
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient modification of PAMAM G1 dendrimer surface with β-cyclodextrin units by CuAAC: impact on the water solubility and cytotoxicity†
Kendra Sorroza-Martínez‡
a,
Israel González-Méndez‡a,
Ricardo D. Martínez-Serranoa,
José D. Solano b,
Andrea Ruiua,
Javier Illescasc,
Xiao Xia Zhud and
Ernesto Rivera*a
b,
Andrea Ruiua,
Javier Illescasc,
Xiao Xia Zhud and
Ernesto Rivera*a
aInstituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, CP 04510, México City, México. E-mail: riverage@unam.mx
bFacultad de Química, Departamento de Biología, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, CP 04510, México City, México
cTecnológico Nacional de México/Instituto Tecnológico de Toluca, Av. Tecnológico S/N, Col. Agrícola Bellavista CP 52149, Metepec, México
dDépartement de Chimie, Université de Montréal, C.P. 6128, Succursale Centre-ville, Montreal, QC H3C 3J7, Canada
First published on 7th July 2020
Abstract
The toxicity of the poly(amidoamine) dendrimers (PAMAM) caused by the peripheral amino groups has been a limitation for their use as drug carriers in clinical applications. In this work, we completely modified the periphery of PAMAM dendrimer generation 1 (PAMAM G1) with β-cyclodextrin (β-CD) units through the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) to obtain the PAMAM G1-β-CD dendrimer with high yield. The PAMAM G1-β-CD was characterized by 1H- and 13C-NMR and mass spectrometry studies. Moreover, the PAMAM G1-β-CD dendrimer showed remarkably higher water solubility than native β-CD. Finally, we studied the toxicity of PAMAM G1-β-CD dendrimer in four different cell lines, human breast cancer cells (MCF-7 and MDA-MB-231), human cervical adenocarcinoma cancer cells (HeLa) and pig kidney epithelial cells (LLC-PK1). The PAMAM G1-β-CD dendrimer did not present any cytotoxicity in cell lines tested which shows the potentiality of this new class of dendrimers.
Introduction
In the last decades, research on the development of new dendrimers has become more relevant and attracted the attention of the scientific community, since these compounds have unique properties that make them ideal carriers for therapeutic and diagnostic agents in the construction of drug delivery systems (DDS).1 One advantage of dendrimers is that between the small molecules and the polymers, dendrimers benefit from the properties of both fields. Given the versatility to modify them (like polymers), it is possible to guarantee a perfectly defined and reproducible structure.2 It has been described that the groups in the periphery are largely responsible for the physicochemical properties of dendrimers and can be modified in a controlled manner to yield monodisperse materials.3One of the most studied dendrimers, is the family of poly(amidoamines) (PAMAM), which is constituted by a core of ethylenediamine or ammonia, repetitive units of polyamides, tertiary amines and a periphery containing primary amines.4 It has been found that low generations (G 0–3) of PAMAM dendrimers tend to form open and amorphous structures, and therefore do not exhibit well defined internal characteristics, while high generations dendrimers (G 4–10) adopt spherical and rigid structures that allow them to encapsulate drug molecules in their branches.5,6
At physiological conditions (pH = 7.4) the primary amines of the PAMAM dendrimers are positively charged,7 they exhibit toxicity, since they interact with the cell membrane, which is negatively charged, compromising its integrity and filtering intracellular components, that finally lead to a cell death. This toxicity is also associated with the increase in generation, due to the augment in the number of terminal amino groups. For this reason, the clinical application of PAMAM dendrimers is quite limited.8,9
In recent years, research has focused on improving the biocompatibility of PAMAM dendrimers by surface engineering, that means the modification of the periphery to neutralize the primary amines that are positively charged on the surface, thereby preserving the ability to deliver drugs. There are several strategies to modify the periphery of PAMAM dendrimers, such as coupling with carbohydrates, amino acids or peptides, acetylations and PEGylations.8 One of the strategies used in surface engineering of PAMAM dendrimers is the coupling with cyclodextrins because it increases the solubility, stability and biocompatibility of dendrimers and therefore causes a synergistic improvement in the administration of drugs.10
Chemically, cyclodextrins (CDs) are cyclic oligosaccharides linked through α-1,4 glucosidic bonds. The nomenclature of the CDs is based on the number of glucose units contained in their structure, so that the CDs containing six units are called alpha-CD (α-CD), seven units, beta-CD (β-CD) and eight units, gamma-CD (γ-CD).11 As a result of the restricted rotation of the bonds in the glucose units by the chair conformation, CDs molecules have a truncated cone shape with a cavity of determined volume. The hydroxyl groups attached to the carbons 3 and 4 are located on the secondary face, and the hydroxyl groups attached to carbon 6 are located on the primary face of the CDs; these hydroxyl groups have a different reactivity, because they are attached to secondary and primary carbons, respectively. The surface of the CDs is coated with hydroxyl groups and the inner part is coated with ether groups of anomeric oxygen atoms. Therefore, CDs have a relatively hydrophobic internal cavity, which contains water molecules and a hydrophilic outer surface. This special conformation allows the inclusion of hydrophobic molecules or parts of molecules within the CD cavity, which must have a compatible size with the cavity of CD, to form a stable inclusion complex.12 β-CD is the most accessible and widely used CD in the design of DDS, due to the adequate size of its cavity to form inclusion complexes with drug molecules.13
On the other hand, the 1,3-dipolar cycloaddition reaction between an azide and a terminal alkyne catalyzed by copper(I) (CuAAC) which produces 1,4-disubstituted 1,2,3-triazoles was reported independently in 2001 by Meldal et al.14 and Sharpless et al.15,16 Meldal used copper iodide and N,N-diisopropylethylamine for the synthesis of peptides, while Sharpless described the use of copper sulfate pentahydrate and sodium ascorbate in aqueous systems and designed a practical procedure for the covalent attachment between any molecule containing an azide group and another molecule containing a terminal alkyne group. The reaction of CuAAC is modular, specific and regioselective, inert byproducts are obtained and offers high yields. In addition, this reaction is carried out under simple conditions and with accessible raw materials.17 During the last decade, the CuAAC reaction has proven to be a reliable and versatile technique for the preparation of new functional materials,18,19 including dendrimers as well as the modification of their periphery.3,20–24
There are several works that describe the conjugation of PAMAM dendrimers (G 2–4) with different CD molecules.25–32 The incorporation from one to five CD units is carried out in the periphery of dendrimers with applications in the delivery of genetic material for the specific treatment of diseases. On the other hand, since low generations of PAMAM dendrimers do not show encapsulating properties of molecules, and particularly β-CD molecules, have a limited water solubility. Therefore, the clinical applications of low generations of PAMAM and of β-CD as single molecules are limited. Based on the previously mentioned properties, we present a novel design and synthesis of a PAMAM G1 conjugate with β-CD units throughout the periphery of dendrimer. This conjugation was achieved by means of a CuAAC reaction with high yields and a simple purification process. Furthermore, we focused on the study of the in vitro cellular behavior of this novel dendrimer as part of the rational evaluation, since our perspective is based on the potential use of this PAMAM G1-β-CD conjugate as a possible carrier of diverse molecules. The advantage of this new modified dendrimer is that it is able to carry a higher number of drug units than a single carrier.
