DOI:
10.1039/D0RA02339F
(Paper)
RSC Adv., 2020,
10, 19346-19352
Synthesis, acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) activities, and molecular docking studies of a novel compound based on combination of flurbiprofen and isoniazide†
Received
12th March 2020
, Accepted 4th May 2020
First published on 20th May 2020
Abstract
Synthesis of a compound with balanced bioactivities against a specific target is always a challenging task. In this study, a novel compound (1) has been synthesized by combination of flurbiprofen and isoniazide and shows ∼2.5 times enhanced acetylcholinesterase (AChE) inhibition activity and ∼1.7 times improved butyrylcholinesterase (BuChE) inhibition activity compared to flurbiprofen and a standard drug (i.e. physostigmine). A comparative AutoDock study has been performed, based on the optimized structure, by the DFT/B3LYP method, which confirmed that compound (1) is more active against AChE and BuChE, with calculated binding energies of −12.9 kcal mol−1 and −9.8 kcal mol−1 respectively as compared to flurbiprofen and an eserine (physostigmine) standard for which the binding energy was calculated to be −10.1 kcal mol−1 and −8.9 kcal mol−1, respectively. A mixed mode of inhibition of AChE and BuChE with compound 1 was confirmed by Lineweaver–Burk plots. AChE and BuChE inhibition activity alongside docking results suggests that compound (1) could be used for treatment of Alzheimer's disease. Moreover, compound (1) also exhibit better α-chymotrypsin activity compared to flurbiprofen. Furthermore, in vitro and in vivo analysis confirmed that compound (1) exhibit more activity and less toxicity than the parent compounds.
Introduction
The history of non-steroidal anti-inflammatory drugs (NSAIDs) dates back thousands of years but the mechanism of NSAID therapies was first fully understood by John Vane in 1971.1–3 Nowadays, NSAIDs (naproxen, indomethacin, aspirin, ibuprofen, flurbiprofen and diclofenac) have been recommended for diseases such as pain, short term fever and inflammation.4–7 Scientists have worked hard to understand the kinetics and structure–activity relationships of these drugs.8,9 In this regard, computer-aided drug design (CADD) is one of the most powerful tools.10–13 It enables us to search and understand the interactions of ligands with potential protein targets.14 Moreover, structure–activity relationships can be established in a better way by comparing the experimental results with theoretical studies. Therefore, CADD has an intrinsic benefit of understanding the phenomenon at the molecular level and providing correct assignments.15
A patient suffering from Alzheimer's disease (AD) experience a progressive and irreversible disorder of brain which slowly destroy thinking and memory skills at large.16 AD is currently ranked as the sixth leading cause of death in United States and there are almost 50 million people around the world suffering from this disease.16–18 It has been observed that change in acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) activity in cerebral cortex and hippocampus contribute to the disease progression.17–19 Increased or unchanged BuChE activity and decreased AChE activity are commonly observed in certain brain regions of patients suffering from AD.20–22 Human AChE active site, a long gorge with overall length of approximately 20 Å, mainly consisting of catalytic active site (CAS) at bottom of gorge while peripheral anionic site (PAS) is situated near the entrance of gorge. These two are linked by a narrow groove. CAS forms the catalytic triad, Ser200, Glu327 and His440, and is responsible for hydrolysis of AChE inside the triad.23,24 PAS consists of several aromatic residues, including Tyr70, Tyr121, and Trp279. Compounds that can interact with both CAS and PAS are believed to exert multiple therapeutic effects.25 The shape and arrangement of the active site of BuChE is similar to that of AChE; however, the volume of the catalytic site in BuChE is much larger than that of AChE.26 Researchers from all over the world are trying to explore new and novel strategies to develop effective methods for treatment of AD and other diseases as well, but it is a challenging task to obtain a specific compound with balanced activities against the specific targets most importantly retaining the drug-like properties.27 Synthesis of novel drug by using existing NSAIDs is an area which is largely unexplored and it has been observed that NSAIDs can be tuned for better lipophilicity, reduced toxicity and better bio-availability.28 Therefore a clever design and synthesis of compound having aforementioned properties is need of the day in order to grasp and improve the pharmacological benefits of NSAIDs.29
Keeping in view of the facts about AD and limitations of existing drugs, a novel compound based on combination of flurbiprofen and isoniazide, N′-(2-(2-fluoro-[1,1′-biphenyl]-4-yl)propanoyl) isonicotinohydrazide (1), has been synthesized. The synthesized compound (1) was subjected to in vitro analysis which shows ∼2.5 times and ∼1.7 times enhanced AChE and BuChE inhibition activity respectively compared to flurbiprofen and standard physostigmine. These experimental results has been supported by comparative AutoDock study of compound (1) with AChE and BuChE which also confirmed that compound (1) is more active against AChE and BuChE with binding energy of −12.9 kcal mol−1 and −9.8 kcal mol−1 respectively as compared to flurbiprofen for which binding energy was calculated to be −8.2 kcal mol−1. Compound (1) showed improved α-chymotrypsin activity as well. Lineweaver–burk plot suggests mixed mode of inhibition of AChE and BuChE with compound 1. Experiments suggests that the compound 1 is slightly more lipophilic compared to flurbiprofen. AChE and BuChE inhibition activities aided with docking results suggested that compound (1) could be used for Alzheimer's disease. To the best of our knowledge, no report has been published yet which describes synthesis, experimental procedures, molecular docking and in vitro studies of the compound (1). Furthermore, the present data as theoretically and experimentally can be helpful for further studies of compounds/derivatives of flurbiprofen.
