicity of the PD hydrogels (PD31, PD21, PD11, PD12 and PD13) and control sample was investigated using HeLa cells incubated with their extract solutions for 1, 3 and 5 days. The quantitative assessments of their cytotoxicity by MTT assay are shown in Fig. 11. The results revealed that there were no significant differences in cytotoxicity among all the PD hydrogel groups. It may be attributed to the biocompatible nature of PGA and dextran, which is consistent with the previous studies.10,63 Furthermore, the mild crosslinking that takes place without any additives and byproducts is also favorable for enhancing the biocompatibility of the hydrogels. The value of cell viability did not show obvious regularity within 5 days, but some samples had values exceeding 100%. The reason may be that the extract solution had dissolved some uncrosslinked polymer chains during the co-culture with the hydrogel. Alternatively, the hydrogels were partially degraded into peptide or oligosaccharide analogs during the co-cultivation process, and these substances had a beneficial effect on cell growth. The calculated value of RGR confirmed the biocompatible nature of PD hydrogels, which meets the requirements of potential materials for drug delivery systems and extracellular matrices.
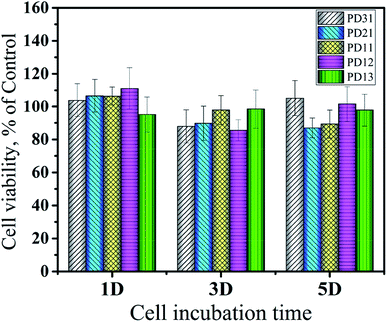 |
| | Fig. 11 Cytotoxicity of five hydrogels (PD31, PD21, PD11, PD12 and PD13) evaluated via MTT assay according to the ISO 10993-5 standard at 37 °C for 5 days. | |
3.12 Cells 3D encapsulation
To further evaluate the PD hydrogels' ability to serve as extracellular matrix mimics effectively, the encapsulation and culture of HeLa cells within PD11 hydrogels were performed. Fig. 12 shows the inverted fluorescence microscopy images of HeLa cells in PD11 hydrogels stained with AO/PI (Beijing Solarbio Science & Technology Co., Ltd.) after culturing for 1, 3, 5 and 7 days. From the images, a significant increase in cell population density could be observed during the 7 days culture, indicating that HeLa cells were able to proliferate in the 3D microenvironment of the hydrogels. The high viability and proliferation of the HeLa cells within PD11 hydrogels may be attributed to the excellent biocompatibility and the reversible hydrazone bond crosslinked networks. Some research groups had also pointed out that the hydrogels with reversible linkages can offer a more biomimetic microenvironment and present many advantages in 3D cell cultures compared with traditional permanent chemically crosslinked hydrogels.64 Through the breaking and reformation of the reversible linkages, the hydrogels could not only promote the exchange of nutrients and metabolites between the cells and external environment but also enable cells to perform some cellular functions (proliferation and migration, etc.).65,66 The above results indicated that the PD11 hydrogels had great potential as a promising 3D cell culture matrix for cell therapy and tissue regeneration.
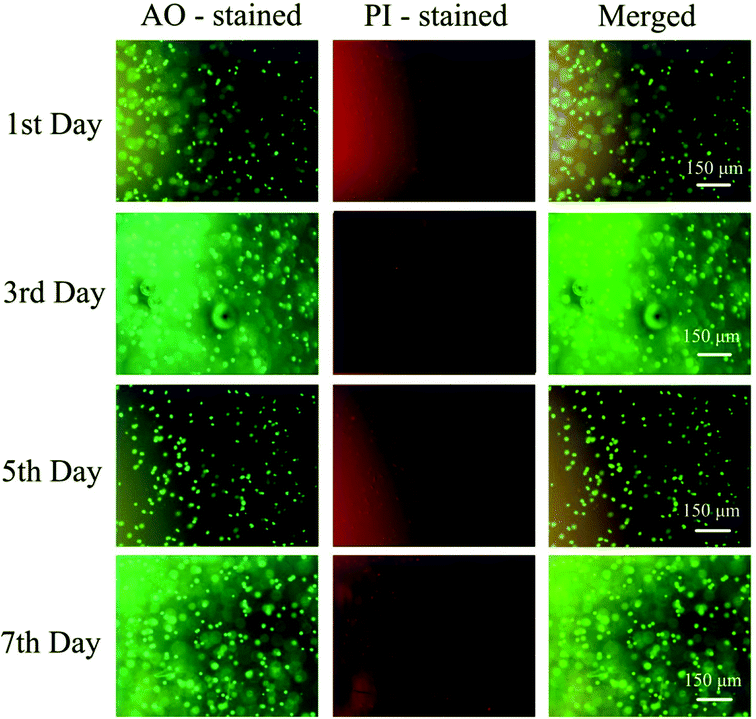 |
| | Fig. 12 Fluorescence images of HeLa cells cultured in PD11 hydrogels for 7 days at 37 °C. The cells were double stained with AO/PI at a concentration of 2 μM. | |
4 Conclusion
In this study, the naturally derived biopolymer poly(γ-glutamic acid) and dextran were successfully modified as two precursors. Utilizing hydrazone chemistry as the crosslinking strategy, a series of biocompatible PD hydrogels were prepared under physiological conditions without any additive or byproducts, which resulted in a gelation time ranging from 15 s to 1 min at room temperature by altering the composition. The PD hydrogels had adjustable mechanical properties, water content and controllable degradation. The drug release experiment of vancomycin-loaded hydrogels indicates that the drug release rate could be effectively regulated by adjusting the precursor content to provide a sustained release or controlled release effect. Herein, isothermal titration calorimetric measurements were also performed to study the thermodynamic interaction between drug and precursors, which provides positive evidence to reveal the drug release mechanism. Also, the hydrogels showed excellent biocompatibility, which was proved by an MTT test and 3D cell encapsulation experiment. All these characteristics provide enough evidence that PD hydrogels have the potential to be used in numerous biomedical applications, e.g. skin fillings, drug delivery vehicles, and tissue regeneration matrices.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the National Science and Technology Major Project of China (Grant No. 2017ZX09201003-013), Natural Science Foundation of Shandong Province, China (Grant No. ZR2016EL04), Open Foundation of the State Key Laboratory of Bioactive Seaweed Substances (SKL-BASS1712), the Open Project of Shandong Collaborative Innovation Center for Antibody Drugs (Grant No. CIC-AD1833), Doctoral Starting up Foundation of Liaocheng University (Grant No. 318051609), and Tai-Shan Scholar Research Fund of Shandong Province of China (Grant No. 319190201). This work was also technically supported by the Engineering Research Center for Nanomedicine and Drug Delivery Systems.
Notes and references
- N. A. Peppas, R. M. Ottenbrite, K. Park and T. Okano, Biomedical applications of hydrogels handbook, Springer, 2010 Search PubMed.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS PubMed.
- Z. Fan, Y. Zhang, J. Ji and X. Li, RSC Adv., 2015, 5, 16740–16747 RSC.
- Z. Fan, P. Cheng, M. Liu, D. Li, G. Liu, Y. Zhao, Z. Ding, F. Chen, B. Wang, X. Tan, Z. Wang and J. Han, New J. Chem., 2017, 41, 8656–8662 RSC.
- Z. Fan, Y. Zhang, S. Fang, C. Xu and X. Li, RSC Adv., 2015, 5, 1929–1936 RSC.
- D. Bairagi, P. Biswas, K. Basu, S. Hazra, D. Hermida-Merino, D. K. Sinha, I. W. Hamley and A. Banerjee, ACS Appl. Bio Mater., 2019, 2, 5235–5244 CrossRef CAS.
- K. Basu, A. Baral, S. Basak, A. Dehsorkhi, J. Nanda, D. Bhunia, S. Ghosh, V. Castelletto, I. W. Hamley and A. Banerjee, Chem. Commun., 2016, 52, 5045–5048 RSC.
- A. Baral, S. Roy, A. Dehsorkhi, I. W. Hamley, S. Mohapatra, S. Ghosh and A. Banerjee, Langmuir, 2014, 30, 929–936 CrossRef CAS PubMed.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2012, 64, 223–236 CrossRef.
- I. BrucknerOtaniBajaj and R. Singhal, Bioresour. Technol., 2011, 102, 5551–5561 CrossRef PubMed.
- S. Nagahara and T. Matsuda, Polym. Gels Networks, 1996, 4, 111–127 CrossRef CAS.
- E. Ruel-Gariépy and J.-C. Leroux, Eur. J. Pharm. Biopharm., 2004, 58, 409–426 CrossRef PubMed.
- H. Park, S. W. Kang, B. S. Kim, D. J. Mooney and K. Y. Lee, Macromol. Biosci., 2009, 9, 895–901 CrossRef CAS PubMed.
- T. J. Sanborn, P. B. Messersmith and A. E. Barron, Biomaterials, 2002, 23, 2703–2710 CrossRef CAS.
- J. L. Ifkovits and J. A. Burdick, Tissue Eng., 2007, 13, 2369–2385 CrossRef CAS PubMed.
- A. K. Burkoth and K. S. Anseth, Biomaterials, 2000, 21, 2395–2404 CrossRef CAS PubMed.
- C. M. Nimmo, S. C. Owen and M. S. Shoichet, Biomacromolecules, 2011, 12, 824–830 CrossRef CAS PubMed.
- A. Gress, A. Völkel and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- N. K. Singha and H. Schlaad, in Functional Polymers by Post-Polymerization Modification, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 65–86, DOI:10.1002/9783527655427.ch3.
- Y. I. Shen, H. E. Abaci, Y. Krupsi, L. C. Weng, J. A. Burdick and S. Gerecht, Biomater. Sci., 2014, 2, 655–665 RSC.
- Y. Li, C. Liu, Y. Tan, K. Xu, C. Lu and P. Wang, Carbohydr. Polym., 2014, 110, 87–94 CrossRef CAS PubMed.
- J. Karvinen, J. T. Koivisto, I. Jönkkäri and M. Kellomäki, J. Mech. Behav. Biomed. Mater., 2017, 71, 383–391 CrossRef CAS PubMed.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- W.-C. Lin, D.-G. Yu and M.-C. Yang, Colloids Surf., B, 2006, 47, 43–49 CrossRef CAS PubMed.
- K. Hoste, E. Schacht and L. Seymour, J. Controlled Release, 2000, 64, 53–61 CrossRef CAS PubMed.
- Z. Fan, Y. Zhang, W. Zhang and X. Li, J. Appl. Polym. Sci., 2015, 132, 42301–42307 Search PubMed.
- S. K. Hahn, J. K. Park, T. Tomimatsu and T. Shimoboji, Int. J. Biol. Macromol., 2007, 40, 374–380 CrossRef CAS PubMed.
- E. J. Oh, S. W. Kang, B. S. Kim, G. Jiang, I. H. Cho and S. K. Hahn, J. Biomed. Mater. Res., Part A, 2008, 86, 685–693 CrossRef PubMed.
- H. Zhang, A. Qadeer and W. Chen, Biomacromolecules, 2011, 12, 1428–1437 CrossRef CAS PubMed.
- H. Zhao and N. D. Heindel, Pharm. Res., 1991, 8, 400–402 CrossRef CAS PubMed.
- R. Jin, L. S. Moreira Teixeira, P. J. Dijkstra, C. A. van Blitterswijk, M. Karperien and J. Feijen, J. Controlled Release, 2011, 152, 186–195 CrossRef CAS PubMed.
- J. Huang, Y. Deng, J. Ren, G. Chen, G. Wang, F. Wang and X. Wu, Carbohydr. Polym., 2018, 186, 54–63 CrossRef CAS PubMed.
- Y. Zhao, X. Zhang, Y. Wang, Z. Wu, J. An, Z. Lu, L. Mei and C. Li, Carbohydr. Polym., 2014, 105, 63–69 CrossRef CAS PubMed.
- W. Zhang, X. Jin, H. Li, R. R. Zhang and C. W. Wu, Carbohydr. Polym., 2018, 186, 82–90 CrossRef CAS PubMed.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- F. Ganji, S. Vasheghani-Farahani and E. Vasheghani-Farahani, Iran. Polym. J., 2010, 19, 375–398 CAS.
- A. R. Kim, S. L. Lee and S. N. Park, Int. J. Biol. Macromol., 2018, 118, 731–740 CrossRef CAS PubMed.
- A. Basu and G. S. Kumar, Food Funct., 2014, 5, 1949–1955 RSC.
- Z. Li, C. He, B. Yuan, X. Dong and X. Chen, Macromol. Biosci., 2017, 17, 1600347 CrossRef PubMed.
- R. Jin, L. S. Moreira Teixeira, P. J. Dijkstra, M. Karperien, C. A. van Blitterswijk, Z. Y. Zhong and J. Feijen, Biomaterials, 2009, 30, 2544–2551 CrossRef CAS PubMed.
- N. Annabi, D. Rana, E. Shirzaei Sani, R. Portillo-Lara, J. L. Gifford, M. M. Fares, S. M. Mithieux and A. S. Weiss, Biomaterials, 2017, 139, 229–243 CrossRef CAS PubMed.
- N. Ahmed, J. Schober, L. Hill and S. P. Zustiak, Tissue Eng., Part C, 2016, 22, 543–551 CrossRef CAS PubMed.
- D. E. Discher, P. Janmey and Y. L. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS PubMed.
- T.-C. Tseng, L. Tao, F.-Y. Hsieh, Y. Wei, I.-M. Chiu and S.-h. Hsu, Adv. Mater., 2015, 27, 3518–3524 CrossRef CAS PubMed.
- H. Sjöberg, S. Persson and N. Caram-Lelham, J. Controlled Release, 1999, 59, 391–400 CrossRef.
- D. C. Coughlan and O. I. Corrigan, Int. J. Pharm., 2006, 313, 163–174 CrossRef CAS PubMed.
- T. Ukmar, U. Maver, O. Planinšek, V. Kaučič, M. Gaberšček and A. Godec, J. Controlled Release, 2011, 155, 409–417 CrossRef CAS PubMed.
- N. A. Peppas and A. R. Khare, Adv. Drug Delivery Rev., 1993, 11, 1–35 CrossRef CAS.
- C. S. Brazel and N. A. Peppas, Biomaterials, 1999, 20, 721–732 CrossRef CAS PubMed.
- J. S. Varghese, N. Chellappa and N. N. Fathima, Colloids Surf., B, 2014, 113, 346–351 CrossRef CAS PubMed.
- A. Nokhodchi, S. Raja, P. Patel and K. Asare-Addo, BioImpacts, 2012, 2, 175–187 CAS.
- Y. Chen, S. Y. H. Abdalkarim, H.-Y. Yu, Y. Li, J. Xu, J. Marek, J. Yao and K. C. Tam, Int. J. Biol. Macromol., 2020, 155, 330–339 CrossRef CAS PubMed.
- P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 23–36 CrossRef CAS.
- P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 37–42 CrossRef CAS.
- N. A. Peppas and J. J. Sahlin, Int. J. Pharm., 1989, 57, 169–172 CrossRef CAS.
- A. P. Rokhade, N. B. Shelke, S. A. Patil and T. M. Aminabhavi, Carbohydr. Polym., 2007, 69, 678–687 CrossRef CAS.
- A. P. Rokhade, S. A. Patil and T. M. Aminabhavi, Carbohydr. Polym., 2007, 67, 605–613 CrossRef CAS.
- S. A. Agnihotri and T. M. Aminabhavi, Drug Dev. Ind. Pharm., 2007, 33, 1254–1262 CrossRef CAS PubMed.
- Y. Shi, M. Liu, H. Yan, C. Cai, Q. Guo, W. Pei, R. Zhang, Z. Wang and J. Han, Spectrochim. Acta, Part A, 2019, 206, 384–395 CrossRef CAS PubMed.
- S. Tunç, O. Duman and B. K. Bozoğlan, J. Lumin., 2013, 140, 87–94 CrossRef.
- M. Liu, Y. Zheng, C. Wang, J. Xie, B. Wang, Z. Wang, J. Han, D. Sun and M. Niu, Food Chem., 2016, 196, 148–154 CrossRef CAS PubMed.
- C. Walsh, Science, 1999, 284, 442–443 CrossRef CAS PubMed.
- C. Liu, X. Liu, C. Liu, N. Wang, H. Chen, W. Yao, G. Sun, Q. Song and W. Qiao, Biomaterials, 2019, 205, 23 CrossRef CAS PubMed.
- H. Wang and S. C. Heilshorn, Adv. Mater., 2015, 27, 3717–3736 CrossRef CAS PubMed.
- D. D. McKinnon, D. W. Domaille, J. N. Cha and K. S. Anseth, Adv. Mater., 2014, 26, 865–872 CrossRef CAS PubMed.
- L. Cao, B. Cao, C. Lu, G. Wang, L. Yu and J. Ding, J. Mater. Chem. B, 2015, 3, 1268–1280 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02218g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Ping Chengb,
Min Liua,
Sangeeta Prakashc,
Jun Han*a,
Zhuang Dinga,
Yanna Zhaoa and
Zhengping Wanga
*a,
Ping Chengb,
Min Liua,
Sangeeta Prakashc,
Jun Han*a,
Zhuang Dinga,
Yanna Zhaoa and
Zhengping Wanga