Results and discussion
Synthesis
In recent years, numerous reports described the advantages of PAMAM dendrimers in various areas of science and technology; however, there are also some disadvantages that limit their use in clinical applications. Firstly, PAMAM dendrimers are toxic and this toxicity can be only resolved by modifying the structure of the periphery of the dendrimer.33 Intense research has focused on the surface engineering of PAMAM dendrimers with the aim of reducing or eliminating their toxicity by neutralizing the amino groups in the periphery by means of coupling of neutral or anionic groups in order to prevent the electrostatic interaction of PAMAM dendrimers with biological membranes to help them to be biocompatible for their use as drug carriers.8 A wide variety of methods have been reported to modify the surface of PAMAM dendrimers, including modification with carbohydrates, specifically with oligosaccharides such as CDs, since it has been reported that a synergy of the properties of PAMAM dendrimers and CDs occurs.34–36 PAMAM and β-CD dendrimers have been individually investigated as promising carriers for clinical applications; however, due to the toxicity of PAMAM dendrimers8,37 and the low water solubility of β-CD,38 their application in the clinical area is limited. In the present study we present a novel and efficient approach to modify the periphery of the PAMAM G1 dendrimer with β-CD units through the CuAAC reaction. All the intermediates and final compounds involved in the synthesis have been fully characterized.Firstly, alkynyl-PAMAM G1 was synthesized (Scheme l) and was used as alkyne source for the CuAAC reaction. A Williamson etherification reaction was carried out between propargyl bromide and methyl 2-(4-hydroxyphenyl)acetate, to obtain intermediate I, whose structure was confirmed by 1H-NMR and 13C-NMR and by ESI mass spectrometry (Fig. S1–S3 in the ESI†). Then, compound I was saponified via a simple hydrolysis with a yield >90% to obtain intermediate II. The structure of this compound was also confirmed by NMR and mass spectrometry (Fig. S4–S6 in ESI†).
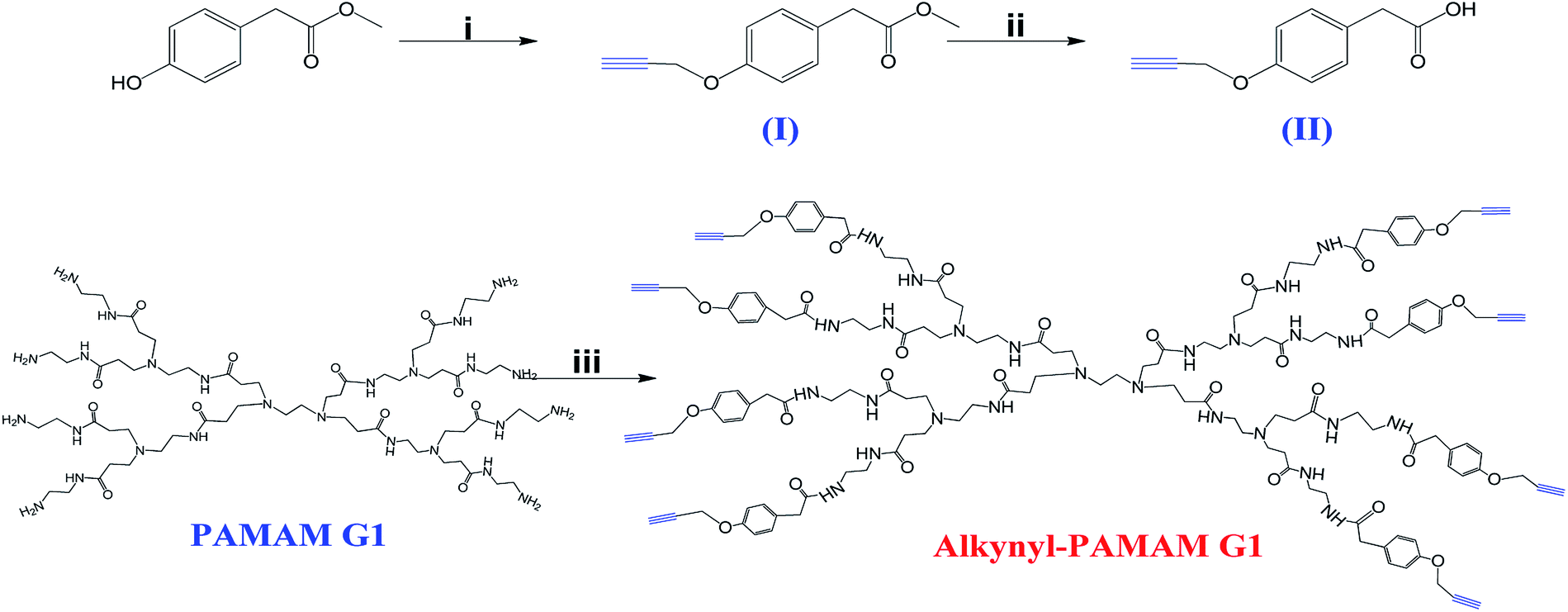 | ||
Scheme 1 Synthesis of alkynyl-PAMAM G1. Conditions: (i) propargyl bromide, K2CO3, anhydrous acetone, 24 h, reflux; (ii) NaOH/HCl, EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O; (iii) EDC, HOBt, DIPEA, DMF, 35 °C, 5 days. :H2O; (iii) EDC, HOBt, DIPEA, DMF, 35 °C, 5 days. | ||
Regarding the synthesis of the PAMAM G1-alkyne compound, we performed a C–N coupling between the amine groups present in the periphery of the PAMAM G1 dendrimer and the carboxylic acid of intermediate II, using HOBt and EDC as coupling agents and DIPEA as acid activator. These coupling agents are frequently employed for the preparation of amides39 in the biomaterials field. It was reported that EDC is a very effective coupling agent for the synthesis of these compounds and when it is used jointly with HOBt, there is an increase in the efficiency of the reaction and the by-products can be easily removed in water.40,41 The modification of the periphery of the PAMAM G1 dendrimer by introducing alkyne groups was confirmed by 1H- and 13C-NMR and by MALDI-TOF mass spectrometry (Fig. S7–S9 in ESI†).
