
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSarcosenones A–C, highly oxygenated pimarane diterpenoids from an endolichenic fungus Sarcosomataceae sp.†
Xintong Houab,
Yang Xub,
Shuaiming Zhuc,
Yang Zhang *c,
Liangdong Guod,
Feng Qiua and
Yongsheng Che*ab
*c,
Liangdong Guod,
Feng Qiua and
Yongsheng Che*ab
aTianjin University of Traditional Chinese Medicine, Tianjin 300193, People's Republic of China
bInstitute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, People's Republic of China. E-mail: cheys@im.ac.cn
cState Key Laboratory of Toxicology & Medical Countermeasures, Beijing Institute of Pharmacology & Toxicology, Beijing 100850, People's Republic of China. E-mail: zhangyang@bmi.ac.cn
dState Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, People's Republic of China
First published on 22nd April 2020
Abstract
Three new highly oxygenated pimarane diterpenoids, sarcosenones A–C (1–3), and the known 9α-hydroxy-1,8(14),15-isopimaratrien-3,7,11-trione (4), were isolated from cultures of an endolichenic fungus Sarcosomataceae sp. Their structures were elucidated based on NMR spectroscopic data and electronic circular dichroism (ECD) calculations. Compound 1 showed moderate cytotoxicity against a small panel of four human tumor cell lines, with IC50 values of 7.5–26.4 μM.
Introduction
Pimarane diterpenoids have been encountered as secondary metabolites of higher plants, fungi, and marine organisms.1 This class of diterpenes can be derived from the original core by substitution, hydroxylation, acetylation, rearrangement, bromination, and ring expansion reactions.2 Since the isolation of pimaric acid, the first example of this class, in 1839,3 pimarane diterpenoids have attracted considerable interest due to their remarkable structural diversity and great antimalarial,4 antibacterial,5 antifungal,6 antiviral,7,8 phytotoxic,6 cytotoxic,9–12 AChE-inhibitory,13 and anti-inflammatory activities.14Sarcosomataceous fungi (Ascomycota), usually known as degraders of wood or as pathogens,15 have been reported to produce spirobisnaphthalenes,16,17 lactones,15,18–21 naphthalones,21 cyclohexenones,22 and isocoumarins.22 Examples include urnucratins A–C with inhibitory effects against methicillin-resistant Staphylococcus aureus (MRSA) isolated from Urnula craterium,16 plecmillin A with potential anticancer effect isolated from a strain of endolichenic fungus (CGMCC 3.1519216),17 and galiellalactone derivatives with potent nematicidal effect, inhibitory activity of IL-6 signalling mediated by SATA3, and cytotoxic activity.15,19
Lichens are combinations of a fungus (the mycobiont) and an algal partner (the photobiont or phycobiont). In addition to fungal mycobionts, some nonobligate microfungi, endolichenic fungi, are also found to live asymptomatically in the bodies (thalli) of lichens.23 Endolichenic fungi have been demonstrated to be a rich source of new bioactive natural products.24 During our continuous search for new cytotoxic metabolites from the endolichenic fungi,23,25–27 the fungus Sarcosomataceae sp. isolated from the lichen Everniastrum sp. (Parmeliaceae), which was collected from Zixi Mountain, Yunnan, People's Republic of China, was subjected to chemical investigation. An ethyl acetate (EtOAc) extract of the culture showed cytotoxic effects towards a small panel of four human tumor cell lines. Fractionation of the extract afforded three new highly oxygenated pimarane diterpenoids, which we named sarcosenones A–C (1–3; Fig. 1), and a known analogue 9α-hydroxy-1,8(14),15-isopimaratrien-3,7,11-trione (4; Fig. 1). Details of the isolation, structure elucidation, and cytotoxicity evaluation of these compounds are reported herein.
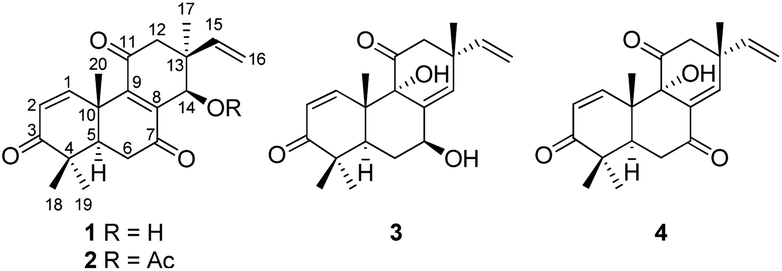 | ||
| Fig. 1 Structures of compounds 1–4. | ||
Results and discussion
