
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFree-standing Li+-conductive films based on PEO–PVDF blends†
Elena E. Ushakovaabd,
Artem V. Sergeev ab,
Artem Morzhukhinc,
Filipp S. Napolskiyc,
Olga Kristavchukd,
Alexander V. Chertovichab,
Lada V. Yashinaab and
Daniil M. Itkis*ab
ab,
Artem Morzhukhinc,
Filipp S. Napolskiyc,
Olga Kristavchukd,
Alexander V. Chertovichab,
Lada V. Yashinaab and
Daniil M. Itkis*ab
aN.N. Semenov Federal Research Center for Chemical Physics, Lab of Electrochemical Energy Conversion, Kosygina str. 4, 119991 Moscow, Russia. E-mail: d.itkis@chph.ras.ru
bLomonosov Moscow State University, Department of Chemistry, Leninskie gory 1, bld. 3, 119991 Moscow, Russia
cDubna State University, Universitetskaya 19, Dubna, 141982, Moscow region, Russia
dJoint Institute for Nuclear Research, FLNR, 141980 Dubna, Moscow region, Russia
First published on 23rd April 2020
Abstract
Solid electrolytes are of high interest for the development of advanced electrochemical energy storage devices with all-solid-state architectures. Here, we report the fabrication of the electrolyte membranes based on LiTFSI (LiN(CF3SO2)2) and PEO–PVDF blends with improved properties. We show that addition of PVDF enables preparation of free-standing films of the compositions within the so called “crystallinity gap” of the LiTFSI–PEO system known to provide high ion conductivity. We show that optimal PVDF content enables preparation of the films with reasonable elastic modulus and high ionic conductivity of about 0.3 mS cm−1 at 60 °C and about 0.1 mS cm−1 at room-temperature. Combining FTIR spectroscopy, XRD and DSC measurements we show that a noticeable fraction of PVDF remains crystalline and enhances the mechanical properties of the material, and at the same time it additionally promotes LiTFSI dissociation and disordering. Density functional theory calculations showed that the Li+–PEO–PVDF complexation energy magnitude is almost as high as that of Li–PEO complexes, thus the salt dissociation ability can be retained in spite of the introduction of the substantial amounts of PVDF required for mechanical stability.
Introduction
All-solid-state Li-ion battery design provides several advantages over conventional cells.1 Those include higher voltage, wider range of operating temperatures, improved durability and safety as the absence of liquid electrolyte minimizes the risk of fire. The solid electrolytes are naturally divided into inorganic and organic ones (both “dry” or solid, and gel-polymer). Currently, encouraging advances in the research and development of inorganic solid electrolytes (both ceramics and glasses) are reported,2,3 which exhibit high lithium ion conductivity of over 10−3 S cm−1 at room temperature and quite high mechanical modulus. However, the problem of maintaining a good sustainable ionic contact between the solid electrolyte particles and active electrode material remains open.2 Solid polymer electrolytes (SPEs) can easily overcome this difficulty due to their plasticity.Polyethylene oxide (PEO) is the most widely used as a basic polymer for SPE fabrication due to its ability to dissolve various alkali metal salts and its relatively high chemical stability.4,5 Oxygen atoms in PEO effectively coordinate alkali metal ions providing a high degree of salt dissociation, while the flexible C–O–C segments enable reasonable chain mobility. The transport of the coordinated cations is carried out due to segmental motion of the chains in amorphous PEO chains above the crystallization temperature.6 Unfortunately, PEO tends to crystallize below 60 °C. Although this transition yields satisfactory mechanical properties, the ionic conductivity sharply reduces since the ion transport in crystalline samples is carried out by hopping mechanism only. It should be noted that addition of salts suppresses PEO crystallinity.7,8 Among various salts including LiClO4, LiBF4 and LiCF3SO3 the latter was found to make a positive impact on the ionic conductivity,9 probably to the larger anion that introduce more disorder in PEO stacking. Lithium bis-(trifluoromethanesulfonyl)imide (LiTFSI) was also shown to be beneficial because of its low lattice energy and high dissociation constant providing higher ion concentration in SPE. In contrast to other available counter-anions (BF4−, ClO4− and others), large TFSI− anions characterized by high degree of charge delocalization prevent the crystallization of PEO–salt mixtures in a certain range of EO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Li ratios (12:1 to 6:1) known as “crystallinity gap”.10 Although such amorphous LiTFSI–PEO electrolytes show attractive ionic conductivity, free-standing films, which are useful for practical application in all-solid-state cells, cannot be obtained.
:Li ratios (12:1 to 6:1) known as “crystallinity gap”.10 Although such amorphous LiTFSI–PEO electrolytes show attractive ionic conductivity, free-standing films, which are useful for practical application in all-solid-state cells, cannot be obtained.
Numerous efforts have been undertaken to achieve a reasonable balance between the conductivity of amorphous PEO-based SPEs and mechanical properties of the films. The most common approaches to suppress crystallization include cross-linking,11,12 synthesis of comb-like SPEs13 and block copolymers,14,15 as well as preparation of polymer blends with PVP,16 PMHS,17 PVC,7,8,18,19 PVDF and its copolymers20,21 or adding ceramic fillers and nanoparticles (e.g. TiO2, SiO2 or Al2O3) with Lewis acid properties.22 The latter group also enables decoupling ion transport and mechanical properties. The experiments combining polymer blending and nano-filler introduction23,24 or preparation of microporous polymer electrolytes based on PVC19 and PVDF12,25,26 blends with PEO and filled by aprotic solvents were also reported.