Results and discussion, experimental
Synthesis
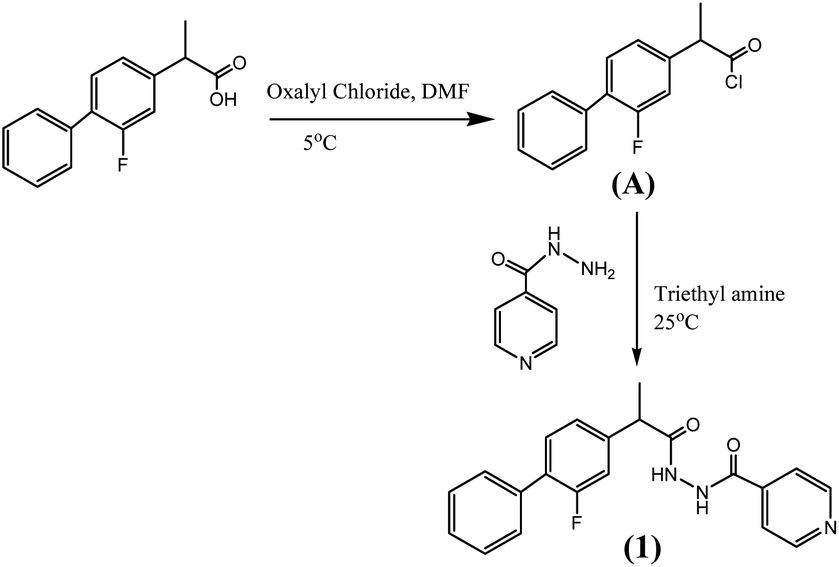
Synthesis of the compound (1) was done by preparing acid chloride of flurbiprofen using oxalyl chloride which was then allowed to react with isoniazide. Scheme 1 (detail in ESI†).
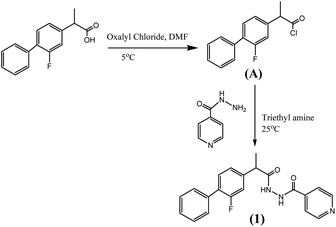 |
| | Scheme 1 Synthesis of novel compound (1) based on combination of flurbiprofen and isoniazide. | |
Biological activities
Toxicity study
The LD50 (oral, rats) values were found to be isoniazide: 645 mg kg−1, flurbiprofen: 115 mg kg−1 and compound (1): 3431 mg kg−1. Toxicity results proved that the compound (1) is much safer to use than the parent analogues.
Lipophilicity studies
In context of drug discovery one of the most important parameter, despite the fact that the drug should show a significant activity against the potential target, is identification of drugs which are more likely to be well absorbed and distributed in human body. It suggests that a drug must be lipophilic enough to penetrate the lipid cores of membranes, but not so lipophilic that they get stuck there.32 Lipophilicity of the compound (1) was calculated by following the recently published method.33 log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P of compound (1) was calculated to be 3.94 which is slightly higher than flurbiprofen (logP 3.82). Flurbiprofen possess carboxylic moiety, therefore it is less lipophilic than compound (1) that contains amide linkage.
P of compound (1) was calculated to be 3.94 which is slightly higher than flurbiprofen (logP 3.82). Flurbiprofen possess carboxylic moiety, therefore it is less lipophilic than compound (1) that contains amide linkage.
Molecular docking studies
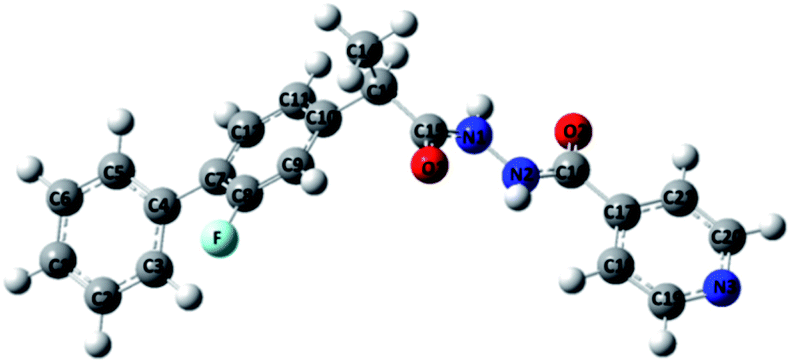
The study was designed for compound (1) against acetylcholinesterase and butyrylcholinesterase enzymes with the following communications; Intel(R) core i7 @ 3.50 GHz system having 8 GB RAM with windows 7 operating platform. Protein–ligand docking was carried out using Autodock Vina software.34 X-ray crystal structures of human BChE (Pdb: 1P0P with 2.30 Å resolution) from the RCSB protein data bank35 was selected as the target protein based on suitable resolution and co-crystallized ligands, i.e. AChE and BuChE. The energy of ligands was minimized using MMFF94x force field. 3D and 2D interactions diagrams were generated through BIOVIA Discovery Studio visualizer V17.2.36 Before proceeding for docking studies structure of compound (1) was optimized with the help of density functional theory (DFT)/B3LYP37–42 method with 6-311G(d, p) as basis sets (Fig. 3). This optimized structure of compound 1 was then used for docking studies with AChE and BuChE enzymes.