Sarcosenone A (1) was assigned a molecular formula of C20H24O4 (9 degrees of unsaturation) by HRESIMS. Its IR absorption bands at 3423 and 1677 cm−1 suggested the presence of hydroxyl and carbonyl groups, respectively. Analysis of its NMR data (Table 1) revealed the presence of one exchangeable proton (δH 4.40), four methyl groups, two methylenes, two methines including one oxymethine (δC 68.0), three sp3 quaternary carbons, six olefinic carbons with four protonated carbons, and three ketone carbons (δC 199.6, 201.7, and 202.2, respectively). These data accounted for six of the nine degrees of unsaturation calculated from the molecular formula, suggesting that 1 was a tricyclic compound. The 1H–1H COSY NMR data of 1 showed two isolated spin-systems of C-1–C-2 and C-5–C-6. The 1H NMR spectrum displayed signals characteristic for a terminal vinyl group (δH 6.09, dd, J = 17.6, 10.9 Hz, H-15; 5.05, d, J = 17.6 Hz, H-16a; 5.08, d, J = 10.9 Hz, H-16b) and four tertiary methyls (δH 0.98, s, H3-17; 1.17, s, H3-18; 1.16, s, H3-19; 1.59, s, H3-20). On the basis of these data, compound 1 should possess the characteristics a pimarane diterpenoid skeleton,28 which was confirmed by relevant HMBC correlations (Fig. 2). HMBC cross-peaks from H3-17 to C-12, C-13, C-14, and C-15, and from H-16 to C-13 indicated that a methyl and a vinyl are both connected to C-13. HMBC correlations from H3-20 to C-9 and from H-14 to C-8 and C-9 suggested the presence of a double bond between C-8 and C-9. While those correlations of H-1, H3-18, H3-19 with C-3, H-5, H-6α, H-6β with C-7, and of H-12α, H-12β with C-11 indicated these three carbonyl groups were attached to C-3, C-7, and C-11, respectively. A free hydroxyl group was located at C-14 by HMBC correlations of H-12α, H-12β and H3-17 to C-14. On the basis of these data, the gross structure of 1 was proposed.| No. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δCa, type | δHb (J in Hz) | δCa, type | δHb (J in Hz) | δCc, type | δHd (J in Hz) | |
| a Recorded in acetone-d6 at 150 MHz.b Recorded in acetone-d6 at 600 MHz.c Recorded in CDCl3 at 150 MHz.d Recorded in CDCl3 at 600 MHz. | ||||||
| 1 | 154.5, CH | 7.78, d (10.5) | 154.1, CH | 7.78, d (10.5) | 152.1, CH | 6.81, d (10.4) |
| 2 | 127.6, CH | 5.92, d (10.5) | 127.7, CH | 5.94, d (10.5) | 129.6, CH | 5.99, d (10.4) |
| 3 | 202.2, qC | 202.1, qC | 203.9, qC | |||
| 4 | 44.6, qC | 44.7, qC | 44.6, qC | |||
| 5 | 46.8, CH | 2.55, dd (15.1, 3.2) | 46.9, CH | 2.55, dd (15.1, 3.3) | 42.1, CH | 2.74, dd (13.1, 2.2) |
| 6α | 35.6, CH2 | 2.62, dd (17.8, 3.2) | 35.5, CH2 | 2.62, dd (17.7, 3.3) | 32.7, CH2 | 2.08, m |
| 6β | 2.80, m | 2.78, m | 1.59, m | |||
| 7 | 199.6, qC | 198.2, qC | 69.4, CH | 4.51, m | ||
| 8 | 145.4, qC | 141.9, qC | 139.9, qC | |||
| 9 | 149.5, qC | 152.1, qC | 79.3, qC | |||
| 10 | 41.6, qC | 41.9, qC | 45.0, qC | |||
| 11 | 201.7, qC | 201.1, qC | 211.6, qC | |||
| 12α | 46.7, CH2 | 3.11, d (14.7) | 46.3, CH2 | 2.34, dd (14.7, 1.2) | 53.2, CH2 | 2.67, d (13.3) |
| 12β | 2.24, d (14.7) | 3.13, d (14.7) | 2.73, d (13.3) | |||
| 13 | 43.8, qC | 43.7, qC | 43.1, qC | |||
| 14 | 68.0, CH | 4.56, s | 68.5, CH | 5.93, s | 129.1, CH | 5.95, s |
| 15 | 144.5, CH | 6.09, dd (17.6, 10.9) | 143.1, CH | 5.86, m | 143.2, CH | 5.62, dd (17.2, 10.4) |
| 16a | 113.3, CH2 | 5.05, d (17.6) | 114.2, CH2 | 5.03, d (17.5) | 114.3, CH2 | 4.91, d (17.2) |
| 16b | 5.08, d (10.9) | 5.07, d (10.8) | 4.96, d (10.4) | |||
| 17 | 22.6, CH3 | 0.98, s | 23.2, CH3 | 1.04, s | 28.9, CH3 | 1.33, s |
| 18 | 21.5, CH3 | 1.17, s | 21.5, CH3 | 1.17, s | 22.6, CH3 | 1.12, s |
| 19 | 26.9, CH3 | 1.16, s | 26.8, CH3 | 1.16, s | 28.0, CH3 | 1.22, s |
| 20 | 23.0, CH3 | 1.59, s | 23.2, CH3 | 1.62, s | 20.9, CH3 | 1.18, s |
| 21 | 169.4, qC | |||||
| 22 | 21.0, CH3 | 1.91, s | ||||
| OH-7 | 3.44, s | |||||
| OH-14 | 4.40, s | |||||
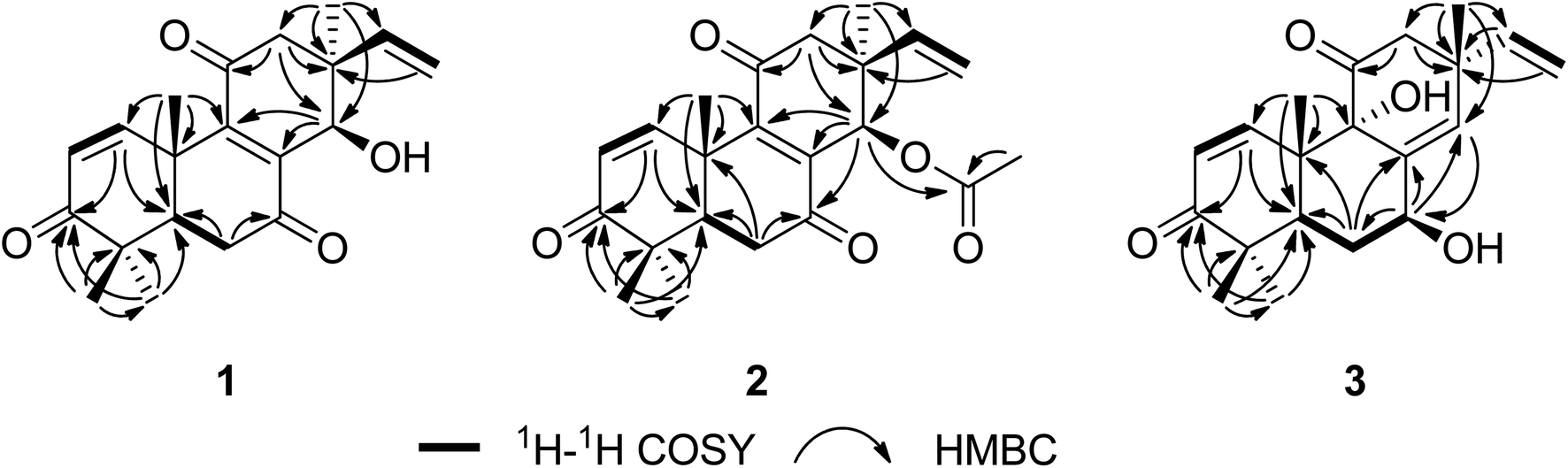 | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations for compounds 1–3. | ||
The relative configuration of 1 was proposed by analysis of NOESY data (Fig. 3). NOESY correlations of H-6β/OH-14 and H3-18/H3-20 implied that OH-14 and Me-20 were both β-oriented, while those of H-5/H3-19, H-6α/H3-19 and H-14/H3-17 revealed α-orientation for these protons, thereby establishing the relative configuration of 1.