Due to its simplicity the blending approach attracted a lot of attention, though only few of the studies reported the measurements of mechanical properties. It was shown that introduction of the second polymer reduces glass transition and melting temperatures and decreases crystallinity of the samples18,23,27,28 that has positive influence on ionic conductivity. According to Marco et al.28 solid-state NMR data indicate significant interchain mixing in the amorphous phase for PVC content ≥40% (that was later confirmed by molecular dynamics simulations).29 Although dry polymer blends were mainly studied with PVC, PVDF might be preferable for battery application due to its higher chemical stability and such blends were also reported as SPEs with LiClO4 or LiCF3SO3 salts.30,31
Herein, we focus on optimization of Li-conducting SPE obtained by blending PEO and PVDF with LiTFSI. Using FTIR spectroscopy, XRD and DSC and additional DFT modelling we uncover the reasons of enhancement of the mechanical properties and ionic conductivity achieved for optimized material composition.
Experimental
Sample preparation
Poly(ethylene oxide) powder with an average molecular weight Mv ≈ 1000000 (Sigma-Aldrich, product 372781), poly(vinylidene fluoride) with molecular weight Mv ≈ 1000000–1100000 (Solvay Solef 5130), propylene carbonate (anhydrous, 99.7%, Sigma-Aldrich) and lithium bis-(trifluoromethanesulfonyl)imide (99.5%, MTI) were used as-received for SPE films preparation.
The films were prepared in a conventional solution-casting way. EO:Li ratio were 20:1, 10:1, 8:1, 6:1 and 3:1. The mass of poly(vinylidene fluoride) was calculated to provide the PVDF weight fraction of 0, 10, 20, 30 and 40% in the resulting mixture. Polymer solutions were prepared in an argon-filled glove box (pH2O < 1 ppm, pO2 < 10 ppm). LiTFSI was dissolved in anhydrous propylene carbonate (PC) at 110 °C under constant stirring. After complete dissolution, PEO and PVDF were consistently added into the above solution under intensive stirring and heating in appropriate proportions so that overall dry polymer content in the solution was 10 wt%. The resulting mixtures were stirred for 24 h. The polymers were dissolved completely in this conditions. Subsequently, the prepared compositions were cast onto a Teflon plates and allowed to evaporate at 110 °C in the glove box for 72 h. Finally, the samples were dried under vacuum at 100 °C for 24 h to obtain dry freestanding films.
Materials characterisation
The XRD measurements were carried out using PANalytical Empyrean Advance X-ray diffractometer equipped with PIXcel3D detector in Bragg–Brentano geometry (CuKα radiation, 0.04 rad Soller slits in incident beam optics). The XRD patterns were collected in 2θ range from 10° to 85° with a step size 0.026°. Diffraction patterns were recorded for the polymer films casted on polyethylene-laminated aluminum foil. To avoid exposure to air the samples were placed into special evacuated cell (10−2 to 10−3 mbar), which was kept at 25 °C.Differential scanning calorimetry measurements were done using Netzsch DSC 204 F1 Phoenix. The samples were weighed and sealed in Al pans in dry-air conditions. DSC curves were recorded from −70 °C to 200 °C at a rate of 10 °C min−1. Netzsch Proteus software was used to define the temperatures of glass transition and melting.
Thermofisher Scientific Nicolet 6700 spectrometer was used to obtain FTIR spectra of PEO, PVDF, LiTFSI powders and the SPE films. The measurements were performed on a horizontal plane monolithic diamond ATR (GladiATR) in spectral range 4000–500 cm−1 with 32 scans for each spectrum and spectral resolution of 4 cm−1.
The elastic modulus of SPE films has been investigated using dynamic mechanical analysis. Netzsch DMA 242 E Artemis analyzer equipped with a shear jaw for film clamping was used for measurements. The rectangular films were exposed to a dynamic strain with an amplitude of 0.3% in the frequency range between 0.05 and 16 Hz. The dynamic strain amplitude of 0.3% at 10 Hz was applied in a temperature range from 25 to 200 °C with a heating rate of 2 °C min−1 for temperature sweeps. To define the optimal measurement parameters the sinusoidal frequency and amplitude of the tensile dynamic load had been preliminary varied until a linear response was achieved (see ESI† for more details).
DFT calculations
Complexation energy calculations were carried out using Gaussian software package. Geometry optimization was performed at B3LYP/6-31G** level followed by single point energy calculation with the 6-311++G** basis set. To recover nuclear motion energy vibrational frequencies were computed analytically within the harmonic approximation using the 6-31G** basis set. Li-polymer complexation enthalpy was calculated as a difference between the enthalpy of gas phase complex and the sum of enthalpies of two separately calculated systems containing Li+ ion and one or two oligomer chains.Electrochemical measurements
To determine the ionic conductivities of PEO–PVDF SPEs the films with a thickness from 0.06 to 0.1 mm were cut into discs of 1.77 cm2, sandwiched between two stainless steel electrodes of the same size and then placed in a sealed glass cell with a built-in micrometer to control the film thickness in situ after cell assembly and heating. Li+ transference numbers were evaluated using the symmetric Li/Li cell assemblies. Lithium foil electrodes and the electrolyte membrane, together with the spacers and compression spring were sealed in 2032-type coin cell. The cells were assembled in an argon-filled glove box (<0.1 ppm moisture and <10 ppm O2).The electrochemical impedance spectroscopy (EIS) measurements were performed using Biologic SP-200 potentiostat/galvanostat with an AC amplitude of 100 mV in the frequency range from 1 MHz to 100 mHz. The impedance spectra were recorded at room temperature (25 °C) and at 60 °C. Measurements at elevated temperature were preceded by keeping the cells for about 5 hours at the temperature set-point to guarantee thermal equilibrium.
The Nyquist plots of AC impedance were fitted by Randles equivalent circuit modified with a constant phase element using EC-Lab software package. The membrane conductivity was calculated according to the equation below as a value inverse to the bulk resistance, which was determined from the intercept of the low frequency signal in the Nyquist plot with the real axis:
| σ = l/ARb |
Bruce–Vincent method was used for transference number determination. To obtain the DC polarization curves constant voltage of 20 mV was applied to the cell until the steady state current was reached. AC impedance spectra were measured before and after polarization; as a result, the initial and steady state resistances were determined. Li+ transference numbers were calculated using Bruce–Vincent equation shown below:32
| tLi+ = Iss(V − I0R0)/I0(V − IssRss) |
Results and discussion
LiTFSI is one of the salts, which are often used for fabrication of PEO-based SPEs, since LiTFSI–PEO system provides the highest conductivity (10−4 to 10−3 S cm−1 at 60 °C) for the composition range within so called “crystallinity gap” (EO:Li from ca. 12:1 to 6:1). It is known that such films cannot be prepared as elastic free-standing films. To resolve this issue, we suggest blending PEO with PVDF homopolymer.
Material properties of the obtained SPE films are summarized in Fig. 1. The schematic diagram illustrating the ranges of compositions (EO:Li and PVDF content), for which free-standing SPE films can be obtained, is shown in Fig. 1a. The ionic conductivity is illustrated by the point color. The conductivity is also shown in Fig. 1b as the dependences on Li salt content for different polymer blends. One can see that the addition of 10 wt% of PVDF does not yield any significant improvement in both ionic conductivity and mechanical modulus, which is presented in Fig. 1c and reaches nearly 40 MPa at room temperature. E′, however, quickly decreases with temperature rise up to 50 °C due to transition from rubbery to viscous flow behavior. An increase of PVDF content results in much better mechanical properties of the material. The films containing 20 wt% PVDF with EO:Li of up to 10:1 can be easily detached from the substrates, while adding 30–40 wt% PVDF enables obtaining the free-standing films with significantly higher lithium salt concentrations (up to EO:Li of 3:1). At the same time, the samples containing 30 wt% PVDF exhibit maximal ionic conductivity at certain salt and PVDF concentration as seen in Fig. 1a and b. The maximal value of total specific conductivity of about 0.3 mS cm−1 at 60 °C is reached at EO:Li ratio around 6:1 that lies in the “crystallinity gap” of LiTFSI–PEO system. At higher LiTFSI concentrations the conductivity goes down, which is most probably due to ion pairing. Adding more PVDF (40 wt%) also results in a drop of conductivity as the charge carrier concentration is decreased (the AC impedance spectra for all SPE films at 25 and 60 °C are shown in Fig. S1 of ESI, while the values are summarized in Table S1†).
 | ||
| Fig. 1 (a) Diagram showing the region in which free-standing LiTFSI–PEO–PVDF films cannot be obtained due to poor mechanical properties. The region with experimental points represents the film compositions that were obtained as free-standing films. Color denotes the ionic conductivity measured at 60 °C. For pure PEO (0% PVDF) “crystallinity gap” region is shown, in which no crystalline complexes of LiTFSI and PEO are formed. (b) The dependence of ionic conductivity measured at 60 °C on EO:Li ratio for various PVDF content, which is denoted by color. (c) Temperature dependencies of the films elastic modulus measured using DMA at 10 Hz and ε = 0.3%. Solid lines denote samples with EO:Li = 20:1, the dotted curve is for EO:Li = 6:1. Curve color denotes PVDF content. The inset shows a photograph of the SPE film (EO:Li 6:1, 30 wt% PVDF), stretched between tweezers. (d) The dependence of ionic conductivity measured at 25 °C on EO:Li ratio for various PVDF content, which is shown by color. | ||
Surprisingly, the mechanical behavior of the blends with high and low EO:Li ratios (low and high LiTFSI concentrations, correspondingly) differs and we observe less pronounced decrease of elastic modulus with temperature for concentrated SPEs. As PEO is expected to be amorphous at room temperature at high LiTFSI concentrations, we can suppose that entanglement length is not decreased due to formation of crystalline lamellas, so PEO and PVDF chains form homogeneous and dense entanglements network preventing viscous flow of the material up to PVDF softening above 100 °C. Even at 60 °C the samples with EO:Li of 6:1 demonstrate 20–30 MPa for PVDF content of 30 and 40 wt%, respectively (see Fig. S2 of ESI† for details on dynamical mechanical analysis). In contrast, in the films with low LiTFSI concentration (EO:Li 20:1) crystallization of PEO and LiTFSI–PEO crystalline complexes result in higher entanglement length and the polymer blend softens at relatively low temperatures.
Room-temperature ionic conductivity was found to decrease along with PVDF addition for low LiTFSI concentration (see the data points for EO:Li of 20:1 in Fig. 1d) as can be expected due to dilution of the system by PVDF and diminishing of charge carrier concentration. However, addition of the salt in higher concentration to the samples containing 30 wt% of PVDF resulted in an order of magnitude increase of room-temperature conductivity. At the same time the sample with PVDF fraction above 30 wt% demonstrated and order of magnitude lower conductivity at both room- and elevated temperature, while the cation concentration decreased slightly while adding additional 10 wt% of PVDF. This fact points out that a percolating network of conducting regions is broken at certain fraction of poly(vinylidene fluoride) in the blend. All-in-all, we conclude that 30 wt% PVDF is an optimal concentration.
To get further insight into the mechanisms providing material properties improvement, we studied the structure of the films of different composition. Analysis of the blended SPEs by X-ray diffraction revealed that PVDF doesn't act as a plasticiser itself showing no effect on PEO crystallinity in as-fabricated films. As seen in Fig. 2a, SPEs with added PVDF demonstrate reflections in XRD patterns originating from crystalline regions and PEO–LiTFSI complexes33,34 as well as PVDF-free sample (reference patterns for polymer powders can be found in Fig. S3 of ESI†). The PVDF films casted from solutions in propylene carbonate, at the same time, are poorly crystalline showing few wide features in the diffraction pattern in Fig. 2b. The PEO–PVDF blends with high PVDF content expectedly demonstrate noticeable intensity of reflection at 20.4°, which we ascribe to PVDF. Adding higher concentration of LiTFSI to PEO–PVDF blends lead to nearly amorphous samples showing no strong diffraction peaks (Fig. 2b). Interestingly, at EO:Li of 3:1 as well the crystal complexes were not detected for the samples containing PVDF, although it can be expected for PEO–LiTFSI system.10
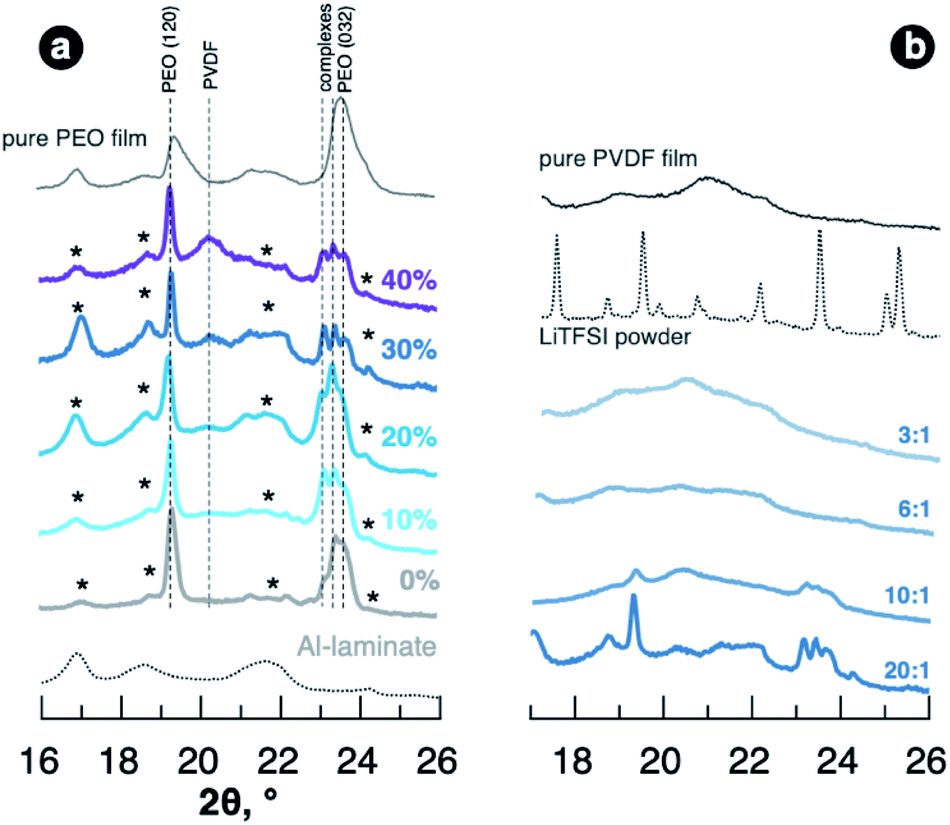 | ||
| Fig. 2 Representative regions of XRD patterns of the films with EO:Li 20:1 and different PVDF fraction (a) and for the films with 30 wt% PVDF and various EO:Li ratio (b). Patterns are collected using CuKα radiation. | ||
