
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanohybrids of reduced graphene oxide and cobalt hydroxide (Co(OH)2|rGO) for the thermal decomposition of ammonium perchlorate †
Gabriel Abarca a,
Paulina L. Ríosb,
Paula Povead,
Christopher Cerda-Cavieresb,
Cesar Morales-Verdejoa,
Juan L. Arroyod and
María B. Camarada*bc
a,
Paulina L. Ríosb,
Paula Povead,
Christopher Cerda-Cavieresb,
Cesar Morales-Verdejoa,
Juan L. Arroyod and
María B. Camarada*bc
aUniversidad Bernardo OHiggins, Escuela de Obstetricia y Puericultura, Centro Integrativo de Biología y Química Aplicada (CIBQA), Santiago, 8370993, Chile
bCentro de Nanotecnología Aplicada, Facultad de Ciencias, Universidad Mayor, Santiago, 8580745, Chile. E-mail: maria.camarada@umayor.cl
cNúcleo de Química y Bioquímica, Facultad de Estudios Interdisciplinarios, Universidad Mayor, Santiago, 8580745, Chile
dLaboratorio de Materiales Energéticos, Instituto de Investigaciones y Control del Ejército de Chile (IDIC), Av. Pedro Montt 2136, 8370899, Santiago, Chile
First published on 17th June 2020
Abstract
The catalytic activity of nanoparticles of cobalt hydroxide supported on reduced graphene oxide, Co(OH)2|rGO, was studied for the decomposition of ammonium perchlorate (AP), the principal ingredient of composite solid propellants. Co(OH)2|rGO was synthesized by an in situ reduction method, which avoided the application of extremely high temperatures and harsh processes. rGO stabilized the nanoparticles effectively and prevented their agglomeration. The performance of Co(OH)2|rGO as a catalyst was measured by differential scanning calorimetry. Co(OH)2|rGO affected the high-temperature decomposition (HTD) of AP positively, decreasing the decomposition temperature of AP to 292 °C, and increasing the energy release to 290 J g−1. The diminution of the HTD of AP by Co(OH)2|rGO is in between the best values reported to date, suggesting its potential application as a catalyst for AP decomposition.
1. Introduction
One of the critical aspects of the outer-space race is the development of a safe, stable, and more efficient fuel. Systems based on liquid fuels are usually more unstable than solid propellants.1,2 In this scenario, composite solid propellants appear as a safer option. However, their energy release is still too low to reach faraway destinations with only one charge. The major ingredient of composite solid propellants is the oxidizer, the source of oxygen, commonly ammonium perchlorate (AP).3 Though, considering the limited loading of AP in a rocket, it is crucial to improve its decomposition efficiency by the production of more significant amounts of energy with a higher combustion rate. The decomposition properties of AP can be tuned by adding a burn rate catalyst,4 which can alter its highest decomposition temperature, and therefore increase the combustion rate. Traditionally, ferrocene derivatives5,6 and fine-grained metal oxides have been used as burn rate catalysts. The effect of metal micro and nanoparticles (NP) on the decomposition of AP has been widely assessed. The high surface area to volume ratio of nanoparticles provides an enormous reactive interface between the nanoparticle and AP. By now, it is well known that NP of transition metals, with a higher surface area, have a better catalytic effect than microparticles.7 Of the transition metals that are typically ferromagnetic, cobalt has technological and theoretical importance because of the existence of two stable structures: uniaxial hexagonal close-packed and face-centered cubic structures.8 Cobalt oxide and cobalt nanoparticles have been widely explored as nanocatalysts. Among the transition metal nanoparticles as catalysts of AP, cobalt has a high impact on the high-temperature decomposition (HTD) process of AP compared to other catalysts.9,10 Hongzhen and coworkers11 published one of the first reports, where cobalt nanoparticles of 42 nm average were produced by DC hydrogen plasma method. The authors found that metallic cobalt was more unstable and active than CoO species and also concluded that the sensitivity is directly connected to the oxidation state of the transition metal nanoparticles; neutral NPs are more sensitive to oxygen and can improve better the thermal decomposition of AP.12,13 In 2009, Liu et al.14 studied the effect of the shape of cobalt nanoparticles on the decomposition mechanism of AP. Independent of the shape, the two decomposition peaks of AP merged into one when the nanoparticles were added. On the other hand, Sharma and coworkers62 reported the synthesis of Co3O4 nanoparticles using leaves extracts of a plant, and as previously reported, cobalt oxide nanoparticles showed lower performance as catalysts than metallic cobalt nanoparticles.15,16 Other authors have produced Co3O4 nanoparticles by calcination of cobalt hydroxides17,18 and oxalates19 at temperatures higher than 350 °C, with positive catalytic effects on the decomposition of AP. Cobalt nanowires of Co3O4 have also been produced by calcination, showing better catalytic activity at 9 wt% than microspheres and nanoparticles of cobalt oxide.20 More recently, Liang and coworkers21 evaluated nanoflowers and micro-rods of Co3O4 synthesized by chemical bath deposition and the hydrothermal method. Owing to the higher specific surface area and larger pore volume, nanoflower Co3O4 had better catalytic performance than micro-rods for AP thermal decomposition. Cheng and coworkers22 tested a cobalt complex based on dinitropyrazine and reported the in situ formation of active CoCO3, Co3O4, CoO, and Co(NH3)6, which accounted for the higher catalytic activity of the complex for the thermal decomposition of AP. Alloys with cobalt have also been explored. Singh et al.23 prepared different nickel alloys; Ni–Co was the best as a catalyst at 1 wt%, surpassing zinc and copper alloys. The catalytic effect of composites of CuO and Co2O3 (ref. 9) revealed that the nanoparticles could easily facilitate the thermal decomposition of AP, while Co2O3 alone has a better catalytic effect at higher temperatures as compared to CuO nanoparticles. Bimetallic nanocatalysts of copper and cobalt24 produced by calcination improved the combustion properties of AP by the synergistic effect of both metals.Most of the production procedures of cobalt nanoparticles require calcination as a final step, which is an energy-consuming process, requires specialized equipment, and has a direct impact on the final price of the catalyst. One approximation to reduce the costs, harsh synthesis pathways, or to decrease the energy demand is the use of cobalt hydroxide nanoparticles as a catalyst. Wang et al.25 have reported the synthesis of flower-like α-Co(OH)2 particles of 300–400 nm diameter. The addition of 3 wt% to AP decreased the decomposition temperature of AP in 158 °C and increased the decomposition heat. The application of Co(OH)2 in the decomposition of AP as nanoscopic burning rate modifiers is problematic because of the agglomeration of the particles, which can decrease the specific surface area and diminish the mass transfer between the catalyst and reactants.26 In this sense, graphene and its derivatives state as a potential support to load NP due to oxygen-containing functional groups.