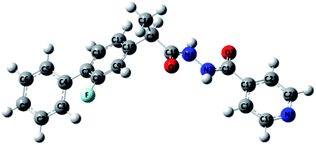 |
| | Fig. 3 Optimized structure of compound (1). | |
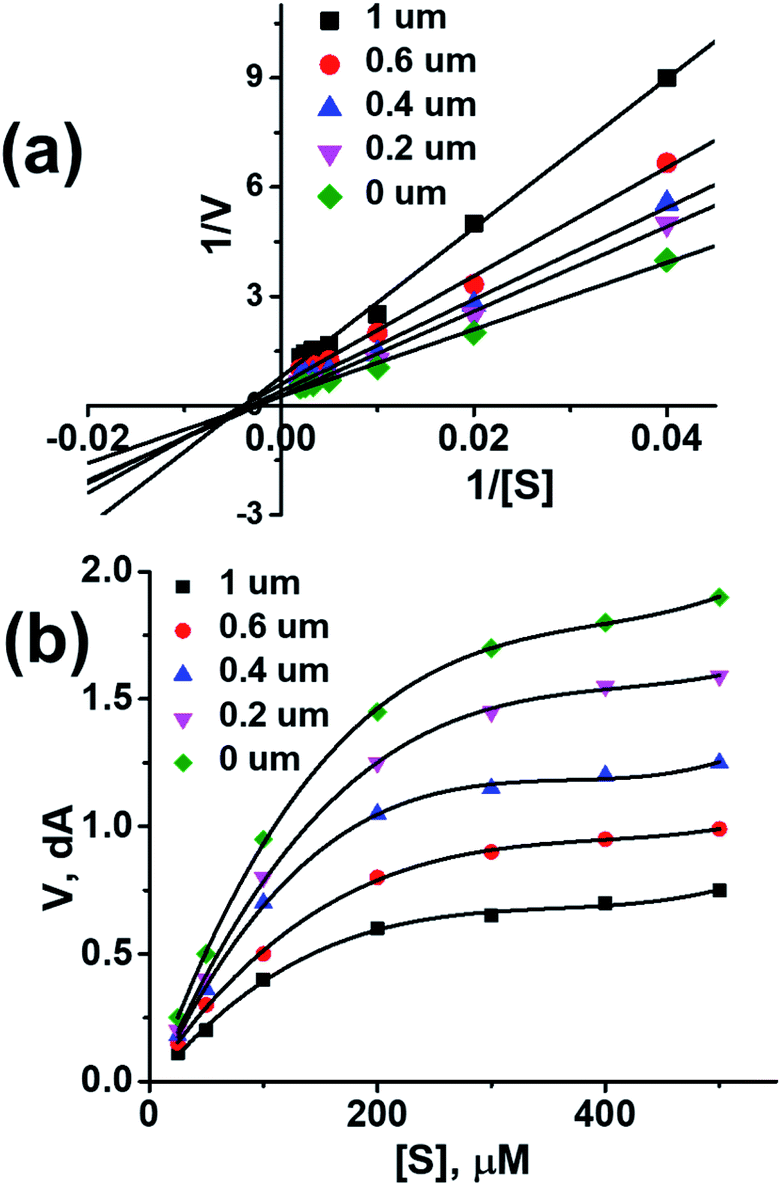
Acetylcholinesterase activity
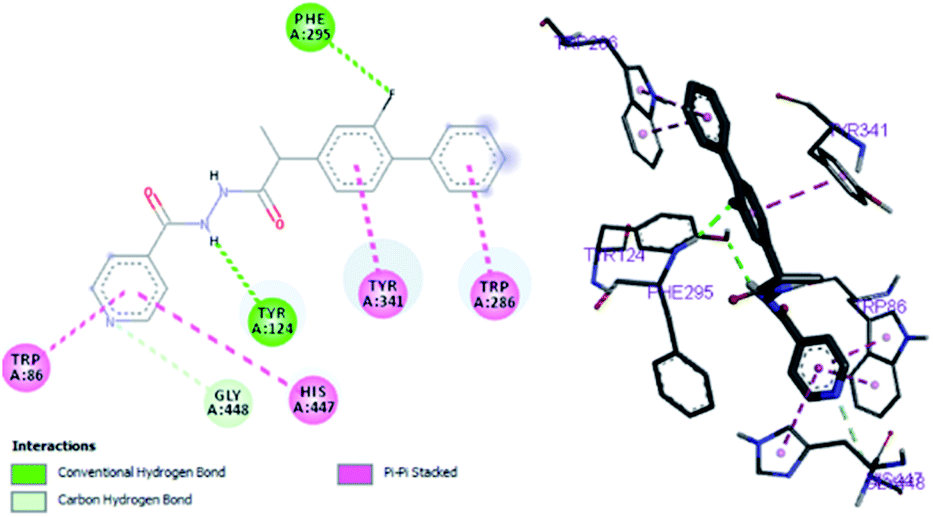
By performing in vitro studies aided with molecular docking simulation it has been observed that compound 1 (% inhibition = 78.92 ± 0.24) is the most active analogue with binding energy −12.9 kcal mol−1 for AChE inhibition activity. In active site, two most important residues Trp286, Trp86, Trp341 and His447 of AChE are frequently involved through hydrogen bonding or π–π interaction and play important inhibitory roles. The compound 1 also involves in hydrogen bonding interaction with Trp124 and Phe295 of AChE. On the basis of docking results it is clear that the compound 1 is able to fit well in active site of acetylcholinesterase and interact with important amino acid residues. The enhanced inhibition activity for compound 1 might be due to the presence of flouro group at ortho position of the extended phenyl ring. The top-ranked docking conformation of compound 1 showed that it is able to