In order to understand the role of PVDF in polymer blend SPEs crystallization in more detail, we further investigated the samples using differential scanning calorimetry. DSC curves obtained for the samples with various PVDF content and revealing endothermic peaks are shown in Fig. 3a. Along with crystalline PEO melting at around 50 °C, melting of crystalline PVDF at about 150 °C becomes more and more pronounced as we increase its fraction in the blends. The crystallinity of PEO was estimated from melting enthalpy using the equation
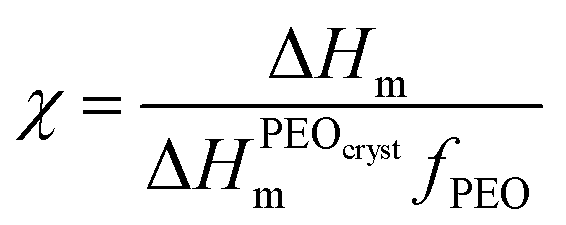
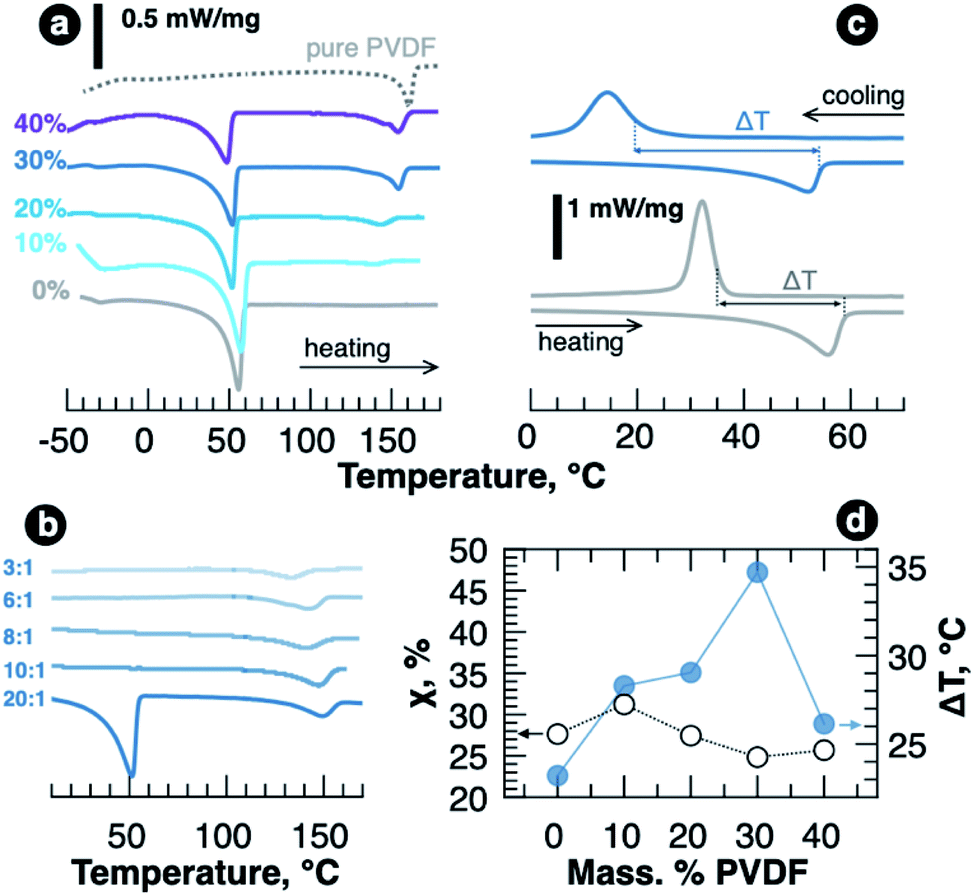 | ||
| Fig. 3 DSC curves for polymer films with EO:Li ratio of 20:1 and various PVDF content (a) and for the samples with 30 wt% PVDF and various EO:Li ratio (b). The data were recorded at 10 °C min−1 heating rate. (c) Heating and cooling DSC curves (10 °C min−1 rate) for the samples with 0 and 30 wt% PVDF and LiTFSI concentration corresponding to EO:Li of 20:1. (d) Dependence of PEO crystallinity (χ) and the temperature range (ΔT), in which PEO is fully amorphous, on the wt. fraction of PVDF for the films with EO:Li of 20:1. | ||
Adding LiTFSI to PEO–PVDF blend resulted in disappearance of PEO melting peak at LiTFSI concentration corresponding to EO:Li of 10:1 as it happens for pure PEO–LiTFSI system.6 However, in contrast to PVDF-free samples, no melting of PEO or PEO–LiTFSI complexes was observed in the region of high concentrations (EO:Li = 3:1), and, as seen in Fig. 3b, the only endothermic peak in DSC curve was detected above 120 °C and it was ascribed to semicrystalline PVDF melting. PVDF melting enthalpy is decreased more than two times (from 12 to 5 J g−1 for 20:1 and 3:1 EO:Li ratios, correspondingly), which might indicate some interaction of PVDF chains with Li+ cation or Li+–PEO complexes. The additional support for this can be found if we analyze the crystallization temperatures for LiTFSI–PEO–PVDF melts. Fig. 3c shows how the temperature range (ΔT), in which LiTFSI–PEO complexes remain amorphous, widens upon PVDF addition (thermograms for all samples are shown in Fig. S4,† while the temperatures and enthalpies are listed in Table S2 of ESI†). The value of ΔT is plotted vs. PVDF weight fraction in Fig. 3d showing that adding 30 wt% PVDF provides the widest temperature range with no crystalline complexes, which spreads down to room-temperature and can enable high conductivities after heating/cooling cycle. The kinetics of crystallization, however, should be studied separately.