Graphene (GR) can influence the propellant burn rate through the acceleration of the electron transfers involved in the combustion.27 Graphene oxide (GO), a chemically modified graphene containing oxygen functional groups,28 offers a less expensive alternative for the obtention of graphene derivatives. Our group has tested previously reduced graphene oxide (rGO) as burning rate catalysts of AP.29 This system catalyzes moderately the decomposition process, but takes part energetically as fuel, contributing significantly to the energy release. Graphene and its derivatives have also been assayed as support and stabilizer of metal nanoparticles.30,31 Yang and coworkers32 successfully synthesized nanosized Co3O4 stabilized by GO for application in high-performance lithium-ion batteries by the pyrolytic decomposition of cobalt phthalocyanine deposited on graphene, reduced by thermal methods. Xu et al.33 reported the synthesis of Co3O4–GO composite by the in situ deposition of the nanoparticles onto the surface of the graphitic surface with an average size of 100 nm. Although the HTD of Co3O4–GO and pure Co3O4 were quite close, Co3O4–GO presented a higher exothermic heat with almost 400 J g−1 more than pure AP. In 2018, Hosseini and coworkers34 prepared cobalt and nickel–cobalt bimetallic nanoparticles supported on nitrogen-doped graphene (Co@3D-(N)G and NiCo@3D-(N)G, respectively) by a chemical reduction method. A good dispersion of the nanoparticles was achieved by their stabilization on the 3D-(N)G structure. The catalytic activity of NiCo@3D-(N)G was better than Co@3D-(N)G due to the synergistic effect between the two metals.
To the best of our knowledge, there is no report about the application of composites of reduced graphene oxide and cobalt hydroxide nanoparticles (Co(OH)2|rGO) as burning rate modifiers for the decomposition of AP. In this work, Co(OH)2 nanoparticles were effectively stabilized by rGO without the application of high temperatures or a calcination process. The supporting effect of rGO reduced the agglomeration of the nanoparticles. An in situ synthesis pathway was followed to ensure the stabilization of the cobalt nanoparticles on rGO and to control the agglomeration by the rGO layers. The effects of Co(OH)2|rGO on the decomposition of AP were compared to other cobalt nanocatalysts synthesized by energy-consuming methods. The material reported in the present work diminished the HTD of AP and increased the energy release. Its performance is in between the best catalyst cobalt-based nanoparticles reported to date.
2. Experimental details
2.1. Reagents and characterization methods
Reagents were of analytical grade or the highest commercially available purity and were used as received. Cobalt dichloride hexahydrate, ethanol, hydrazine hydrate, and sodium hydroxide were purchased from Merck. Aqueous solutions were prepared with in-house ultrapure water of resistivity not less than 18 MΩ cm (Milli-Q, USA). Fourier transform infrared spectroscopy (FT-IR) spectra were measured using a Perkin Elmer Spectrum two UATR spectrometer in the frequency range 450–4000 cm−1. UV-visible spectra were recorded with a Merck Spectroquant Prove 300 spectrophotometer. All measurements were taken at room temperature. Differential scanning calorimetry (DSC) analyses were performed on a Perkin Elmer DSC 4000 instrument at a heating rate of 5 °C min−1 under a nitrogen blanket in the range of 140–500 °C. The thermogravimetric analysis (TGA) was performed on a Perkin Elmer TGA 4000 equipment, at a heating rate of 5 °C min−1 between 50 and 450 °C. The catalytic performance of the compounds for the thermal decomposition of ammonium perchlorate (AP) was investigated by adding the catalysts to AP in 1, 3, and 5 wt% TEM images were collected on a Hitachi HT7700 equipment. The samples were prepared by directly applying the sample powder onto standard TEM grids. The structural crystallite of the samples was examined by X-ray diffraction (XRD) using a Bruker D2 equipment with cobalt radiation at a scan rate of 0.02° s−1. XPS measurements were carried out using a Kratos AXIS Ultra DLD instrument. The chamber pressure during the measurements was 5 × 10−9 torr. Wide energy range survey scans were collected at pass energy of 80 eV in hybrid slot lens mode and a step size of 0.5 eV. High-resolution XPS data on the C 1s, O 1s, and Co 2p photoelectron peaks were collected at pass energy of 20 eV over energy ranges suitable for each peak, collection times of 5 min, and step sizes of 0.1 eV. The X-ray source was a monochromated Al Kα emission run at 10 mA and 12 kV (120 W). The energy range for each pass energy (resolution) was calibrated using the Kratos Cu 2p3/2, Ag 3d5/2, and Au 4f7/2 three-point calibration method. The data was charge corrected to the reference carbon adventitious signal at 284.8 eV. Micro Raman spectroscopy was performed using a Horiba Jobin Yvon LabRAM HR Raman microscope. Spectra were acquired using a 532 nm laser (at ∼0.2 mW power), a 100× objective, a 600 lines per mm rotatable diffraction grating, and a 200 μm confocal pinhole. The spectral resolution in this configuration is better than 1.7 cm−1. Single point spectra were collected over the range 800–3500 cm−1 (2 spectral windows) using 30 s integration time, repeated once, to automatically remove the spikes due to cosmic rays and improve the signal-to-noise ratio, from five random locations. Temperature programmed reduction (TPR) of the catalysts was performed using a Micromeritics 3Flex instrument. Typically, 15–30 mg of the catalyst in a quartz reactor was heated gradually at 10 °C min−1 until 800 °C in a mixture of 10% H2/Ar (100 mL min−1). A cold trap (liquid nitrogen with isopropanol) was placed between the reactor and the TCD to remove water formed during reduction.2.2. Synthesis of cobalt hydroxide nanoparticles supported on reduced graphene oxide (Co(OH)2|rGO)