form potential π–π interaction with residue Tyr286 and hydrophobic interactions with other residues within the esteratic pocket of the active site (Fig. 4). On the other hand, several amino acid residues appear to stabilize the ground state binding orientation of the phenyl rings by engaging in hydrophobic interaction with the side chains of residues Try124, Gly448 and Phe295 (Table 6).
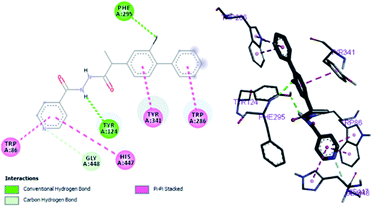 |
| | Fig. 4 Binding position for compound (1) in the active site of AChE. | |
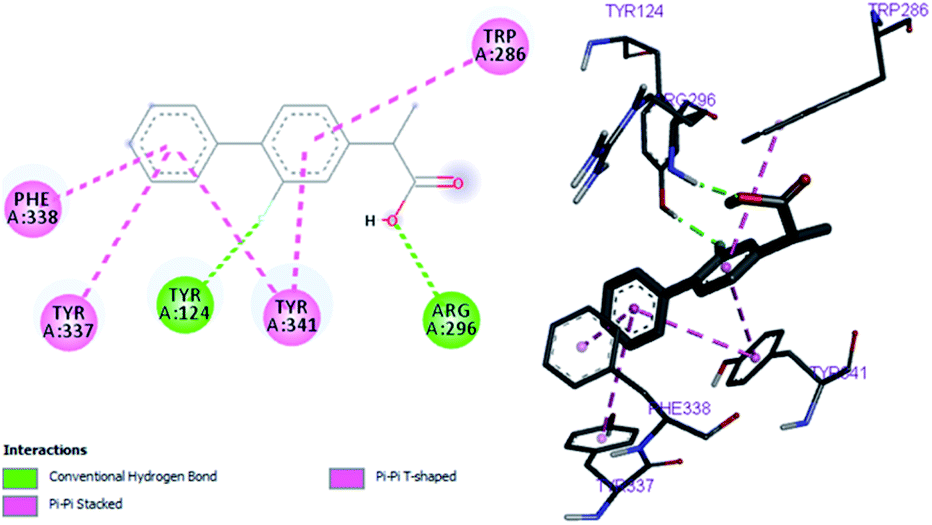
Table 6 Important interactions of compound 1, flurbiprofen and serine with AChE
| Compound code |
Binding energies kcal mol−1 |
H-bonds interactions |
Hydrophobic interaction (π–π) |
| Compound 1 |
−12.9 |
Tyr124, Phe295 |
Trp286, Trp86, Tyr341, His447 |
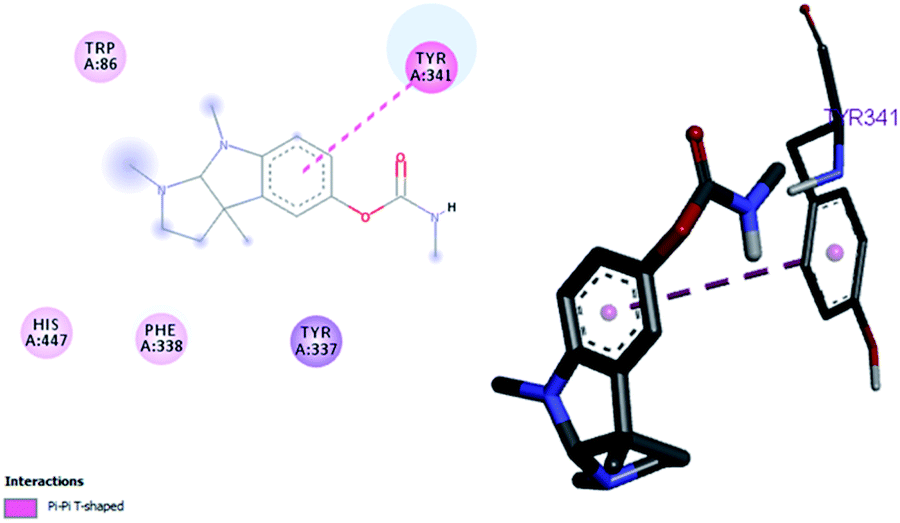
| Eserine |
−8.9 |
— |
Tyr341, |
| Flurbiprofen |
−8.3 |
Phe298 |
Phe338, Tyr341, Tyr337 |
Flurbiprofen and eserine were observed to be least active compound having binding score of −10.1 kcal mol−1 and −8.9 kcal mol−1. Binding interactions are shown in Fig. 5 and 6.
