
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile, practical and metal-free microwave-assisted protocol for mono- and bis-[1,2,4]triazolo[1,5-a]pyridines synthesis utilizing 1-amino-2-imino-pyridine derivatives as versatile precursors†
Hamada Mohamed Ibrahim *a,
Haider Behbehani*b and
Wael Abdelgayed Ahmed Arafaac
*a,
Haider Behbehani*b and
Wael Abdelgayed Ahmed Arafaac
aChemistry Department, Faculty of Science, Fayoum University, P.O. Box 63514, Fayoum, Egypt. E-mail: hmi00@fayoum.edu.eg; hamadaaldeb@yahoo.com
bChemistry Department, Faculty of Science, Kuwait University, P.O. Box 5969, Safat 13060, Kuwait. E-mail: drhaider.b@gmail.com
cChemistry Department, College of Science, Jouf University, P.O. Box 2014, Sakaka, Aljouf, Kingdom of Saudi Arabia
First published on 20th April 2020
Abstract
A facile and effective assembly of several substituted functionalized mono- and bis-[1,2,4]triazolo[1,5-a]pyridines from conveniently attainable 1-amino-2-imino-pyridines has been established. Using microwave irradiation speeds up the reaction efficiently, proceeding with a higher rate and yields than with conventional heating. In the presented protocol, a broad variety of carboxylic acids could be employed effectively to synthesize the respective derivatives via direct metal-free C–N bond construction. Interestingly, other substrates such as aldehydes (or their arylidene malononitriles), phenyl isothiocyanate, glyoxalic acid, and acrylonitriles could also provide the corresponding 1,2,4-triazolo[1,5-a]pyridines successfully. This versatile and convergent approach performs well with both deactivating and activating substrates in an environmentally benign manner compared with other already reported protocols. Other notable merits of the current strategy involve no need for column chromatography, no tedious work-up, and a direct pathway for the fast design of triazolopyridine frameworks. The identity of the newly synthesized compounds was established using several spectroscopic techniques, and X-ray single-crystal tools were employed to authenticate the suggested structures of some representative samples.
Introduction
N-Fused heteroaromatic frameworks are an essential structural moiety in several effective pharmacological compounds and natural products.1 Among them, 1,2,4-triazolo[1,5-a]pyridines which are considered as a unique category of N-bridged 5,6-bicyclic compounds have received substantial consideration for either their potential utility as bioactive precursors or for other industrial applications.2,3 For example, they exhibit several pharmaceutical behaviors including, mGlu modulation,4 PHD-1 inhibition,5 PDE10 inhibition,6 and acting as an antioxidant.7 In addition examples of these compounds have been utilized as herbicidal agents8 and for treatment of diabetes (type-II),9,10 cardiovascular disorders,11 and hyperproliferative disorders.12 Besides, such derivatives have been included in a variety of pharmaceutically effective compounds as dipeptidomimetics13 and have been employed as effective ligands for various transition metals.14–16 As a consequence of the above-mentioned applications, several approaches for the triazolopyridine assembly have been established over the past decades. The reported synthetic strategies for assembling triazolopyridines could be classified into three approaches depending on the reactants: triazoles, pyridines, and multiple components. The oxidative cyclization of N-(2-pyridyl)amidines is amongst the most simple protocols for developing the 1,2,4-triazolo[1,5-a]pyridines that have been accomplished via employing oxidants including Pb(OAc)4,17 NaClO/base18 and MnO2.19 Nonetheless, there are several drawbacks associated with these procedures such as restricted scopes, lower yields, lack of regioselectivity, and multi-step synthetic strategies. Moreover, Ueda and Nagasawa20 developed a method for achieving 2-aryl-1,2,4-triazolo[1,5-a]pyridines from a copper-catalyzed cyclization reaction of aryl nitriles and 2-aminopyridines. Similarly, Zhao et al.21 described a Cu–Zn/Al–Ti reusable catalyst for the same conversion. Further, Jianguang and co-workers successfully synthesized triaryl[1,2,4]triazolo[1,5-a]pyridine derivatives via copper-catalyzed radical cyclization reaction of benzylidenmalononitriles and azines.22 More recently, Xia et al. presented the preparation of triazolopyridines through the copper-catalyzed oxidative cyclization of amidines or 2-aminopyridines with several nitriles.23 Notwithstanding, these reactions are followed by certain drawbacks, such as using the catalysts in higher loads (5–20 mol%) and not being recyclable or reusable. Moreover, these metal catalysts could be interacted conveniently with the obtained products, since [1,2,4]triazolo[1,5-a]pyridines are reported to be coordinated effectively with transition metals producing stable complexes.24,25 Such challenges are of particular economic and environmental concerns, limiting the utility of such protocols in large-scale productions, and hence in industrial purposes. Furthermore, access to [1,2,4]triazolo[1,5-a]pyridines has also been documented for certain metal-free synthetic procedures. For instance, Alizadeh et al. reported the synthesis of [1,2,4]triazolo[1,5-a]pyridine derivatives via iodine-catalyzed one-pot four-component reaction comprising malononitrile, dimethyl acetylenedicarboxylate, aromatic aldehydes, and benzylidenehydrazines.26 Even though, this approach could be implemented only to alkynes with powerful electron-deficient moieties. Also, Zhao and co-workers27 recorded a PIFA-mediated conversion of amidines into 2-substituted triazolopyridine derivatives. On the other hand, Chang et al.28 published a method for the preparation of 1,2,4-triazolo[1,5-a]pyridine derivatives employing I2/KI as a catalyst. Ashish et al.29 also recorded the contraction of 1,2,4-triazolo[1,5-a]pyridines through oxidative cyclization of trichloroisocyanuric acid and benzamidine derivatives. In addition to more reported protocols,30 however, in one aspect or another, the above reported protocols acquire several disadvantages, including the utility of costly catalysts, a restricted substrates scope, lower regioselectivity, and the isolation of by-products. Consequently, the development of creative and generalized protocols to access these unique heterocyclic categories is still in demand. Following on with our reported works31–33 herein we describe an alternative, more effective, and environmentally friendly metal-free approach for assembling 1,2,4-triazolo[1,5-a]pyridines from 1-amino-2-imino-4-aryl-1,2-dihydropyridine-3-carbonitrile precursors via the formation of C–N bonds employing the microwave irradiation as a sustainable energy source.Results and discussion
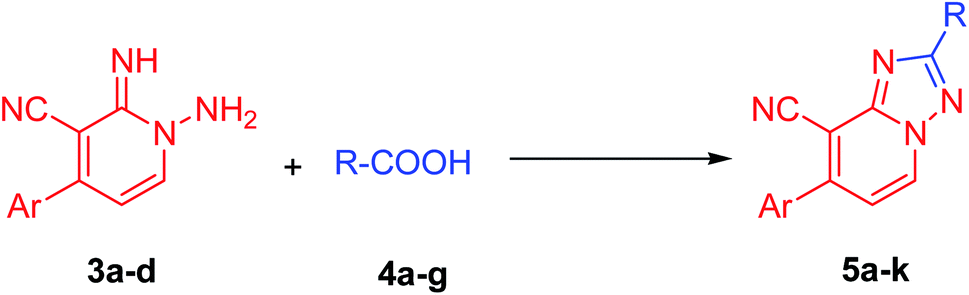
The strategy utilized to construct 1-amino-2-imino-pyridine derivatives 3a–e includes two sequentially steps that start with the synthesis of enaminonitriles 2a–e, by reacting dimethylformamide dimethylacetal (DMF–DMA) with the respective ethylidenemalononitriles 1a–e (Scheme 1). Enaminonitrile derivatives (2a–e) could then be converted to their corresponding targets 3a–e through thermally mediated reaction with hydrazine hydrate in EtOH (Scheme 1).33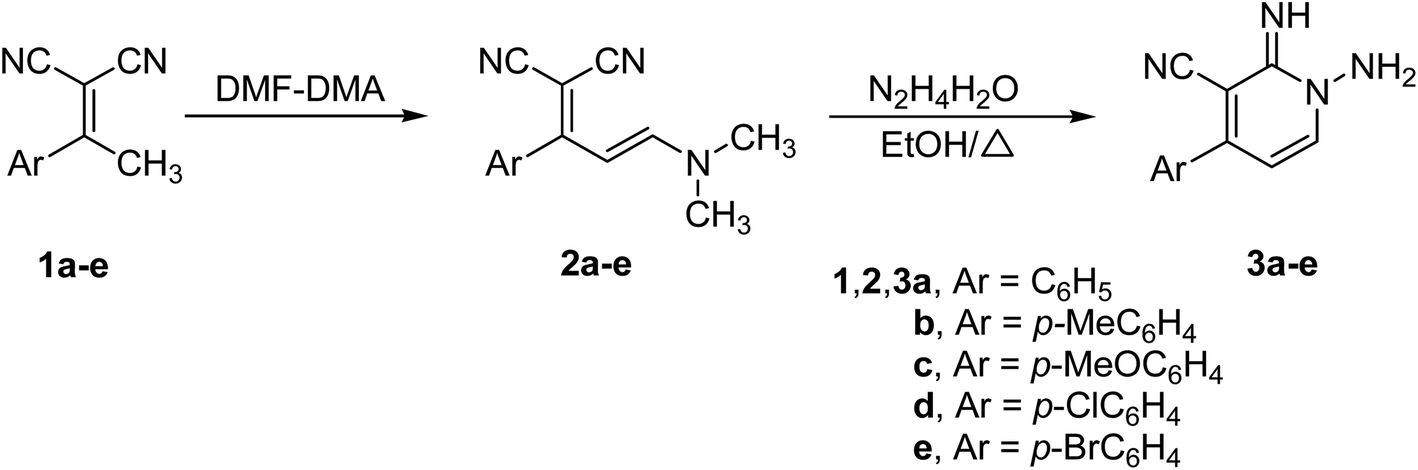 | ||
| Scheme 1 Synthesis of 1-amino-2-imino-pyridines 3a–e.33 | ||
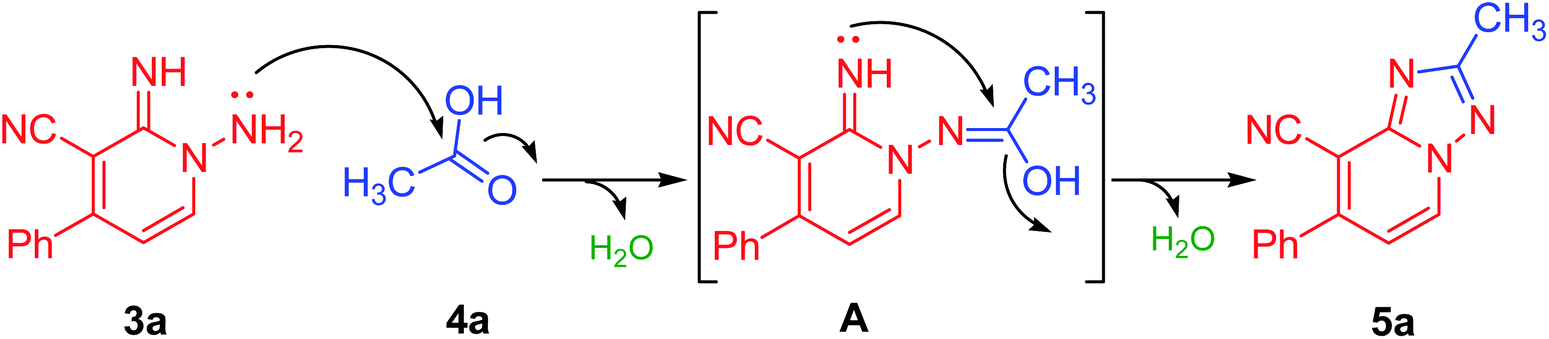
Our initial investigation commenced by the reaction of 1-amino-2-imino-pyridine derivative (3a) with acetic acid (as a solvent and reactant) at reflux for 3 h. Interestingly, the formation of the required product (5a) has been detected, albeit in a good yield of 74%, as a preliminary endeavor (Table 1, entry 1). Encouraged by the obtained results, the reaction conditions such as solvents, energy sources, and temperature have been evaluated to improve both reaction rate and yield (Table 1). Thereby, the reaction of 3a with acetic acid (1 equiv.) was investigated utilizing various solvents such as polar protic (EtOH, MeOH, and propanol), polar aprotic (CH3CN, and dioxane), and nonpolar (toluene) solvents at different times using various energy sources (heating, US, and MW irradiations). Unfortunately, none of them could afford the required product (Table 1, entries 2–13). Then the model reaction (Table 1) was also examined in 10.0 mL of ethanol comprising 5 equiv. of acetic acid to investigate its efficiency in the production of derivative 5a. Interestingly, compound 5a was crystalline out from the reaction mixture in 3 h of refluxing in 80% yield (Table 1, entry 14). Next, the molar ratio of acetic acid employed has also assessed. It was found that employing a mixture of ethanol/acetic acid (10 equiv.), not only improved the reaction yield to 85% but also provided a cleaner and lighter color product (Table 1, entry 15). Increasing the equivalents of acetic acid more than 10 equiv., neither improves the reaction rate nor the yield (Table 1, entry 16). Due to the versatility, efficiency, and selectivity of microwave irradiation, the model reaction (Scheme 1) was also investigated under the microwave irradiation conditions. The required product 5a was obtained in 89% in 25 min under microwave irradiation at 80 °C (Table 1, entry 17). On reducing the reaction duration to 15 min and increasing the temperature to 100 °C, the isolated yield was enhanced to be 92% (Table 1, entry 18). Increasing the temperature more than 100 °C did not improve the reaction (Table 1, entry 19). Thus, employing the model reaction (Table 1) using 3.0 mmol of derivative 3a in ethanol (10.0 mL) containing acetic acid (10 equiv.) and irradiation under microwave for 15 min at 100 °C has been identified as the optimal conditions (Table 1, entry 18). A reasonable reaction mechanism for the synthesis of 1,2,4-triazole derivative (5a) was described in Table 1, based on experimental evidences and our reported studies.31–33 As outlined in Table 1, the transformation of the non-isolable intermediate (A) to the target compound (5a) occurred under metal-free conditions.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Method | Time | Yield (%) |
| a Reaction conditions: a mixture of 1-amino-2(1H)-pyridine-2-imine derivatives (3a) (3.0 mmol) and acetic acid 4a (as reported) in solvent (10.0 mL) was heated or irradiated by microwave or ultrasound for the given time.b NR: no reaction. | ||||
| 1 | AcOH | Heating | 3 h | 74 |
| 2 | EtOH | Heating | 12 h | NRb |
| 3 | EtOH | MW | 45 min | NR |
| 4 | MeOH | Heating | 12 h | NR |
| 5 | MeOH | MW | 45 min | NR |
| 6 | CH3CN | Heating | 12 h | NR |
| 7 | CH3CN | MW | 45 min | NR |
| 8 | Propanol | Heating | 12 h | NR |
| 9 | Propanol | MW | 45 min | NR |
| 10 | 1,4-Dioxane | Heating | 12 h | NR |
| 11 | 1,4-Dioxane | MW | 45 min | NR |
| 12 | Toluene | Heating | 12 h | NR |
| 13 | Toluene | US | 45 min (80 °C) | NR |
| 14 | EtOH/AcOH (5 equiv.) | Heating | 3 h | 80 |
| 15 | EtOH/AcOH (10 equiv.) | Heating | 3 h | 85 |
| 16 | EtOH/AcOH (15 equiv.) | Heating | 3 h | 85 |
| 17 | EtOH/AcOH (10 equiv.) | MW | 25 min (80 °C, 250 W) | 89 |
| 18 | EtOH/AcOH (10 equiv.) | MW | 15 min (100 °C, 250 W) | 92 |
| 19 | EtOH/AcOH (10 equiv.) | MW | 15 min (120 °C, 250 W) | 92 |
Now, the limitations and scope of the aforesaid reaction have then investigated. Therefore the reaction of 1-amino-2(1H)-pyridine-2-imines (3a–d) with various carboxylic acid derivatives (4a–g, 10 equiv.) in EtOH (10.0 mL) was scrutinized under microwave irradiation. It was observed that these reactions did not proceed smoothly without using additives, as in the case of acetic acid. After several optimization trials, the optimal reaction condition for acids (4b–g) other than acetic acid was established to be 3.0 mmol of 1-amino-pyridine-2-imines 3a–d with 4.0 mmol of carboxylic acid derivatives (4b–g) in EtOH (10.0 mL) containing acetic acid (5 equiv.) as catalyst, under microwave irradiation (Table 2). It is worth mentioning that, the amount of acetic acid should not exceed 5 equiv., otherwise the reaction between 1-amino-pyridine-2-imines (3a–d) and acetic acid will have occurred. As displayed in Table 2, the summarized results demonstrate that all the proposed reactions yielded their corresponding products (5a–k) in outstanding isolated yields without detecting by-products. Also, all the reactions were effectively afforded the desired products regardless of the substitution pattern of the aromatic moiety (Ar, Table 2). Further, the influence of the R-groups on reaction efficiency was also been investigated (Table 2). In this regard, electron-donating and electron-deficient groups are both acceptable in the present process. For instance, the substrates comprising cyano groups (Table 2, entries 4–6) were easily converted to the corresponding products in excellent yields. Besides, the current protocol has shown a good tolerance for both aromatic and aliphatic carboxylic acids (Table 2).