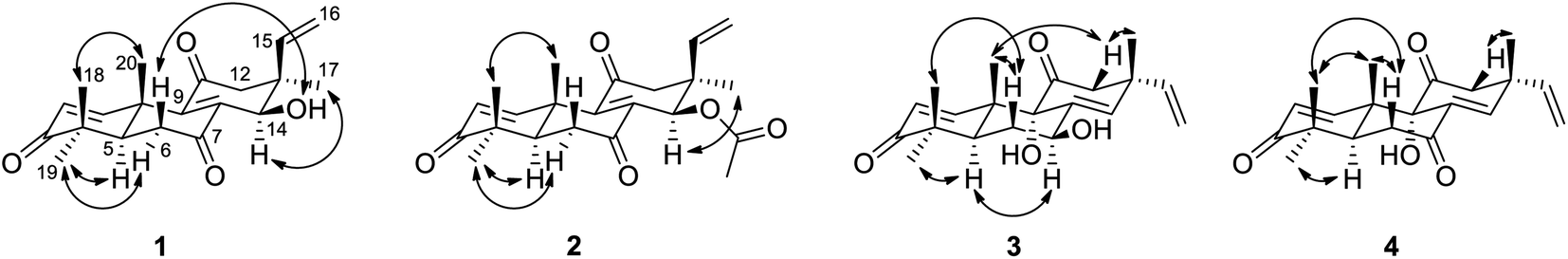 | ||
| Fig. 3 Key NOESY correlations for compounds 1–4. | ||
The absolute configurations of 1 were deduced by comparison of the experimental and simulated electronic circular dichroism (ECD) spectra calculated using the time-dependent density functional theory (TDDFT).29 The ECD spectra of the four possible enantiomers 1a–d (Fig. S11†) were calculated. A random conformational analysis was performed for 1a–d using the MMFF94 force field followed by reoptimization at the B3LYP/6-311G(2d,2p) level, affording the lowest energy conformers (Fig. S11†). The overall calculated ECD spectra of 1a–d were then generated according to Boltzmann weighting of their lowest energy conformers by their relative energies (Fig. 4). The experimental CD spectrum of 1 correlated well to the calculated ECD curve of (5R, 10S, 13S, 14R)-1 (1a; Fig. 4), suggesting the 5R, 10S, 13S, 14R absolute configuration for 1.
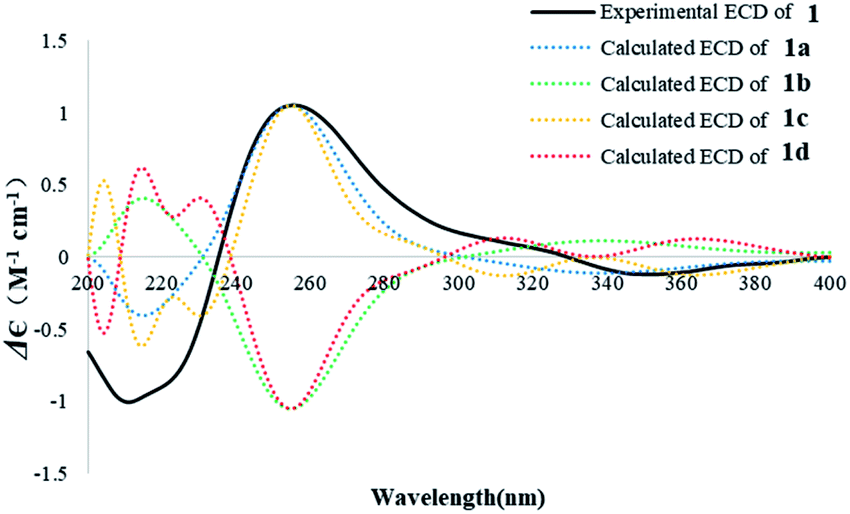 | ||
| Fig. 4 Experimental ECD spectrum of 1 in MeOH and the calculated ECD spectra of 1a–d. | ||
The molecular formula of sarcosenone B (2) was determined to be C22H26O5 (10 degrees of unsaturation) based on HRESIMS and the NMR data (Table 1), which is 42 mass units higher than that of 1. Analysis of the 1H and 13C NMR data for 2 revealed the presence of structural features similar to those found in 1, except that the oxygenated methine proton (H-14) at 4.56 ppm was significantly downfield (δH 5.93). In addition, the NMR resonances corresponding to an acetyl group (δH/δC 1.91/21.0, 169.4) were observed, indicating that the C-14 oxygen of 2 is acylated, which was supported by HMBC cross-peak from H-14 to the carboxylic carbon at 169.4 ppm. On the basis of these data, 2 was determined as the C-14 monoacetate of 1.
The relative configuration of 2 was assigned by analysis of NOESY data and comparison of its 1H NMR data with those of sarcosenone A (1). NOESY correlation of H3-18/H3-20 implied that Me-18 and Me-20 were β-oriented, while those of H-5/H3-19, H-6α/H3-19, H-12α/H3-17, and H-14/H3-17 revealed that the protons were all α-oriented, establishing the relative configuration of 2.
The absolute configuration of 2 was similarly deduced by comparison of the experimental CD spectrum with the simulated ECD spectra predicted using the TDDFT at the B3LYP/6-311G(2d,2p) level. The ECD spectra of the four possible isomers 2a–d (Fig. S22†) were calculated to represent all possible configurations. The experimental CD spectrum of 2 was nearly identical to that calculated for 2a (Fig. 5), suggesting that 2 has the 5R, 10S, 13S, 14R absolute configuration.