GO and rGO synthesis and characterization was previously performed and described by our group.29 The synthesis protocol of Co(OH)2|rGO is based on Bai's method35 with some modifications and simplifications. Briefly, previously synthesized GO (32.7 mg) was dispersed in cobalt dichloride hexahydrate ethanol solution (5 mL, 0.111 M) with sonication in heat (68 °C, 60 min). A hydrazine hydrate (5 mL, 3.53 M) and sodium hydroxide (1.11 M) ethanol solution were added drop-wise, and the mixture was kept in stirring heat (68 °C) overnight. The nanoparticles samples were kept in solution.3. Results and discussion
3.1. Characterization of GO, rGO, and Co(OH)2|rGO
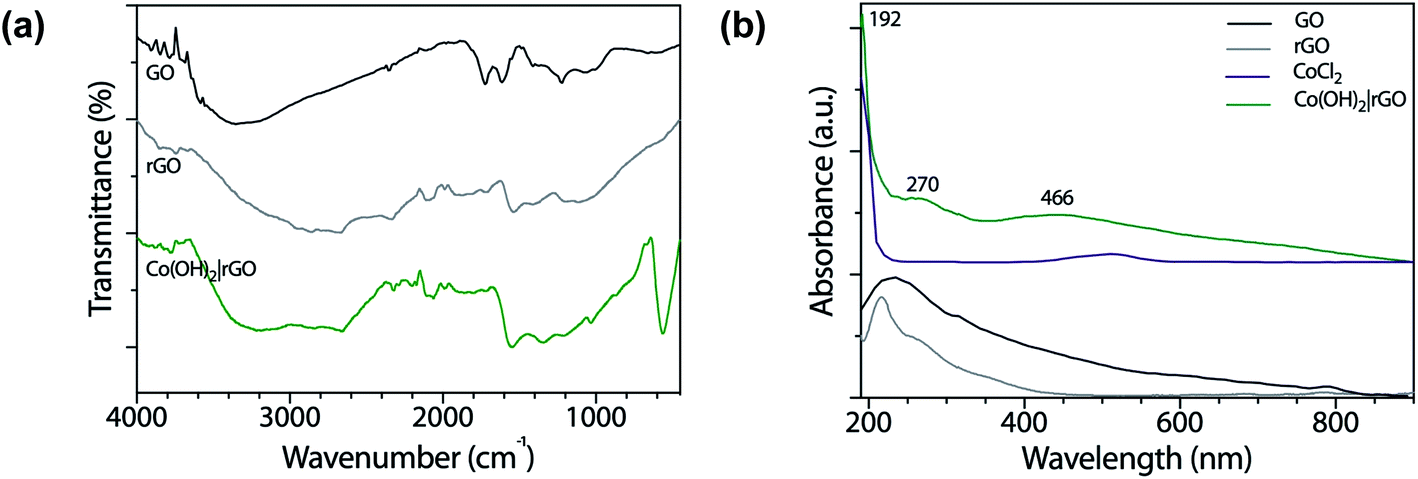
Fig. 1a shows the FT-IR spectra of GO and rGO. The spectrum of GO revealed oxygen functionalities at 3280 (O–H stretching vibrations), 1720 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations), and 1230 cm−1 (C–O stretching vibrations), characteristic of the interruptions of the graphene lattice by oxygen-containing groups like alcohols, epoxides, and carbonyls. At 1615 cm−1 appeared the CC skeletal vibrations from un-oxidized graphitic diamonds. The spectrum of pure graphite had absorption bands at 3440 and 1040 cm−1 corresponding to the O–H and C–O stretching vibrations, respectively.36 The absorption peak at 1640 cm−1 was attributed to the skeletal vibrations of CC.37 The O–H stretching vibrations diminished in the rGO sample as a result of the deoxygenation process and the removal of most oxygen functionalities.
O stretching vibrations), and 1230 cm−1 (C–O stretching vibrations), characteristic of the interruptions of the graphene lattice by oxygen-containing groups like alcohols, epoxides, and carbonyls. At 1615 cm−1 appeared the CC skeletal vibrations from un-oxidized graphitic diamonds. The spectrum of pure graphite had absorption bands at 3440 and 1040 cm−1 corresponding to the O–H and C–O stretching vibrations, respectively.36 The absorption peak at 1640 cm−1 was attributed to the skeletal vibrations of CC.37 The O–H stretching vibrations diminished in the rGO sample as a result of the deoxygenation process and the removal of most oxygen functionalities.
 | ||
| Fig. 1 (a) FT-IR spectra of GO, rGO, and Co(OH)2|rGO, and (b) UV-vis spectra of GO, rGO, CoCl2, and Co(OH)2|rGO in water. | ||
In the case of Co(OH)2|rGO (Fig. 1a), we detected the presence of water, as well as the typical peaks associated with rGO. Cobalt hydroxide has two stable structures characterized previously by FT-IR.38 The α-structure has two strong signals at 667 and 489 cm−1, while the β-cobalt hydroxide has only one peak at ca. 590 cm−1. The synthesized Co(OH)2|rGO sample presents an energetic vibration at 585 cm−1, attributed to the Co–O stretching vibrations39 of the β-cobalt hydroxide structure.
We also obtained UV-vis spectra of the reagents and product samples. As Fig. 1b shows, GO has two bands, one assigned to the π → π* transition of the aromatic rings and the other to the n → π* of the carbonyl groups at approximately 230 nm and 318 nm, respectively.40 After the reduction of GO with hydrazine, rGO exhibits its characteristic band at 270 nm. The bathochromic shifting of the bands is related to the increase of electronic conjugation after the removal of the oxygen groups of GO and partial restoration of conjugation.41 The higher conjugation degree, the less energy needs to be applied to produce the electronic transition. The CoCl2 spectrum presents a broad absorption band at 510 nm assigned to the weakly allowed d → d transition, forbidden by Laporte rule, which disappeared after the addition of hydrazine. The Co(OH)2|rGO sample presents the characteristic peak of rGO at 270 nm and a second band at 466 nm. Jayashree and coworkers reported the UV-vis spectra for the two structures of cobalt hydroxide.42 The α-form profile shows a signature absorption of tetrahedral Co2+ above 620 nm, whereas the β-hydroxide exhibits an absorption maximum at 470 nm, which agrees with the peak registered in this work. The following information and the data from the FT-IR analysis confirm the β-hydroxide form in the Co(OH)2|rGO sample.
The morphology and size distribution of the Co(OH)2|rGO sample was assessed by Transmission Electron Microscopy (TEM). Fig. S1† shows the distribution of Co(OH)2 at 1 μm scale. Most of the nanoparticles are near the rGO layers. Fig. 2 shows two images at a smaller scale and the histogram of the Co(OH)2|rGO nanoparticles. As other authors have reported, the nanoparticles have cauliflower-like shape.14
 | ||
| Fig. 2 Particle size distribution and TEM images of the synthesized Co(OH)2|rGO nanoparticles. | ||
The mean size of the Co(OH)2 nanoparticles is 30 nm. The aggregation of the sample is low, with a size distribution centered between 25 and 35 nm. rGO stabilizes Co(OH)2 nanoparticles by the remaining functional groups after reduction, like epoxy and hydroxyl moieties.43 The layers of rGO might also hinder the aggregation of the nanoparticles physically.
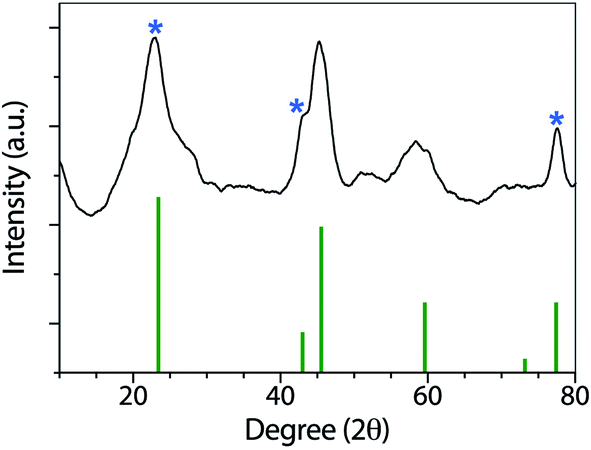
Fig. 3 shows the XRD patterns of the synthesized nanoparticles. GO, and rGO have been widely characterized by XRD. As described by Fan and coworkers,44 GO has a sharp and intense diffraction peak at 10.3°, associated with the (001) lattice plane.
 | ||
| Fig. 3 XRD pattern of Co(OH)2|rGO, showing the typical signals of rGO (marked with an asterisk) and cobalt hydroxide. | ||
After reduction, the interlayer distance decreases due to the removal of some functional groups. rGO presents a sharp peak at 23.4° related to the C (002) plane, and two smaller peaks at 42.9 °C (100) and 77.4 °C (100).45 In the Co(OH)2|rGO sample, we detected all the rGO peaks and assigned the rest of the signals to Co(OH)2 (ICSD collection code 010357). FT-IR, UV-vis, and XRD analysis confirmed cobalt hydroxide in the Co(OH)2|rGO catalyst. However, the presence of Co3O4 cannot be entirely discarded in this sample by the XRD analysis. The peaks of cobalt oxide could be hidden by the signals of rGO and Co(OH)2 at similar places in the XRD. To get more insights into the composition of the sample, we conducted XPS and RAMAN analysis.