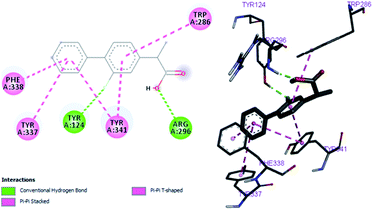 |
| | Fig. 5 Binding position for fIurbiprofen in the active site of AChE. | |
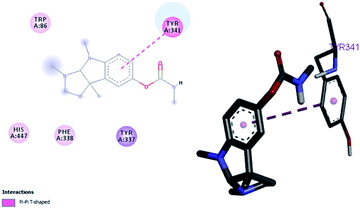 |
| | Fig. 6 Binding position for eserine in the active site of AChE. | |
Butyrylcholinesterase activity
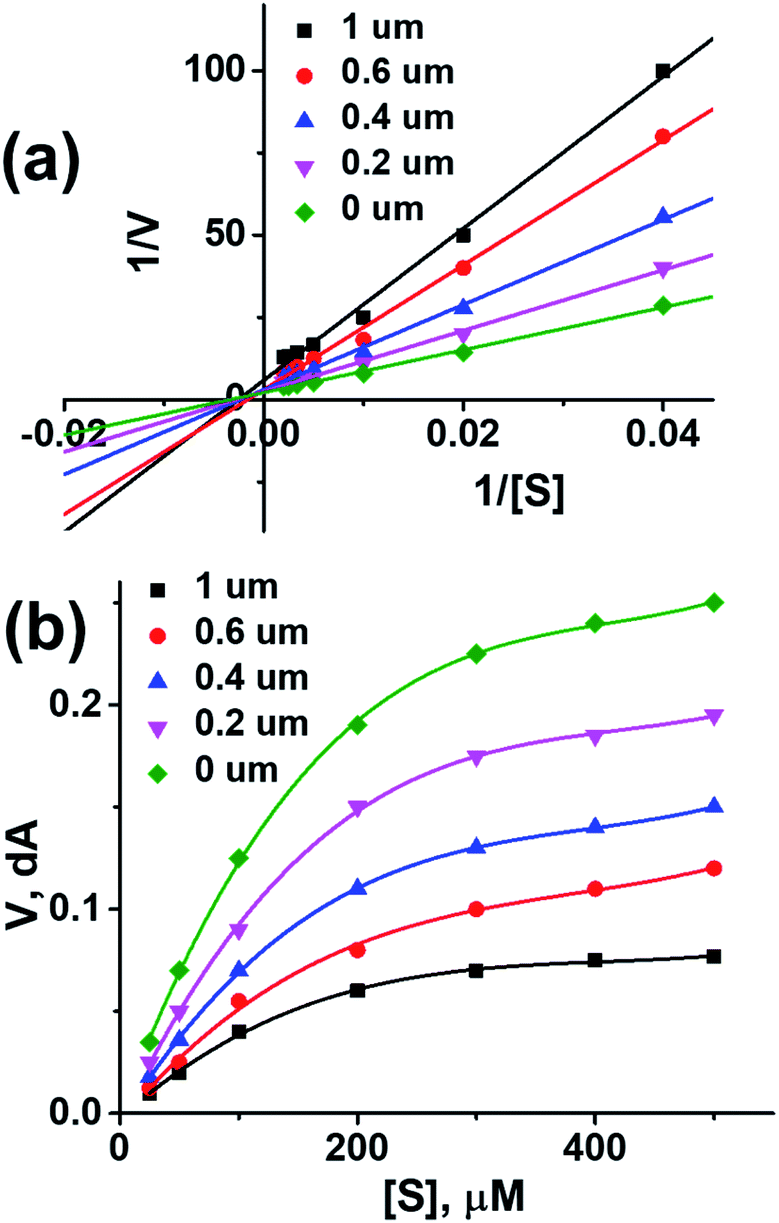
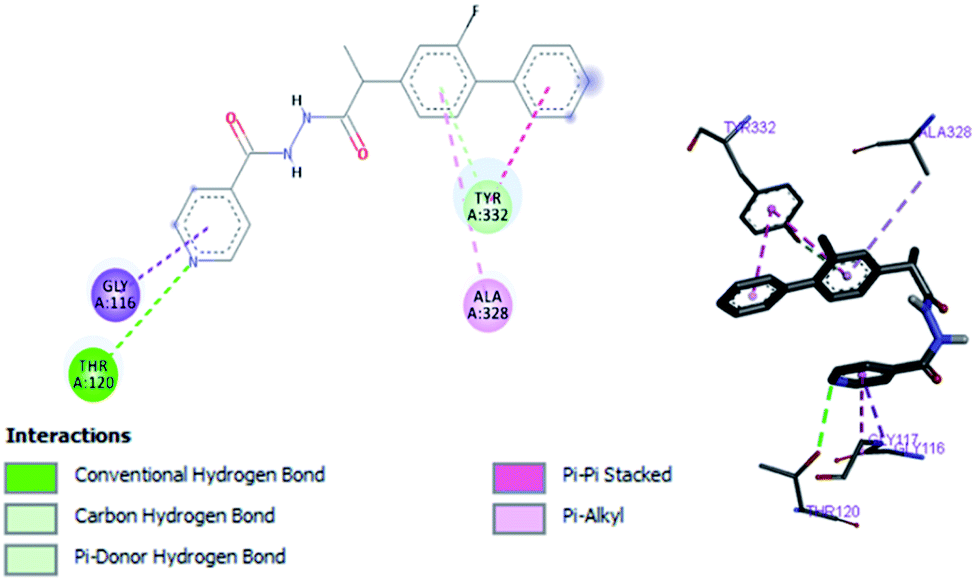
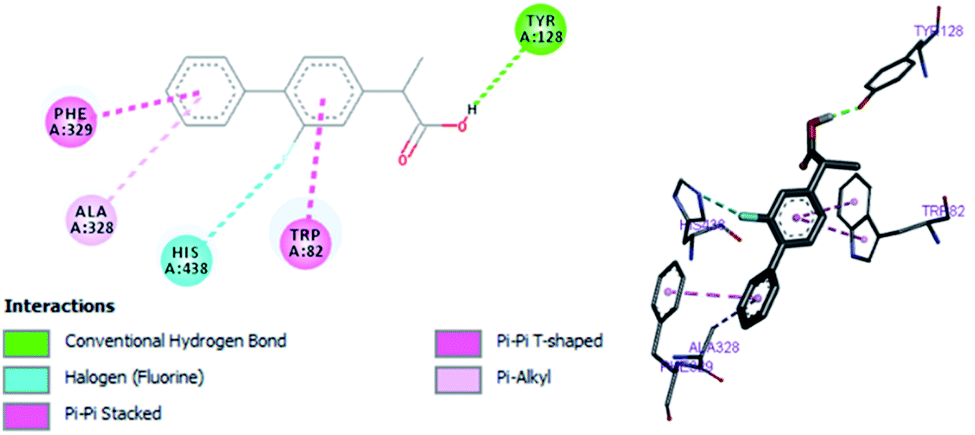
In vitro studies aided with molecular docking simulation revealed that compound 1 (% inhibition = 61.25 ± 0.85) is the active compound with binding energy −9.8 kcal mol−1 compared to eserine (−8.5 kcal mol−1) which is used as standard drug for BuChE inhibition activity and flurbiprofen (−8.2 kcal mol−1). In active site there are two most important residues Trp82 and Tyr332 of BuChE involving through hydrogen bonding or π–π interaction which play an important inhibitory role.
The docking results suggest that compound 1 is able to fit well in active site of butyrylcholinesterase and interact with important amino acid residues. The inhibition activity for compound 1 might be due to the presence of flouro group at ortho position of the extended phenyl ring. Compound 1 showed hydrogen bond interaction with the Thr120 amino acid residue. The top-ranked docking conformation of compound 1 showed that it is able to form potential π–π interaction with residue Tyr332 and hydrophobic interactions with other residues within the esteratic pocket of the active site (Fig. 7). On the other hand, several amino acid residues appear to stabilize the ground state binding orientation of the phenyl rings by engaging in hydrophobic interaction with the side chains of residues Tyr332, Ala328 and Gly116 (Table 7).