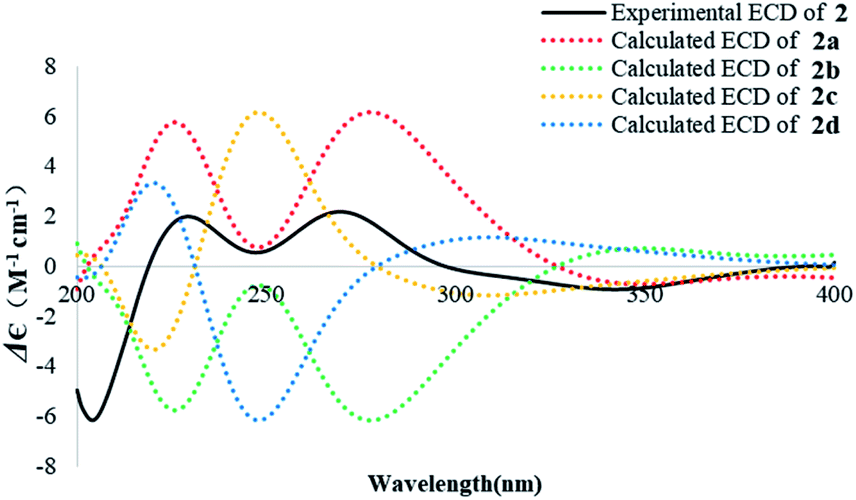 | ||
| Fig. 5 Experimental ECD spectrum of 2 in MeOH and the calculated ECD spectra of 2a–d. | ||
Sarcosenone C (3) was assigned a molecular formula of C20H26O4 (eight degrees of unsaturation) by HRESIMS. Analysis of its NMR data (Table 1) revealed the presence of one exchangeable proton (δH 3.44), four methyl groups, two methylenes, two methines including one oxymethine (δC 69.4), one oxygenated tertiary carbon (δC 79.3), three sp3 quaternary carbons, six olefinic carbons with five protonated, and two ketone carbons (δC 203.9 and 211.6, respectively). These data accounted for five of the eight degrees of unsaturation calculated from the molecular formula, suggesting that 3 was a tricyclic compound. The 1H and 13C NMR data of 3 revealed structural features closely related to those of a known compound, 9α-hydroxy-1,8(14),15-isopimaratrien-3,7,11-trione (4).28 Comparison of the 1H and 13C NMR spectroscopic data of 3 with those of 4 revealed a resonance for one more oxymethine (δH/δC 4.51/69.4) and the absence of signals for a ketone functionality (δC 198.4),28 suggesting that the carbonyl group at C-7 in 4 was reduced to a hydroxy group. This observation was supported by HMBC correlations from the newly observed H-7 to C-6, C-8, and C-14. Therefore, the gross structure of sarcosenone C was proposed as 3.
The relative configuration of 3 was also proposed by analysis of NOESY data (Fig. 3). NOESY correlations of H-6β/H3-18, H-6β/H3-20, H-12β/H3-17 and H-12β/H3-20 suggested that Me-17, Me-18, and Me-20 were both β-oriented, while those of H-5/H-7, H-5/H3-19 and H-6α/H-7 revealed that the protons were all α-oriented. NOESY correlations of H3-20 with H-12β and of H-12β with H3-17 indicated that these protons are on the same face of the ring system and necessitates α-orientation for OH-9.28 Therefore, the relative configuration of 3 was deduced as shown.
The absolute configuration of 3 was also assigned by comparison of the experimental CD spectrum with the simulated ECD spectra generated by excited state calculation using TDDFT. The ECD spectra of the four enantiomers 3a–d (Fig. S33†) were calculated to represent all possible configurations. The MMFF94 conformational search followed by reoptimization at the B3LYP/6-311G(2d,2p) level afforded the lowest-energy conformers (Fig. S33†). The overall ECD spectra were then generated according to Boltzmann weighting of each conformer. The CD spectrum of 3 correlated well to the calculated curve of 3a (Fig. 6), suggesting the 5R, 7S, 9S, 10S, 13R absolute configuration.
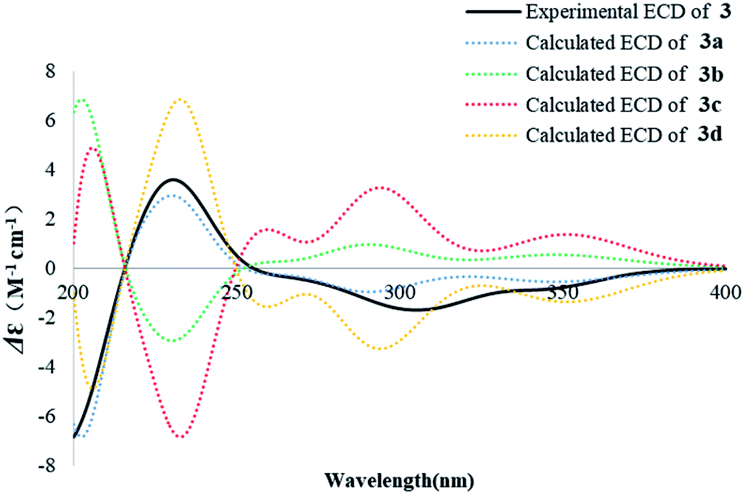 | ||
| Fig. 6 Experimental ECD spectrum of 3 in MeOH and the calculated ECD spectra of 3a–d. | ||
The known compound 4 was identified as 9α-hydroxy-1,8(14),15-isopimaratrien-3,7,11-trione by comparison of its MS and NMR data with those reported.28 Although its relative configuration was assigned by NOE correlations, the absolute configuration remained unsolved. Therefore, the absolute configuration was deduced by comparison of the experimental and calculated ECD spectra for the eight possible enantiomers 4a–h. The MMFF94 conformational search followed by TDDFT reoptimization at the B3LYP/6-31G(2d,2p) basis set level afforded the lowest energy conformers (Fig. S36†). The experimental CD spectrum of 4 was nearly identical to the calculated ECD curve of 4a (Fig. 7), suggesting the 5R, 9S, 10S, 13R absolute configuration.