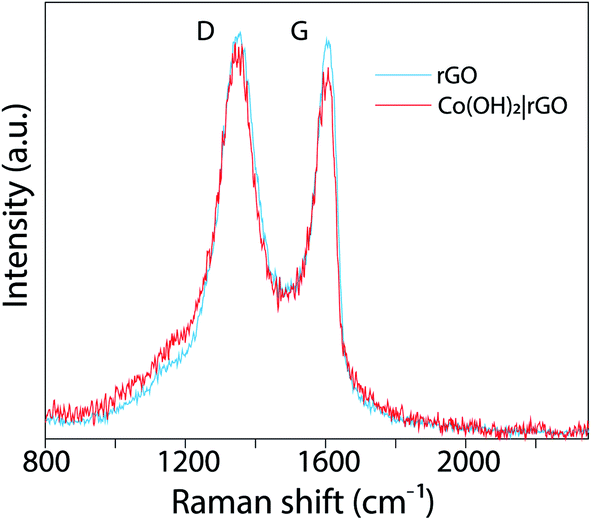
With XPS, we evaluated the chemical composition of the surface of GO, rGO, and Co(OH)2|rGO. The C/O atomic percentage was calculated from XPS wide-scan (see Fig. S2†), revealing an increase from 2.1 for GO to 6.1 for rGO, which was consistent with FT-IR and XRD results.46 Fig. S3† presents the high-resolution C 1s spectra of as-made GO and rGO, with binding energies referred to literature.47 With GO, a large amount of oxygen functional groups was detected (C–O/CO and O–CO), that could be associated with the destruction of the sp2 structure of graphene.48 rGO exhibits the same oxygen functional groups, but with reduced intensities. These features are consistent with the Raman analysis. Fig. 4 presents the Raman spectrum of rGO (light blue) and Co(OH)2|rGO (red), with two Raman peaks; at ∼1350 cm−1 and other at ∼1607 cm−1 which are D and G bands, respectively. The intensity of D and G signals (ID/IG) indicate more structural defects after incorporating the Co(OH)2 nanoparticles, reducing the sp2 hybridized carbon sites in the matrix, which confirms the XPS results.49,50
 | ||
| Fig. 4 Raman analyses of rGO and Co(OH)2|rGO. | ||
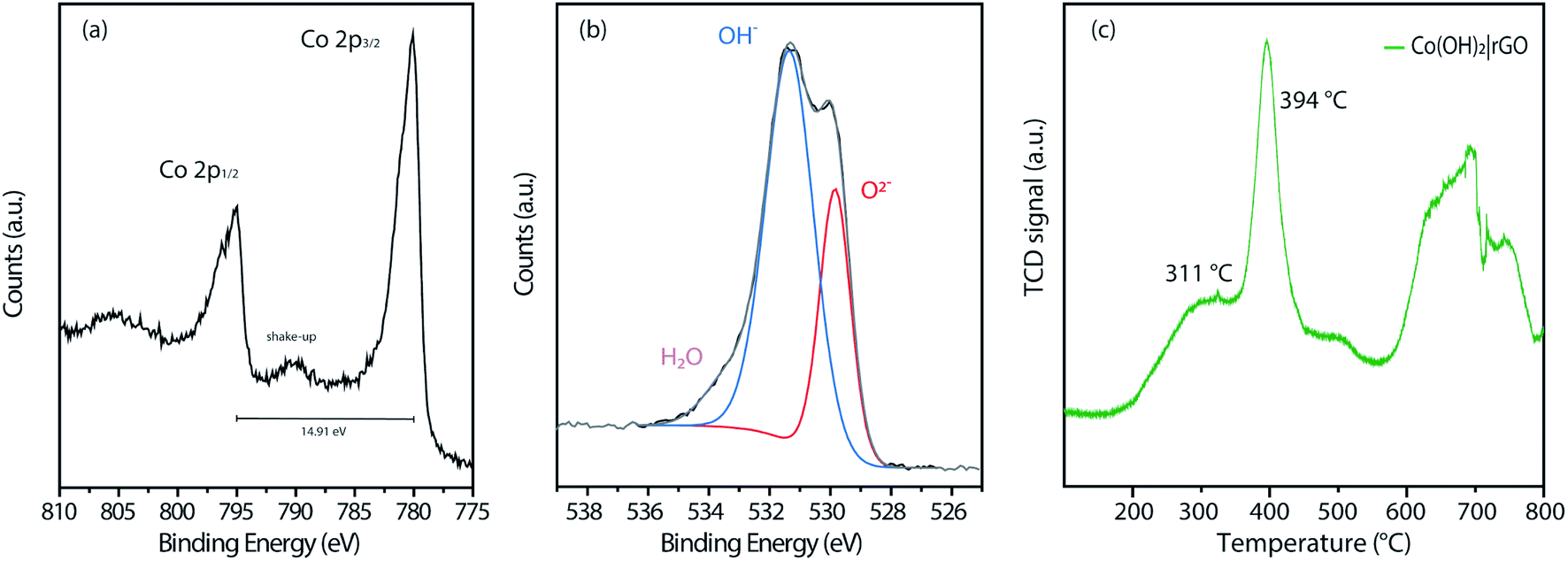
Fig. S4† displays the complete wide-scan XPS spectra of the Co(OH)2|rGO sample, indicating Co, O, and C, as expected. Furthermore, Fig. 5a shows the high-resolution Co 2p XPS region of the Co(OH)2|rGO, which presents two broad peaks at 780.0 eV and 794.9 eV associated with Co 2p3/2 and Co 2p1/2, respectively. As it can be noticed, these values correspond to cobalt in different oxidation states (Co2+ and Co3+),51 which can be related to the presence of Co(OH)2 and Co3O4 in the Co(OH)2|rGO sample. Moreover, Co(OH)2 nanoparticles present a significant contribution of Co–OH and Co–O species, supported by the O 1s XPS spectra (Fig. 5b). In fact, the peak at 531.4 eV implies that Co(OH)2 is the predominant species of Co(OH)2|rGO.52 Presumably, the surface of the Co(OH)2 nanoparticles is easily oxidized to Co3O4 during the sample storage analysis, where the majority of the oxide is placed in the outermost layer of the nanoparticle.53 We relate the oxidative process to the incident photon energy of 1486 eV, where the inelastic mean free path of photoelectrons ejected for Co(OH)2 is around 2 nm.54 Indeed, if we considered that Co(OH)2NP possesses a mean particle diameter of 30 nm, it is expected a small quantity of Co3O4, surrounding and passivating the surface of the Co(OH)2NP as a thin layer.