|
||||
|---|---|---|---|---|
| Entry | Ar | R | Products | Yielda (%) |
| a Isolated yield.b Reaction conditions: a mixture of 1-amino-2(1H)-pyridine-2-imine derivatives (3a–d) (3.0 mmol) and acetic acid 4a (10 equiv.) in ethanol (10.0 mL) was charged in the glass tube of the microwave tube and irradiated at 100 °C for 15 min.c Reaction conditions: a mixture of 1-amino-2(1H)-pyridine-2-imine derivatives (3a–d) (3.0 mmol) and different carboxylic acids (4b–g) (4.0 mmol) in ethanol (10.0 mL), acetic acid (5 equiv.), was charged in the glass tube of the microwave tube and irradiated at 80 °C for 15 min. | ||||
| 1 | C6H5 | CH3 | 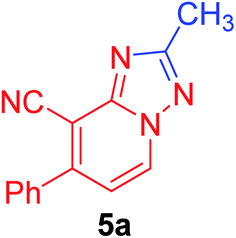 |
92b |
| 2 | p-ClC6H4 | CH3 | 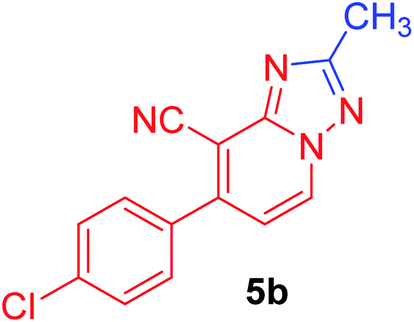 |
87b |
| 3 | p-ClC6H4 | Cl–CH2 | 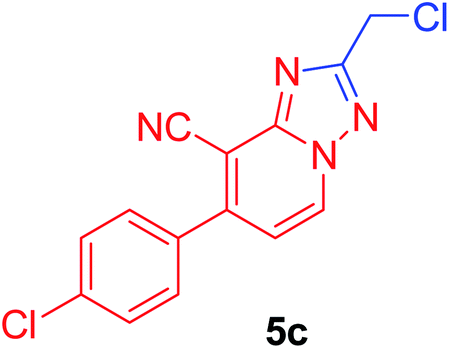 |
91c |
| 4 | C6H5 | NC–CH2 | 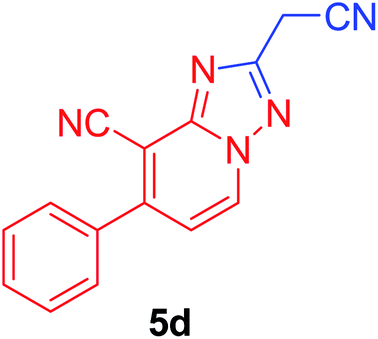 |
96c |
| 5 | p-ClC6H4 | NC–CH2 | 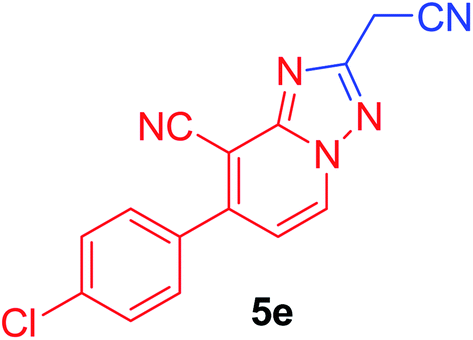 |
97c |
| 6 | p-MeOC6H4 | NC–CH2 | 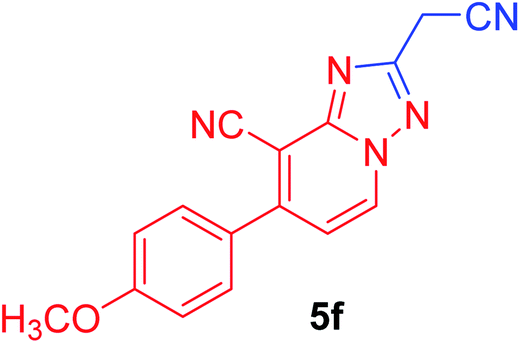 |
94c |
| 7 | p-MeOC6H4 | 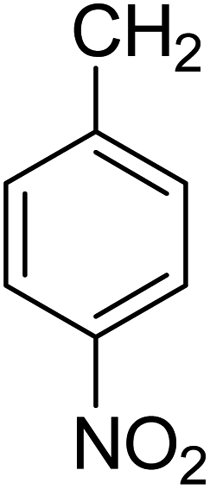 |
 |
93c |
| 8 | p-MeOC6H4 | C6H5 | 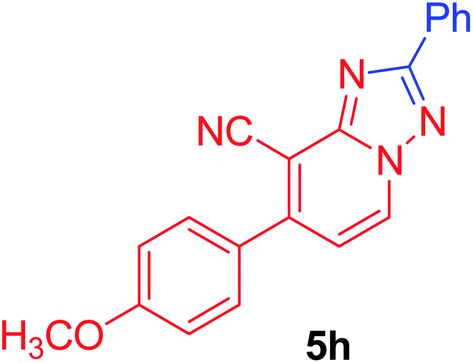 |
83c |
| 9 | p-ClC6H4 | C6H5 | 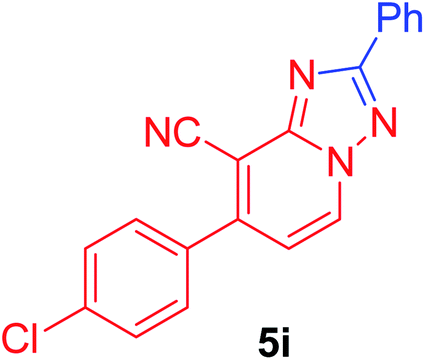 |
86c |
| 10 | p-ClC6H4 | 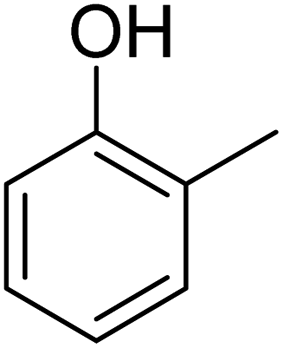 |
 |
87c |
| 11 | p-ClC6H4 | 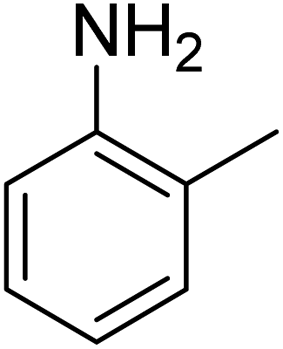 |
 |
84c |
Moreover, the proposed approach could also be successfully applied for carboxylic acid esters. For example when the diethyl oxalate (3.0 mmol) allowed to react with N-amino-2-iminopyridines (3a–e, 3.0 mmol) using 5 equiv. of acetic acid in EtOH (10.0 mL) under microwave irradiation at 100 °C for 15 min, the desired products (5l–p) were received in excellent yields (85–93%, Scheme 2). In these cyclization reactions, both electron-deficient and electron-rich Ar groups are also applicable. The cyclization reaction of electron-rich bearing substrates and diethyl oxalate, proceeded smoothly to produce the corresponding products (5m and 5n) in good yields (87 and 85% yield, respectively, Scheme 2). Similarly, in comparison to the unsubstituted aromatic derivative (5l), electron-deficient derivatives provided the respective products (5o and 5p) in excellent yields (92 and 93% yield, respectively, Scheme 2, Fig. 1 and 2).
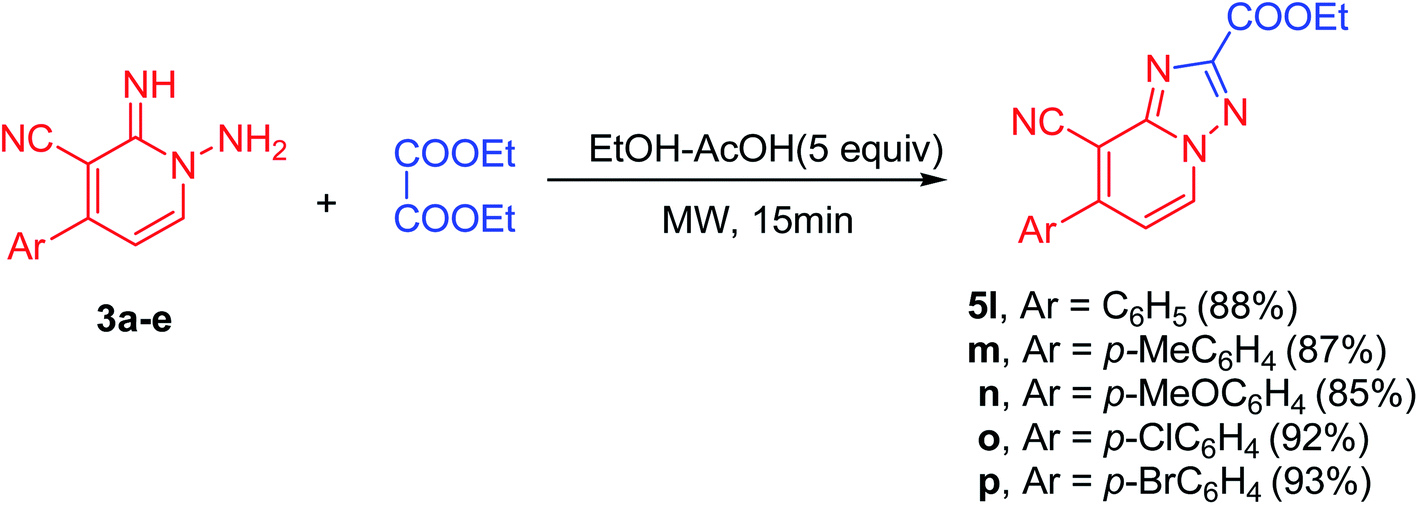 | ||
| Scheme 2 Substrate scope for the reaction of 1-amino-2-imino-pyridine derivatives (3a–e) with diethyl oxalate. | ||
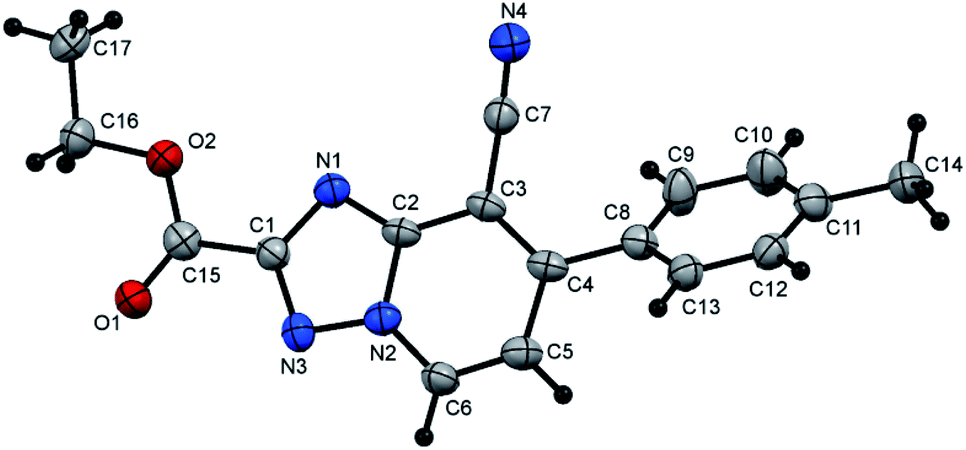 | ||
| Fig. 1 X-ray single crystal data determined for 5m. | ||
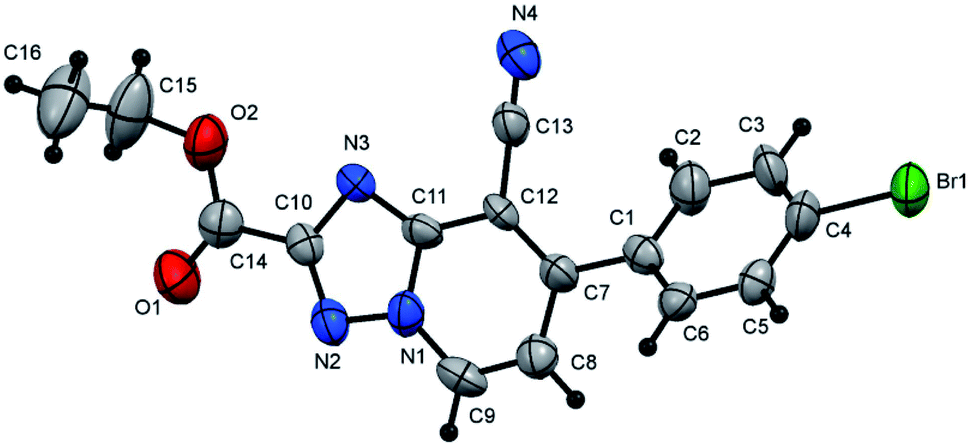 | ||
| Fig. 2 X-ray single crystal data determined for 5p. | ||
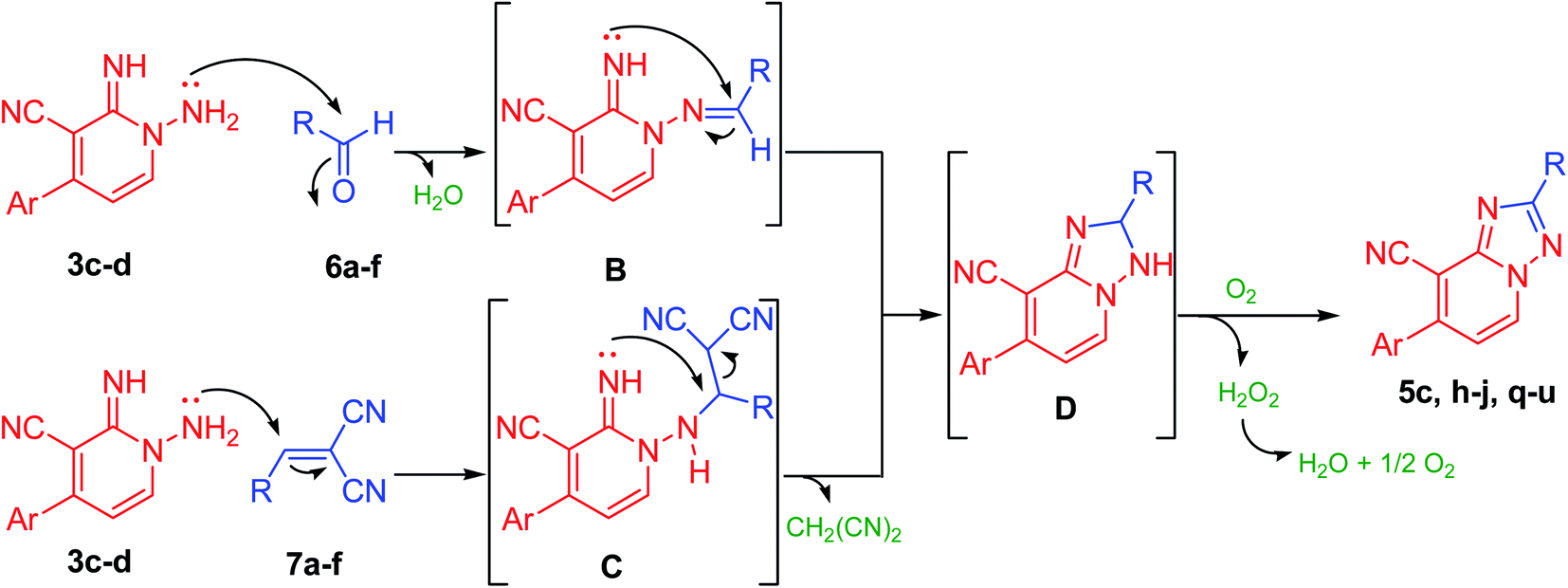
Notably, 1,2,4-triazolo[1,5-a]pyridine-8-carbonitrile derivatives (5) could also be obtained through the cyclization reaction of derivatives 3c–d with either the corresponding aldehydes (6a–f) or with their arylidene malononitriles (7a–f) (Table 3, Fig. 3 and 4). These reactions were effectively performed with several aromatic aldehydes and their arylidenes comprising electron-withdrawing or electron-donating groups and afforded the corresponding products in comparable yields (Table 3). Also, aliphatic aldehydes such as chloroacetaldehyde yielded the targeted product in slightly lower yield in parallel to aromatic aldehydes (Table 3, entry 1). In comparison to carboxylic acids, the aldehydes or their arylidene malononitriles underwent the cyclization reaction at a fast rate with much more yields. Moreover, derivative 5t could be also obtained via refluxing of (E)-1-methyl-4-(2-nitrovinyl)benzene (8) with derivative 3d in CH3CN/DMF mixture (Scheme 3).