On the other hand, m-N3-β-CD was synthesized as a source of azide for the CuAAC reaction (see Scheme 2). Firstly, the intermediary m-Ts-β-CD was obtained by functionalizing a single hydroxyl group of the primary face of the β-CD via a tosylation reaction, which was further reacted with β-CD.42 With this proposed method it was possible to modify a single position of the primary face of the β-CD with a high yield, compared to the previously reported procedures.43,44 Tosylation of a single group of the β-CD was confirmed by 1H-NMR, DEPTQ-NMR and MALDI-TOF mass spectrometry (Fig. S10–S12 in ESI†). Then a bimolecular nucleophilic substitution reaction was carried out to replace the tosyl group of m-Ts-β-CD with the azido group on the primary face of the β-CD to afford m-N3-β-CD, whose structure was confirmed by 1H-NMR, DEPTQ-NMR and mass spectrometry (Fig. S13–S15 in ESI†).
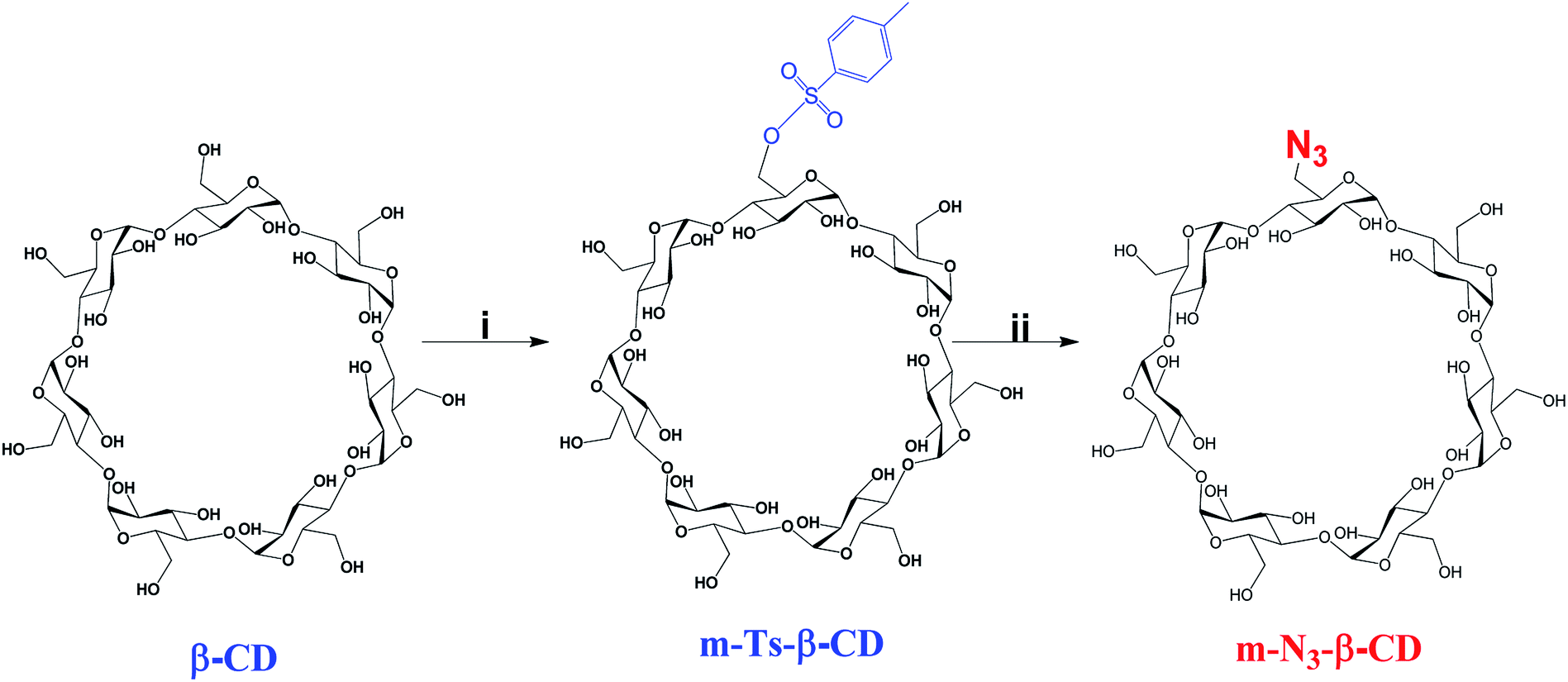 | ||
| Scheme 2 Synthesis of m-N3-β-CD. Conditions: (i) TsO2/NaOH, H2O, 2 h, RT; (ii) NaN3, DMF, 80 °C, 48 h. | ||
The CuAAC reaction allows a successful functionalization of biomaterials in a selective way45–53 and has been often used to modify the surface of PAMAM dendrimers54–56 because it offers high yields with a single reaction product obtained with great purity and harmless by-products easily removable.16,57 In this work, we used the CuAAC reaction to efficiently functionalize the entire periphery of the PAMAM G1 dendrimer with β-CD units to build a novel dendritic structure, which can act as a potential drug carrier. Once the necessary intermediates for the reaction of CuAAC (Scheme 3), the alkyne (alkynyl-PAMAM G1) and the azide (m-N3-β-CD) were obtained, the coupling was carried out using CuSO4·5H2O and H2Asc to generate in situ Cu(I), which is the main catalyst in this reaction. To ensure the functionalization of the 8 alkyne groups of the modified periphery of the PAMAM G1 dendrimer, an excess of m-N3-β-CD was employed. The PAMAM G1-β-CD dendrimer was obtained with 98% yield and its structure was established by 1H-NMR, DEPTQ-NMR, elemental analysis by X-ray fluorescence (Fig. S16–S17 in ESI†). Dendrimer purification was performed by size exclusion chromatography (SEC) using water as eluent. This technique of purification proved to be highly efficient to remove the remaining copper and ascorbic acid, and thereby avoiding the classic method of dialysis.58
 | ||
| Scheme 3 Synthesis of PAMAM G1-β-CD. Conditions: “Click” CuSO4·5H2O, H2Asc, DMSO, 80 °C, 48 h. | ||
Characterization
Full characterization of all the synthetic intermediates and final compounds was carried out. Regarding the characterization of intermediate I, in the 1H- and 13C-NMR spectra, the signals corresponding to the alkyne group at 2.54 ppm and 75.54 ppm are observed, respectively. In addition, the disappearance of the signal corresponding to the proton of the phenol is observed. The molecular ion at 204.08 m/z, corresponding to the molecular weight of the compound, is observed in the ESI spectrum. In the 1H-NMR spectrum of intermediate II, we can observe a signal at 12.25 ppm, due to the proton of the carboxylic acid, produced by the hydrolysis of intermediate I. The molecular ion is observed at 191.07 m/z in the ESI spectrum, which corresponds to the molecular weight of this compound. On the other hand, in the 1H-NMR spectrum of alkynyl-PAMAM G1, the signals corresponding to the alkyne group at 3.55 ppm are observed, and the proton signal of the 8 amide groups appear at 8.48 ppm, thereby confirming that the C–N coupling took place. In addition, the signals due to the protons of the aromatic ring appear at 7.18 and 6.90 ppm, exhibiting an up-field shift. Finally, the signals corresponding to the protons of the PAMAM G1 dendrimer are observed. In the MALDI-TOF spectrum, the molecular ion is observed at 2810.819 m/z, which