 | ||
| Fig. 5 XPS measurements of (a) Co 2p and (b) O 1s of the Co(OH)2|rGO sample. (c) H2-TPR profiles of the synthesized catalysts. | ||
To explore the thermal reduction behavior Co(OH)2|rGO and correlate the presence of Co3O4, we performed a TPR analysis. Fig. 5c shows the H2-TPR profile of Co(OH)2|rGO, with a typical reduction behavior: a low-temperature peak (311 °C) associated with Co3+/Co2+ reduction, and a high-temperature peak (394 °C) associated to Co2+/Co0 reduction, respectively. Finally, it is clearly observed the stretched reduction peak between 581 °C and 793 °C, corresponding to rGO reduction.55 All the analyses performed above confirm the presence of Co3O4 in the Co(OH)2|rGO sample, which consists mainly of cobalt hydroxide and cobalt oxide in a lesser amount.
The synthesis procedure of Co(OH)2|rGO was almost equivalent to the synthesis of CoNP,56 with mainly two modifications: the introduction of GO as a stabilizing agent and the use of a higher amount of hydrazine to reduce GO. These modifications affected the final oxidation state of the nanoparticles, resulting in cobalt hydroxide. This difference can be related to the stabilizing effect of the functional groups of GO. According to the in situ reduction path, in the first step of the reaction, the cations and GO get in contact through stirring or sonication to favor the interaction of the GO electron-rich groups with the ions.29 This step is critical to allow direct contact between both materials and generate an effective stabilization of the ions before adding the reducing agent. Unlike the in situ procedure, the direct mixing of rGO and metal nanoparticles neglects the initial stabilization procedure, and therefore, the close contact of the nanoparticles with the rGO surface is not assured. Finally, when hydrazine is added to the solution, its strong reducing potential (−1.49 V vs. NHE)57 leads first to the partial reduction of GO (reduction potential of −0.79 V vs. NHE).58 The reduction reaction of Co2+ to Co0 has a reduction potential of −0.28 V vs. NHE, and it is expected after the reduction of GO. However, the cobalt ions are previously stabilized by the functional groups and surface of GO, and thus, the energy required to reduce them is higher, favoring the partial reduction of GO instead of the cobalt ions. The hydroxyl groups coming from the hydrazine hydrolysis and the sodium hydroxide promote the precipitation of the cobalt centers as Co(OH)2 on the rGO surface.
3.2. Application as propellant catalysts
In this work, we tested nanostructured materials derived from cobalt in the combustion of ammonium perchlorate, the primary component of composite solid propellants. We obtained TG analysis (Fig. 6a) and differential scanning calorimetry curves (Fig. 6b) for the synthesized catalyst, mixed with AP in 1, 3, and 5 wt%.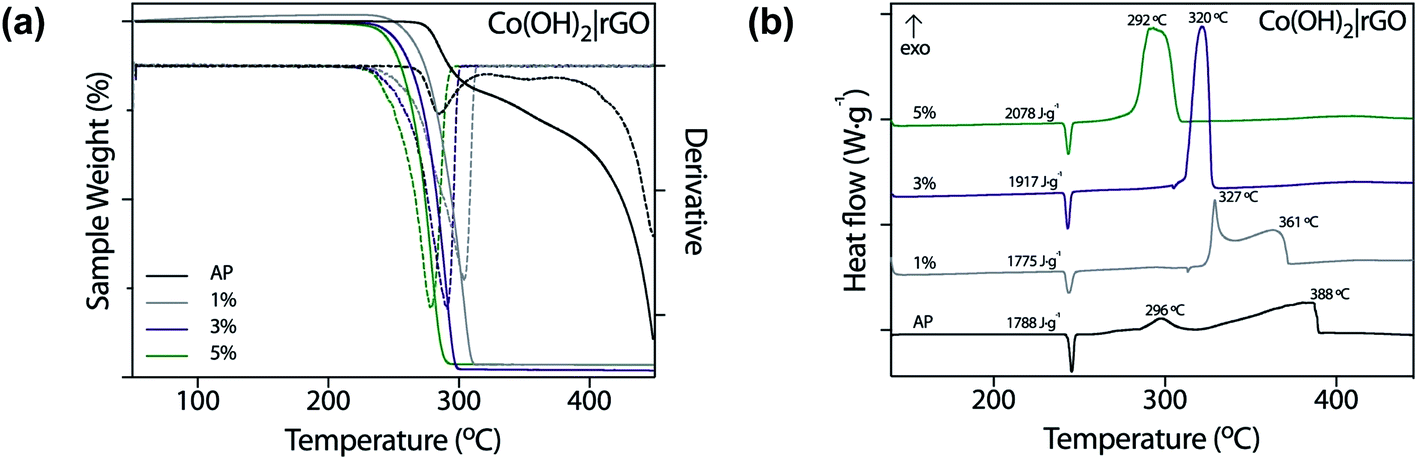 | ||
| Fig. 6 Thermal decomposition of pure AP with 1, 3, and 5 wt% of Co(OH)2|rGO measured by (a) TGA and (b) DSC. | ||
From Fig. 6a, it is evident that Co(OH)2|rGO affects the thermal decomposition of AP. As shown in the TG curves, the thermal decomposition of AP has two decomposition processes. By incorporating the catalyst, the two-step decomposition of AP changed into a rapid single-step decomposition. The highest weight percentage of catalyst diminished the most the decomposition temperature of AP.
Fig. 6b shows the DSC profiles of the thermal decomposition of pure AP and AP with cobalt-based catalysts in different percentages. We also measured the performance of rGO and Co(OH)2 nanoparticles (Fig. S5†) as catalysts of AP. Co(OH)2 nanoparticles were synthesized following the same Co(OH)2|rGO methodology, without adding rGO as support. The thermal decomposition of pure AP showed three stages: an endothermal peak related to the classical first endothermic crystal phase transition from orthorhombic to cubic phase (240–250 °C),59 and two further exothermic processes. The first one, the low-temperature decomposition (LTD), is associated with an electron transfer between both ions (298 °C), which is the most accepted mechanism.60 The second peak corresponds to the high-temperature decomposition (HTD) step.
As already pointed, a burning rate additive could significantly reduce the HTD. As Fig. 6b shows, Co(OH)2|rGO does not influence the crystallographic transition temperature of AP, while we observed significant modifications in the exothermic peaks. At the highest concentration of catalyst, i.e., 5 wt%, both LTD and HTD peaks merged into a single exothermic peak, observed at 292 °C. An increase in the additive loading, lowered the HTD peak and increased much more the heat release of AP in 290 J g−1. Although at 5 wt% Co(OH)2 had better performance as burning rate catalyst (Fig. S5a†) compared to Co(OH)2|rGO (the HTD is 17 °C lesser), only Co(OH)2|rGO increased positively the heat release of AP. At 3 wt% Co(OH)2 the heat release is comparable to the one of Co(OH)2|rGO at 5 wt%. However, the HTD increased from 275 °C to 304 °C. Therefore, a combination of high exothermic heat and low HTD can only be achieved by the use of the Co(OH)2|rGO catalysts.
As published before by our group,29 rGO has no significant influence on the diminution of the high-temperature decomposition of AP. As Fig. S5b† shows, rGO acts mainly as fuel during the combustion, increasing the energy release.
It is useful to compare the catalytic activity of the Co(OH)2|rGO with cobalt nanoparticles and cobalt oxide nanoparticles that have been previously tested as burning rate catalysts of AP. Duan et al.11 reported the introduction of cobalt nanoparticles at 2 wt%, diminishing the decomposition temperature of AP to 298 °C, while Co3O4 nanoparticles presented a HTD peak at 337 °C. The shape has also been described in the catalytic activity of nanoparticles;14 snow-flake like nanoparticles decreased the HTD to 284 °C and merged the LTD and HTD into one peak. Li and coworkers15 studied CoO nanocrystals synthesized by a solvothermal method using hydrazine (44 nm). The HTD process disappeared utterly, and the catalyst diminished the maximum decomposition temperature of AP to approximately 308 °C. Mixtures of cobalt oxide and neutral cobalt nanoparticles have also been tested.61 Ultra-long cobalt chains diminished the highest thermal decomposition of AP to 303 °C. Xu et al.33 reported the synthesis of Co3O4–GO composite by the in situ deposition of the nanoparticles onto the surface of the graphitic surface, decreasing the HTD to 303 °C and increasing the exothermic heat of AP in almost 400 J g−1. Wang and coworkers explored the use of α-Co(OH)2 NP.25 The HTD decreased to 281 °C, 11 °C less than Co(OH)2|rGO. However, the authors do not inform the energy release increase, which is probably lower than the value reported in this study, due to the absence of rGO. Table 1 summarizes this information. The catalytic activities are compared based on peak temperature corresponding to the HTD (DSC) and the percentage of additive. The lower the value of the peak, the higher the catalytic activity.
Most of the nanoparticles cited in Table 1 were obtained by hydrothermal or solvothermal methods, using synthesis temperatures above 100 °C, and requiring calcination as a final step, with temperatures above 300 °C. These methodologies are energy-consuming and increase the final cost of the propellant. In the present work, we synthesized the nanoparticles with lower temperatures and, thus, lower energy demands. Co(OH)2|rGO catalyst is in between the cobalt catalysts with the highest activity. The higher activity of CoNP as a reducer of the HTD of AP can be related to the superior activity of neutral cobalt nanoparticles compared to the oxidized species.11 The oxidation state of the transition metal nanoparticles is directly connected to the sensitivity: neutral NPs are much more unstable to oxygen and thus, can improve better the thermal decomposition of AP.12,13 The influence of Co(OH)2|rGO on the decomposition mechanism of AP will be further discussed in the next section.
3.3. Catalytic mechanism of Co(OH)2|rGO on AP
Following the traditional electron-transfer decomposition theory of AP,60 there are two controlling steps: the electron transfer from ClO4− to NH4+ and the transformation from oxygen (O2) to superoxide (O2−). In the first stage, the ions NH4+ and ClO4− are transformed into NH3 and HClO4. During the second stage, oxygen is formed from HClO4 and then produces O2−. These ions with other gaseous products help in the final decomposition of NH3.The addition of any material that supports the electronic transfers will speed up the decomposition of AP. In the case of Co(OH)2|rGO, rGO can accelerate the electron transfer reactions due to its high conductivity; the movement of electrons in this material is rather faster than in metals and travel a much longer distance without being scattered.
The catalytic performance of Co(OH)2|rGO shows the hybrid material can promote both LTD and HTD process. As before mentioned, Co(OH)2|rGO has a high surface and adsorption capacity to support gaseous intermediates on the surface, which is traduced as an increase in the contact area and better catalytic influence. The exothermic and fast reaction of cobalt with gaseous species like Cl2 and O2 may accelerate the decomposition of AP.
According to the experimental characterization analysis, Co(OH)2|rGO has an oxidized thin layer of Co3O4, mixed with cobalt hydroxide. The mixture of Co2+ and Co3+ oxidation states provides alternative channels for the redox decomposition reaction of AP:64
| Co2+ + NH4+ → Co3+ + NH3 + H˙ |
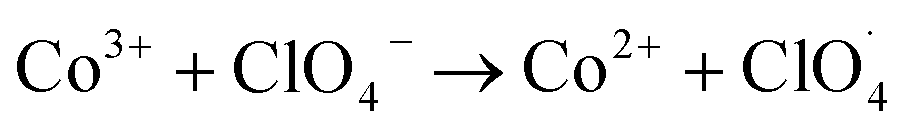
The partially filled orbitals of cobalt can benefit the electron transfer by acting as an electron acceptor of AP ions and intermediate products.65 With the accelerated electron flow, NH4+ and ClO4− can be easily transformed into NH3, H˙, and 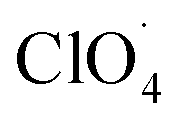 . Subsequently, the radicals react to produce perchloric acid (
. Subsequently, the radicals react to produce perchloric acid (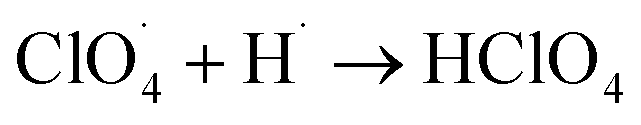 ), which further decomposes like NH3 into small molecules, such as H2O, NOx, Cl2, N2, HCl, H2 and O2. O2 is subsequently transformed into O2− that could help and accelerate the NH3 decomposition (Fig. 7).66 Unlike traditional CoO systems, Co(OH)2|rGO provides two alternative paths of reaction, favoring tandem catalysis and facilitating the decomposition of AP.
), which further decomposes like NH3 into small molecules, such as H2O, NOx, Cl2, N2, HCl, H2 and O2. O2 is subsequently transformed into O2− that could help and accelerate the NH3 decomposition (Fig. 7).66 Unlike traditional CoO systems, Co(OH)2|rGO provides two alternative paths of reaction, favoring tandem catalysis and facilitating the decomposition of AP.
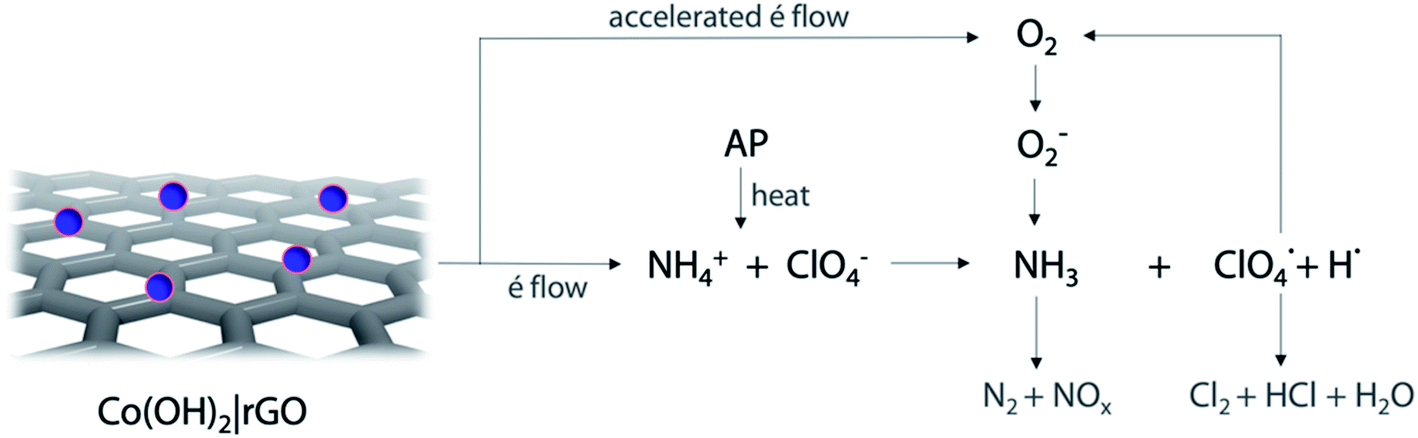 | ||
| Fig. 7 Scheme of the catalytic thermal decomposition process of AP accelerated by Co(OH)2|rGO. | ||
4. Conclusions
In this work, we followed an in situ synthesis of cobalt hydroxide nanoparticles supported on reduced graphene oxide (Co(OH)2|rGO) without high energy-consuming steps or special equipment. rGO stabilized the nanoparticles effectively and avoided their agglomeration, as pointed by the TEM analysis. Cobalt hydroxide nanoparticles with an average size of 30 nm were mixed with a fine layer of cobalt oxide, detected by XPS analysis. The catalytic activity of Co(OH)2|rGO was assessed by DSC. The top activity of Co(OH)2|rGO as a reducer of the HTD of AP can be related to the superior reactivity of the cobalt centers of different oxidation states, Co2+ and Co3+. Co(OH)2|rGO affected the energy release due to the presence of rGO as fuel. The diminution of the HTD of AP by Co(OH)2|rGO is in between the best values reported to date, suggesting its potential application as catalysts in composite solid propellants.Conflicts of interest
There are no conflicts to declare.Acknowledgements
US Army CCDC Award supported this work (W911NF-18-1-0398). M. B. C. is grateful to Fondecyt Project Regular 1180023. C. M. V. thanks Fondecyt Chile, Project Regular 1161297. G. A. A. thanks Fondecyt Iniciación 11170879.References
- B. S. Cho and S. T. Noh, J. Appl. Polym. Sci., 2011, 121, 3560–3568 CrossRef CAS.
- J. Humphries, Rockets and Guided Missiles, Macmillan, 1957 Search PubMed.
- K. K. Kuo and M. Summerfield, Fundamentals of solid-propellant combustion, American Institute of Aeronautics and Astronautics New York, 1984 Search PubMed.
- J. Gao, L. Wang, Y.-l. Tai, J. Wang, J. Huo, A. M. Amin, H. Yu and W. Ding, J. Propul. Power, 2011, 27, 1143–1145 CrossRef CAS.
- C. Morales-Verdejo, M. B. Camarada, J. L. Arroyo, P. Povea, G. Carreño and J. M. Manriquez, J. Therm. Anal. Calorim., 2018, 131, 353–361 CrossRef CAS.
- P. Povea, J. L. Arroyo, G. Carreño, Á. Norambuena, P. L. Rios, M. B. Camarada, I. Chavez, J. M. Manriquez and C. Morales-Verdejo, Thermochim. Acta, 2018, 666, 181–189 CrossRef CAS.
- V. V. Boldyrev, Thermochim. Acta, 2006, 443, 1–36 CrossRef CAS.
- A. Longo, L. Sciortino, F. Giannici and A. Martorana, J. Appl. Crystallogr., 2014, 47, 1562–1568 CrossRef CAS.
- Z. Ma, F. Li and A. Chen, Nanoscience, 2006, 11, 142–145 CAS.
- G. Singh, I. Kapoor, S. Dubey and P. F. Siril, 2009.
- D. Hongzhen, L. Xiangyang, L. Guanpeng, X. Lei and L. Fengsheng, Chin. J. Chem. Eng., 2008, 16, 325–328 CrossRef.
- S. Vyazovkin and C. A. Wight, Chem. Mater., 1999, 11, 3386–3393 CrossRef CAS.
- D. Survase, M. Gupta and S. Asthana, Prog. Cryst. Growth Charact. Mater., 2002, 45, 161–165 CrossRef CAS.
- Z.-T. Liu, X. Li, Z.-W. Liu and J. Lu, Powder Technol., 2009, 189, 514–519 CrossRef CAS.
- L. Li, X. Sun, X. Qiu, J. Xu and G. Li, Inorg. Chem., 2008, 47, 8839–8846 CrossRef CAS PubMed.
- X. Xu, Y. Zhao, J. Li, H. Jin, Y. Zhao and H. Zhou, Mater. Res. Bull., 2015, 72, 7–12 CrossRef CAS.
- S. Lu, X. Jing, J. Liu, J. Wang, Q. Liu, Y. Zhao, S. Jamil, M. Zhang and L. Liu, J. Solid State Chem., 2013, 197, 345–351 CrossRef CAS.
- X. Xu, Y. Zhao, Y. Zhao, H. Zhou, F. Rehman, J. Li and H. Jin, CrystEngComm, 2015, 17, 8248–8255 RSC.
- L.-N. Jin, J.-G. Wang, X.-Y. Qian, D. Xia and M.-D. Dong, J. Nanomater., 2015, 2015, 854310 CrossRef.
- C. Yu, W. Zhang, Y. Gao, Y. Chen, K. Ma, J. Ye, R. Shen and Y. Yang, Mater. Res. Bull., 2018, 97, 483–489 CrossRef CAS.
- Y. Liang and G. Li, Mater. Res. Express, 2019, 6, 0850e0858 Search PubMed.
- J. Cheng, Y. Zheng, Z. Li, Z. Liu, L. Li, F. Zhao and S. Xu, J. Therm. Anal. Calorim., 2017, 129, 1875–1885 CrossRef CAS.
- G. Singh, I. Kapoor and S. Dubey, J. Alloys Compd., 2009, 480, 270–274 CrossRef CAS.
- D. C. K. Rao, N. Yadav and P. C. Joshi, Def. Technol., 2016, 12, 297–304 CrossRef.
- M. Wang and Y. Ma, Chin. J. Explos. Propellants, 2017, 40, 27–31 CrossRef.
- J. Wang, W. Zhang, Z. Zheng, Y. Gao, K. Ma, J. Ye and Y. Yang, J. Alloys Compd., 2017, 724, 720–727 CrossRef CAS.
- J. L. Sabourin, D. M. Dabbs, R. A. Yetter, F. L. Dryer and I. A. Aksay, ACS Nano, 2009, 3, 3945–3954 CrossRef CAS PubMed.
- W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. Yang, A. Velamakanni, S. J. An and M. Stoller, Science, 2008, 321, 1815–1817 CrossRef CAS PubMed.
- P. L. Ríos, P. Povea, C. Cerda-Cavieres, J. L. Arroyo, C. Morales-Verdejo, G. Abarca and M. B. Camarada, RSC Adv., 2019, 9, 8480–8489 RSC.
- N. Li, Z. Geng, M. Cao, L. Ren, X. Zhao, B. Liu, Y. Tian and C. Hu, Carbon, 2013, 54, 124–132 CrossRef CAS.
- Y. Yuan, W. Jiang, Y. Wang, P. Shen, F. Li, P. Li, F. Zhao and H. Gao, Appl. Surf. Sci., 2014, 303, 354–359 CrossRef CAS.
- S. Yang, G. Cui, S. Pang, Q. Cao, U. Kolb, X. Feng, J. Maier and K. Müllen, ChemSusChem, 2010, 3, 236–239 CrossRef CAS PubMed.
- C. Xu, X. Wang, J. Zhu, X. Yang and L. Lu, J. Mater. Chem., 2008, 18, 5625–5629 RSC.
- S. G. Hosseini, S. Gholami and M. Mahyari, Res. Chem. Intermed., 2019, 45, 1527–1543 CrossRef CAS.
- S. Bai, X. Shen, G. Zhu, M. Li, H. Xi and K. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 2378–2386 CrossRef CAS PubMed.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS.
- P. Khanra, C.-N. Lee, T. Kuila, N. H. Kim, M. J. Park and J. H. Lee, Nanoscale, 2014, 6, 4864–4873 RSC.
- Y. Zhu, H. Li, Y. Koltypin and A. Gedanken, J. Mater. Chem., 2002, 12, 729–733 RSC.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2009, 114, 111–119 CrossRef.
- Z. Luo, Y. Lu, L. A. Somers and A. T. C. Johnson, J. Am. Chem. Soc., 2009, 131, 898–899 CrossRef CAS PubMed.
- D. Li, M. B. Mueller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- R. Jayashree and P. V. Kamath, J. Mater. Chem., 1999, 9, 961–963 RSC.
- R. Rozada, J. I. Paredes, M. J. López, S. Villar-Rodil, I. Cabria, J. A. Alonso, A. Martínez-Alonso and J. M. Tascón, Nanoscale, 2015, 7, 2374–2390 RSC.
- Z. Fan, K. Wang, T. Wei, J. Yan, L. Song and B. Shao, Carbon, 2010, 48, 1686–1689 CrossRef CAS.
- B. Rezaei, A. R. T. Jahromi and A. A. Ensafi, Int. J. Hydrogen Energy, 2017, 42, 16538–16546 CrossRef CAS.
- A. L. Higginbotham, D. V. Kosynkin, A. Sinitskii, Z. Sun and J. M. Tour, ACS Nano, 2010, 4, 2059–2069 CrossRef CAS PubMed.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Carbon, 2012, 50, 914–921 CrossRef CAS.
- W. Yan, Y. Huang, Y. Xu, L. Huang and Y. Chen, J. Nanosci. Nanotechnol., 2012, 12, 2270–2277 CrossRef CAS PubMed.
- N. M. Galdino, G. S. Brehm, R. Bussamara, W. D. Gonçalves, G. Abarca and J. D. Scholten, J. Mater. Chem. B, 2017, 5, 9482–9486 RSC.
- N. Zhou, L. Li, S. Chen, X. Peng, W. Niu and Y. Qu, J. Mater. Sci., 2019, 54, 4168–4179 CrossRef CAS.
- M. Qorbani, N. Naseri and A. Z. Moshfegh, ACS Appl. Mater. Interfaces, 2015, 7, 11172–11179 CrossRef CAS PubMed.
- D. Wu, Y. Wei, X. Ren, X. Ji, Y. Liu, X. Guo, Z. Liu, A. M. Asiri, Q. Wei and X. Sun, Adv. Mater., 2018, 30, 1705366 CrossRef PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1994, 21, 165–176 CrossRef CAS.
- F. Yang, J. Mao, S. Li, J. Yin, J. Zhou and W. Liu, Catal. Sci. Technol., 2019, 9, 1329–1333 RSC.
- F. Guo, H. Zheng, Z. Yang and Y. Qian, Mater. Lett., 2002, 56, 906–909 CrossRef CAS.
- T. Shimada, A. Tamaki, H. Nakai and T. Homma, Electrochemistry, 2007, 75, 45–49 CrossRef CAS.
- H.-C. Hsu, I. Shown, H.-Y. Wei, Y.-C. Chang, H.-Y. Du, Y.-G. Lin, C.-A. Tseng, C.-H. Wang, L.-C. Chen and Y.-C. Lin, Nanoscale, 2013, 5, 262–268 RSC.
- R. Fitzgerald and M. Brewster, Combust. Flame, 2004, 136, 313–326 CrossRef CAS.
- H. Kumar, P. N. Tengli, V. K. Mishra, P. Tripathi, A. Bhushan and P. K. Mishra, RSC Adv., 2017, 7, 36594–36604 RSC.
- Y. Zhao, X. Zhang, X. Xu, Y. Zhao, H. Zhou, J. Li and H. Jin, Mater. Chem. Phys., 2017, 201, 235–240 CrossRef CAS.
- J. Sharma, P. Srivastava, G. Singh, M. S. Akhtar and S. Ameen, Mater. Sci. Eng., B, 2015, 193, 181–188 CrossRef CAS.
- S. Jamil, M. R. S. A. Janjua and T. Ahmad, Solid State Sci., 2014, 36, 73–79 CrossRef CAS.
- W. Zhang, B. Jiang, X. Ma, J. Wang, J. Liu, R. Wu, Z. Zheng, J. Liu and K. Ma, RSC Adv., 2019, 9, 23888–23893 RSC.
- S. Chaturvedi and P. N. Dave, J. Saudi Chem. Soc., 2013, 17, 135–149 CrossRef CAS.
- T. Chen, P. Du, W. Jiang, J. Liu, G. Hao, H. Gao, L. Xiao, X. Ke, F. Zhao and C. Xuan, RSC Adv., 2016, 6, 83838–83847 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02853c |
| This journal is © The Royal Society of Chemistry 2020 |