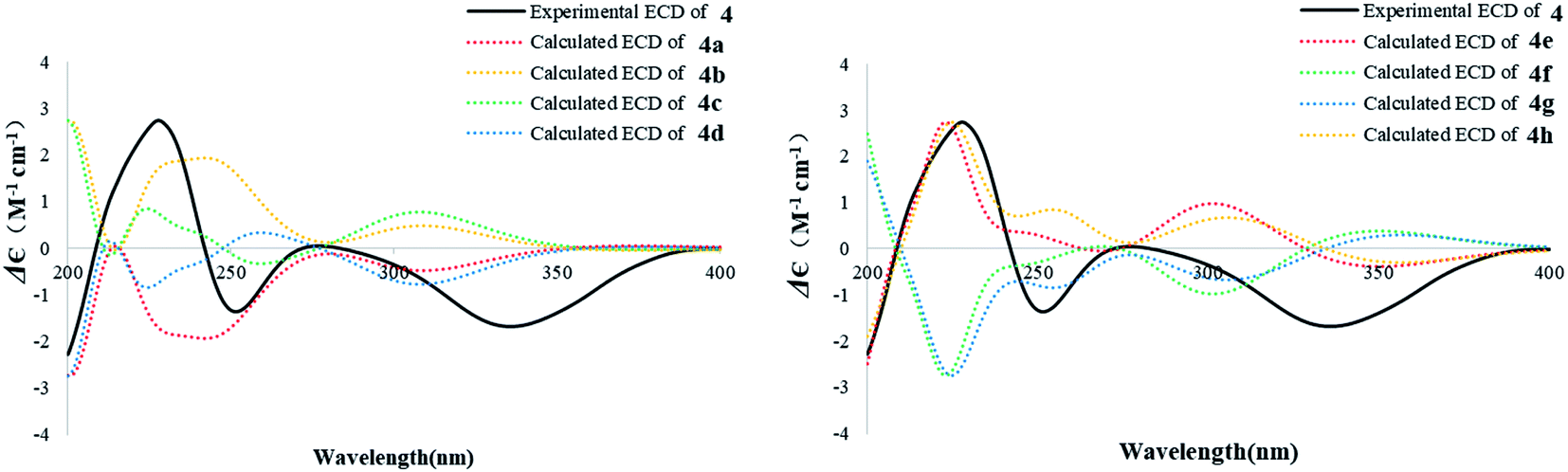 | ||
| Fig. 7 Experimental ECD spectrum of 4 in MeOH and the calculated ECD spectra of 4a–h. | ||
Compounds 1–4 were tested for cytotoxicity against a panel of four human tumor cell lines, MCF-7 (breast cancer), HeLa (cervical carcinoma), HepG2 (hepatocellular carcinoma), and 786-O (renal cell adenocarcinoma). Compound 1 showed moderate cytotoxic effects (Table 2), with IC50 values of 7.5–26.4 μM (the positive control cisplatin showed IC50 values of 24.2–29.2 μM). However, compounds 2–4 did not show detectable activity at 50 μM.
Experimental
General experimental procedures
Optical rotations were measured on a Rudolph Research Analytical automatic polarimeter, and UV data were recorded on a Shimadzu Biospec-1601 spectrophotometer. CD spectra were recorded on a JASCO J-815 spectropolarimeter. IR data were recorded using a Nicolet Magna-IR 750 spectrophotometer. 1H and 13C NMR spectra were acquired with Bruker Avance III-600 spectrometers using solvent signals (acetone-d6: δH 2.05/δC 29.8, 206.1; CDCl3: δH 7.26/δC 77.2) as references. The HSQC and HMBC experiments were optimized for 145.0 and 8.0 Hz, respectively. ESIMS and HRESIMS data were obtained on an Agilent Accurate-Mass-Q-TOF LC/MS G6550 instrument equipped with an ESI source. HPLC analysis and separation were performed using an Agilent 1260 instrument equipped with a variable-wavelength UV detector.Fungal material
The culture of Sarcosomataceae sp. was isolated from the lichen Everniastrum sp. (Parmeliaceae) collected from Zixi Mountain, Yunnan, People's Republic of China, in November 2006. The fungus was identified by L. G. and assigned the Accession no. 65-7-1-2 in L. G.'s culture collection at the Institute of Microbiology, Chinese Academy of Sciences, Beijing. The fungal strain was cultured on slants of potato dextrose agar (PDA) at 25 °C for 10 days. Agar plugs were cut into small pieces (about 0.5 × 0.5 × 0.5 cm3) under aseptic conditions, and 25 pieces were used to inoculate in five 250 mL Erlenmeyer flasks, each containing 50 mL of media (0.4% glucose, 1% malt extract, and 0.4% yeast extract), and the final pH of the media was adjusted to 6.5 and sterilized by autoclave. Five flasks of the inoculated media were incubated at 25 °C on a rotary shaker at 170 rpm for 5 days to prepare the seed culture. Fermentation was carried out in 10 Fernbach flasks (500 mL) each containing 80 g of rice. Distilled H2O (120 mL) was added to each flask, and the contents were soaked overnight before autoclaving at 15 psi for 30 min. After cooling to room temperature, each flask was inoculated with 5.0 mL of the spore inoculum and incubated at 25 °C for 40 days.Extraction and isolation
The fermentation material was extracted repeatedly with EtOAc (4 × 4.0 L), and the organic solvent was evaporated to dryness under vacuum to afford 2.7 g of crude extract. The crude extract was fractionated by silica gel vacuum liquid chromatography (VLC) using petroleum ether–EtOAc–MeOH gradient elution. The fraction (107.5 mg) eluted with 6.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 petroleum ether–EtOAc was separated by Sephadex LH-20 column chromatography (CC) eluting with 1:1 CH2Cl2–MeOH and the resulting subfractions were combined and purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 47% CH3CN in H2O for 30 min; 2 mL min−1) to afford 2 (1.0 mg, tR 20.0 min). The fraction (165.8 mg) eluted with 3.5:1 petroleum ether–EtOAc was separated by reversed-phase silica gel column chromatography (CC) eluting with a MeOH–H2O gradient. The subfraction (18 mg) eluted with 40% MeOH–H2O was purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 37% CH3CN in H2O for 30 min; 2 mL min−1) to afford 3 (4.0 mg, tR 22.5 min). The subfraction (23 mg) eluted with 50% MeOH–H2O was purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 55% MeOH in H2O for 90 min; 2 mL min−1) to afford 4 (2.7 mg, tR 67.5 min) and 1 (3.4 mg, tR 82.5 min).
ε) 206 (3.82), 223 (3.83) nm; CD (c 2.0 × 10−4 M, MeOH) λmax (Δε) 211 (−1.01), 256 (+1.05), 352 (−0.12) nm; IR (neat) νmax 3423, 2969, 2926, 1677, 1612, 1395, 1043, 593 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 600 MHz) H-1 → C-3, 5; H-2 → C-4, 10; H-5 → C-4, 7, 10, 20; H-6α → C-7, 10; H-6β → C-5, 7, 10; H-12α → C-11, 13, 14, 15, 17; H-12β → C-9, 11, 13, 14, 15, 17; H-14 → C-8, 9, 12; H-16a → C-13, 15; H-16b → C-13, 15; H3-17 → C-12, 13, 14, 15; H3-18 → C-3, 4, 19; H3-19 → C-3, 4, 5, 18; H3-20 → C-1, 5, 9, 10; NOESY correlations (acetone-d6, 600 MHz) H-5 ↔ H3-19; H-6α ↔ H3-19; H-6β ↔ OH-14; H-14 ↔ H3-17; H3-18 ↔ H3-20; HRESIMS m/z 329.1749 [M + H]+ (calcd for C20H24O4, 329.1747).ε) 223 (3.93), 246 (3.86) nm; CD (c 8.0 × 10−4 M, MeOH) λmax (Δε) 202 (−5.85), 229 (+1.99), 269 (+2.18), 350 (−0.92) nm; IR (neat) νmax 3480, 2972, 2935, 1756, 1684, 1370, 1221, 1021, 599, 499 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 600 MHz) H-1 → C-3, 5, 10; H-2 → C-10; H-5 → C-10; H-6α → C-5, 7, 10; H-6β → C-5, 7, 10; H-12α → C-9, 11, 13, 14, 17; H-12β → C-11, 13, 14, 17; H-14 → C-7, 8, 9, 12, 21; H-16a → C-13, 15; H-16b → C-13, 15; H3-17 → C-12, 13, 14, 15; H3-18 → C-3, 4, 5, 19; H3-19 → C-3, 4, 5, 10, 18; H3-20 → C-1, 5, 9, 10; H3-22 → C-21; NOESY correlations (acetone-d6, 600 MHz) H-5 ↔ H3-19; H-6α ↔ H3-19; H-12α ↔ H-17; H-14 ↔ H3-17; H3-18 ↔ H3-20; HRESIMS m/z 393.1670 [M + Na]+ (calcd for C22H26O5, 393.1672).ε) 205 (3.82), 219 (3.79) nm; CD (c 2.4 × 10−4 M, MeOH) λmax (Δε) 230 (+3.58), 305 (−1.69) nm; IR (neat) νmax 3440, 2963, 2877, 1710, 1666, 1374, 1057, 1035 cm−1; 1H and 13C NMR data see Table 1; HMBC data (CDCl3, 600 MHz) H-1 → C-3, 5, 9; H-2 → C-4; H-5 → C-6, 7, 18, 20; H-6α → C-5, 7, 8, 10; H-6β → C-5, 7, 8, 10; H-7 → C-6, 8, 14; H-12α → C-9, 11, 13, 14, 17; H-12β → C-11, 13, 14, 15, 17; H-14 → C-7, 9, 12, 13, 17; H-15 → C-12, 13, 14, 17; H-16a → C-13, 15; H-16b → C-13; H3-17 → C-11, 12, 13, 14, 15; H3-18 → C-3, 4, 5, 19; H3-19 → C-3, 5, 18; H3-20 → C-1, 5, 9, 10; NOESY correlations (CDCl3, 600 MHz) H-5 ↔ H-7; H-5 ↔ H3-19; H-6α ↔ H-7; H-6β ↔ H3-18; H-6β ↔ H3-20; H-12β ↔ H-17; H-12β ↔ H-20; HRESIMS m/z 331.1908 [M + H]+ (calcd for C20H26O4, 331.1904).
:1 petroleum ether–EtOAc was separated by Sephadex LH-20 column chromatography (CC) eluting with 1:1 CH2Cl2–MeOH and the resulting subfractions were combined and purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 47% CH3CN in H2O for 30 min; 2 mL min−1) to afford 2 (1.0 mg, tR 20.0 min). The fraction (165.8 mg) eluted with 3.5:1 petroleum ether–EtOAc was separated by reversed-phase silica gel column chromatography (CC) eluting with a MeOH–H2O gradient. The subfraction (18 mg) eluted with 40% MeOH–H2O was purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 37% CH3CN in H2O for 30 min; 2 mL min−1) to afford 3 (4.0 mg, tR 22.5 min). The subfraction (23 mg) eluted with 50% MeOH–H2O was purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 55% MeOH in H2O for 90 min; 2 mL min−1) to afford 4 (2.7 mg, tR 67.5 min) and 1 (3.4 mg, tR 82.5 min).