corresponds to the molecular weight of the multiprotonated species, which confirms the obtainment of the alkynyl-PAMAM G1 compound.On the other hand, m-Ts-β-CD was characterized by 1H-NMR spectroscopy, the signals corresponding to the aromatic ring protons at 7.75 and 7.44 ppm and the methyl protons at 2.42 ppm of the tosyl group can be observed. In the MALDI-TOF spectrum the appearance of the peak at 1311.591 m/z corresponding to the expected molecular weight of the compound plus a sodium ion confirmed the mono-substitution of the primary face of the β-CD. After the modification of m-Ts-β-CD, compound m-N3-β-CD was obtained. In the 1H-NMR spectrum, the disappearance of the signals corresponding to the protons of the aromatic ring and the methyl protons of the tosyl group confirms its replacement by the azide group. The signal at 1182.764 m/z in the MALDI-TOF spectrometry corresponds to the molecular weight of the compound, plus a sodium ion.
Once the CuAAC reaction was achieved, the PAMAM G1-β-CD dendrimer was obtained and further characterized. In the 1H-NMR spectrum in DMSO-d6 (Fig. 1), a signal due to the triazole group appears at 8.16 ppm, followed by the signals of the aromatic ring at 7.17 and 6.97 ppm. The signals corresponding to the alkynyl-PAMAM G1 and the m-N3-β-CD exhibited a clear change in the chemical environment due to the conjugation between both moieties. The PAMAM G1-β-CD dendrimer was also characterized by MALDI-TOF, using the following conditions: 2,5-dihydroxybenzoic acid (DHB) in relation (2/5) sample/matrix; ditranol (DIT) in relation (2/5) sample/matrix; DHB and AgNO3 as a cationizer agent, in ratios (2/5/1) sample/matrix/cationizer; DHB dissolved in H2O/ACN/TFA in ratios (60/40/0.1); and 2-acetylphloroglucinol (THAP) dissolved in methanol in ratios (1/1), (3/4), (1/9) sample/matrix. Only the signals corresponding to the matrices were observed in the range 1000 to 20000 m/z. Result of the foregoing, it was not possible to characterize PAMAM G1-β-CD dendrimer by MALDI-TOF. It has been reported in the literature40,59,60 that PAMAM dendrimers are difficult to ionize and to be detected by mass spectrometry. This dendrimer was therefore characterized by elemental analysis and X-ray fluorescence, where the following values were found for C = 44.62, H = 6.65, N = 5.28, and O = 43.53, which is related to a molecular formula of C486H744N50O300 + 56H2O corresponding to the PAMAM G1-β-CD dendrimer with 56 molecules of water, this result is consistent with previous reports,61 which indicates that in the cavities of the β-CD units water molecules are trapped.
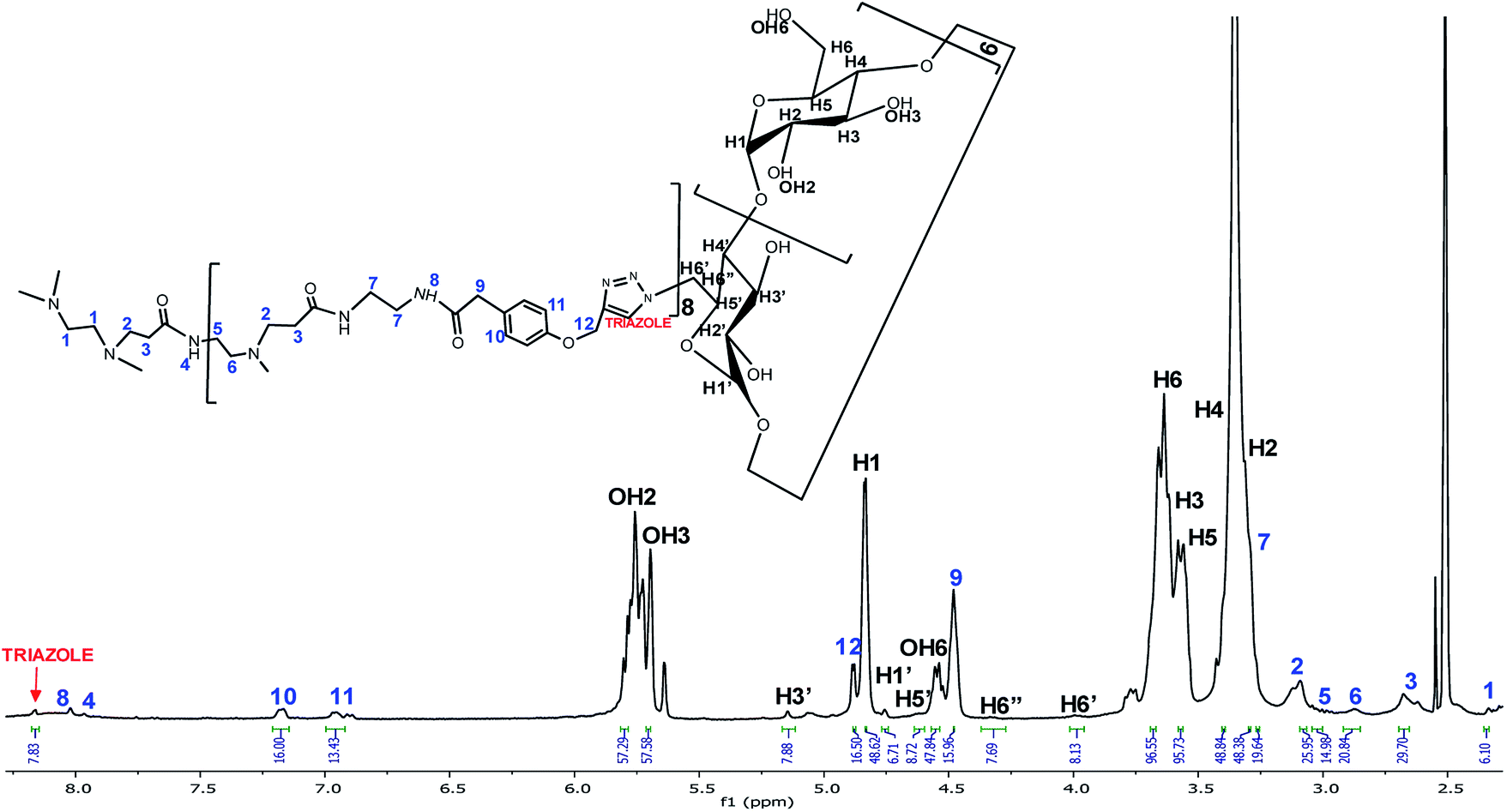 | ||
| Fig. 1 1H NMR spectrum of PAMAM G1-β-CD in DMSO-d6. | ||
Determination of the water solubility of the PAMAM G1-β-CD dendrimer
The solubility of dendrimers is determined by the functional groups located in the periphery, by the generation of the dendrimer, by the repetitive units and even by the core. Their high water solubility guarantees their application as enhancers of the solubility of drugs with hydrophobic nature, as well as their application in the clinic area as drug carriers.62 The conjugation of dendrimers with hydrophilic molecules, such as CDs, significantly increases their solubility in water and their stability, prolonging the blood circulation of these platforms.63 The quantitative solubility in water of the PAMAM G1-β-CD dendrimer was determined by the method reported by Jozwiakowski and Connors64 and a result of 620.5 ± 0.0004 mg mL−1 was obtained, which is considerably higher, compared to the solubility value of the native β-CD (18.5 mg mL−1).65 This result can be explained by the union of the PAMAM G1 dendrimer with the β-CD units, which generates a new dendritic structure with higher number of β-CD groups on the periphery, and is able to form hydrogen bonds with the aqueous