|
||||
|---|---|---|---|---|
| Entry | Ar | R | Products | Yield (%) |
| 1 | p-ClC6H4 | Cl–CH2 | 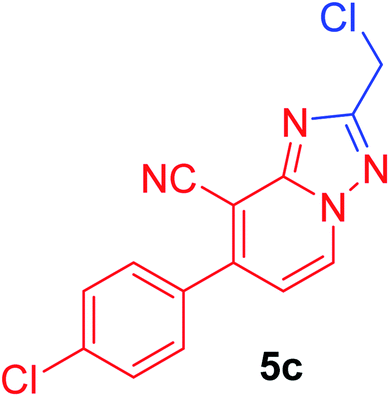 |
85 |
| 2 | p-MeOC6H4 | C6H5 | 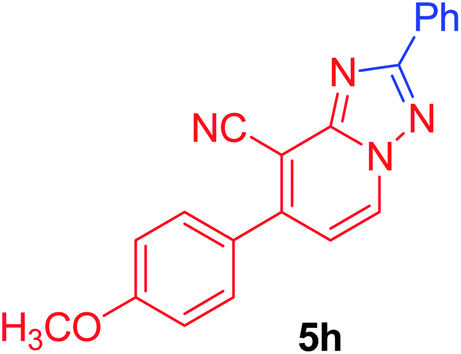 |
91 |
| 3 | p-ClC6H4 | C6H5 | 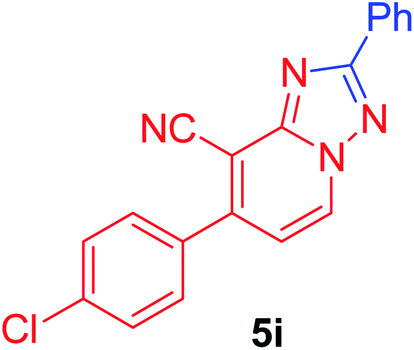 |
94 |
| 4 | p-ClC6H4 |  |
 |
93 |
| 5 | p-MeOC6H4 | p-ClC6H4 | 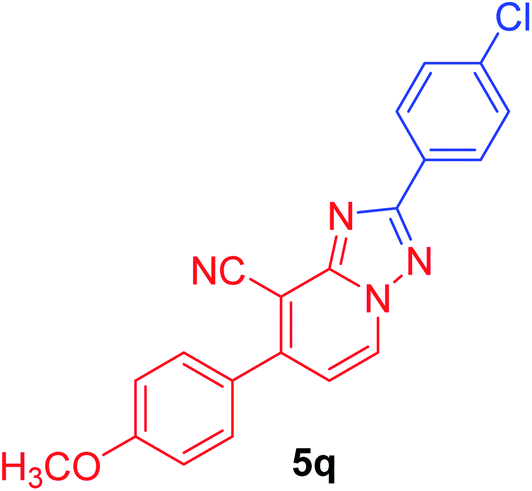 |
90 |
| 6 | p-MeOC6H4 | p-MeOC6H4 | 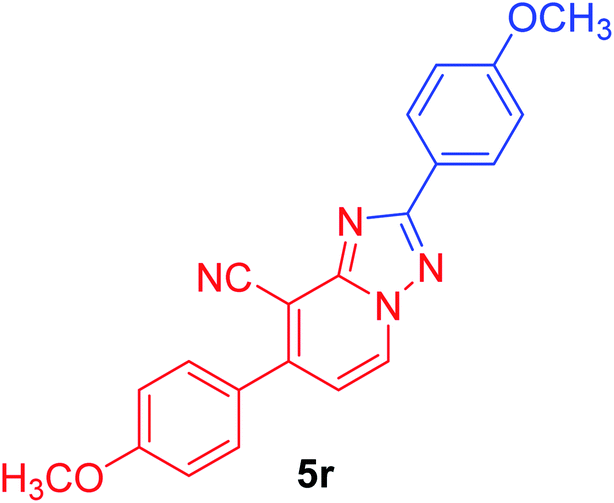 |
93 |
| 7 | p-ClC6H4 | p-ClC6H4 | 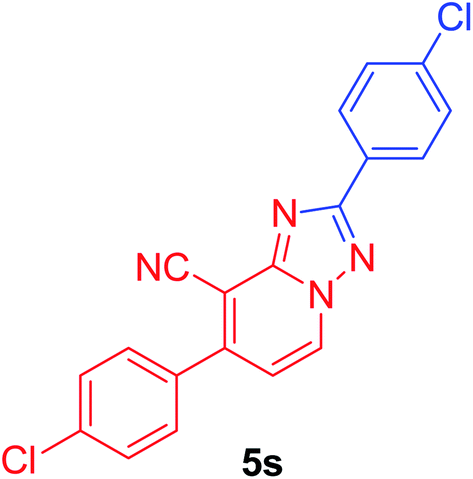 |
94 |
| 8 | p-ClC6H4 | p-MeC6H4 | 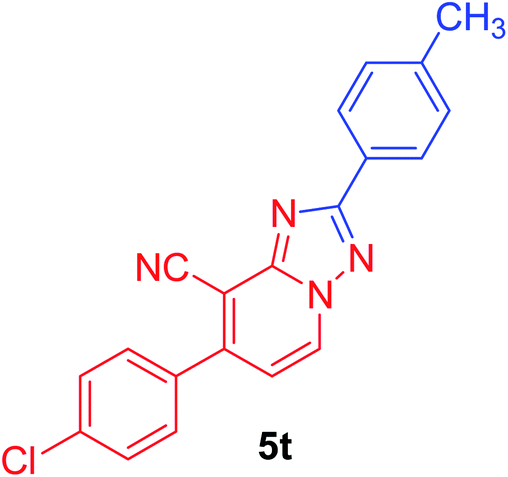 |
91 |
| 9 | p-ClC6H4 | Ph–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH CH |
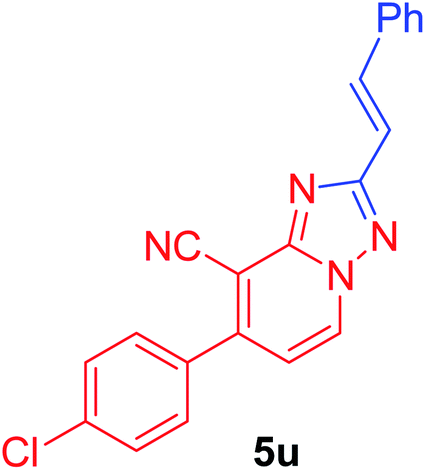 |
88 |
 | ||
| Fig. 3 X-ray single crystal data determined for 5q. | ||
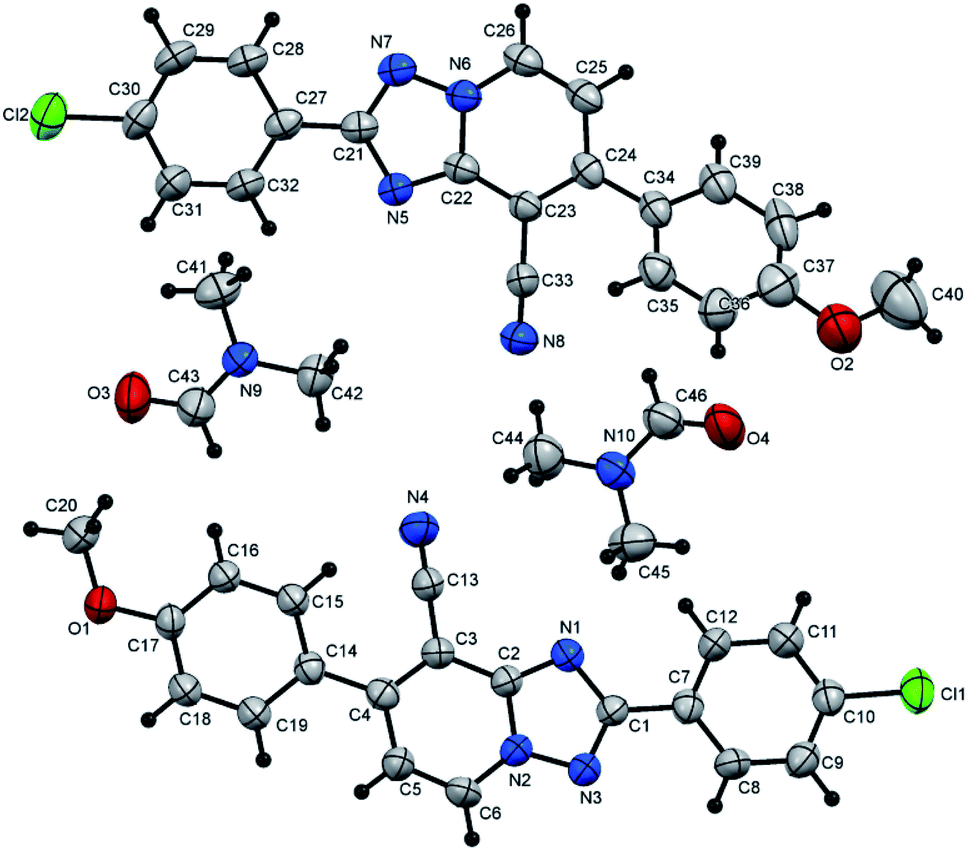
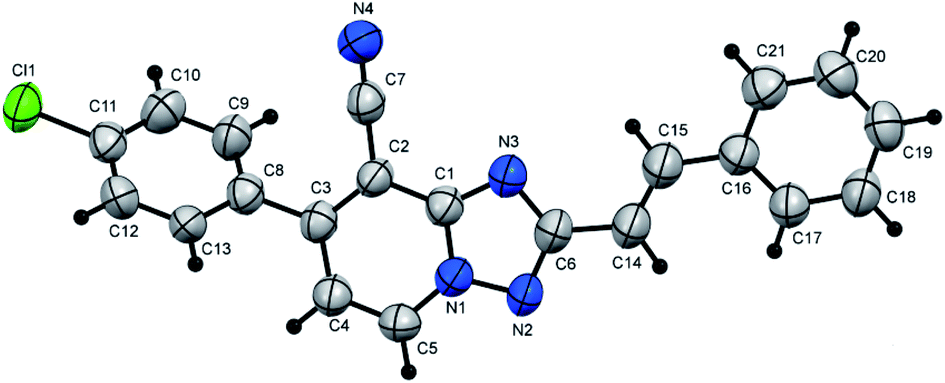 | ||
| Fig. 4 X-ray single crystal data determined for 5u. | ||
 | ||
| Scheme 3 Alternative route for 5t. | ||
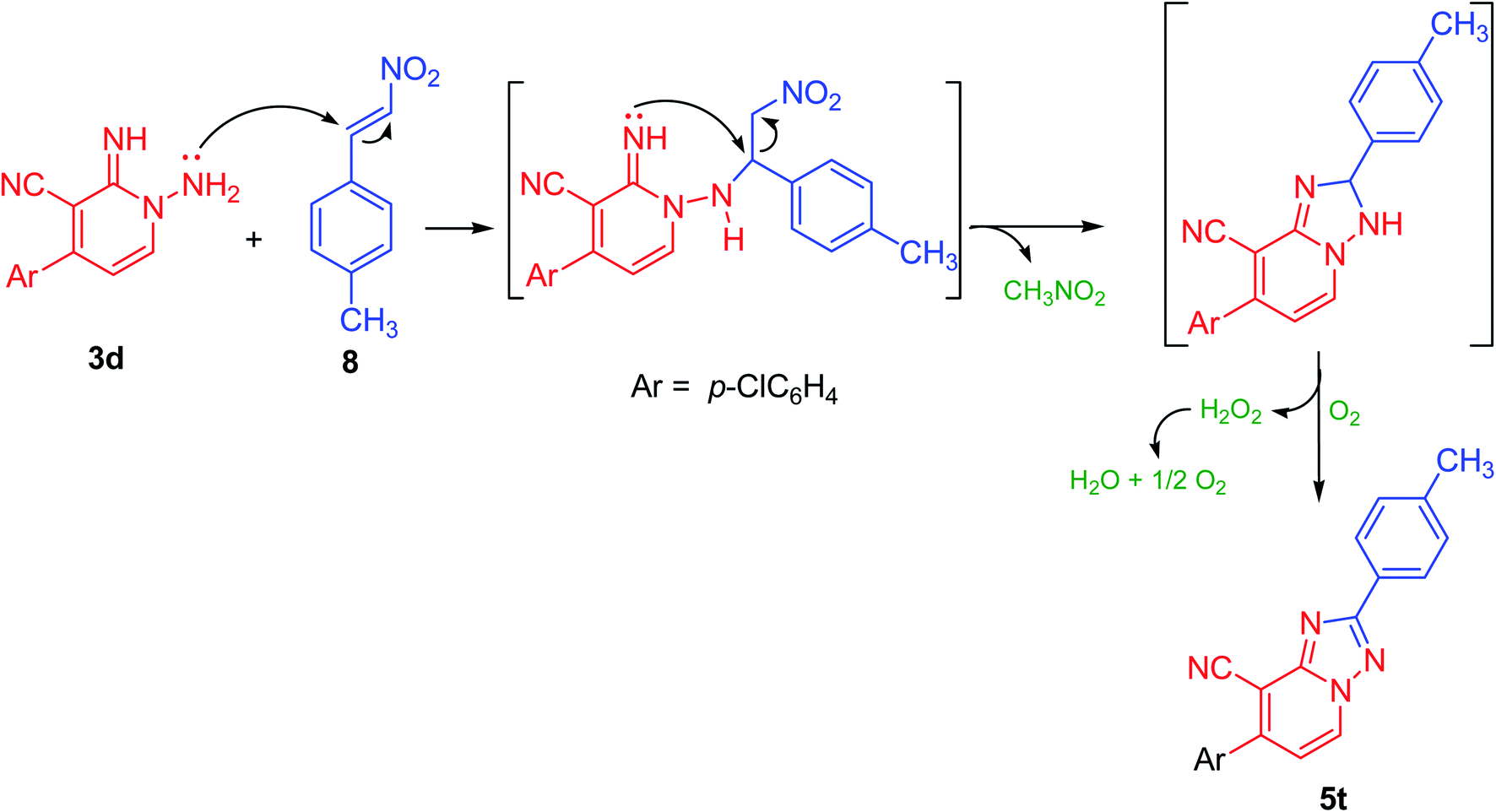
Likewise, compounds 5d–f could be also acquired via the cyclization reaction of derivatives 3a,c,d with (E)-3-(piperidin-1-yl)acrylonitrile (9) or with (E)-3-(dimethylamino)acrylonitrile (10) in superb yield (Scheme 4). Besides, this active methylene derivatives 5d–f underwent condensation reaction either with DMF–DMA or benzaldehyde easily to afford the isolable enamines 11a,b and arylidenes 12a,b, respectively (Scheme 4).