ε) 206 (3.82), 223 (3.83) nm; CD (c 2.0 × 10−4 M, MeOH) λmax (Δε) 211 (−1.01), 256 (+1.05), 352 (−0.12) nm; IR (neat) νmax 3423, 2969, 2926, 1677, 1612, 1395, 1043, 593 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 600 MHz) H-1 → C-3, 5; H-2 → C-4, 10; H-5 → C-4, 7, 10, 20; H-6α → C-7, 10; H-6β → C-5, 7, 10; H-12α → C-11, 13, 14, 15, 17; H-12β → C-9, 11, 13, 14, 15, 17; H-14 → C-8, 9, 12; H-16a → C-13, 15; H-16b → C-13, 15; H3-17 → C-12, 13, 14, 15; H3-18 → C-3, 4, 19; H3-19 → C-3, 4, 5, 18; H3-20 → C-1, 5, 9, 10; NOESY correlations (acetone-d6, 600 MHz) H-5 ↔ H3-19; H-6α ↔ H3-19; H-6β ↔ OH-14; H-14 ↔ H3-17; H3-18 ↔ H3-20; HRESIMS m/z 329.1749 [M + H]+ (calcd for C20H24O4, 329.1747).ε) 223 (3.93), 246 (3.86) nm; CD (c 8.0 × 10−4 M, MeOH) λmax (Δε) 202 (−5.85), 229 (+1.99), 269 (+2.18), 350 (−0.92) nm; IR (neat) νmax 3480, 2972, 2935, 1756, 1684, 1370, 1221, 1021, 599, 499 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 600 MHz) H-1 → C-3, 5, 10; H-2 → C-10; H-5 → C-10; H-6α → C-5, 7, 10; H-6β → C-5, 7, 10; H-12α → C-9, 11, 13, 14, 17; H-12β → C-11, 13, 14, 17; H-14 → C-7, 8, 9, 12, 21; H-16a → C-13, 15; H-16b → C-13, 15; H3-17 → C-12, 13, 14, 15; H3-18 → C-3, 4, 5, 19; H3-19 → C-3, 4, 5, 10, 18; H3-20 → C-1, 5, 9, 10; H3-22 → C-21; NOESY correlations (acetone-d6, 600 MHz) H-5 ↔ H3-19; H-6α ↔ H3-19; H-12α ↔ H-17; H-14 ↔ H3-17; H3-18 ↔ H3-20; HRESIMS m/z 393.1670 [M + Na]+ (calcd for C22H26O5, 393.1672).ε) 205 (3.82), 219 (3.79) nm; CD (c 2.4 × 10−4 M, MeOH) λmax (Δε) 230 (+3.58), 305 (−1.69) nm; IR (neat) νmax 3440, 2963, 2877, 1710, 1666, 1374, 1057, 1035 cm−1; 1H and 13C NMR data see Table 1; HMBC data (CDCl3, 600 MHz) H-1 → C-3, 5, 9; H-2 → C-4; H-5 → C-6, 7, 18, 20; H-6α → C-5, 7, 8, 10; H-6β → C-5, 7, 8, 10; H-7 → C-6, 8, 14; H-12α → C-9, 11, 13, 14, 17; H-12β → C-11, 13, 14, 15, 17; H-14 → C-7, 9, 12, 13, 17; H-15 → C-12, 13, 14, 17; H-16a → C-13, 15; H-16b → C-13; H3-17 → C-11, 12, 13, 14, 15; H3-18 → C-3, 4, 5, 19; H3-19 → C-3, 5, 18; H3-20 → C-1, 5, 9, 10; NOESY correlations (CDCl3, 600 MHz) H-5 ↔ H-7; H-5 ↔ H3-19; H-6α ↔ H-7; H-6β ↔ H3-18; H-6β ↔ H3-20; H-12β ↔ H-17; H-12β ↔ H-20; HRESIMS m/z 331.1908 [M + H]+ (calcd for C20H26O4, 331.1904).Computational details
Conformational analyses for 1–4 were performed via the Molecular Operating Environment (MOE) version 2009.10 (Chemical Computing Group, Canada) software package with LowModeMD at the MMF94 force field. The MMF94 conformers were further optimized using TDDFT at the B3LYP/6-311G(2d,2p) basis set level in MeOH with the IEFPCM model. The stationary points have been checked as the true minima of the potential energy surface by verifying that they do not exhibit vibrational imaginary frequencies. The 25 lowest electronic transitions were calculated, and the rotational strengths of each electronic excitation were given using both dipole length and velocity representations. ECD spectra were simulated in SpecDis23 (ref. 30) using a Gaussian function with half-bandwidths of 0.30 eV. The overall ECD spectra were then generated according to Boltzmann weighting of each conformer. The systematic errors in the prediction of wavelength and excited-state energies are compensated by employing UV correlation. All quantum computations were performed using the Gaussian 09 package.31Cytotoxicity assays
The cytotoxic activity of compounds 1–4 were tested using 96 well plates according to a literature MTS method with slight modification.32 Cells were seeded in 96-well plates at a density of 1.0 × 104 cells per well in 100 μL of complete culture medium. After cell attachment overnight, the medium was removed, and each well was treated with 100 μL of medium containing 0.1% DMSO or appropriate concentrations of the test compounds and the positive control cisplatin and incubated with cells at 37 °C for 48 h in a 5% CO2-containing incubator. Proliferation was assessed by adding 20 μL of MTS (Promega) to each well in the dark, after 90 min of incubation at 37 °C. The optical density was recorded on a microplate reader at 490 nm. Three duplicate wells were used for each concentration, and all the tests were repeated three times.Conclusions
In summary, three new highly oxygenated pimarane diterpenoids, sarcosenones A–C (1–3), and a structurally related known compound (4) were isolated from cultures of Sarcosomataceae sp., an endolichenic fungus found in the lichen Everniastrum sp. (Parmeliaceae). Their structures were elucidated based on NMR spectroscopic data and electronic circular dichroism (ECD) calculations. The absolute configuration of known compound 4 was determined for the first time. To our knowledge, this is the first report of pimarane diterpenoids isolated from the family Sarcosomataceae. Compound 1 showed moderate cytotoxicity, with IC50 values of 7.5–26.4 μM. Theoretically, 1–4 could be generated from the precursor (E,E,E)-geranylgeranyl diphosphate (GGDP).33 GGPP was first cyclized by copalyl diphosphate (CPP) synthases, and then CPP was completely cyclized by pimaradiene synthases, leading to the formation of these metabolites (Scheme S1†).Conflicts of interest
The authors declare no conflict interest.Acknowledgements
X. H., S. Z., and Y. Z. were supported in part by the National Program of Drug Research and Development (2012ZX09301-003); Y. X. and Y. C. was supported by the CAMS Innovation Fund for Medical Sciences (2018-I2M-3-005).Notes and references
- P. Reveglia, A. Cimmino, M. Masi, P. Nocera, N. Berova, G. Ellestad and A. Evidente, Chirality, 2018, 30, 1–20 CrossRef PubMed.
- X. Wang, H. Yu, Y. Zhang, X. Lu, B. Wang and X. Liu, Chem. Biodiversity, 2008, 15, e1700276 CrossRef PubMed.
- S. Palkin, J. Chem. Educ., 1935, 12, 35–40 CrossRef CAS.
- S. Thongnest, C. Mahidol, S. Sutthivaiyakit and S. Ruchirawat, J. Nat. Prod., 2005, 68, 1632–1636 CrossRef CAS PubMed.
- A. Ulubelen, G. Topcu and C. B. Johansson, J. Nat. Prod., 1997, 60, 1275–1280 CrossRef CAS PubMed.
- A. Andolfi, L. Maddau, S. Basso, B. T. Linalddeddu, A. Cimmino, B. Scanu, A. Deidda, A. Tuzi and A. Evidente, J. Nat. Prod., 2014, 77, 2352–2360 CrossRef CAS PubMed.
- Y. P. Tan, Y. Xue, A. I. Savchenko, S. D. Houston, N. Modhiran, C. L. D. Mcmillan, G. M. Boyle, P. V. Bernhardt, P. R. Young, D. Watterson and C. M. Williams, J. Nat. Prod., 2019, 82, 2828–2834 CrossRef CAS PubMed.
- S. Huang, Q. Ma, W. Fang, F. Xu, H. Peng, H. Dai, J. Zhou and Y. Zhao, J. Asian Nat. Prod. Res., 2013, 7, 750–755 CrossRef PubMed.
- L. Sun, D. Li, M. Tao, Y. Chen, F. Dan and W. Zhang, Mar. Drugs, 2012, 10, 539–550 CrossRef CAS PubMed.
- H. Yu, X. Wang, Y. Zhang, W. Xu, J. Zhang, X. Zhou, X. Lu, X. Liu and B. Jiao, J. Nat. Prod., 2018, 81, 1553–1560 CrossRef CAS PubMed.
- U. K. Karmakar, N. Ishikawa, M. A. Arai, F. Ahmed, T. Koyano, T. Kowithayakorn and M. Ishibashi, J. Nat. Prod., 2016, 79, 2075–2082 CrossRef CAS PubMed.
- X. Wang, B. P. Bashyal, E. M. K. Wijeratne, J. M. U'Ren, M. X. Liu, M. K. Gunatilaka, A. E. Arnold and A. A. L. Gunatilaka, J. Nat. Prod., 2011, 74, 2052–2061 CrossRef CAS PubMed.
- S. Huang, H. Luo, Q. Ma, H. Peng, H. Dai, J. Zhou and Y. Zhao, Chem. Biodiversity, 2014, 11, 1406–1416 CrossRef CAS PubMed.
- R. Chokchaisiri, W. Chaichompoo, W. Chunklok, S. Cheenpracha, L. Ganranoo, N. Phutthawong, S. Bureekaew and A. Suksamrarn, J. Nat. Prod., 2020, 83, 14–19 CrossRef CAS PubMed.
- B. Köpcke, R. W. S. Weber and H. Anke, Phytochemistry, 2002, 60, 709–714 CrossRef.
- X. Liu, W. R. Schwan, T. J. Volk, M. Rott, M. Liu, P. Huang, Z. Liu, Y. Wang, N. C. Zitomer, C. Sleger, S. Hartsel, A. Monte and L. Zhang, J. Nat. Prod., 2012, 75, 1534–1538 CrossRef CAS PubMed.
- J. Li, R. Ding, H. Gao, L. Guo, X. Yao, Y. Zhang and J. Tang, RSC Adv., 2019, 9, 39082–39089 RSC.
- M. Johansson, B. Köpcke, H. Anke and O. Sterner, J. Antibiot., 2002, 55, 104–106 CrossRef CAS PubMed.
- J. Tian, R. Yu, X. Li, H. Gao, L. Guo, Y. Li, J. Li, J. Tang and X. Yao, Fitoterapia, 2015, 101, 92–98 CrossRef CAS PubMed.
- J. Tian, R. Yu, X. Li, H. Gao, L. Guo, J. Tang and X. Yao, Magn. Reson. Chem., 2015, 53, 861–871 CrossRef PubMed.
- W. A. Ayer, L. S. Trifonov, L. J. Hutchison and P. Chakravarty, Nat. Prod. Lett., 2000, 14, 405–410 CrossRef CAS.
- J. Tian, R. Yu, X. Li, H. Gao, D. Hu, L. Guo, J. Tang and X. Yao, J. Asian Nat. Prod. Res., 2015, 17, 550–558 CrossRef CAS PubMed.
- F. Zhang, L. Li, S. Niu, Y. Si, L. Guo, X. Jiang and Y. Che, J. Nat. Prod., 2012, 75, 230–237 CrossRef CAS PubMed.
- J. J. Kellogg and H. A. Raja, Phytochem. Rev., 2017, 16, 271–293 CrossRef CAS.
- Y. Wang, S. Niu, S. Liu, L. Guo and Y. Che, Org. Lett., 2010, 12, 5081–5083 CrossRef CAS PubMed.
- F. Zhang, S. Liu, X. Lu, L. Guo, H. Zhang and Y. Che, J. Nat. Prod., 2009, 72, 1782–1785 CrossRef CAS PubMed.
- Y. Wang, Z. Zheng, S. Liu, H. Zhang, E. Li, L. Guo and Y. Che, J. Nat. Prod., 2010, 73, 920–924 CrossRef CAS PubMed.
- J. A. Findlay, G. Li and P. E. Penner, J. Nat. Prod., 1995, 58, 197–200 CrossRef CAS.
- S. Zhu, F. Ren, Z. Guo, J. Liu, X. Liu, G. Liu and Y. Che, J. Nat. Prod., 2019, 82, 462–468 CrossRef CAS PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev D.01, Gaussian, Inc.: Wallingford, CT, 2009 Search PubMed.
- N. Zhang, Y. Chen, R. Jiang, E. Li, X. Chen, Z. Xi, Y. Guo, X. Liu, Y. Zhou, Y. Che and X. Jiang, Autophagy, 2011, 7, 598–612 CrossRef CAS PubMed.
- M. Xu, M. L. Hillwig, M. S. Tiernan and R. J. Peters, J. Nat. Prod., 2017, 80, 328–333 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV, IR, CD, HRESIMS, NMR spectra of compounds 1–3; ECD calculations of compounds 1–4. See DOI: 10.1039/d0ra02485f |
| This journal is © The Royal Society of Chemistry 2020 |