medium. Another explanation for the enhanced solubility of PAMAM G1-β-CD is that once the β-CD is linked to the PAMAM G1 dendrimer the intramolecular hydrogen bonds formed in the β-CD molecules alone, which are the cause of their low solubility,66 are broken by mono-modification, giving rise to a better solubility in the new dendrimer. If the solubility of our dendrimer is compared to the solubility values of some commercial β-CD derivatives, such as sulfobutylether-β-CD (>500 mg mL−1), O-methyl-β-CD (>500 mg mL−1) and 2-hydroxypropyl-β-CD (600 mg mL−1),65 our dendrimer was found to have a higher solubility than all of them.Cytotoxicity studies
To study the in vitro behavior of PAMAM G1-β-CD dendrimer and corroborate that the design does not produce an undesirable toxic carrier, the cellular cytotoxicity of the PAMAM G1 and PAMAM G1-β-CD dendrimers was evaluated in three different cancer cell lines; MCF-7, MDA-MB-231 and HeLa. Furthermore, and in accordance with our systematic evaluation, we also analyzed cytotoxic activity in a non-carcinogenic cell line, therefore LLC-PK1 was selected for this study. It has been reported that low-generation PAMAM dendrimers (G 0–2) do not present cellular toxicity.67–72 To corroborate this behavior, we tested the cytotoxic activity of PAMAM G1 dendrimer in the four cell lines mentioned above. The results presented in Fig. S18† show that the PAMAM G1 dendrimer does not present any toxicity in the tested cell lines. Regarding the PAMAM G1-β-CD dendrimer, in the carcinogenic and non-carcinogenic cell lines, six different concentrations from 2 μM to 100 μM were tested, taking a 100% of viability to untreated cells. In Fig. 2, we present the percentage of cell viability (%) versus the concentration of the tested PAMAM G1-β-CD dendrimer; it is shown that in cancer cell lines and kidney cells, the PAMAM G1-β-CD dendrimer does not show measurable cytotoxicity even at the highest evaluated concentration 100 μM. This can be due to the fact that the primary amino groups on the periphery of the PAMAM G1 dendrimer are neutralized by the conjugation with the β-CD units and therefore no longer interact with the cell membrane, preventing it from compromising and eventually causing cellular toxicity. If we compare Fig. 2 and S18,† we can assert that the complete modification of the periphery of the PAMAM G1 dendrimer with β-CD molecules produces a new non-toxic dendrimer.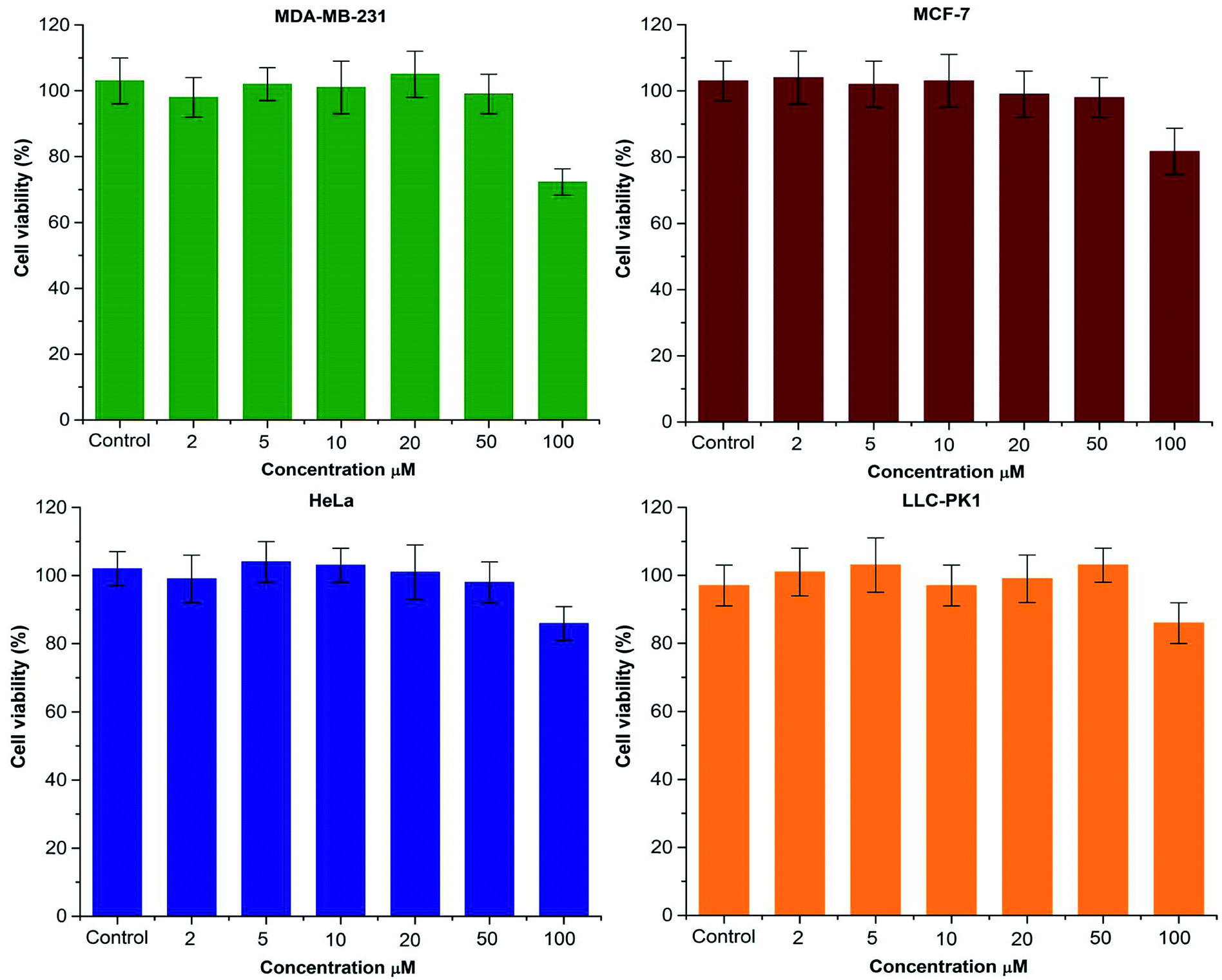 | ||
| Fig. 2 Cell viability in different cell lines of PAMAM G1-β-CD dendrimer. The treated cells were cultured in six wells by concentration, and the experiments were performed three times independently. Data are means ± S.D. | ||
Conclusions
In this work, the complete modification of the periphery of the PAMAM G1 dendrimer with monofunctionalized β-CD molecules was successfully carried out. These macromolecules were coupled by a CuAAC reaction, giving the desired product with yields higher than 80% and a simple purification. The PAMAM G1-β-CD dendrimer has been characterized by 1H- and 13C-NMR and elemental analysis by X-ray fluorescence as a complementary way to characterize the modified PAMAM dendrimers. The water solubility of the PAMAM G1-β-CD dendrimer was determined and resulted to be 620.5 mg mL−1, considerably higher than native β-CD and even than other commercial CDs. Finally, we demonstrated that our PAMAM G1-β-CD dendrimer does not present any cytotoxicity in the analyzed cell lines. Thus, this novel PAMAM G1-β-CD dendrimer is a promising candidate to be used in clinical applications as a possible drug carrier, which has the possibility of carrying larger units of drug molecules per carrier unit, with the advantage of having the synergy of the individual properties of the PAMAM dendrimer and the β-CD units.Experimental section
Materials