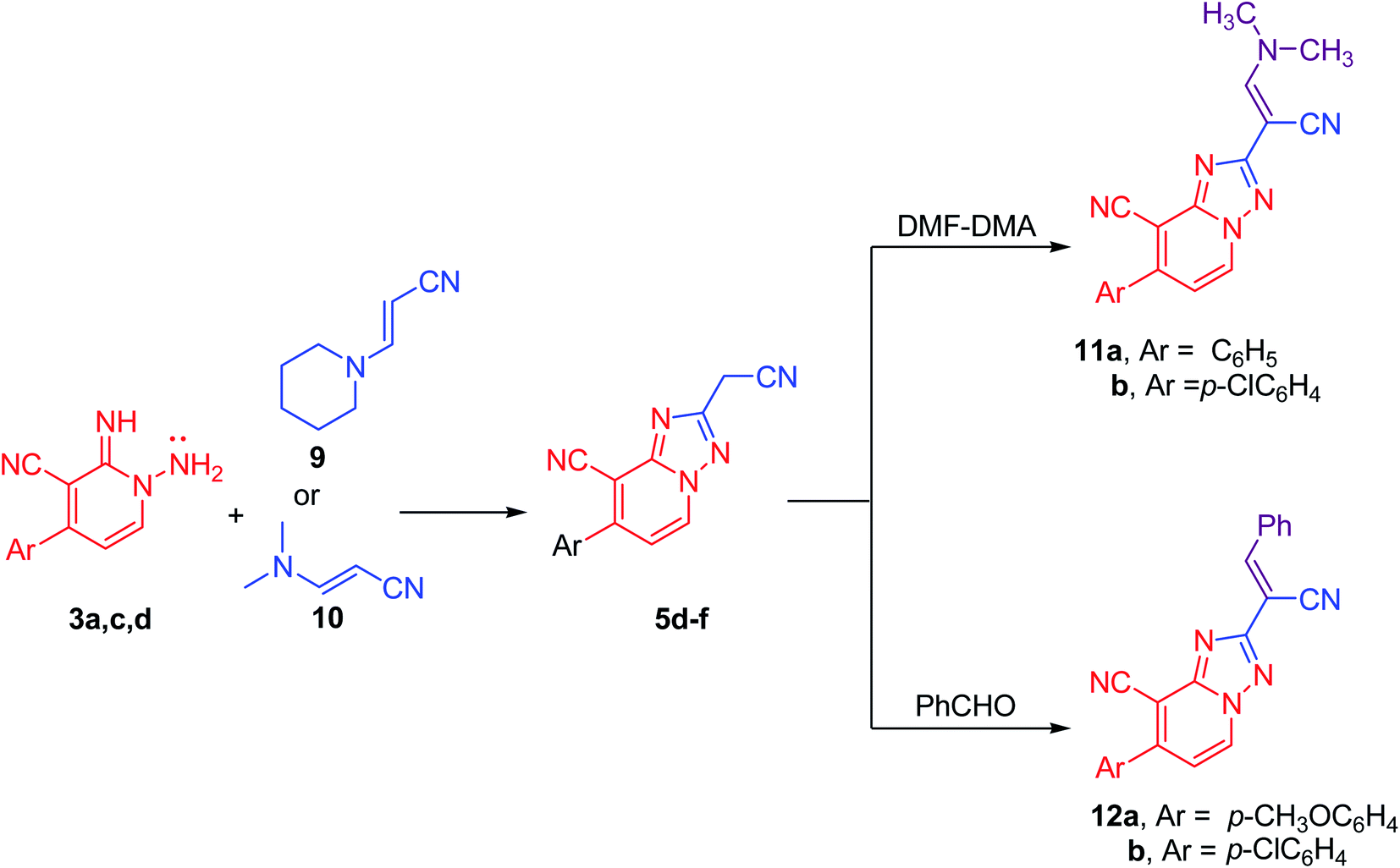 | ||
| Scheme 4 Alternative preparation of 5d–f and their reactions with DMF–DMA and PhCHO. | ||
Further, the present approach was effectively applied also to isothiocyanate derivatives under moderate conditions. Thus, under microwave irradiation, 1,2-dihydropyridine-3-carbonitrile derivative (3d) underwent cyclization reaction when treated with phenyl isothiocyanate providing the unreported [1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile derivative (5v) in excellent yield (90%) (Scheme 5). In the course of this reaction, the sulfur of the isothiocyanate moiety gets lost presumably in the form of hydrogen sulfide gas. Therefore, the reaction may be started by the nucleophilic addition of the amino group of derivative 3d onto azomethine motif of phenyl isothiocyanate. Then, hydrogen sulfide was removed, possibly via an addition–elimination reaction, which results in a [1,2,4]triazolo[1,5-a]pyridine ring formation (Scheme 5, Fig. 5). Moreover, by boiling pyridine derivatives (3a,b,d) in DMF or glyoxalic acid, the unsubstituted triazole derivatives (5w–y) have been achieved in superb yields (89–91%, Scheme 5). In the latter reactions, the aldehydic group might be involved in cyclization reaction followed by the loss of dimethylamine (in case of DMF) or carbon dioxide (in the case of glyoxalic acid).
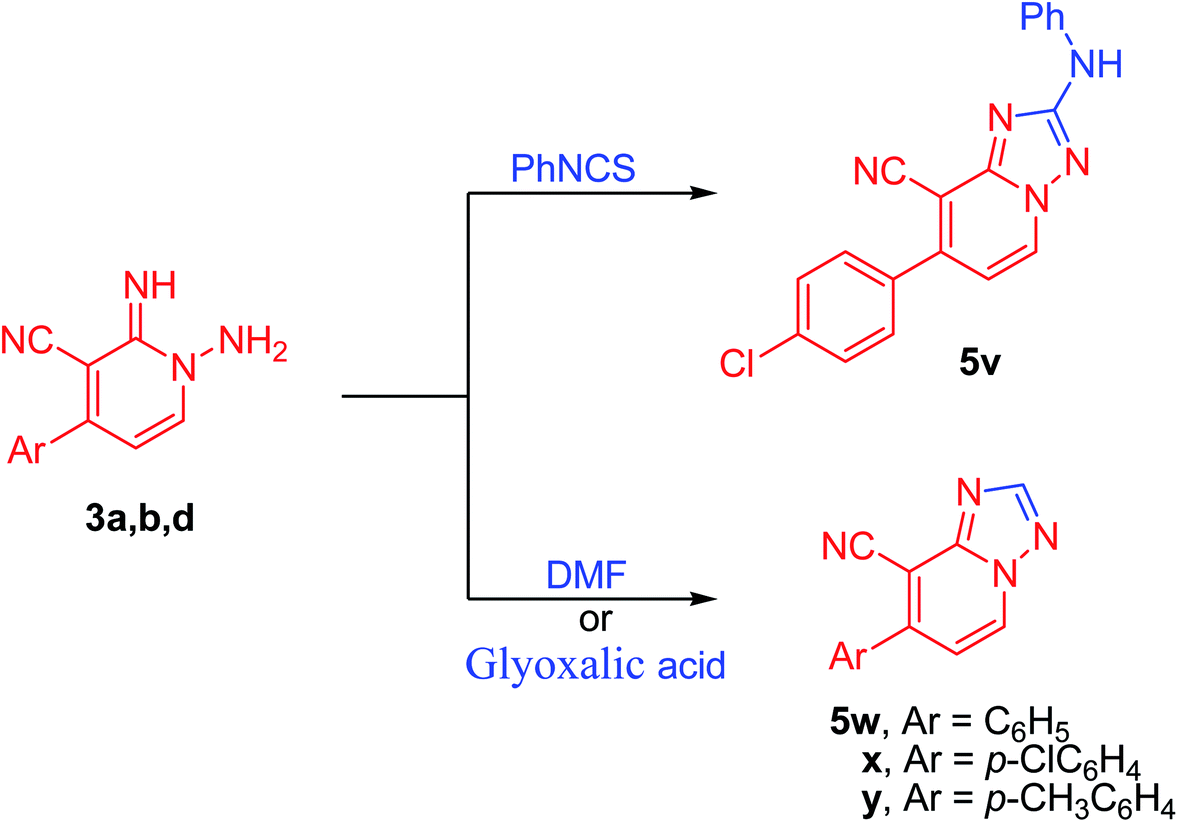 | ||
| Scheme 5 Reaction of 1-amino-2-imino-pyridine with PhNCS and DMF. | ||
 | ||
| Fig. 5 X-ray single crystal data determined for 5v. | ||
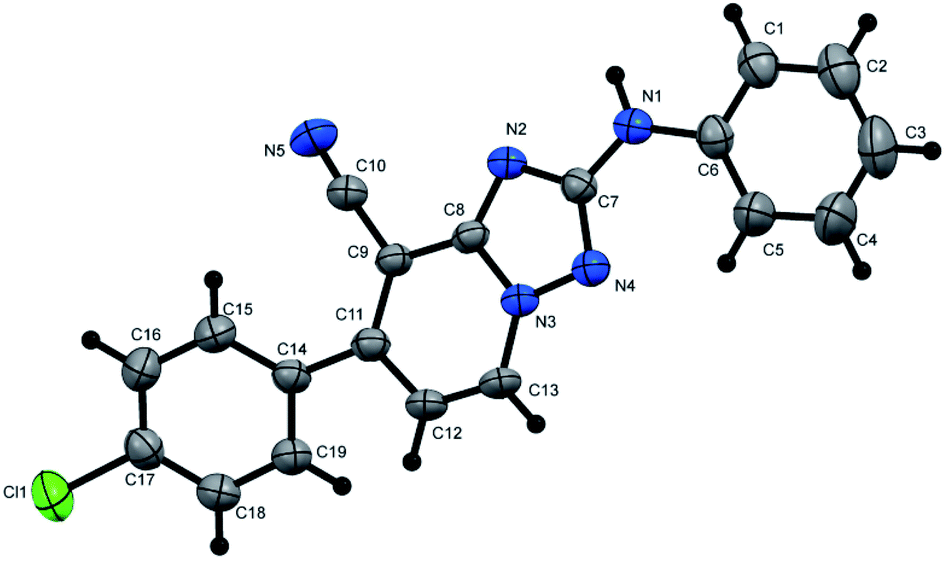
The aforesaid protocol also applied successfully for the bi-function aromatic aldehydic compounds. For example, the synthesis of bis-triazolopyridine derivatives (13a–c) was achieved through the cyclization reaction of the commercially available terephthalaldehyde with N-amino-2-imino-pyridine derivatives (3b,d,e) in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio (Scheme 6). Whereas, the mono-triazolopyridine derivative (5z), could be received on conducting the reaction between terephthalaldehyde and derivative 3d in 1:1 molar ratio (Scheme 6). Interestingly, the bis-derivative (13b) could be also synthesized via the reaction of the mono-derivative (5z), with another batch of 3d (1.0 mmol) (13b, Scheme 6).
:1 molar ratio (Scheme 6). Whereas, the mono-triazolopyridine derivative (5z), could be received on conducting the reaction between terephthalaldehyde and derivative 3d in 1:1 molar ratio (Scheme 6). Interestingly, the bis-derivative (13b) could be also synthesized via the reaction of the mono-derivative (5z), with another batch of 3d (1.0 mmol) (13b, Scheme 6).
 | ||
| Scheme 6 Reaction of 1-amino-2-imino-pyridines with terephthalaldehyde. | ||
Ultimately, the nicotinonitrile derivative (14) was produced under microwave irradiation in excellent yield (98%, Scheme 7, Fig. 6) on boiling compound 3d in EtOH containing a catalytic amount of TEA (triethylamine) or DBU (1,8-diazabicyclo[5.4.0]undec-7-ene).
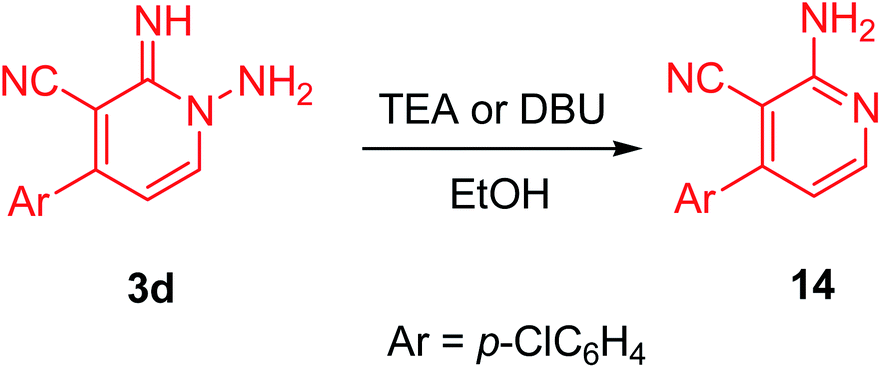 | ||
| Scheme 7 Conversion 1-amino-2-imino-pyridine derivative (3d) to 2-aminopyridine derivative (14). | ||
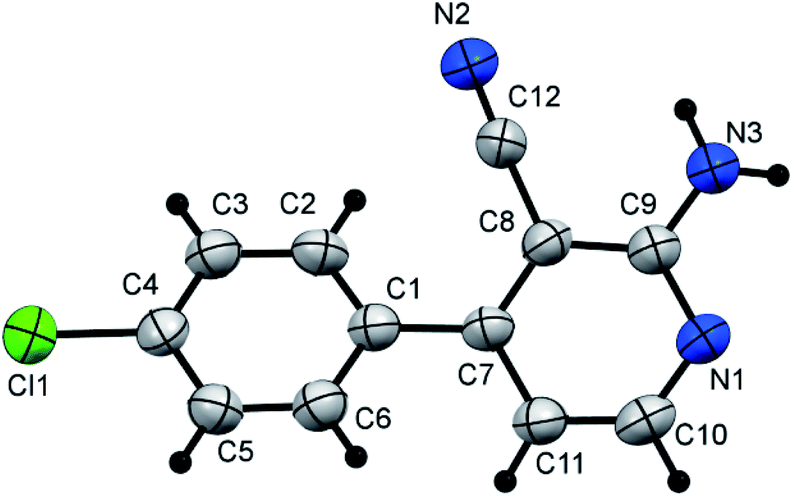 | ||
| Fig. 6 X-ray single crystal data determined for 14. | ||
The suggested structures of the synthesized mono- and bis-triazolopyridines have been verified based on several techniques of spectrometric analyses including 1H NMR and 13C NMR, in addition to the mass and accurate mass assignment. Moreover, the above structures were assured without any doubt through the X-ray single-crystal structure determination in some representative examples.