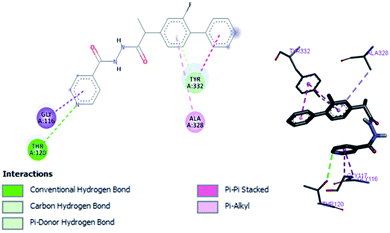 |
| | Fig. 7 Binding position for compound (1) in the active site of BuChE. | |
Table 7 Important interactions of compound 1, flurbiprofen and eserine with BuChE
| Compound code |
Binding energies kcal mol−1 |
H-bonds interactions |
Hydrophobic interaction (π–π) |
| Compound 1 |
−9.8 |
Trp82, Trp332, Thr120 |
Trp82, Trp332, Ala332, Gly116, Ala328 |
| Eserine |
−8.5 |
Tyr128, His438, Phe329 |
Trp82, Phe329 |
| Flurbiprofen |
−8.2 |
Tyr128, His438 |
Trp82, Ala328, Phe329 |
Flurbiprofen was observed to be least active compound having binding score −8.2 kcal mol−1. Binding interactions are shown in Fig. 8. Docking and BuChE inhibition activity results revealed that compound 1 could be used for Alzheimer's disease.43
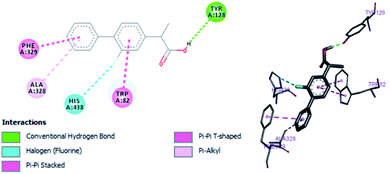 |
| | Fig. 8 Binding position for fIurbiprofen in the active site of BuChE. | |
Conclusions
A novel compound (1) based on combination of flurbiprofen and isoniazide has been designed and synthesized. AChE and BuChE inhibition activities of compound (1) were found to be ∼2.5 and ∼1.7 folds higher respectively as compare to standard physostigmine and parent drug i.e. flurbiprofen. Molecular docking studies prove that compound (1) is more active against AChE and BuChE having calculated binding energies of −12.9 kcal mol−1 and −9.8 kcal mol−1 respectively, as compared to flurbiprofen and eserine (physostigmine) for which binding energy was calculated to be −10.1 kcal mol−1 and −8.9 kcal mol−1 respectively. Lineweaver–Burk plot suggests mixed mode of inhibition of AChE and BuChE with compound 1. AChE and BuChE inhibition activities aided with docking results suggested that compound (1) could be used for Alzheimer's disease. Moreover, compound (1) also exhibit improved α-chymotrypsin activity as well. Compound (1) was found to be more active and less toxic than the parent analogues in various in vitro and in vivo tests.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge University of Education, Township, Lahore, Pakistan for providing laboratory facilities to accomplish this project.
Notes and references
- J. R. Vane, J. Physiol. Pharmacol., 2000, 51, 573–586 CAS.
- J. R. Vane, Nat. New Biol., 1971, 43, 232–235 CrossRef PubMed.
- W. L. Smith, Metal ions in Biological systems, ed. H. Siegel and A. Siegel, Marcel Dekker, New York, 1994 Search PubMed.
- J. C. Otto and W. L. Smith, J. Lipid Mediat. Cell Signal, 1995, 12, 139–156 CrossRef CAS PubMed.
- A. Inotai, B. Hanko and A. Meszaro, Pharmacoepidemiol. Drug Saf., 2010, 19, 183–190 CrossRef PubMed.
- J. J. Talley, Prog. Med. Chem., 1999, 36, 201–234 CrossRef CAS PubMed.
- W. C. Black, Annu. Rep. Med. Chem., 2004, 39, 125–138 CAS.
- T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347–1365 CrossRef CAS PubMed.
- P. Prasit, Z. Wang, C. Brideau, C. C. Chan, S. Charleson, W. Cromlish, D. Ethier, J. F. Evans, A. W. Ford-Hutchinson, J. Y. Gauthier, R. Gordon, J. Guay, M. Gresser, S. Kargman, B. Kennedy, Y. Leblanc, S. Léger, J. Mancini, G. P. O'Neill, M. Ouellet, M. D. Percival, H. Perrier, D. Riendeau, I. Rodger and R. Zamboni, Bioorg. Med. Chem. Lett., 1999, 9, 1773–1778 CrossRef CAS PubMed.
- B. G. Johnson, P. M. W. Gill and J. A. Pople, J. Chem. Phys., 1993, 98, 5612–5626 CrossRef CAS.
- N. Oliphant and R. T. Bartlett, J. Chem. Phys., 1994, 100, 6550 CrossRef CAS.
- Y. Zhang, Z. J. Guo and X. Z. You, J. Am. Chem. Soc., 2001, 123, 9378 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, et. al., Gaussian 03, Revision E.01, Gaussian Inc., Wallingford, CT, 2004 Search PubMed.
- G. Fitzgerald and J. Andzelm, J. Phys. Chem., 1991, 95, 10531–10534 CrossRef CAS.
- A. A. Seliman, M. Altaf, A. T. Onawole, S. Ahmad, M. Y. Ahmed, A. A. Al-Saadi, S. Altuwaijri, G. Bhatia, J. Singh and A. A. Isab, J. Organomet. Chem., 2017, 848, 175–183 CrossRef CAS.