All starting materials were commercially available reagent grade and were used without any further purification. β-Cyclodextrin (β-CD), PAMAM dendrimer G1 (PAMAM G1), propargyl bromide solution (80% by weight in toluene), p-toluenesulfonyl chloride (Cl-Ts), p-toluenesulfonic acid (OH-Ts), methyl 2-(4-hydroxyphenyl)acetate, N,N-dimethylformamide anhydrous (DMF), anhydrous potassium carbonate (K2CO3), dimethylsulfoxide (DMSO), ascorbic acid (H2Asc), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl), Bio-Gel P-10 medium from BIO-RAD, 1-hydroxybenzotriazole hydrochloride (HOBt·HCl), N,N-diisopropylethylamina (DIPEA), potassium iodide (KI), sodium azide (NaN3), sodium hydroxide (NaOH), copper sulfate pentahydrate (CuSO4·5H2O), dichloromethane (CH2Cl2), methanol (MeOH), ethanol (EtOH) and acetone (CH3COCH3) were purchased from Sigma-Aldrich.Synthetic procedures
:acetone (1:1) and allowed to dry under vacuum. The yield of this reaction was 88% (1.7 g, 0.0014 mol) and a white solid was obtained. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 5.74 (m, 7H) OH2, 5.67 (m, 6H) OH3, 5.62 (d, J = 2.4 Hz, 1H) OH3′, 4.88 (d, J = 3.5 Hz, 1H) H1′, 4.83 (m, 6H) H1, 4.48 (m, 6H) OH6, 3.77 (m, 2H) H6′, 3.68 (m, 12H) H6, 3.60 (m, 7H) H3, 3.55 (m, 7H) H5, 3.39 (m, 7H) H4, 3.29 (m, 7H) H2. 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 102.38, 102.04, 83.41, 81.99, 73.50, 73.30, 72.85, 72.67, 72.46, 70.63, 60.30, 51.53. MALDI-TOF (m/z): 1182.764 [M + Na]+.Determination of solubility in water of PAMAM G1-β-CD dendrimer
The determination of the solubility of the PAMAM G1-β-CD dendrimer was carried out according to the method reported by Jozwiakowski and Connors64,75 with some modifications. Excess amounts of the compound were placed in three amber vials of 5 mL with a screw cap, which were sealed with parafilm to avoid water evaporation. They were stirred in an oil bath at a constant temperature of 25 ± 0.01 °C for 48 h. The supernatant was separated from the solid phase by filtration through a Milli-Q membrane (pore size of 0.45 μm) by injection of the mixture into disposable plastic syringes of 3 mL at 25 °C. The supernatant of each sample was placed in three different vials. The samples were lyophilized for 48 h and the obtained solid was weighed on a scale with an uncertainty of ±0.0001 g.Characterization
1H- and 13C-NMR measurements were carried out on a Bruker spectrometer (400 MHz), using DMSO-d6 and CDCl3 as solvent. Chemical shifts are reported in ppm (δ) and the signals are described as singlet (s), doublet (d), triplet (t), quartet (q), broad (br), multiplet (m), and doublet of doublets (dd); coupling constants (J) are reported in Hz. Mass measurements, ESI-MS and MALDI-TOF were performed on a JEOL JMS-AX505-HA instrument and on a Bruker Daltonics Flex Analysis instrument, respectively, matrices of 2,5-dihydroxybenzoic acid (DHB), dithranol (DIT) and 2-acetylphloroglucinol (THAP), AgNO3 and TFA as cationizing agents for the case of the PAMAM G1-β-CD conjugate, were used for MALDI-TOF. The elemental analyses were recorded in an Ultra High Vacuum (UHV) of Physical Electronics, scanning XPS microprobe PHI 5000 VersaProbe II, with a MCD detector of 16 channel electron multipliers.Biological procedures
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The financial support for a Quebec-Mexico Bilateral Research Program from FQRNT of Quebec and CONACYT (Project 279380) of Mexico is gratefully acknowledged. KS-M (No. 604166/771826), IG-M (No. 510108/288832) and RDM-S are very grateful with Posgrado en Ciencias Químicas UNAM and CONACyT for the granted fellowship. We also thank M.Sc. Gerardo Cedillo and M.Sc. Lucero Ríos for their assistance with NMR and mass spectrometry measurements, respectively.Notes and references
- A. K. Sharma, A. Gothwal, P. Kesharwani, H. Alsaab, A. K. Iyer and U. Gupta, Drug Discovery Today, 2017, 22, 314 CrossRef CAS PubMed.