To analyze the possibility of PVDF interactions with lithium ions we performed DFT calculations and estimated the enthalpy of Li+ cation complexation (solvation) by both polymers. For simplicity we modelled polymers by relatively small oligomer chains – 3, 4 or 5 monomer units for PEO (labeled as H(EO)nCH3, where n = 3, 4 or 5) and 3 monomer units in α-configuration for PVDF. The oligomer molecules were terminated by –CH3 for both polymers. We considered complexation of a single Li+ cation by a single or by two oligomer chains, and also estimated enthalpy of solvation by two different oligomer chains (PEO + PVDF).
The calculated complexation enthalpy values for all of the aforementioned systems are collected in Table 1. The simulation results for Li+–PEO systems is in accordance with the fact that the PEO chains tend to fold around the cation and the complexation enthalpy increases in absolute magnitude with the number of O atoms coordinating Li+ with a typical interatomic distance of 1.9–2.2 Å. In case of a single PEO oligomer chain the maximum coordination number is 5 due to the steric limitations; such configuration yields solvation enthalpy of −4.5 eV. The possibility of 6-fold coordination was tested using the initial configuration of two H(EO)3CH3 chains in a form of crossed half-arcs with Li+ ion between them (see Fig. 4a). However, the geometry optimization resulted in the shift of one oxygen atom apart from Li+ to the distance longer than 3 Å, while in all the other cases the Li–O distance was in the range 1.9–2.2 Å. Therefore, for two PEO chains the maximum coordination number was found to be 5 as well, though the complexation enthalpy is almost 0.2 eV greater in absolute value against the case with single linear H(EO)3CH3 chain.
| System | Complexation enthalpy, eV |
|---|---|
| Li+ + H(EO)3CH3 | −3.61 |
| Li+ + H(EO)4CH3 | −4.09 |
| Li+ + H(EO)5CH3 | −4.50 |
| Li+ + 2H(EO)3CH3 | −4.69 |
| Li+ + H(CH2CF2)3CH3 | −2.31 |
| Li+ + 2H(CH2CF2)3CH3 | −3.38 |
| Li+ + H(EO)3CH3 + H(CH2CF2)CH3 | −4.37 |
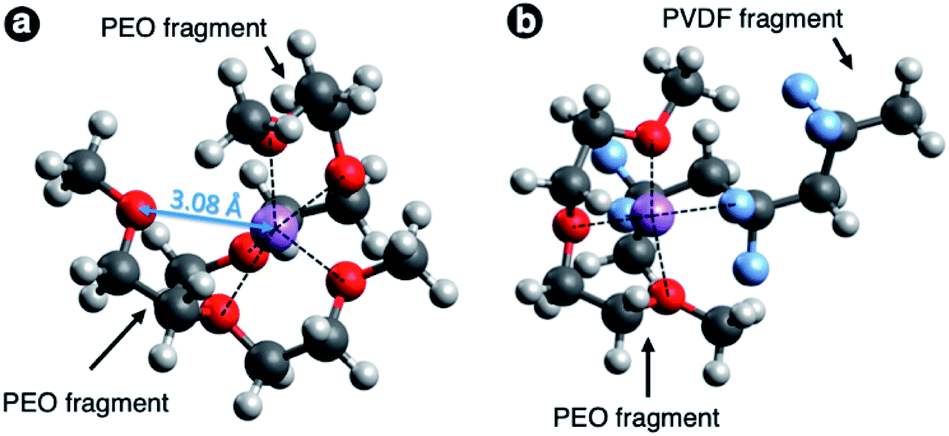 | ||
| Fig. 4 Optimized geometries of Li+ complexes with two H(EO)3CH3 chains (a) and with one H(EO)3CH3 chain + one H(CH2CF2)3CH3 chain (b). Black dash lines denote coordinating atoms (distance is within in the range 1.9–2.2 Å). Oxygen are shown in red, lithium – in purple, fluorine – in blue, carbon – in dark grey, hydrogen – in light grey. | ||
The complexation enthalpy in the case of Li+ coordinated by two PVDF chains was calculated to be about −3.4 eV, i.e. even lower than in the case of Li+ coordination by the shortest of considered PEO chains (H(EO)3CH3). This result indicates much lower solvation ability of PVDF towards Li+ ions as compared to PEO, and hence much lower solubility of Li salts. One can expect that the admixing PVDF to PEO-based electrolyte leads to a proportional decreased of free charge carrier concentration. However, simulation of H(EO)3CH3 + H(CH2CF2)CH3 configuration gave us unexpectedly high magnitude of solvation enthalpy. Optimized geometry for this system is shown in Fig. 4b. In addition to coordination by three oxygen atoms Li+ ion interacts with PVDF, and complexation enthalpy becomes almost as high as in case of coordination by five oxygens of PEO. This makes us believe that the effect of lower salt solubility in the blends with PVDF, which can be expected, won't be realized in the case of a proper polymer mixing at molecular level, while the benefit of higher mechanical stability would be preserved.
The hypothesis of possible participation of PVDF chains in cation solvation by PEO–PVDF blends was further checked using FTIR spectroscopy. The representative regions of FTIR spectra of the SPEs with 30 wt% of PVDF and various concentration of LiTFSI are shown in Fig. 5a (full spectral range and also the spectra for the samples with other PVDF content are shown in Fig. S5 in ESI†). Addition of the salt to the blends results in appearance of well-resolved bands of TFSI− anion vibrations in 500–675, 730–830 and 1170–1235 cm−1 ranges, in which there is no or little interference with PEO or PVDF bands. In the blends δsCF3 vibration band at 1205 cm−1 is shifted by ca. 15 cm−1 to lower wavenumbers most probably due to LiTFSI dissociation. We also observe typical signs of Li+ complexation by PEO.36,37 The peak in the 2800 to 3000 cm−1 region, which is assigned to ναC–H vibrations in pure PEO, widens upon salt addition. The intensities of the components of characteristic three peaks in 1050–1150 cm−1 region, which are connected with C–O–C vibrations, are rearranged, and the components are slightly shifted (by ca. 6–8 cm−1) to lower wavenumbers. At the same time it seems that PVDF is also involved in the interaction as the band at 880 cm−1 ascribed to asymmetric stretching vibrations of CF2 groups in PVDF (ναCF2) is shifted towards higher wavenumbers. The wagging CH2 vibration band at ca. 1400 cm−1 also loses it intensity with an increase of LiTFSI concentration. The interactions of PVDF chains can also inhibit crystallization of Li–PEO complexes at low EO:Li ratios.
 | ||
| Fig. 5 (a) FTIR spectra of SPE films with 30 wt% PVDF and various EO:Li ratios. FTIR spectra of pure PEO, PVDF and crystalline LiTFSI are shown for the reference. Regions are colored to denote the wavenumber ranges representative for TFSI− anion vibrations (grey), PEO (blue) and PVDF (violet). Cation transference numbers for the samples with different fraction of PVDF and 20:1 EO:Li ratio (b) and for the samples with different EO:Li ratio (c). | ||
While PVDF seems to assist in LiTFSI dissolution that is supported by DFT calculations and FTIR spectroscopy, we should note that blending with PVDF doesn't lead to any negative effect on cation transference number for a certain composition range. On the contrary, we see nearly no effect on tLi+ for PVDF fraction below 40 wt% in the blends (Fig. 5b). The samples with optimal salt concentration possess cation transference number over 30% that is seen in Fig. 5c (current transients and AC impedance spectra used for transference number determination are shown in Fig. S6 of ESI, values are summarized in Table S3†). The basic reasons for the transference number decrease at EO:Li of 3:1 are not clear and require more detailed studies.
Conclusions
Summarizing all of above, we have shown that addition of PVDF to LiTFSI/PEO solutions in propylene carbonate enables casting of the free-standing SPE films in a wide range of EO:Li and PVDF weight fractions. PVDF not only helps to maintain mechanical properties of the films but also doesn't inhibit LiTFSI dissolution, which can be expected. On the contrary, as can be deducted from FTIR data and DFT calculations, PVDF participates in Li+ coordination by fluorine atoms and promote LiTFSI dissociation making it possible to achieve reasonable conductivity values of about 0.3 mS cm−1 at 60 °C. As confirmed by XRD and DSC, the films with 30 wt% of PVDF and EO:Li of 6:1 maintain amorphous state of LiTFSI–PEO–PVDF complexes being cooled down even to room temperature and the conductivity remains high. Even as-casted films of the mentioned composition exhibit ionic conductivity of 0.1 mS cm−1 at room-temperature, which is not so high, however, it can be enough for battery to start up, while typical operation is accompanied by self-heating and temperature rise to 40–60 °C.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by Russian Science Foundation (grant # 19-73-00312). D. M. I. and L. V. Y. acknowledge the governmental funding of N.N. Semenov Federal research center for chemical physics (topic 47.25). The authors are grateful to Dr I. Bobrikov and S. Sumnikov for XRD patterns acquisition.Notes and references
- J. G. Kim, B. Son, S. Mukherjee, N. Schuppert, A. Bates, O. Kwon, M. J. Choi, H. Y. Chung and S. Park, J. Power Sources, 2015, 282, 299–322 CrossRef CAS
.
- F. Zheng, M. Kotobuki, S. Song, M. O. Lai and L. Lu, J. Power Sources, 2018, 389, 198–213 CrossRef CAS
- J. C. Bachman, S. Muy, A. Grimaud, H.-H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC
- S. Tung, S. Ho, M. Yang, R. Zhang and N. Kotov, Nat. Commun., 2015, 6, 6152 CrossRef CAS PubMed
- Z. Gadjourova, Y. G. Andreev, D. P. Tunstall and P. G. Bruce, Nature, 2001, 412, 520–523 CrossRef CAS PubMed
- N. Reddeppa, A. K. Sharma, V. V. R. N. Rao and W. Chen, Microelectron. Eng., 2013, 112, 57–62 CrossRef CAS
- R. Nadimicherla, R. Kalla, R. Muchakayala and X. Guo, Solid State Ionics, 2015, 278, 260–267 CrossRef CAS
- S. Rajendran, R. S. Babu and M. U. Rani, Bull. Mater. Sci., 2011, 34, 1525–1530 CrossRef CAS
- S. Lascaud, M. Perrier, A. Vallee, S. Besner, J. Prud'homme and M. Armand, Macromolecules, 1994, 27, 7469–7477 CrossRef CAS
- T. Sakakibara, M. Kitamura, T. Honma, H. Kohno, T. Uno, M. Kubo, N. Imanishi, Y. Takeda and T. Itoh, Electrochim. Acta, 2019, 296, 1018–1026 CrossRef CAS
- Y. Song, S. Wu, X. Jing, J. Sun and D. Chen, Radiat. Phys. Chem., 1997, 49, 541–546 CrossRef CAS
- M. Guo, M. Zhang, D. He, J. Hu, X. Wang, C. Gong, X. Xie and Z. Xue, Electrochim. Acta, 2017, 255, 396–404 CrossRef CAS
- W.-S. Young, W.-F. Kuan and T. H. Epps, J. Polym. Sci., Part B: Polym. Phys., 2013, 52, 1–16 CrossRef
- Q. Lu, J. Fang, J. Yang, G. Yan, S. Liu and J. Wang, J. Membr. Sci., 2013, 425–426, 105–112 CrossRef CAS
- K. Sundaramahalingam, M. Muthuvinayagam and N. Nallamuthu, Polym. Sci., Ser. A, 2019, 61, 565–576 CrossRef CAS
- Y.-J. Li, C.-Y. Fan, J.-P. Zhang and X.-L. Wu, Dalton Trans., 2018, 47, 14932–14937 RSC
- S. Ramesh, A. H. Yahaya and A. K. Arof, Solid State Ionics, 2002, 148, 483–486 CrossRef CAS
- S. Ramesh, T. Winie and A. K. Arof, Eur. Polym. J., 2007, 43, 1963–1968 CrossRef CAS
- I. Elashmawi, N. Elsayed and F. Altalhi, J. Alloys Compd., 2014, 617, 877–883 CrossRef CAS
- S. Jayanthi, Adv. Compos. Hybrid Mater., 2019, 2, 351–360 CrossRef CAS
- P. Yao, H. Yu, Z. Ding, Y. Liu, J. Lu, M. Lavorgna, J. Wu and X. Liu, Front. Chem., 2019, 7, 522 CrossRef CAS PubMed
- H. W. Han, W. Liu, J. Zhang and X.-Z. Zhao, Adv. Funct. Mater., 2005, 15, 1940–1944 CrossRef CAS
- L. Lee, S.-J. Park and S. Kim, Solid State Ionics, 2013, 234, 19–24 CrossRef CAS
- F. Deng, X. Wang, D. He, J. Hu, C. Gong, Y. S. Ye, X. Xie and Z. Xue, J. Membr. Sci., 2015, 491, 82–89 CrossRef CAS
- J. Xi, X. Qiu, J. Li, X. Tang, W. Zhu and L. Chen, J. Power Sources, 2006, 157, 501–506 CrossRef CAS
- I. Nicotera, G. A. Ranieri, M. Terenzi, A. V. Chadwick and M. I. Webster, Solid State Ionics, 2002, 146, 143–150 CrossRef CAS
- C. Marco, M. A. Gömez, J. G. Fatou, A. Etxeberria, M. M. Elorza and J. J. Iruin, Eur. Polym. J., 1993, 29, 1477–1481 CrossRef CAS
- Z. Luo and J. Jiang, Polymer, 2010, 51, 291–299 CrossRef CAS
- M. M. E. Jacob, S. R. S. Prabaharan and S. Radhakrishna, Solid State Ionics, 1997, 104, 267–276 CrossRef CAS
- P. Dhatarwal and R. J. Sengwa, Ionics, 2019, 1–17 Search PubMed
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef CAS
- M. Marzantowicz, J. R. Dygas, F. Krok, J. L. Nowiński, A. Tomaszewska, Z. Florjańczyk and E. Zygadło-Monikowska, J. Power Sources, 2006, 159, 420–430 CrossRef CAS
- Y. Takahashi and H. Tadokoro, Macromolecules, 1973, 6, 672–675 CrossRef CAS
- R. S. Porter, Macromolecular physics, volume 3—crystal melting, ed. B. Wunderlich, Academic Press, New York, 1980, vol. 18, 1980, p. 363 Search PubMed
- S. J. Wen, T. J. Richardson, D. I. Ghantous, K. A. Striebel, P. N. Ross and E. J. Cairns, J. Electroanal. Chem., 1996, 408, 113–118 CrossRef
- I. Rey, J. C. Lassègues, J. Grondin and L. Servant, Electrochim. Acta, 1998, 43, 1505–1510 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02325f |
| This journal is © The Royal Society of Chemistry 2020 |