Conclusion
A microwave metal-free protocol towards the assembly of mono- and bis-[1,2,4]triazolo[1,5-a]pyridines has been established from the cyclization reaction of 1-amino-2-imino-pyridine derivatives with readily available carboxylic acids and diesters. Moreover, the utility and versatility of the present procedure are also established for a diverse range of other substrates, such as mono-aldehydes, di-aldehydes, phenyl isothiocyanate, acrylonitriles, and glyoxalic acid. The essential strengths of the current procedure are operational efficiency, conveniently accessible substrates, inexpensive reagents, good to excellent yields, broad functional group tolerance, and chromatography-free procedure. Therefore, we believe that such an environmentally friendly strategy paves the way for the design of biologically important scaffolds and provides practical alternatives to the design of these hybrid molecules.Experimental
General
Melting points were recorded on a Griffin melting point apparatus and are uncorrected. IR spectra were recorded using KBr disks using Jasco FT-IR-6300 spectrophotometer. 1H NMR (400 MHz) or (600 MHz) and 13C NMR (100 MHz) or (150 MHz) spectra were recorded at 25 °C using DMSO-d6 (or CDCl3) as a solvent with TMS as internal standard on a Bruker DPX 400 or 600 super-conducting NMR spectrometer. Chemical shifts are reported in ppm. Low-resolution electron impact mass spectra [MS (EI)] and high-resolution electron impact mass spectra [HRMS (EI)] were performed using a high-resolution GC-MS (DFS) thermo spectrometer at 70.1 eV and a magnetic sector mass analyzer. Follow up of the reactions and checking homogeneity of the prepared compounds was made by using thin-layer chromatography (TLC). Microwave heating was carried out with a single mode cavity Explorer Microwave synthesizer (CEM Corporation, NC, USA), producing continuous irradiation and equipped with simultaneous external air-cooling system. The X-ray crystal structures were determined by using a Rigaku R-AXIS RAPID diffractometer and Bruker X8 Prospector and the collection of single-crystal data was made at room temperature by using Cu-Kα radiation. The structures were solved by using direct methods and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. The structures were solved and refined using the Bruker SHELXTL Software Package (Structure solution program-SHELXS-97 and Refinement program-SHELXL-97).34 Data were corrected for the absorption effects using the multi-scan method (SADABS). The N-amino-2-iminopyridines 3a–e were prepared according to the literature procedure.331-Amino-2-imino-4-phenyl-1,2-dihydropyridine-3-carbonitrile (3a). Yellow crystals; yield: 3.7 g (89%); m.p. 165–166 °C, IR (KBr): ν/cm−1 3318, 3226 (NH2), 3137 (NH), 2211 (C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N); 1H NMR (400 MHz, DMSO-d6): δ 5.90 (d, J = 7.2 Hz, 1H, C–H6), 6.16 (s, 2H, NH2), 6.53 (brs, 1H, imine NH), 7.52–7.59 (m, 5H, Ar–H), 7.81 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (100 MHz, DMSO-d6): δ 97.2, 101.6, 117.1, 127.8, 128.8, 130.1, 136.3, 143.1, 154.6, 155.1 ppm; MS (EI): m/z (%) 211 (M+ + 1, 18.25), 210 (M+, 100); HRMS (EI): m/z calcd for C12H10N4 (M+) 210.0899, found 210.0899.
N); 1H NMR (400 MHz, DMSO-d6): δ 5.90 (d, J = 7.2 Hz, 1H, C–H6), 6.16 (s, 2H, NH2), 6.53 (brs, 1H, imine NH), 7.52–7.59 (m, 5H, Ar–H), 7.81 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (100 MHz, DMSO-d6): δ 97.2, 101.6, 117.1, 127.8, 128.8, 130.1, 136.3, 143.1, 154.6, 155.1 ppm; MS (EI): m/z (%) 211 (M+ + 1, 18.25), 210 (M+, 100); HRMS (EI): m/z calcd for C12H10N4 (M+) 210.0899, found 210.0899.
1-Amino-2-imino-4-p-tolyl-1,2-dihydropyridine-3-carbonitrile (3b). Yellow crystals; yield: 4.3 g (90%); m.p. 223–224 °C, IR (KBr): ν/cm−1 3315, 3262 (NH2), 3171 (NH), 2207 (C
N); 1H NMR (400 MHz, DMSO-d6): δ 2.38 (s, 3H, CH3), 5.88 (d, J = 7.2 Hz, 1H, C–H6), 6.13 (s, 2H, NH2), 6.58 (brs, 1H, imine NH), 7.34 (d, J = 8.0 Hz, 2H, Ar–H), 7.48 (d, J = 8.0 Hz, 2H, Ar–H), 7.79 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (100 MHz, DMSO-d6): δ 20.9 (CH3), 101.6, 117.2, 127.7, 129.3, 133.3, 140.0, 142.6, 143.0, 154.6, 155.0 ppm; MS (EI): m/z (%) 225 (M+ + 1, 13.19), 224 (M+, 72.89); HRMS (EI): m/z calcd for C13H12N4 (M+) 224.1056, found 224.1055.
1-Amino-2-imino-4-(4-methoxyphenyl)-1,2-dihydropyridine-3-carbonitrile (3c). Yellow crystals; yield: 4.2 g (88%); m.p. 225–226 °C, IR (KBr): ν/cm−1 3316, 3248 (NH2), 3167 (NH), 2206 (C
N); 1H NMR (400 MHz, DMSO-d6): δ 3.83 (s, 3H, OCH3), 5.89 (d, J = 6.8 Hz, 1H, C–H6), 6.11 (s, 2H, NH2), 6.55 (brs, 1H, imine NH), 7.08 (d, J = 8.4 Hz, 2H, Ar–H), 7.56 (d, J = 8.4 Hz, 2H, Ar–H), 7.76 ppm (d, J = 6.8 Hz, 1H, C–H5); 13C{1H} NMR (100 MHz, DMSO-d6): δ 55.84 (OCH3), 101.99, 102.02, 114.65, 117.83, 128.66, 129.91, 143.27, 155.06, 155.23, 161.23 ppm; MS (EI): m/z (%) 241 (M+ + 1, 19.27), 240 (M+, 100); HRMS (EI): m/z calcd for C13H12N4O (M+) 240.1005, found 240.1005.
1-Amino-4-(4-chlorophenyl)-2-imino-1,2-dihydropyridine-3-carbonitrile (3d). Bright yellow crystals; yield: 4.35 g (89%); m.p. 234–235 °C, IR (KBr): ν/cm−1 3314, 3267 (NH2), 3178 (NH), 2210 (C
N); 1H NMR (400 MHz, DMSO-d6): δ 5.89 (d, J = 6.8 Hz, 1H, C–H6), 6.16 (s, 2H, NH2), 6.61 (brs, 1H, imine NH), 7.61–7.63 (m, 4H, Ar–H), 7.81 ppm (d, J = 6.8 Hz, 1H, C–H5); 13C{1H} NMR (100 MHz, DMSO-d6): δ 101.3, 116.8, 128.8, 129.7, 134.8, 135.0, 143.1, 153.9, 154.3 ppm; MS (EI): m/z (%) 246 (M+ + 2, 34.29), 245 (M+ + 1, 17.94), 244 (M+, 100); HRMS (EI): m/z calcd for C12H9N4Cl (M+) 244.0510, found 244.0510.
1-Amino-4-(4-bromophenyl)-2-imino-1,2-dihydropyridine-3-carbonitrile (3e). Yellow crystals; yield: 5.3 g (92%); m.p. 239–240 °C, IR (KBr): ν/cm−1 3311, 3263 (NH2), 3176 (NH), 2208 (C
N); 1H NMR (400 MHz, DMSO-d6): δ 5.90 (d, J = 6.8 Hz, 1H C–H6), 6.17 (s, 2H, NH2), 6.73 (brs, 1H, imine NH), 7.54 (d, J = 8.4 Hz, 2H, Ar–H), 7.74 (d, J = 8.4 Hz, 2H, Ar–H), 7.82 ppm (d, J = 6.8 Hz, 1H, C–H5); 13C{1H} NMR (100 MHz, DMSO-d6): δ 102.2, 116.6, 123.7, 130.0, 131.8, 135.3, 143.4, 143.4, 154.2, 154.3 ppm; MS (EI): m/z (%) 290 (M+ + 2, 97.06), 289 (M+ + 1, 18.49), 288 (M+, 100); HRMS (EI): m/z calcd for C12H9N4Br (M+) 288.0005, found 288.0005.
For acetic acid. Independent mixtures of 1-amino-2-imino-pyridine 3a,d (3.0 mmol), and acetic acid (10 equiv.) in EtOH (10 mL).
For other acids. Independent mixtures of 1-amino-2-imino-pyridine 3a–d (3.0 mmol), and the appropriate carboxylic acids (4a–g) (4.0 mmol), in EtOH (10.0 mL) containing acetic acid (0.90 g, 5 equiv.), were charged in the glass tube of the microwave tube and irradiated by focused microwave using a single-mode cavity explorer microwave synthesizer (CEM Corporation, NC, USA) for 15 min at 100 °C, and 250 W. The build-up of pressure in the closed reaction vessel was carefully monitored. After the irradiation, the reaction tube was cooled through an inbuilt system in the instrument until the temperature had fallen below 50 °C. The reaction was controlled by TLC and continued until the starting substrates were completely consumed. The solid products that formed on standing at room temperature were collected by filtration, washed with ethanol and recrystallized from the proper solvent (see below), to give 5a–k as pure products.
2-Methyl-7-phenyl[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5a). Recrystallized from EtOH/dioxane mixture (5
:1), as yellowish white crystals, yield: 0.65 g (92%), m.p. 155–156 °C; IR (KBr): ν/cm−1 2218 (CN); 1H NMR (600 MHz, DMSO-d6): δ 2.55 (s, 3H, CH3), 7.37 (d, J = 6.8 Hz, 1H, C–H6), 7.59–7.63 (m, 3H, Ar–H), 7.75 (d, J = 8.4 Hz, 2H, Ar–H) 9.19 ppm (d, J = 6.8 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 14.13 (CH3), 96.41, 114.61, 114.64, 128.78, 128.99, 130.18, 132.45, 135.47, 149.06, 150.30, 165.01 ppm; MS (EI): m/z (%) 235 (M+ + 1, 26.78), 234 (M+, 100). HRMS (EI): m/z calcd for C14H10N4 (M+) 234.0899, found 234.0898.
7-(4-Chlorophenyl)-2-methyl[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5b). Recrystallized from EtOH/dioxane mixture (3
:1), as buff crystals, yield: 0.70 g (87%), m.p. 225–226 °C; IR (KBr): ν/cm−1 2223 (CN); 1H NMR (600 MHz, DMSO-d6): δ 2.54 (s, 3H, CH3), 7.36 (d, J = 6.8 Hz, 1H, C–H6), 7.67 (d, J = 8.4 Hz, 2H, Ar–H), 7.76 (d, J = 8.4 Hz, 2H, Ar–H), 9.20 ppm (d, J = 6.8 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 14.1, 96.6, 114.5, 128.7, 129.1, 130.7, 132.6, 134.3, 135.2, 147.8, 150.2, 165.1 ppm; MS (EI): m/z (%) 270 (M+ + 2, 29.65), 269 (M+ + 1, 14.89), 268 (M+, 100). HRMS (EI): m/z calcd for C14H10ClN4 (M+) 268.1147, found 268.1147.
2-(Chloromethyl)-7-(4-chlorophenyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5c). Recrystallized from EtOH/dioxane mixture (3
:1), as beige crystals, yield: 0.80 g (91%) in case of acid, 0.75 g (85% in case of chloroacetaldehyde), m.p. 179–180 °C; IR (KBr): ν/cm−1 2239 (CN); 1H NMR (600 MHz, CDCl3): δ 4.86 (s, 2H, CH2), 7.22 (d, J = 7.2 Hz, 1H, C–H6), 7.56 (d, J = 8.4 Hz, 2H, Ar–H), 7.64 (d, J = 8.4 Hz, 2H, Ar–H), 8.77 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, CDCl3): δ 37.36, 99.14, 113.49, 115.25, 129.70, 129.97, 131.50, 133.49, 137.26, 148.92, 151.00, 165.10 ppm; MS (EI): m/z (%) 304 (M+ + 2, 64.82), 303 (M+ + 1, 17.68), 302 (M+, 100). HRMS (EI): m/z calcd for C14H8Cl2N4 (M+) 302.01205, found 302.01205.
2-(Cyanomethyl)-7-phenyl-[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5d). Recrystallized from dioxane as buff crystals, yield: 0.75 g (96%), m.p. 245–246 °C; IR (KBr): ν/cm−1 2261, 2231 (2C
N); 1H NMR (400 MHz, DMSO-d6): δ 4.56 (s, 2H, CH2), 7.50 (d, J = 7.2 Hz, 1H, C–H6), 7.60–7.64 (m, 3H, Ar–H), 7.74–7.78 (m, 2H, Ar–H), 9.33 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 17.86, 97.44, 114.04, 115.70, 116.14, 128.62, 128.96, 130.26, 132.71, 135.39, 150.04, 150.74, 159.59 ppm; MS (EI): m/z (%) 260 (M+ + 1, 20.54), 259 (M+, 100). HRMS (EI): m/z calcd for C15H9N5 (M+) 259.0852, found 259.0853.
7-(4-Chlorophenyl)-2-(cyanomethyl)-[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5e). Recrystallized from dioxane as buff crystals, yield: 0.85 g (97%), m.p. 229–230 °C; IR (KBr): ν/cm−1 2264, 2231 (2C
N); 1H NMR (400 MHz, DMSO-d6): δ 4.56 (s, 2H, CH2), 7.52 (d, J = 7.2 Hz, 1H, C–H6), 7.72 (d, J = 8.4 Hz, 2H, Ar–H), 7.81 (d, J = 8.4 Hz, 2H, Ar–H), 9.36 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 17.83, 97.36, 114.26, 115.60, 116.61, 129.12, 130.78, 133.09, 134.08, 135.43, 148.67, 150.56, 159.65 ppm; MS (EI): m/z (%) 295 (M+ + 2, 75.08), 294 (M+ + 1, 61.59), 293 (M+, 100). HRMS (EI): m/z calcd for C15H8ClN5 (M+) 293.0462, found 293.0462.
2-(Cyanomethyl)-7-(4-methoxyphenyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5f). Recrystallized from dioxane as buff crystals, yield: 0.82 g (94%), m.p. 270–271 °C; IR (KBr): ν/cm−1 2261, 2230 (2C
N); 1H NMR (600 MHz, DMSO-d6): δ 3.86 (s, 3H, OCH3), 4.52 (s, 2H, CH2), 7.17 (d, J = 9.0 Hz, 2H, Ar–H), 7.47 (d, J = 7.2 Hz, 1H, C–H6), 7.75 (d, J = 9.0 Hz, 2H, Ar–H), 9.26 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 17.79, 55.47, 96.12, 114.55, 114.70, 115.58, 116.62, 127.25, 130.52, 132.70, 149.60, 150.83, 159.38, 161.06 ppm; MS (EI): m/z (%) 290 (M+ + 1, 17.36), 289 (M+, 100). HRMS (EI): m/z calcd for C16H11N5O (M+) 289.0958, found 289.289.0957.
2-(4-Nitrobenzyl)-7-(4-methoxyphenyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5g). Recrystallized from dioxane as white crystals, yield: 1.05 g (93%), m.p. 159–160 °C; IR (KBr): ν/cm−1 2233 (C
N); 1H NMR (600 MHz, DMSO-d6): δ 3.87 (s, 3H, OCH3), 4.57 (s, 2H, CH2), 7.18 (d, J = 9.0 Hz, 2H, Ar–H), 7.40 (d, J = 7.2 Hz, 1H, C–H6), 7.67 (d, J = 9.0 Hz, 2H, Ar–H), 7.74 (d, J = 9.0 Hz, 2H, Ar–H), 8.21 (d, J = 9.0 Hz, 2H, Ar–H), 9.21 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 33.99, 55.45, 95.94, 114.51, 114.90, 115.02, 123.56, 127.43, 130.34, 130.46, 132.53, 145.45, 146.36, 149.18, 150.65, 160.95, 166.03 ppm; MS (EI): m/z (%) 386 (M+ + 1, 24.83), 385 (M+, 100). HRMS (EI): m/z calcd for C21H15N5O3 (M+) 385.1169, found 385.1169.
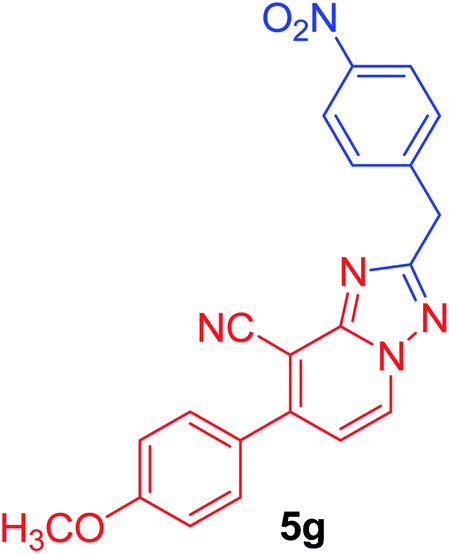
7-(4-Methoxyphenyl)-2-phenyl[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5h). Recrystallized from EtOH/dioxane mixture (1
:3), as creamy white crystals, yield: 0.80 g (83%), m.p. 199–200 °C; IR (KBr): ν/cm−1 2221 (CN); 1H NMR (600 MHz, DMSO-d6): δ 3.88 (s, 3H, OCH3), 7.18 (d, J = 9.0 Hz, 2H, Ar–H), 7.43 (d, J = 7.2 Hz, 1H, C–H6), 7.56–7.59 (m, 3H, Ar–H), 7.77 (d, J = 9.0 Hz, 2H, Ar–H), 8.23–8.25 (m, 2H, Ar–H), 9.28 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 55.46, 96.09, 114.50, 114.99, 115.28, 127.03, 129.02, 129.63, 130.50, 130.74, 132.63, 149.15, 151.04, 160.94, 164.13 ppm; MS (EI): m/z (%) 327 (M+ + 1, 23.79), 326 (M+, 100). HRMS (EI): m/z calcd for C20H14N4O (M+) 326.1162, found 326.1162.
7-(4-Chlorophenyl)-2-phenyl[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5i). Recrystallized from dioxane as creamy white crystals, yield: 0.85 g (86%), m.p. 210–212 °C; IR (KBr): ν/cm−1 2224 (C
N); 1H NMR (600 MHz, DMSO-d6): δ 7.48 (d, J = 7.2 Hz, 1H, C–H6), 7.57–7.60 (m, 3H, Ar–H), 7.71 (d, J = 8.4 Hz, 2H, Ar–H), 7.81 (d, J = 8.4 Hz, 2H, Ar–H), 8.25–8.26 (m, 2H, Ar–H), 9.37 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 97.29, 114.53, 115.29, 127.10, 129.08, 129.54, 130.73, 130.87, 133.00, 134.25, 135.30, 148.22, 150.81, 164.54 ppm; MS (EI): m/z (%) 332 (M+ + 2, 40.03), 331 (M+ + 1, 30.11), 330 (M+, 100). HRMS (EI): m/z calcd for C19H11ClN4 (M+) 330.0666, found 330.0667.
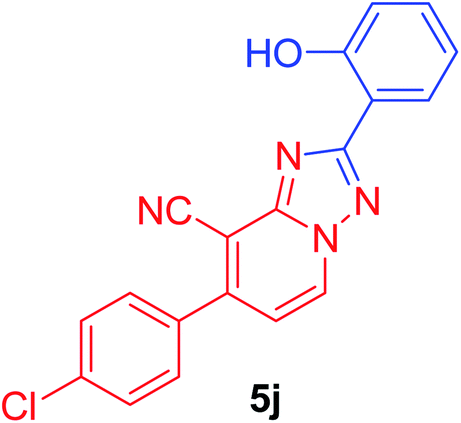
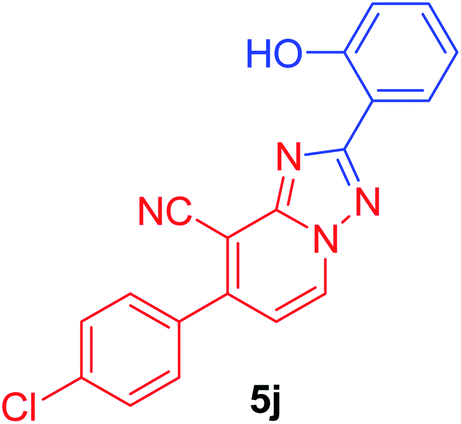
7-(4-Chlorophenyl)-2-(2-hydroxyphenyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5j). Recrystallized from dioxane as yellowish white crystals, yield: 0.90 g (87%), m.p. 244–245 °C; IR (KBr): ν/cm−1 3385 (OH), 2226 (C
N); 1H NMR (600 MHz, DMSO-d6): δ 6.99–7.04 (m, 2H, Ar–H), 7.41 (t, J = 7.8 Hz, 1H, Ar–H), 7.53 (d, J = 7.2 Hz, 1H, C–H6), 7.71 (d, J = 8.4 Hz, 2H, Ar–H), 7.81 (d, J = 8.4 Hz, 2H, Ar–H), 8.06 (d, J = 7.8 Hz, 1H, Ar–H), 9.37 (d, J = 7.2 Hz, 1H, C–H5), 10.87 ppm (s, 1H, OH); 13C{1H} NMR (150 MHz, DMSO-d6): δ 96.71, 112.76, 113.44, 115.39, 116.85, 119.28, 127.50, 128.65, 130.11, 132.10, 132.29, 133.65, 135.16, 148.29, 149.08, 156.82, 163.16 ppm; MS (EI): m/z (%) 348 (M+ + 2, 34.08), 347 (M+ + 1, 23.17), 346 (M+, 100). HRMS (EI): m/z calcd for C19H11ClN4O (M+) 346.0615, found 346.0614.
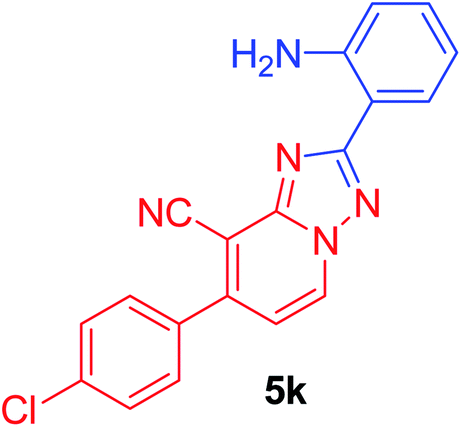
2-(2-Aminophenyl)-7-(4-chlorophenyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5k). Recrystallized from EtOH/dioxane mixture (1
:2), as creamy white crystals, yield: 0.86 g (84%), m.p. 215–216 °C; IR (KBr): ν/cm−1 3364, 3292 (NH2), 2221 (CN); 1H NMR (600 MHz, DMSO-d6): δ 5.01 (brs, 2H, NH2), 6.59 (d, J = 7.8 Hz, 1H, Ar–H), 6.73 (d, J = 7.2 Hz, 1H, C–H6), 6.95 (t, J = 7.8 Hz, 1H, Ar–H), 7.32 (t, J = 7.8 Hz, 1H, Ar–H), 7.43 (d, J = 7.8 Hz, 2H, Ar–H), 7.65 (d, J = 7.8 Hz, 2H, Ar–H), 7.95 (d, J = 7.8 Hz, 1H, Ar–H), 8.22 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 97.10, 110.26, 113.63, 114.80, 120.38, 121.30, 128.39, 129.58, 131.39, 131.89, 131.98, 141.44, 146.24, 147.34, 155.09, 158.35, 168.96 ppm; MS (EI): m/z (%) 347 (M+ + 2, 3.01), 346 (M+ + 1, 1.78), 345 (M+, 9.65). HRMS (EI): m/z calcd for C19H12ClN5 (M+) 345.0775, found 345.0776.
Ethyl 8-cyano-7-phenyl[1,2,4]triazolo[1,5-a]pyridine-2-carboxylate (5l). Recrystallized from EtOH/dioxane mixture (1
:1), as white crystals, yield: 0.77 g (88%), m.p. 234–235 °C; IR (KBr): ν/cm−1 2231 (CN), 1722 (CO); 1H NMR (400 MHz, DMSO-d6): δ 1.39 (t, J = 7.2 Hz, 3H, CH3CH2), 4.45 (q, J = 7.2 Hz, 2H, CH3CH2), 7.61–7.66 (m, 4H, pyridine C–H6 and 3 Ar–H), 7.79–7.82 (m, 2H, Ar–H), 9.42 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (100 MHz, CDCL3): δ 14.19 (CH3), 62.79 (CH2), 100.15, 113.13, 117.18, 128.65, 129.34, 130.86, 131.64, 134.82, 150.81, 151.08, 157.46, 159.49 ppm; MS (EI): m/z (%) 293 (M+ + 1, 3.24), 292 (M+, 16.04). HRMS (EI): m/z calcd for C16H12N4O2 (M+) 292.0954, found 292.0954.
Ethyl 8-cyano-7-p-tolyl-[1,2,4]triazolo[1,5-a]pyridine-2-carboxylate (5m). Recrystallized from EtOH/dioxane mixture (1
:1), as white crystals, yield: 0.80 g (87%), m.p. 180–181 °C; IR (KBr): ν/cm−1 2230 (CN), 1726 (CO); 1H NMR (400 MHz, DMSO-d6): δ 1.38 (t, J = 7.2 Hz, 3H, CH3CH2), 2.43 (s, 3H, CH3), 4.45 (q, J = 7.2 Hz, 2H, CH3CH2), 7.45 (d, J = 8.0 Hz, 2H, Ar–H), 7.61 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.70 (d, J = 8.0 Hz, 2H, Ar–H), 9.38 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (100 MHz, DMSO-d6): δ 14.06, 20.96 (2CH3), 61.99 (CH2), 97.99, 114.41, 117.32, 128.86, 129.74, 132.23, 133.47, 140.73, 150.49, 150.60, 156.19, 159.37 ppm; MS (EI): m/z (%) 307 (M+ + 1, 7.09), 306 (M+, 37.28). HRMS (EI): m/z calcd for C17H14N4O2 (M+) 306.1111, found 306.1111. Crystal data, moiety formula: C17H14N4O2, M = 306.32, monoclinic, a = 16.043(2) Å, b = 9.9874(9) Å, c = 18.960(2) Å, V = 2972.1(5) Å3, α = γ = 90°, β = 101.956(8)°, space group: P21/c (#14), Z = 8, Dcalc = 1.369 g cm−3, no. of reflection measured = 5132, 2θmax = 50.10°, R1 = 0.0638 (CCDC 1982378†).35
Ethyl 8-cyano-7-(4-methoxyphenyl)[1,2,4]triazolo[1,5-a]pyridine-2-carboxylate (5n). Recrystallized from EtOH/dioxane mixture (1
:1), as white crystals, yield: 0.82 g (85%), m.p. 184–185 °C; IR (KBr): ν/cm−1 2231 (CN), 1732 (CO); 1H NMR (400 MHz, DMSO-d6): δ 1.38 (t, J = 7.2 Hz, 3H, CH3CH2), 3.86 (s, 3H, OCH3), 4.43 (q, J = 7.2 Hz, 2H, CH3CH2), 7.17 (d, J = 8.8 Hz, 2H, Ar–H), 7.59 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.76 (d, J = 8.8 Hz, 2H, Ar–H), 9.32 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (100 MHz, DMSO-d6): δ 14.47, 55.96 (2CH3), 62.40 (CH2), 97.66, 115.03, 117.65, 127.45, 131.05, 133.67, 150.61, 150.97, 156.55, 159.77, 161.65 ppm; MS (EI): m/z (%) 323 (M+ + 1, 8.65), 322 (M+, 43.39). HRMS (EI): m/z calcd for C17H14N4O3 (M+) 322.1060, found 322.1060.
Ethyl 7-(4-chlorophenyl)-8-cyano[1,2,4]triazolo[1,5-a]pyridine-2-carboxylate (5o). Recrystallized from EtOH/dioxane mixture (1
:3), as white crystals, yield: 0.90 g (92%), m.p. 218–219 °C; IR (KBr): ν/cm−1 2231 (CN), 1735 (CO); 1H NMR (600 MHz, DMSO-d6): δ 1.38 (t, J = 7.2 Hz, 3H, CH3CH2), 4.44 (q, J = 7.2 Hz, 2H, CH3CH2), 7.65 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.72 (d, J = 8.4 Hz, 2H, Ar–H), 7.82 (d, J = 8.4 Hz, 2H, Ar–H), 9.43 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 14.05 (CH3), 62.03 (CH2), 98.69, 114.13, 117.23, 129.22, 130.85, 133.68, 133.91, 135.64, 149.33, 150.30, 156.28, 159.30 ppm; MS (EI): m/z (%) 328 (M+ + 2, 6.34), 327 (M+ + 1, 3.57), 326 (M+, 17.29). HRMS (EI): m/z calcd for C16H11ClN4O2 (M+) 326.0565, found 326.0565.
Ethyl 7-(4-bromophenyl)-8-cyano[1,2,4]triazolo[1,5-a]pyridine-2-carboxylate (5p). Recrystallized from EtOH/dioxane mixture (1
:3), as white crystals, yield: 1.00 g (92%), m.p. 229–230 °C; IR (KBr): ν/cm−1 2231 (CN), 1733 (CO); 1H NMR (400 MHz, DMSO-d6): δ 1.38 (t, J = 7.2 Hz, 3H, CH3CH2), 4.45 (q, J = 7.2 Hz, 2H, CH3CH2), 7.65 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.75 (d, J = 8.4 Hz, 2H, Ar–H), 7.87 (d, J = 8.4 Hz, 2H, Ar–H), 9.43 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 14.07 (CH3), 62.05 (CH2), 98.66, 114.16, 117.18, 124.48, 131.05, 132.17, 133.72, 134.31, 149.44, 150.33, 156.30, 159.33 ppm; MS (EI): m/z (%) 372 (M+ + 2, 24.53), 371 (M+ + 1, 5.12), 370 (M+, 24.85). HRMS (EI): m/z calcd for C16H11BrN4O2 (M+) 370.0059, found 370.0058. Crystal data, moiety formula: C16H11BrN4O2, M = 371.19, orthorhombic, a = 13.845(2) Å, b = 7.528(1) Å, c = 30.459(4) Å, V = 3174.7(8) Å3, α = β = γ = 90°, space group: Pbca (#61), Z = 8, Dcalc = 1.553 g cm−3, no. of reflection measured = 2742, 2θmax = 49.9°, R1 = 0.0807 (CCDC 1982379†).35
2-(4-Chlorophenyl)-7-(4-methoxyphenyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5q). Recrystallized from dioxane/DMF mixture (2
:1), as creamy white crystals, yield: 0.95 g (90%), m.p. 224–225 °C; IR (KBr): ν/cm−1 2229 (CN); 1H NMR (600 MHz, DMSO-d6): δ 3.89 (s, 3H, OCH3), 7.17 (d, J = 8.4 Hz, 2H, Ar–H), 7.39 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.60 (d, J = 8.4 Hz, 2H, Ar–H), 7.75 (d, J = 8.4 Hz, 2H, Ar–H), 8.22 (d, J = 8.4 Hz, 2H, Ar–H), 9.17 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 55.10 (OCH3), 96.03, 114.14, 114.24, 114.98, 127.15, 128.28, 128.40, 128.60, 129.88, 131.97, 135.12, 148.95, 150.75, 160.77, 163.21 ppm; MS (EI): m/z (%) 362 (M+ + 2, 30.89), 361 (M+ + 1, 20.56), 360 (M+, 100.00). HRMS (EI): m/z calcd for C20H13ClN4O (M+) 360.0772, found 360.0772. Crystal data, moiety formula: C20H13ClN4O, C3H7NO, sum formula: C23H20ClN5O2, M = 433.90, orthorhombic, a = 22.8101(9) Å, b = 7.5155(3) Å, c = 24.720(2) Å, V = 4237.7(4) Å3, α = β = γ = 90°, space group: Pca21 (#29), Z = 8, Dcalc = 1.360 g cm−3, no. of reflection measured = 3817, 2θmax = 50.1°, R1 = 0.0428 (CCDC 1982380†).35
2,7-Bis(4-methoxyphenyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5r). Recrystallized from dioxane as creamy white crystals, yield: 1.00 g (93%), m.p. 226–227 °C; IR (KBr): ν/cm−1 2232 (C
N); 1H NMR (600 MHz, DMSO-d6): δ 3.86 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 7.12 (d, J = 8.4 Hz, 2H, Ar–H), 7.18 (d, J = 9.0 Hz, 2H, Ar–H), 7.40 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.78 (d, J = 8.4 Hz, 2H, Ar–H), 8.18 (d, J = 9.0 Hz, 2H, Ar–H), 9.25 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 55.33, 55.46 (2OCH3), 95.80, 114.45, 114.51, 114.96, 115.07, 122.05, 127.52, 128.71, 130.45, 132.50, 148.97, 151.07, 160.93, 161.30, 164.36 ppm; MS (EI): m/z (%) 357 (M+ + 1, 24.09), 356 (M+, 100.00). HRMS (EI): m/z calcd for C21H16N4O2 (M+) 356.1267, found 356.1266.
2,7-Bis(4-chlorophenyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5s). Recrystallized from dioxane as creamy white crystals, yield: 1.02 g (94%), m.p. 279–280 °C; IR (KBr): ν/cm−1 2230 (C
N); 1H NMR (600 MHz, DMSO-d6): δ 7.49 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.66 (d, J = 7.8 Hz, 2H, Ar–H), 7.71 (d, J = 7.8 Hz, 2H, Ar–H), 7.81 (d, J = 7.8 Hz, 2H, Ar–H), 8.26 (d, J = 7.8 Hz, 2H, Ar–H), 9.37 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 97.22, 113.87, 115.10, 128.24, 128.57, 128.73, 128.82, 130.27, 132.55, 133.96, 135.06, 135.33, 148.10, 150.60, 163.46 ppm; MS (EI): m/z (%) 366 (M+ + 2, 59.97), 365 (M+ + 1, 24.36), 364 (M+, 100.00). HRMS (EI): m/z calcd for C20H13ClN4O (M+) 360.0772, found 360.0774.
7-(4-chlorophenyl)-2-p-tolyl[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5t). Recrystallized from dioxane as creamy white crystals, yield: 0.95 g (91%), m.p. 227–228 °C; IR (KBr): ν/cm−1 2228 (C
N); 1H NMR (600 MHz, DMSO-d6): δ 2.39 (s, 3H, CH3), 7.37 (d, J = 8.4 Hz, 2H, Ar–H), 7.44 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.71 (d, J = 8.4 Hz, 2H, Ar–H), 7.80 (d, J = 8.4 Hz, 2H, Ar–H), 8.12 (d, J = 8.4 Hz, 2H, Ar–H), 9.32 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 21.05 (CH3), 97.12, 114.56, 115.12, 126.79, 127.07, 129.08, 129.65, 130.73, 132.92, 134.28, 135.27, 140.73, 148.09, 150.78, 164.66 ppm; MS (EI): m/z (%) 346 (M+ + 2, 33.47), 345 (M+ + 1, 27.19), 344 (M+, 100.00). HRMS (EI): m/z calcd for C20H13ClN4 (M+) 344.0823, found 344.0823.
(E)-7-(4-Chlorophenyl)-2-styryl[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5u). Recrystallized from dioxane as creamy white crystals, yield: 0.94 g (88%), m.p. 243–244 °C; IR (KBr): ν/cm−1 2231 (C
N); 1H NMR (600 MHz, DMSO-d6): δ 7.39–7.47 (m, 5H, Ar–H), 7.72 (d, J = 8.4 Hz, 2H, Ar–H), 7.81–7.89 (m, 5H, Ar–H), 9.29 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 96.83, 114.59, 114.98, 116.97, 127.54, 128.87, 129.08, 129.26, 130.73, 132.78, 134.30, 135.28, 135.43, 137.41, 148.23, 150.43, 164.45 ppm; MS (EI): m/z (%) 358 (M+ + 2, 17.05), 357 (M+ + 1, 38.14), 356 (M+, 47.89), 355 (M+ − 1, 100.00). HRMS (EI): m/z calcd for C21H13ClN4 (M+) 356.0823, found 356.0823. Crystal data, moiety formula: C21H13ClN4, M = 356.81, monoclinic, a = 7.653(1) Å, b = 6.928(9) Å, c = 32.82(4) Å, V = 1734(4) Å3, α = γ = 90°, β = 94.79(3)°, space group: P21/n (#14), Z = 4, Dcalc = 1.367 g cm−3, no. of reflection measured = 3041, 2θmax = 50.10°, R1 = 0.0800 (CCDC 1982381†).35
7-(4-Chlorophenyl)-2-(phenylamino)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5v). Recrystallized from EtOH/dioxane mixture (1
:2), as yellow crystals, yield: 0.95 g (91%), m.p. 216–218 °C; IR (KBr): ν/cm−1 3362 (NH), 2225 (2CN); 1H NMR (400 MHz, DMSO-d6): δ 6.92 (t, J = 7.8 Hz, 1H, Ar–H), 7.17 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.31 (t, J = 7.8 Hz, 2H, Ar–H), 7.63 (d, J = 8.4 Hz, 2H, Ar–H), 7.68 (d, J = 7.8 Hz, 2H, Ar–H), 7.74 (d, J = 8.4 Hz, 2H, Ar–H), 9.07 (d, J = 7.2 Hz, 1H, C–H5), 10.09 ppm (s, 1H, NH); 13C{1H} NMR (150 MHz, DMSO-d6): δ 93.95, 112.72, 114.76, 116.86, 120.75, 128.77, 128.95, 130.52, 131.82, 134.49, 134.98, 140.42, 146.95, 149.72, 163.23 ppm; MS (EI): m/z (%) 347 (M+ + 2, 33.07), 346 (M+ + 1, 48.39), 345 (M+, 100.00). HRMS (EI): m/z calcd for C19H12ClN5 (M+) 345.0775, found 345.0775. Crystal data, moiety formula: C19H12ClN5, M = 345.79, triclinic, a = 8.9886(8) Å, b = 12.8468(10) Å, c = 15.1372(12) Å, V = 1635.2(2) Å3, α = 71.352(4)°, β = 80.896(4)°, γ = 86.548(5)°, space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 4, Dcalc = 1.405 g cm−3, no. of reflection measured = 5570, θmax = 66.470°, R1 = 0.0441 (CCDC 1982382†).35
, Z = 4, Dcalc = 1.405 g cm−3, no. of reflection measured = 5570, θmax = 66.470°, R1 = 0.0441 (CCDC 1982382†).35
7-Phenyl[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5w). Recrystallized from EtOH/dioxane mixture (4
:1), as yellow crystals, yield: 0.55 g (84%), m.p. 159–160 °C; IR (KBr): ν/cm−1 2221 (CN); 1H NMR (400 MHz, DMSO-d6): δ 7.49 (d, J = 7.2 Hz, 1H, C–H6), 7.62–7.67 (m, 3H, Ar–H), 7.79 (d, J = 8.4 Hz, 2H, Ar–H), 8.75 (s, 1H, C–H) 9.35 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 97.60, 114.41, 115.56, 128.85, 129.03, 130.28, 133.27, 135.40, 149.60, 149.68, 155.40 ppm; MS (EI): m/z (%) 221 (M+ + 1, 17.65), 220 (M+, 100). HRMS (EI): m/z calcd for C13H8N4 (M+) 220.0743, found 220.0743.
7-(4-Chlorophenyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5x). Recrystallized from EtOH/dioxane mixture (3
:1), as yellow crystals, yield: 0.65 g (85%), m.p. above 300 °C; IR (KBr): ν/cm−1 2226 (CN); 1H NMR (600 MHz, DMSO-d6): δ 7.48 (d, J = 7.2 Hz, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.80 (d, J = 8.4 Hz, 2H), 8.75 (s, 1H), 9.35 ppm (d, J = 7.2 Hz, 1H); 13C{1H} NMR (150 MHz, DMSO-d6): δ 97.8, 114.4, 115.4, 129.1, 130.8, 133.2, 134.2, 135.3, 148.3, 149.6, 155.5 ppm; MS (EI): m/z (%) 256 (M+ + 2, 33.19), 255 (M+ + 1, 16.29), 254 (M+, 100). HRMS (EI): m/z calcd for C13H7ClN4 (M+) 254.1147, found 254.1147.
7-p-Tolyl-[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5y). Recrystallized from EtOH/dioxane mixture (3
:1), as yellowish white crystals, yield: 0.60 g (83%), m.p. 172–173 °C; IR (KBr): ν/cm−1 2224 (CN); 1H NMR (600 MHz, DMSO-d6): δ 2.40 (s, 3H, CH3), 7.40 (d, J = 7.8 Hz, 2H, Ar–H), 7.42 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.65 (d, J = 7.8 Hz, 2H, Ar–H), 8.70 (s, 1H, C–H2), 9.28 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 20.87 (CH3), 97.15, 114.63, 115.46, 128.73, 129.58, 132.47, 132.94, 140.29, 149.55 149.73, 155.31 ppm; MS (EI): m/z (%) 235 (M+ + 1, 13.94), 234 (M+, 100). HRMS (EI): m/z calcd for C14H10N4 (M+) 234.0899, found 234.0898.
7-(4-Chlorophenyl)-2-(4-formylphenyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (5z). Recrystallized from dioxane/DMF mixture (4
:1), as orange crystals, yield: 1.00 g (92%), m.p. 276–277 °C; IR (KBr): ν/cm−1 2231 (CN), 1695 (CO); 1H NMR (600 MHz, DMSO-d6): δ 7.54 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.74 (d, J = 8.4 Hz, 2H, Ar–H), 7.84 (d, J = 8.4 Hz, 2H, Ar–H), 8.12 (d, J = 8.4 Hz, 2H, Ar–H), 8.48 (d, J = 8.4 Hz, 2H, Ar–H), 9.41 (d, J = 7.2 Hz, 1H, C–H5), 10.12 ppm (s, 1H, CHO); 13C{1H} NMR (150 MHz, DMSO-d6): δ 97.41, 113.55, 115.20, 127.32, 128.56, 129.44, 130.06, 132.40, 133.81, 134.41, 135.01, 137.31, 148.15, 150.54, 163.29, 191.82 ppm; MS (EI): m/z (%) 360 (M+ + 2, 31.05), 359 (M+ + 1, 41.58), 358 (M+, 100). HRMS (EI): m/z calcd for C20H11ClN4O (M+) 358.0615, found 358.0615.
(E)-2-(1-Cyano-2-(dimethylamino)vinyl)-7-phenyl-[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (11a). Recrystallized from EtOH/dioxane mixture (1
:3), as yellowish white crystals, yield: 0.85 g (90%), m.p. 279–280 °C; IR (KBr): ν/cm−1 2227, 2202 (2CN); 1H NMR (600 MHz, DMSO-d6): δ 3.36 (s, 6H, 2CH3), 7.29 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.60–7.62 (m, 3H, Ar–H), 7.73–7.76 (m, 2H, Ar–H), 7.98 (s, 1H, enamine C–H), 9.14 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 30.55, 49.89 (2CH3), 65.96, 95.00, 113.45, 114.04, 117.91, 128.11, 128.39, 129.48, 131.31, 135.31, 148.43, 150.07, 153.27, 165.03 ppm; MS (EI): m/z (%) 315 (M+ + 1, 47.38), 314 (M+, 100.00). HRMS (EI): m/z calcd for C18H14N6 (M+) 314.1274, found 314.1274.
(E)-7-(4-Chlorophenyl)-2-(1-cyano-2-(dimethylamino)vinyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (11b). Recrystallized from dioxane, as yellowish white crystals, yield: 0.90 g (87%), m.p. 21–292 °C; IR (KBr): ν/cm−1 2228, 2203 (2C
N); 1H NMR (400 MHz, DMSO-d6): δ 3.36 (s, 6H, 2CH3), 7.31 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.69 (d, J = 8.4 Hz, 2H, Ar–H), 7.78 (d, J = 8.4 Hz, 2H, Ar–H), 7.99 (s, 1H, enamine C–H), 9.17 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 30.42, 50.07 (2CH3), 66.32, 95.57, 113.79, 114.40, 118.40, 128.96, 130.49, 131.91, 134.56, 135.19, 147.58, 150.46, 153.78, 165.62 ppm; MS (EI): m/z (%) 350 (M+ + 2, 31.57), 349 (M+ + 1, 29.98), 348 (M+, 100.00). HRMS (EI): m/z calcd for C18H13ClN6 (M+) 348.0884, found 348.0884.
(E)-2-(1-Cyano-2-phenylvinyl)-7-(4-methoxyphenyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (12a). Recrystallized from dioxane/DMF mixture (3
:1), as yellowish white crystals, yield: 1.00 g (89%), m.p. 257–258 °C; IR (KBr): ν/cm−1 2225, 2208 (2CN); 1H NMR (600 MHz, DMSO-d6): δ 3.88 (s, 3H, OCH3), 7.20 (d, J = 8.4 Hz, 2H, Ar–H), 7.52 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.61–7.62 (m, 3H, Ar–H), 7.79 (d, J = 8.4 Hz, 2H, Ar–H), 8.13–8.14 (m, 2H, Ar–H), 8.58 (s, 1H, arylidene C–H), 9.34 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, TFA-d): δ 57.46 (OCH3), 68.17, 95.42, 95.65, 117.90, 122.64, 128.22, 131.88, 133.19, 133.22, 133.58, 135.79, 137.64, 148.82, 157.48, 159.06, 159.79, 165.53 ppm; MS (EI): m/z (%) 378 (M+ + 1, 53.67), 377 (M+, 53.67), 376 (M+ − 1, 100.00). HRMS (EI): m/z calcd for C23H15N5O (M+) 377.1271, found 377.1270.
(E)-7-(4-Chlorophenyl)-2-(1-cyano-2-phenylvinyl)[1,2,4]triazolo[1,5-a]pyridine-8-carbonitrile (12b). Recrystallized from DMF as yellowish white crystals, yield: 1.05 g (93%), m.p. 282–283 °C; IR (KBr): ν/cm−1 2226, 2210 (2C
N); 1H NMR (600 MHz, DMSO-d6): δ 7.49 (d, J = 7.2 Hz, 1H, pyridine C–H6), 7.60–761 (m, 3H, Ar–H), 7.69 (d, J = 8.4 Hz, 2H, Ar–H), 7.80 (d, J = 8.4 Hz, 2H, Ar–H), 8.10–8.11 (m, 2H, Ar–H), 8.57 (s, 1H, arylidene C–H), 9.30 ppm (d, J = 7.2 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 97.20, 101.09, 113.50, 115.28, 115.43, 128.61, 128.66, 129.48, 130.13, 131.55, 132.09, 132.52, 133.72, 135.12, 148.06, 148.67, 150.25, 160.90 ppm; MS (EI): m/z (%) 383 (M+ + 2, 62.87), 382 (M+ + 1, 36.81), 381 (M+, 100), HRMS (EI): m/z calcd for C22H12ClN5 (M+) 381.0775, found 381.0775.
7-(p-Tolyl)-2-{4-(7-(p-tolyl)-8-cyano[1,2,4]triazolo[1,5-a]pyridin-2-yl)phenyl}-8-cyano[1,2,4]triazolo[1,5-a]pyridine (13a). Recrystallized from DMF as orange crystals, yield: 1.50 g (92%), m.p. above 300 °C; IR (KBr): ν/cm−1 2231, 2175 (C
N); 1H NMR (600 MHz, TFA-d): δ 2.59 (s, 6H. 2CH3), 7.60 (d, J = 8.4 Hz, 4H, Ar–H), 7.83 (d, J = 8.4 Hz, 4H, Ar–H), 8.07 (d, J = 7.2 Hz, 2H, C–H5), 8.69 (s, 4H, Ar–H), 9.26 ppm (d, J = 7.2 Hz, 2H, C–H6); 13C{1H} NMR (150 MHz, TFA-d): δ 21.05 (CH3), 94.85, 112.61, 122.40, 128.34, 129.79, 130.52, 130.94, 131.79, 135.20, 146.78, 156.93, 160.24 ppm; MS (EI): m/z (%) 543 (M+ + 1, 37.19), 542 (M+, 100). HRMS (EI): m/z calcd for C34H22N8 (M+) 542.1961, found 542.1961.
7-(4-Chlorophenyl)-2-{4-(7-(4-chlorophenyl)-8-cyano[1,2,4]triazolo[1,5-a]pyridin-2-yl)phenyl}-8-cyano[1,2,4]triazolo[1,5-a]pyridine (13b). Recrystallized from DMF as orange crystals, yield: 1.70 g (97%), m.p. above 300 °C; IR (KBr): ν/cm−1 2231, 2173 (C
N); 1H NMR (600 MHz, TFA-d): δ 7.68 (d, J = 8.4 Hz, 4H, Ar–H), 7.79 (d, J = 8.4 Hz, 4H, Ar–H), 7.79 (d, J = 7.2 Hz, 2H, C–H5), 8.62 (s, 4H, Ar–H), 9.24 ppm (d, J = 7.2 Hz, 2H, C–H6); 13C{1H} NMR (150 MHz, TFA-d): δ 95.88, 112.17, 122.16, 128.41, 130.55, 130.99, 131.42, 132.20, 135.56, 141.71, 146.73, 157.40, 158.56 ppm; MS (EI): m/z (%) 584 (M+ + 2, 71.23), 583 (M+ + 1, 42.09), 582 (M+, 100). HRMS (EI): m/z calcd for C32H16Cl2N8 (M+) 582.0869, found 582.0869.
7-(4-Bromophenyl)-2-{4-(7-(4-bromophenyl)-8-cyano[1,2,4]triazolo[1,5-a]pyridin-2-yl)phenyl}-8-cyano[1,2,4]triazolo[1,5-a]pyridine (13c). Recrystallized from DMF, as orange crystals, yield: 1.90 g (95%), m.p. above 300 °C; IR (KBr): ν/cm−1 2231, 2177 (C
N); 1H NMR (600 MHz, TFA-d): δ 7.75 (d, J = 8.4 Hz, 4H, Ar–H), 7.90 (d, J = 8.4 Hz, 4H, Ar–H), 8.01 (d, J = 7.2 Hz, 2H, C–H5), 8.66 (s, 4H, Ar–H), 9.29 ppm (d, J = 7.2 Hz, 2H, C–H6); 13C{1H} NMR (150 MHz, TFA-d): δ 95.87, 122.06, 128.48, 129.88, 130.58, 130.97, 132.62, 134.57, 135.56, 146.85, 157.52, 158.68 ppm; MS (EI): m/z (%) 672 (M+ + 2, 56.78), 671 (M+ + 1, 15.94), 670 (M+, 27.15). HRMS (EI): m/z calcd for C32H16Br2N8 (M+) 669.9859, found 669.9859.
2-Amino-4-(4-chlorophenyl)nicotinonitrile (14). Recrystallized from EtOH/dioxane mixture (3
:1), as yellow crystals, yield: 0.70 g (98%), m.p. 217–218 °C; IR (KBr): ν/cm−1 3462, 3317 (NH2), 2210 cm−1 (CN); 1H NMR (600 MHz, DMSO-d6): δ 6.70 (d, J = 5.4 Hz, 1H, pyridine C–H6), 6.99 (s, 2H, NH2), 7.60 (s, 4H, Ar–H), 8.23 ppm (d, J = 5.4 Hz, 1H, C–H5); 13C{1H} NMR (150 MHz, DMSO-d6): δ 87.58, 112.26, 116.50, 128.81, 130.05, 134.49, 135.47, 152.56, 152.78, 161.07 ppm; MS (EI): m/z (%) 231 (M+ + 2, 28.16), 230 (M+ + 1, 16.32), 229 (M+, 100). HRMS (EI): m/z calcd for C12H8ClN3 (M+) 229.0401, found 229.0401, Crystal data, moiety formula: C12H8ClN3, M = 229.67, monoclinic, a = 3.8851(9) Å, b = 19.350(4) Å, c = 14.116(3) Å, V = 1055.5(4) Å3, α = γ = 90°, β = 95.964(7)°, space group: P21/c (#14), Z = 4, Dcalc = 1.445 g cm−3, no. of reflection measured = 1863, 2θmax = 50.0°, R1 = 0.0782 (CCDC 1982383†).35
Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The facilities of Analab/SAF supported by research grants GS01/01, GS01/05, GS01/03 and GS03/08 are gratefully acknowledged.References
- J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley, New York, 5th edn, 2010 Search PubMed.
- Y. Chai, C. Li, L. Meng, X. Zhou, Z. Xia, W. Li and Y. He, The diverse supramolecular synthons formed by 2-substituted 5-morpholinomethylphenyl triazolo[1,5-a]pyridines in solid state, J. Mol. Struct., 2019, 1176, 149–156 CrossRef CAS.
- H. Oloney, N. A. Magnus, J. Y. Buser and M. C. Embry, Cyclization of methylcoumalate-derived methyl 1-benzamido-6-oxo-1,6-dihydropyridine-3-carboxylates: assembly of the [1,2,4]triazolo[1,5-a]pyridine ring system, J. Org. Chem., 2017, 82, 6279–6288 CrossRef PubMed.
- J. M. Cid, G. Tresadern, J. A. Vega, A. I. de Lucas, E. Matesanz, L. Iturrino, M. L. Linares, A. Garcia, J. I. Andres, G. J. Macdonald, D. Oehlrich, H. Lavreysen, A. Megens, A. Ahnaou, W. Drinkenburg, C. Mackie, S. Pype, D. Gallacher and A. A. Trabanco, Discovery of 3-cyclopropylmethyl-7-(4-phenylpiperidin-1-yl)-8-trifluoromethyl[1,2,4]triazolo[4,3-a]pyridine (JNJ-42153605): a positive allosteric modulator of the metabotropic glutamate 2 receptor, J. Med. Chem., 2012, 55, 8770–8789 CrossRef CAS PubMed.
- S. Ahmed, A. Ayscough, G. R. Barker, H. E. Canning, R. Davenport, R. Downham, D. Harrison, K. Jenkins, N. Kinsella, D. G. Livermore, S. Wright, A. D. Ivetac, R. Skene, S. J. Wilkens, N. A. Webster and A. G. Hendrick, 1,2,4-Triazolo-[1,5-a]pyridine HIF prolylhydroxylase domain-1 (PHD-1) inhibitors with a novel monodentate binding interaction, J. Med. Chem., 2017, 60, 5663–5672 CrossRef CAS PubMed.
- G. D. Ho, E. M. Smith, E. Y. Kiselgof, K. Basu, T. Zheng, B. Mckittrick and D. Tulshian, Substituted Triazolopyridines and Analogs Thereof, WO 2010117926A1, 2010.
- R. A. Mekheimer, A. A. R. Sayed and E. A. Ahmed, Novel 1,2,4-triazolo[1,5-a]pyridines and their fused ring systems attenuate oxidative stress and prolong lifespan of caenorhabiditis elegans, J. Med. Chem., 2012, 55, 4169–4177 CrossRef CAS PubMed.
- Y. Nakaya, D. Tanima, M. Inaba, Y. Miyakado, T. Furuhashi and K. Maeda, Heterocyclic Amide Compound, PCT/JP2014/064492, 2014 Search PubMed.
- C. Hamdouchi and P. Maiti, Phenyl-triazolo-pyridine compounds, WO 2015105786A1, 2015.
- S. D. Edmondson, A. Mastracchio, R. J. Mathvink, J. He, B. Harper, Y.-J. Park, M. Beconi, J. D. Salvo, G. J. Eiermann, H. He, B. Leiting, J. F. Leone, D. A. Levorse, K. Lyons, R. A. Patel, S. B. Patel, A. Petrov, G. Scapin, J. Shang, R. S. Roy, A. Smith, J. K. Wu, S. Xu, B. Zhu, N. A. Thornberry and A. E. Weber, (2S,3S)-3-Amino-4-(3,3-difluoropyrrolidin-1-yl)-N,N-dimethyl-4-oxo-2-(4-[1,2,4]triazolo[1,5-a]-pyridin-6-ylphenyl)butanamide: a selective a-amino amide dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes, J. Med. Chem., 2006, 49, 3614–3627 CrossRef CAS PubMed.
- H. Englert, D. Mania, D. Wettlaufer, E. Klaus, Arylcarbonylaminoalkyl-dihydrooxo-pyridines, their production and their use, US Pat., 5360808, 1994.
- J. Blake, J. P. Lyssikatos, A. L. Marlow, J. Seo, E. Wallace, H. W. Yang, Heterocyclic inhibitors of MEK and methods of use thereof, WO2005051301, 2006.
- Y. Hitotsuyanagi, S. Motegi, H. Fukaya and K. Takeya, A cis amide bond surrogate incorporating 1,2,4-trihazole, J. Org. Chem., 2002, 67, 3266–3271 CrossRef CAS PubMed.
- K. Liu, W. Shi and P. Cheng, The coordination chemistry of Zn(II), Cd(II) and Hg(II) complexes with 1,2,4-triazole derivatives, Dalton Trans., 2011, 40, 8475–8490 RSC.
- P. L. Wu, X. J. Feng, H. L. Tam, M. S. Wong and K. W. Cheah, Efficient Three-Photon Excited Deep Blue Photoluminescence and Lasing of Diphenylamino and 1,2,4-Triazole Endcapped Oligofluorenes, J. Am. Chem. Soc., 2009, 131, 886–887 CrossRef CAS PubMed.
- Y. Tao, Q. Wang, L. Ao, C. Zhong, C. Yang, J. Qin and D. Ma, Highly Efficient Phosphorescent Organic Light-Emitting Diodes Hosted by 1,2,4-Triazole-Cored Triphenylamine Derivatives: Relationship between Structure and Optoelectronic Properties, J. Phys. Chem. C, 2010, 114, 601–609 CrossRef CAS.
- K. T. Potts, H. R. Burton and J. Bhattacharyya, 1,2,4-Triazoles. XIII. Derivatives of the s-Triazolo[1,5-a]pyridine Ring System, J. Org. Chem., 1966, 31, 260–265 CrossRef CAS.
- V. J. Grenda, R. E. Jones, G. Gal and M. Sletzinger, Novel Preparation of Benzimidazoles from N-Arylamidines. New Synthesis of Thiabendazole, J. Org. Chem., 1965, 30, 259–261 CrossRef CAS.
- J. P. Raval and K. R. Desai, Synthesis and antimicrobial activity of new triazolopyridinyl phenothiazines, ARKIVOC, 2005, 13, 21–28 Search PubMed.
- S. Ueda and H. Nagasawa, Facile synthesis of 1,2,4-triazoles via a copper-catalyzed tandem addition-oxidative cyclization, J. Am. Chem. Soc., 2009, 131, 15080–15081 CrossRef CAS PubMed.
- X. Meng, C. Yu and P. Zhao, An efficient and recyclable heterogeneous catalytic system for the synthesis of 1,2,4-triazoles using air as the oxidant, RSC Adv., 2014, 4, 8612–8616 RSC.
- L. Jianguang, H. Zhiqing, Z. Jianmin, G. Yuwei, H. Ziwei and B. Xinhua, One-pot synthesis of [1,2,4]triazolo[1,5-a]pyridines from azines and benzylidenemalononitriles via copper-catalyzed tandem cyclization, Tetrahedron, 2018, 74, 3996–4004 CrossRef.
- J. Xia, X. Huang and M. Cai, Heterogeneous copper(I)-catalyzed cascade addition-oxidative cyclization of nitriles with 2-aminopyridines or amidines: efficient and practical synthesis of 1,2,4-triazoles, Synthesis, 2019, 51, 2014–2022 CrossRef CAS.
- J.-P. Zhang, Y.-Y. Lin, X.-C. Huang and X.-M. Chen, Copper(I) 1,2,4-triazolates and related complexes: studies of the solvothermal ligand reactions, network topologies, and photoluminescence properties, J. Am. Chem. Soc., 2005, 127, 5495–5506 CrossRef CAS PubMed.
- M. H. Klingele and S. Brooker, The coordination chemistry of 4-substituted 3,5-di(2-pyridyl)-4H-1,2,4-triazoles and related ligands, Coord. Chem. Rev., 2003, 241, 119–132 CrossRef CAS.
- A. Alizadeh, V. Sabegri and J. Mokhtari, A Simple one-pot procedure for the synthesis of 1,2,4-triazolo[1,5-a]pyrid-ines via pseudo five-component reactions catalyzed by molecular iodine, Synlett, 2013, 14, 1825–1829 CrossRef.
- Z. Zheng, S. Ma, L. Tang, D. Zhang-Negrerie, Y. Du and K. Zhao, PhI(OCOCF3)2-mediated intramolecular oxidative N–N bond formation: metal-free synthesis of 1,2,4-triazolo[1,5-a]pyridines, J. Org. Chem., 2014, 79, 4687–4693 CrossRef CAS PubMed.
- L. Song, X. Tian, Z. Lv, E. Li, J. Wu, Y. Liu, W. Yu and J. Chang, I2/KI-mediated oxidative N–N bond formation for the synthesis of 1,5-fused 1,2,4-triazoles from N-aryl amidines, J. Org. Chem., 2015, 80, 7219–7225 CrossRef CAS PubMed.
- B. Ashish, K. S. Rajesh and K. S. Bhupendra, Trichloroisocyanuric acid-mediated synthesis of 1,5-fused 1,2,4-triazoles from N-heteroaryl benzamidines via intramolecular oxidative N–N bond formation, Tetrahedron Lett., 2019, 60, 151026 CrossRef.
- (a) E. Gómez, C. Avendaño and A. McKillop, Ethyl carboethoxyformimidate in heterocyclic chemistry, Tetrahedron, 1986, 42, 2625–2634 CrossRef; (b) G. Hajós, G. Timári, A. Messmer, A. Zagyva, I. Miskolczi and J. G. Schantl, A new synthesis of the s-triazolo[1,5-a]pyridine ring system, Monatsh. Chem., 1995, 126, 1213–1215 CrossRef; (c) K. Hirota, Y. Nakazawa, Y. Kitade and H. Sajiki, Convenient synthesis of pyrido[4,3-d]pyrimidine-2,4(1H,3H)-diones, Heterocycles, 1998, 47, 871–882 CrossRef CAS; (d) F. Al-Omran, A.-Z. A. Elassar and A. Abou El-Khair, Synthesis and biological effects of new derivatives of azines incorporating coumarin, J. Heterocycl. Chem., 2003, 40, 249–254 CrossRef CAS.
- H. Behbehani and H. M. Ibrahim, Synthetic Strategy for pyrazolo[1,5-a]pyridine and pyrido[1,2-b]indazole derivatives through AcOH and O2-promoted cross-dehydrogenative coupling reactions between 1,3-dicarbonyl compounds and N-amino-2-iminopyridines, ACS Omega, 2019, 4, 15289–15303 CrossRef CAS PubMed.
- H. M. Ibrahim, H. Behbehani and N. S. Mostafa, Scalable sonochemical synthetic strategy for pyrazolo[1,5-a]pyridine derivatives: first catalyst free concerted [3+2] cycloaddition of alkyne and alkene derivatives to 2-imino-1H-pyridin-1-amines, ACS Omega, 2019, 4, 7182–7193 CrossRef CAS PubMed.
- H. M. Ibrahim and H. Behbehani, Sustainable synthetic approach for (pyrazol-4-ylidene)pyridines by metal catalyst-free aerobic C(sp2)–C(sp3) coupling reactions between 1-Amino-2-imino-pyridines and 1-aryl-5-pyrazolones, ACS Omega, 2019, 4, 11701–11711 CrossRef CAS PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- The crystallographic data for 5m (ref. CCDC 1982378), 5p (ref. CCDC 1982379), 5q (ref. CCDC 1982380), 5u (ref. CCDC 1982381), 5v (ref. CCDC 1982382) and 14 (ref. CCDC 1982383) can be obtained on request from the director, Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EW, UK.†.
Footnote |
| † CCDC 1982378–1982383 and crystal data for compounds 5m (CIF), 5p (CIF), 5q (CIF), 5u (CIF), 5v (CIF), 14 (CIF). For crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra02256j |
| This journal is © The Royal Society of Chemistry 2020 |