- A. M. Caminade, Chem. Commun., 2017, 53, 9830 RSC.
- B. K. Nanjwade, H. M. Bechra, G. K. Derkar, F. V. Manvi and V. K. Nanjwade, Eur. J. Pharm. Sci., 2009, 38, 185 CrossRef CAS PubMed.
- C. Diaz, C. Benitez, F. Vidal, L. F. Barraza, V. A. Jiménez, L. Guzman, J. Fuentealba, G. E. Yevenes and J. B. Alderete, Nanomedicine, 2018, 14, 2227 CrossRef CAS PubMed.
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294 CrossRef CAS.
- P. Kesharwani, K. Jain and N. K. Jain, Prog. Polym. Sci., 2014, 39, 268 CrossRef CAS.
- P. K. Maiti, T. Çaǧin, S. T. Lin and W. A. Goddard, Macromolecules, 2005, 38, 979 CrossRef CAS.
- D. Luong, P. Kesharwani, R. Deshmukh, M. C. I. Mohd Amin, U. Gupta, K. Greish and A. K. Iyer, Acta Biomater., 2016, 43, 14 CrossRef CAS PubMed.
- L. Albertazzi, M. Serresi, A. Albanese and F. Beltram, Mol. Pharm., 2010, 7, 680 CrossRef CAS PubMed.
- J. Yang, Q. Zhang, H. Chang and Y. Cheng, Chem. Rev., 2015, 115, 5274 CrossRef CAS PubMed.
- F. Gómez-Galván, L. Pérez-Álvarez, J. Matas, A. Álvarez-Bautista, J. Poejo, C. M. Duarte, L. Ruiz-Rubio, J. L. Vila-Vilela and L. M. León, Carbohydr. Polym., 2016, 142, 149 CrossRef PubMed.
- D. Duchêne and A. Bochot, Int. J. Pharm., 2016, 514, 58 CrossRef PubMed.
- G. Varan, C. Varan, N. Erdoğar, A. A. Hıncal and E. Bilensoy, Int. J. Pharm., 2017, 531, 457 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS PubMed.
- H. T. Le, H. M. Jeon, C. W. Lim and T. W. Kim, J. Pharm. Sci., 2014, 103, 3183 CrossRef CAS PubMed.
- X. Wang, B. Huang, X. Liu and P. Zhan, Drug Discovery Today, 2016, 21, 118 CrossRef CAS.
- N. M. Meghani, H. H. Amin and B.-J. Lee, Drug Discovery Today, 2017, 22, 1604 CrossRef CAS PubMed.
- J. W. Lee, B.-K. Kim, H. J. Kim, S. C. Han, W. S. Shin and S.-H. Jin, Macromolecules, 2006, 39, 2418 CrossRef CAS.
- X. H. Dai, W. H. Yang, W. L. Yan, J. M. Hu, Y. R. Dai, J. M. Pan and Y. S. Yan, Colloids Surf., A, 2017, 520, 222 CrossRef CAS.
- G. Franc and A. Kakkar, Chem. Commun., 2008, 5267 RSC.
- Y. Kunsang, P. Goyal and M. Weck, Org. Lett., 2007, 9, 2051 CrossRef.
- M. Malkoch, P. Wu, V. V. Fokin, E. Drockenmuller, C. J. Hawker, K. Schleicher and T. P. Russell, Macromolecules, 2005, 38, 3663 CrossRef CAS.
- I. González-Méndez, A. Hameau, R. Laurent, C. Bijani, V. Bourdon, A. M. Caminade, E. Rivera and K. I. Moineau-Chane Ching, Eur. J. Org. Chem., 2020, 1114 CrossRef.
- H. Arima, F. Kihara, F. Hirayama and K. Uekama, Bioconjugate Chem., 2001, 12, 476 CrossRef CAS PubMed.
- A. F. A. Mohammed, T. Higashi, K. Motoyama, A. Ohyama, R. Onodera, K. A. Khaled, H. A. Sarhan, A. K. Hussein and H. Arima, AAPS J., 2019, 21, 1 CrossRef CAS PubMed.
- H. Arima and K. Motoyama, Sensors, 2009, 9, 6346 CrossRef CAS PubMed.
- A. F. Abdelwahab, A. Ohyama, T. Higashi, K. Motoyama, K. A. Khaled, H. A. Sarhan, A. K. Hussein and H. Arima, J. Drug Targeting, 2014, 22, 927 CrossRef CAS PubMed.
- H. Wang, N. Shao, S. Qiao and Y. Cheng, J. Phys. Chem. B, 2012, 116, 11217 CrossRef CAS PubMed.
- S. Shah, A. Solanki, P. K. Sasmal and K. B. Lee, J. Am. Chem. Soc., 2013, 135, 15682 CrossRef CAS PubMed.
- H. Arima, Y. Chihara, M. Arizono, S. Yamashita, K. Wada, F. Hirayama and K. Uekama, J. Controlled Release, 2006, 116, 64 CrossRef CAS PubMed.
- M. Labieniec-Watala and C. Watala, J. Pharm. Sci., 2015, 104, 2 CrossRef CAS PubMed.
- H. Arima, K. Motoyama and T. Higashi, Pharmaceutics, 2012, 4, 130 CrossRef CAS PubMed.
- H. Arima, K. Motoyama and T. Higashi, Adv. Drug Delivery Rev., 2013, 65, 1204 CrossRef CAS.