- M. Prince, A. Comas-Herrera, M. Knapp, M. Guerchet and M. Karagiannidou, Alzheimer's Disease International. World Alzheimer Report 2016. September 2016. http://www.alz.co.uk/research/WorldAlzheimerReport2016.pdf Search PubMed.
- E. K. Perry, R. H. Perry, G. Blessed and B. E. Tomlinson, Neuropathol. Appl. Neurobiol., 1978, 4, 273–277 CrossRef CAS PubMed.
- P. Davies and A. J. Maloney, Lancet, 1976, 8000, 1403 CrossRef.
- P. J. Whitehouse, D. L. Price, R. G. Struble, A. W. Clark, J. T. Coyle and M. R. Delon, Science, 1982, 215, 1237–1239 CrossRef CAS PubMed.
- A. Ciro, J. Park, G. Burkhard, Y. Nicole and G. Changiz, Curr. Alzheimer Res., 22012, 9, 138–143 CrossRef PubMed.
- G. Mushtaq, N. H. Greig, J. A. Khan and M. A. Kamal, CNS Neurol. Disord.: Drug Targets, 2014, 13, 1432–1439 CrossRef CAS PubMed.
- A. Morsy and P. C. Trippier, J. Alzheimer's Dis., 2019, 72, S145–S176 CAS.
- Y. Bourne, P. Taylor, Z. Radic and P. Marchot, EMBO J., 2003, 22, 1–12 CrossRef CAS PubMed.
- J. L. Sussman, M. Harel, F. Frolow, C. Oefner, A. Goldman, L. Toker and I. Silman, Science, 1991, 253, 872–879 CrossRef CAS PubMed.
- M. Bajda, N. Guzior, M. Ignasik and B. Malawska, Curr. Med. Chem., 2011, 18, 4949–4975 CrossRef CAS PubMed.
- Y. Chen, W. Li, H. Lin, L. Wu, H. Yang, Y. Pei, R. Tan and H. Sun, RSC Adv., 2017, 7, 3429 RSC.
- D. Panek, A. Więckowska, J. Jończyk, J. Godyń, M. Bajda, T. Wichur, A. Pasieka, D. Knez, A. Pišlar, J. Korabecny, O. Soukup, V. Sepsova, R. Sabaté, J. Kos, S. Gobec and B. Malawska, ACS Chem. Neurosci., 2018, 9, 1074–1094 CrossRef CAS PubMed.
- A. Asghar, M. Yousuf, H. Mubeen, R. Nazir, K. Haruna, A. T. Onawole and L. Rasheed, Bioorg. Med. Chem., 2019, 27, 2397–2404 CrossRef CAS PubMed.
- A. Jaramillo, P. Bhattacherjee, G. Sonnenfold and G. Paterson, Curr. Eye Res., 1992, 11, 571–579 CrossRef CAS PubMed.
- P. Bacalhau, A. A. S. Juan, C. S. Marques, D. Peixoto, A. Goth, C. Guarda, M. Silva, S. Arantes, A. T. Calderia, R. Martins and A. J. Burke, Bioorg. Chem., 2016, 67, 1–8 CrossRef CAS PubMed.
- E. K. Ulleberg, I. Comi, H. Holm, E. B. Herud, M. Jacobsen and G. E. Vegarud, Food Dig., 2011, 2, 52–61 CrossRef CAS PubMed.
- A. Andrés, M. Rosés, C. Ràfols, E. Bosch, S. Espinosa, V. Segarra and J. M. Huerta, Eur. J. Pharm. Sci., 2015, 76, 181–191 CrossRef PubMed.
- A. Czyrski, J. Chem., 2019, 3407091, 1–6 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- https://www.rcsb.org/structure/1p0p.
- K. Stierand and M. Rarey, ACS Med. Chem. Lett., 2010, 9, 540–545 CrossRef PubMed.
- R. Dennington, T. Keith and J. Millam, Gauss View, Version 4.1.2, Semichem Inc., Shawnee Mission, KS, 2007 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- I. Dennington, R. T. Keith and J. Millam, GaussView, Semichem, Inc., Shawnee Mission, KS, 2003 Search PubMed.
- M. Haroon, T. Akhtar, M. Yousuf, M. W. Baig, M. N. Tahir and L. Rasheed, J. Mol. Struct., 2018, 1167, 154–160 CrossRef CAS.
- M. Yousuf, I. S. Youn, J. Yun, L. Rasheed, R. Valero, G. Shi and K. S. Kim, Chem. Sci., 2016, 7(6), 3581–3588 RSC.
- P. Bacalhau, A. A. S. Juan, C. S. Marques, D. Peixoto, A. Goth, C. Guarda, M. Silva, S. Arantes, A. T. Calderia, R. Martins and A. J. Burke, Bioorg. Chem., 2016, 67, 1–8 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02339f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Ghulam Fareedd,
Rabia Nazire,
Abida Hassanf,
Aneela Maalik
c,
Ghulam Fareedd,
Rabia Nazire,
Abida Hassanf,
Aneela Maalik