- S. Menuel, S. Fontanay, I. Clarot, R. E. Duval, L. Diez and A. Marsura, Bioconjugate Chem., 2008, 19, 2357 CrossRef CAS PubMed.
- N. A. Stasko, C. B. Johnson, M. H. Schoenfisch, T. A. Johnson and E. L. Holmuhamedov, Biomacromolecules, 2007, 8, 3853 CrossRef CAS PubMed.
- T. Loftsson and M. E. Brewster, J. Pharm. Sci., 1996, 85, 1017 CrossRef CAS PubMed.
- Y. Zeng, P. Li, X. Liu, T. Yu, J. Chen, G. Yang and Y. Li, Polym. Chem., 2014, 5, 5978 RSC.
- T. I. Al-Warhi, H. M. A. Al-Hazimi and A. El-Faham, J. Saudi Chem. Soc., 2012, 16, 97 CrossRef CAS.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557 CrossRef CAS PubMed.
- N. Zhong, H. S. Byun and R. Bittman, Tetrahedron Lett., 1998, 39, 2919 CrossRef.
- G. Tripodo, C. Wischke, A. T. Neffe and A. Lendlein, Carbohydr. Res., 2013, 381, 59 CrossRef CAS PubMed.
- Y. Toomari, H. Namazi and E. A. Akbar, Carbohydr. Polym., 2015, 132, 205 CrossRef CAS PubMed.
- K. S. Anseth and H.-A. Klok, Biomacromolecules, 2016, 17, 1 CrossRef CAS PubMed.
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713 CrossRef CAS PubMed.
- J. Fang, S. H. Ye, J. Wang, T. Zhao, X. Mo and W. R. Wagner, Biomacromolecules, 2015, 16, 1622 CrossRef CAS PubMed.
- R. T. Chen, S. Marchesan, R. A. Evans, K. E. Styan, G. K. Such, A. Postma, K. M. McLean, B. W. Muir and F. Caruso, Biomacromolecules, 2012, 13, 889 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046 CrossRef CAS PubMed.
- G. Yi, J. Son, J. Yoo, C. Park and H. Koo, Biomater. Res., 2018, 22, 1 CrossRef PubMed.
- S. Srinivasachari, K. M. Fichter and T. M. Reineke, J. Am. Chem. Soc., 2008, 130, 4618 CrossRef CAS PubMed.
- B. Parrish, R. B. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2005, 127, 7404 CrossRef CAS PubMed.
- C. Zhang, D. Pan, K. Luo, N. Li, C. Guo, X. Zheng and Z. Gu, Polym. Chem., 2014, 5, 5227 RSC.
- A. Ghilardi, D. Pezzoli, M. C. Bellucci, C. Malloggi, A. Negri, A. Sganappa, G. Tedeschi, G. Candiani and A. Volonterio, Bioconjugate Chem., 2013, 24, 1928 CrossRef CAS PubMed.
- J. H. Lee, Y. B. Lim, J. S. Choi, M. U. Choi, C. H. Yang and J. S. Park, Bull. Korean Chem. Soc., 2003, 24, 1637 CrossRef CAS.
- E. Arnáiz, L. I. Doucede, S. García-Gallego, K. Urbiola, R. Gómez, C. Tros De Ilarduya and F. J. De La Mata, Mol. Pharm., 2012, 9, 433 CrossRef PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC.
- L. J. López-Méndez, I. González-Méndez, R. Aguayo-Ortiz, L. Dominguez, S. L. Alcaraz-Estrada, Y. Rojas-Aguirre and P. Guadarrama, Carbohydr. Polym., 2018, 184, 20 CrossRef PubMed.
- J. Peterson, V. Allikmaa, J. Subbi, T. Pehk and M. Lopp, Eur. Polym. J., 2003, 39, 33 CrossRef CAS.
- S. M. Weidner, U. Just, W. Wittke, F. Rittig, F. Gruber and J. F. Friedrich, Int. J. Mass Spectrom., 2004, 238, 235 CrossRef CAS.
- D. Harries, D. C. Rau and V. A. Parsegian, J. Am. Chem. Soc., 2005, 127, 2184 CrossRef CAS PubMed.
- Y. Cheng, Z. Xu, M. Ma and T. Xu, J. Pharm. Sci., 2008, 97, 123 CrossRef CAS PubMed.
- S. Mignani, S. El Kazzouli, M. Bousmina and J. P. Majoral, Prog. Polym. Sci., 2013, 38, 993 CrossRef CAS.
- M. J. Jozwiakowski and K. A. Connors, Carbohydr. Res., 1985, 143, 51 CrossRef CAS.
- M. E. Brewster and T. Loftsson, Adv. Drug Delivery Rev., 2007, 59, 645 CrossRef CAS PubMed.
- T. Loftsson and D. Duchêne, Int. J. Pharm., 2007, 329, 1 CrossRef CAS PubMed.
- N. Malik, R. Wiwattanapatapee, R. Klopsch, K. Lorenz, H. Frey, J. Weener, E. Meijer, W. Paulus and R. Duncan, J. Controlled Release, 2000, 65, 133 CrossRef CAS PubMed.
- R. Dong, L. Zhou, J. Wu, C. Tu, Y. Su, B. Zhu, H. Gu, D. Yan and X. Zhu, Chem. Commun., 2011, 47, 5473 RSC.
- M. El-Sayed, C. A. Rhodes, M. Ginski and H. Ghandehari, Int. J. Pharm., 2003, 265, 151 CrossRef CAS PubMed.
- F. Tajarobi, M. El-Sayed, B. D. Rege, J. E. Polli and H. Ghandehari, Int. J. Pharm., 2001, 215, 263 CrossRef CAS PubMed.
- J. Auclair, E. Morel, K. J. Wilkinson and F. Gagné, EC Pharmacol. Toxicol. 3.5, 2017, 138–145 Search PubMed.
- R. Duncan and L. Izzo, Adv. Drug Delivery Rev., 2005, 57, 2215 CrossRef CAS PubMed.
- S. Rahmani and A. S. zadeh Mahani, Macromol. Res., 2015, 23, 1018 CrossRef CAS.
- N. Zhong, H. S. Byun and R. Bittman, Tetrahedron Lett., 1998, 39, 2919 CrossRef.
- E. Sabadini, T. Cosgrove and F. D. C. Egídio, Carbohydr. Res., 2016, 341, 270 CrossRef PubMed.
- I. González-Méndez, J. D. Solano, P. Porcu, A. Ruiu, Y. Rojas-Aguirre and E. Rivera, J. Mol. Struct., 2019, 1177, 143 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02574g |
| ‡ These people contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |