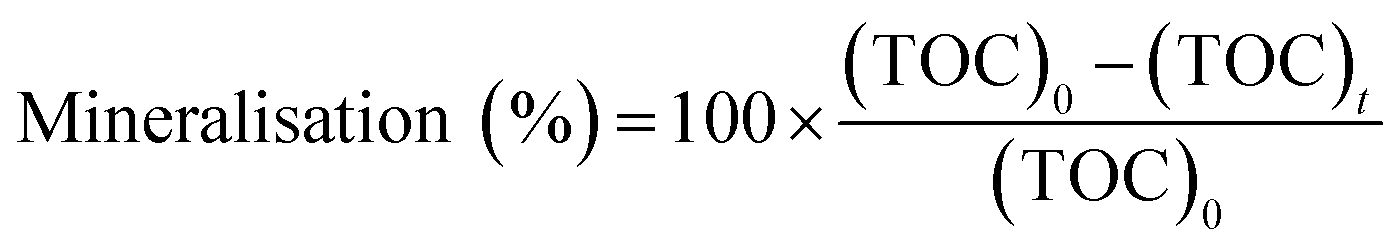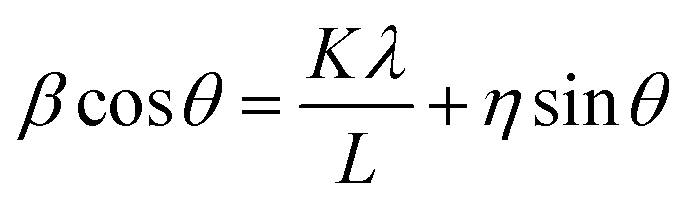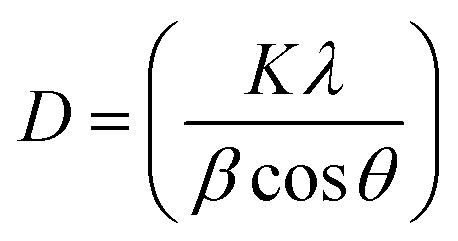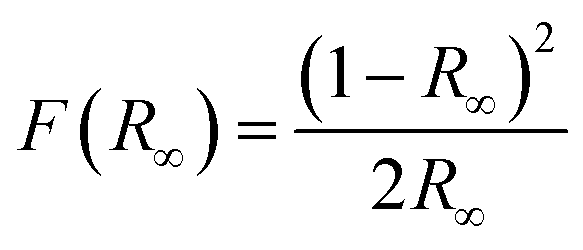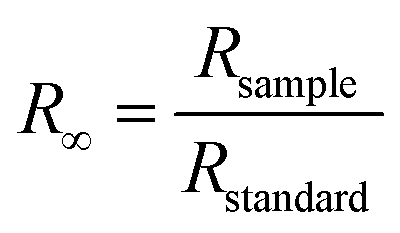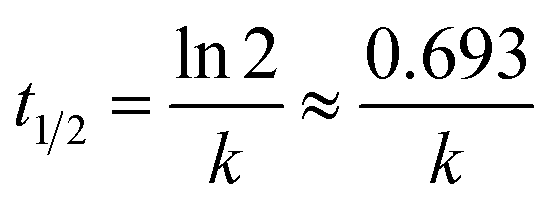DOI:
10.1039/D0RA03157G
(Paper)
RSC Adv., 2020,
10, 19490-19500
C-Dot TiO2 nanorod composite for enhanced quantum efficiency under direct sunlight†
Received
8th April 2020
, Accepted 7th May 2020
First published on 21st May 2020
Abstract
C-Dots obtained from a sustainable route (melon-rinds were used as the source) were combined with TiNRs to create a composite via a simple hydrothermal technique. The obtained materials were subjected to analyses viz. FESEM, XRD, Raman spectroscopy, XPS, PL, FTIR and UV-vis-DRS for understanding their intrinsic nature. The solar photocatalytic performance was evaluated by the degradation of methyl orange (MO) as a model pollutant. The optical properties indicated that there was a clear redshift in the composite with a band gap of 2.49 eV, while the XRD results corresponded to a calculated crystalline size of 24.80 nm. The PL analysis proved the role of C-dots as an electron surge to the TiNRs. The photocatalytic reaction was faster with C-dots as compared to the solo TiNR with a higher degradation of 93.3% within 150 min obeying pseudo-first order kinetics with a rate constant k = 0.01723 min−1. The charge carrier scavenging investigation showed the role of numerous reactive oxygen species (ROS) on the degradation of MO. The formulated composite has demonstrated its ability in effectively handling the contaminants in water. Thus, this study establishes the two-step thermal method as an easy, facile, environmentally-friendly, and low-cost synthesis method for the large-scale production of a photocatalyst.
1. Introduction
In the last era, the scarcity of potable water has been observed at an alarming rate across the several nations of the world. In general, developed countries consume more water resources as compared to developing countries.1 However, the presence of large population in a specific nation makes its water demand equivalent to that of developed nations. On the other hand, the industrialization of fast-growing economy nations overburdens the water utilization and generates an enormous amount of wastewater as effluent.2 Such disposed effluent poses a serious threat to the quality of life due to the presence of numerous classifications of organic and inorganic pollutants. Also, the whole ecosystem is unbalanced and placed at risk with the presence of effluent. In order to improve the negative impacts of these scenarios, the sustainable recycling of domestic and industrial effluents becomes essential and is achieved through cutting edge technologies. The most practiced approaches of the past and present (coagulation, flocculation, adsorption, filtration, ion-exchange and chlorine dosing) do not have enough potential to maximize the gap. Amongst the sustainable cleanest technologies, solar energy is one that has the ability to address the mentioned gap.3 This source of energy can be used in the field of light-driven energy materials with diversified applications, such as water and wastewater treatment,4 solar cells,5 artificial photosynthesis,6 air purification,7 and solar water heating.8 Though the solar spectrum contains photons with numerous different energy levels, the spectrum that is obtained at the near-earth surface is in the UV-visible-infrared range in the ratio ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9:10.9 The task for materials researchers is to utilize the maximum range of the spectrum i.e., the visible-infrared region, through the use of light-driven nanomaterials. Materials that are quantum-confined can generate charge carriers that further react with water molecules to generate reactive oxygen species (ROS), a surface-active agent that eliminates the pollutants present in water.10,11
:9:10.9 The task for materials researchers is to utilize the maximum range of the spectrum i.e., the visible-infrared region, through the use of light-driven nanomaterials. Materials that are quantum-confined can generate charge carriers that further react with water molecules to generate reactive oxygen species (ROS), a surface-active agent that eliminates the pollutants present in water.10,11
In nanomaterial syntheses, the manipulation of atoms through interfacial modification can enhance the properties of the materials, such as specific surface area and pore volume, where larger pore sizes improve the electrical, optical, chemical, and physical properties.12,13 Thus, they help the materials to become robust and reliable towards their specified applications.14 Nanomaterials like TiO2, unmodified or doped with Fe, Co, Cu, Mn, Mg, Zn, etc., have been widely used to maximize the work function. However, a cut-off wavelength (maximum) near 600 nm was achieved as against 380 nm, allowing only a portion of the solar spectrum to be utilized. This restricts the resulting quantum efficiency of the material, resulting in the inefficient treatment of aquatic pollutants. Hence, scientists have developed numerous combinations of nanomaterials, such as TiO2–WO3,15 TiO2–V2O5,16 g-C3N4/TiO2 (ref. 17) and co-catalysts supported with noble metals to improve the heterojunction and electron sink reservoir for maximizing the quantum efficiency.18,19
In the present contribution, carbon dots (C-dots) were fused with TiO2 for a similar quantum efficiency enhancement. C-Dots are basically carbon nanoparticles with sizes less than 10 nm. They exhibit remarkable photoluminescence behavior that utilizes the right extreme of the solar spectrum (500 ≤ λ ≤ 1000 nm), thus delivering an efficient work function and improved charge transfer characteristics.20 C-Dots effectively absorb visible light and promote photoelectron transfer to enhance the visible light photocatalytic performance. Some of the recent reports have examined the mentioned unique characteristics in functional materials. Feizpoor et al. (2019) synthesized particles that exhibited high stability and durability in degrading a wide range of dyes (RhB, MO, and MB).21 On the other hand, Roshni V et al. (2019) obtained C-dots derived from agricultural waste and used them for sensing applications (Cr(VI) and bio-imaging of MCF-7 cells).22 Yousaf et al. (2018) showed that Cr(VI) could be removed from an aqueous medium using rosin-biochar-coated (RBC) TiO2@C with a 95% removal in 60 s and further observed that a negative Gibbs free energy governed its spontaneous and favourable conditions.23 Liu et al. (2019) reported a TiO2/C composite loaded with Pt (0.04–0.1 wt%) for the oxidation of formaldehyde (HCHO) at room temperature.24 The electron sink behaviour of the C-dots was revealed by Li et al. (2018) through the ternary combination of C-dots/g-C3N4/TiO2 that showed enhanced H2 production obtained through the classical water splitting mechanism.25 Some natural materials, including graphite oxide, citric acid obtained from citrus, Gingko biloba, coffee grounds, soya milk, grass and eggs, have been explored for the synthesis of C-dots.26,27 However, it is still desirable to rapidly synthesize high-quality C-dots by an easy and environmentally benign method with low-costs and readily available starting materials. Here, we focused on natural organic watermelon rinds for making C-dots for the first time.
A rapid route to synthesize highly luminescent novel C-dots/TiO2nanorods (C-dots/TiNR) by a simple hydrothermal treatment is presented here. Watermelon rind (Citrullus lanatus) contains about 95% of nutrients and a portion of citrulline (α-amino acid).28 It has a unique light absorption characteristic from the C-dots that is enhanced in the presence of TiNRs. The as prepared C-dots are combined with the TiNRs for better UV-vis light absorption capacity and better surface area. The characterization of the C-dots/TiNR was carried out through FESEM, XRD, XPS, FTIR, Raman spectroscopy, PL, and UV-vis-DRS. The light function experiments were performed that verified the ROS interaction with the pollutant in their course of degradation along with a recyclable study. The work function for the enhanced quantum efficiency of the composite and its interaction with the pollutant were analyzed and discussed.
2. Materials and methods
2.1. Materials
Raw watermelon was obtained from the local market of Dhanbad City, India. Titanium tetrachloride (TiCl4, 99.9%), sodium hydroxide (NaOH, ≥98%), ammonia (NH3, 25%), hydrochloric acid (HCl, 36%), absolute ethanol (C2H5OH) and methyl orange with analytical grade were purchased from Merck (India) and they were used as received. Double de-ionized water was used throughout this study and was prepared by the ion exchange method.
2.2. Synthesis
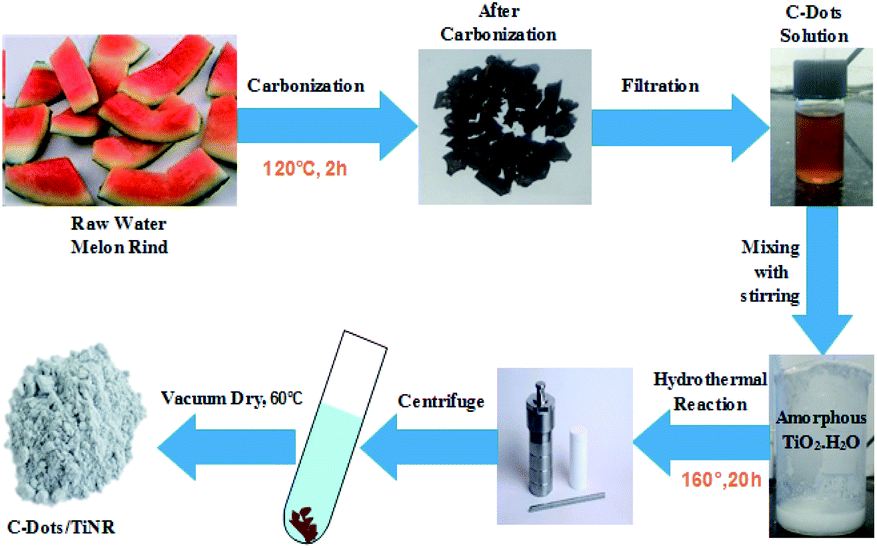
2.2.1. Preparation of C-dots. The C-dots were prepared by obtaining watermelon peel through a low-temperature pyrolysis and filtration technique. First, the watermelon (WM) was peeled into small pieces and washed thoroughly with the distilled water (DI). The WM peel was weighed and put into a silica crucible and kept in the muffle furnace at 220 °C for 2 h under ambient conditions. Finally, carbonization of the WM rind took place and the color changed to black. The obtained product was transferred into DI water and sonicated for 20 min followed by filtering with a 0.21 μm filter membrane. The filtrate product was centrifuged at 12000 rpm for 30 min, resulting in the formation of a supernatant containing C-dots.
2.2.2. Preparation of C-dots/TiNR. C-Dots/TiNR (2.15% w/w) was synthesized by a hydrothermal process. In a beaker, anhydrous titanium tetrachloride was added drop-wise in hydrochloric acid in the molar ratio of 2.1:1 under inert N2 gas. This led to the formation of H2TiCl4. Then, 25 mL of H2TiCl4 and 75 mL of DI water were added under vigorous stirring to make the solution 100 mL. An excess 100 mL NH3 solution was added into the H2TiCl6 solution to complete the reaction. Due to hydrolysis, a raw amorphous gel (TiO2·nH2O) of titanium was formed. The obtained gel was filtered with the help of DI water until residual chlorine was removed; this was tested by reaction with a AgNO3 solution. The amorphous TiO2·nH2O gel (∼1 g) was mixed with 10 mL of C-dots (using a serological pipet) with NaOH (10 mol L−1) aqueous solution and then placed in a poly(tetrafluoroethylene)-lined autoclave with a volume capacity of 100 mL and filled with the capacity of 80%. Hydrothermal syntheses were performed at 160 °C for 20 h in the muffle furnace. The obtained powder was centrifuged at 8000 rpm and thoroughly washed with ethanol and DI water, then dried overnight at 60 °C in the vacuum oven. The detailed synthesis is shown in Scheme 1.
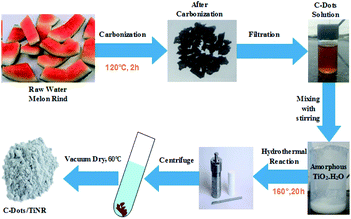 |
| | Scheme 1 Schematic illustration of C-dot/TiNR synthesis. | |
2.2.3. Characterization. The morphologies of C-dots and C-dot/TiNRs were analyzed by a field emission scanning electron microscope (FE-SEM, Supra 55, Zeiss, Germany) with a voltage of 15 kV. X-ray diffraction (XRD, X'Pert PRO; Pananlytical; Netherlands) patterns were recorded with Cu Kα radiation (λ = 0.15418). The measurements were performed over the 2θ range of 20–90°, with the scan speed 20°/h. X-ray photoelectron spectroscopy (XPS) was measured with a KratosXPS system with an Al Kα (1486.6 eV) source. The functional groups were characterized by FTIR spectroscopy (Model: Cary 660, Agilent, USA) in the range of 4000 to 400 cm−1 using KBr pellets. Raman spectra were recorded (LabRAM HR, Horiba, France) in at 325 nm with a 514 nm laser using a CCD detector. The recombination of electron–hole pairs in the photocatalysts was determined photoluminescence (PL) emission spectroscopy (PTI QM400, Horiba) at room temperature. UV-visible diffuse reflectance spectroscopy (DRS) was recorded on a Cary 5000, Agilent, USA.
2.3. Photocatalytic degradation experiment
The photocatalytic performance of C-dots/TiNR was assessed through methyl orange (MO) photodegradation using direct sunlight on a sunny day from 11AM to 2PM IST (Indian Standard Time) at the IIT (ISM) campus on the roof of the Environmental Engineering Department (latitude −23°48′ N, longitude −86°26′ E) on May 25, 2019. The solar irradiation intensity was measured through a Net radiometer (LPNET14, Delta Ohm, Italy) with the range of 0–2000 Wm−2. The average illumination intensity over each iteration of run was noted. The conversion of incoming photon flux (lx) into power intensity (Wm−2) depends on the sun intensity on a particular day. The other corresponding metrological parameters were recorded and the mean are reported in Table 1. In the experiment, a beaker contained 50 mL of MO solution (10 ppm). Prior to solar irradiation, the synthesized photocatalyst (50 mg) was put into the beaker containing MO solution and magnetically stirred in the dark for 1 h to establish the adsorption–desorption equilibrium. After that, it was exposed to the direct sunlight irradiation with continuous magnetic stirring and circulation of water through the water jacket was done to maintain the solution temperature. At regular intervals of time, an aliquot (about 5 mL) of the mixed solution was collected and centrifuged to remove the composite; the residual MO concentration in the supernatant was analyzed by UV-visible spectroscopy at 465 nm.
Table 1 Meteorological conditions during experiment
| Ambient temp. (°C) |
Solution temp. (°C) |
Humidity (%) |
Solar intensitya (W m−2) |
Wind speed (km h−1) |
Wind direction |
| The solar intensity was measured for the direct sunlight from 1100 h to 1400 h during the experiment. |
| 38.8 |
27 |
40 |
480 |
10 |
NE |
The degradation rate largely depends on the structure of organic dye, light intensity, illumination source, dye concentration, catalyst morphology, and the pH of the medium. In all experiments, the concentration of organic dyes was keep constant, which is 5 mg L−1 and pH was maintained at 7. The degradation efficiency of organic dyes was calculated using the following formula:29
| | |
Degradation ratio (%) = [(C0 − Ct)/C0] × 100
| (1) |
Here,
C0 represents the initial concentration of organic dye and
Ct represents the total concentration of organic dye degradation after time interval
t.
2.4. Radical scavenging study
The degradation of MO was performed under direct sunlight irradiation. In a typical experiment, 50 mg of as-synthesized photocatalyst was added to a beaker containing 50 mL of MO. The mixture was then stirred in the dark until it achieved adsorption–desorption equilibrium. The standard radical scavenging agents viz. isopropyl alcohol (IPA, 1 mM), benzoquinone (BQ, 1 mM), and methyl alcohol (MeOH, 1 mM) were added for trapping hydroxyl radical (˙OH), superoxide anion radical (˙O2−) and holes (h+), respectively. The procedures of the radical trapping species experiments were analogous to that of the photodegradation experiment.30 An inert atmosphere was maintained during the superoxide anion radical (˙O2−) test by purging with argon gas (Ar).
2.5. Catalyst reusability
The consecutive cyclic reuses of the catalysts were carried out in a recycling test.31 In this test, the catalyst C-dots/TiNR was examined. When the experiment was complete, the solution was centrifuged, filtered and washed with DI water to obtain the catalysts and dried at 80 °C for 12 h. The recovered catalysts were used for the degradation of MO solution.
2.6. Mineralization
In this experiment, 50 mL of the MO solution was added into a beaker, the solution was maintained at pH 4, and 10 mg of catalyst was added. The photocatalytic degradation experiment was performed after the beaker was kept in the dark for 40 min. The photocatalytic reaction in one reactor was terminated after 0 min, 20 min, 40 min, 60 min, 90 min, 120 min, and 150 min. The solid catalyst in the system was removed by centrifugation, and the liquid was placed in a TOC analyzer to measure the TOC. The mineralization rate was evaluated using the following equation:32| |
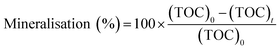 | (2) |
where, (TOC)0 is the initial TOC value of the solution (mg L−1), and (TOC)t is the TOC value of the solution at treatment time t (mg L−1).
3. Result and discussion
3.1. FESEM morphology and XRD pattern
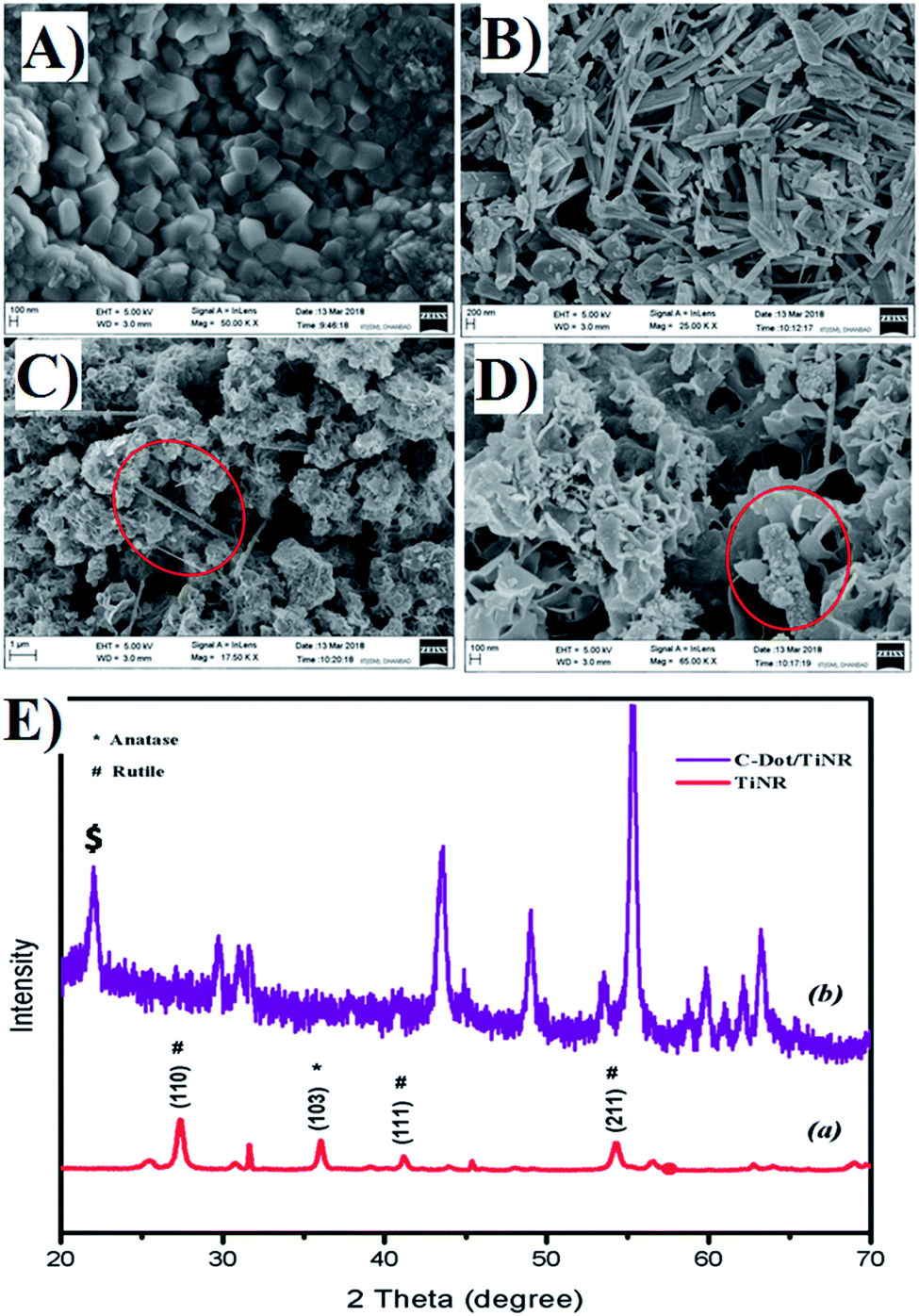
Fig. 1A and B shows the representative topography of the as-prepared TiNR before and after calcination (Fig. 1B). The non-calcined (air dried at room temperature) materials exhibited small cubes to elongated rod shapes with the length of ∼0.96 nm. It was clear that the thermal treatment transformed the cubic structure to rod shapes with different orientations. The composite of C-dots with TiO2 nanorods is depicted in Fig. 1C and D. The image showed the strong adhesion of C-dots on to the periphery of the nanorods (see the red circle). The flake-like structure indicates that the residual formation of the carbon precursor took place.
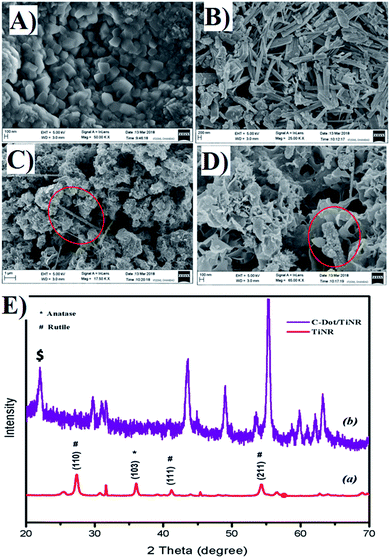 |
| | Fig. 1 FESEM image of (A) nucleation of TiNR, (B) final structure of TiNR, (C and D) C-dots/TiNR [red circle shows the C-dots/TiNR] and (E) crystallographic pattern of (a) TiNRs and (b) C-dots/TiNR. | |
The corresponding XRD pattern of obtained TiNR and C-dots/TiNR compared with JCPDS card no. 21-1272 for anatase and 21-1276 for rutile are shown in Fig. 1E. A strong peak was observed at 36.11° (103), denoting the presence of an anatase phase, while similar prominent peaks at 27.44° (110), 41.18° (111) and 54.27° (211) are attributed to the rutile phase. The crystallographic peaks of TiNR imply the existence of an anatase and rutile mixed phase. However, the C-dots/TiNR shows a zigzag pattern, resulting in the superimposed positions of anatase onto rutile at 43° onwards. A broad sharp peak at 22.59° (marked with $) of the (103) plane, indicates an amorphous phase is present in the composite (Fig. 1E) and confirms the presence of C-dots in the composite.81
Using the Williamson–Hall method, one can calculate the average crystallite size and the strain:33
| |
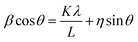 | (3) |
where,
β is the full width at half maximum of the high intensity diffraction peaks,
η is strain,
L is the crystallite size,
λ is the X-ray wavelength and
K is the shape factor (0.9).
The average crystallite grain size was estimated using Debye–Scherrer's equation as follows:34,35
| |
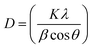 | (4) |
where,
D is the average crystallite size,
K is a constant (0.9),
λ is the X-ray wavelength,
β is the corrected full width at half maximum (FWHM) of the high intensity peaks.
The intercept on the Y-axis and the slope of a linearly fitted plot between βcosθ and sinθ (size–strain plot) is shown in Fig. S1.† The different intensity peaks as recorded in the diffraction pattern gives the crystallite strain with respect to the FWHM intensities. The average crystallite size and strain were found to be 42.43 nm and 0.0033, respectively, while the Debye–Scherrer's equation calculated the size to be 24.80 nm. It is obvious that there is a clear deviation in the calculated grain size of C-dots/TiNR. The former method includes all the obtained crystallographic peaks whereas the latter adopts only the highest intensity diffraction peaks, which may lead to an underestimated crystallite size.36 Also, the crystallographic structure of the composite was revealed through the obtained Williamson–Hall (W–H) plot. The deformation of the crystal structure including the influence of all the grains appears in the material. A lower strain indicates that the composite has a non-deformative nature and good structural stability.37
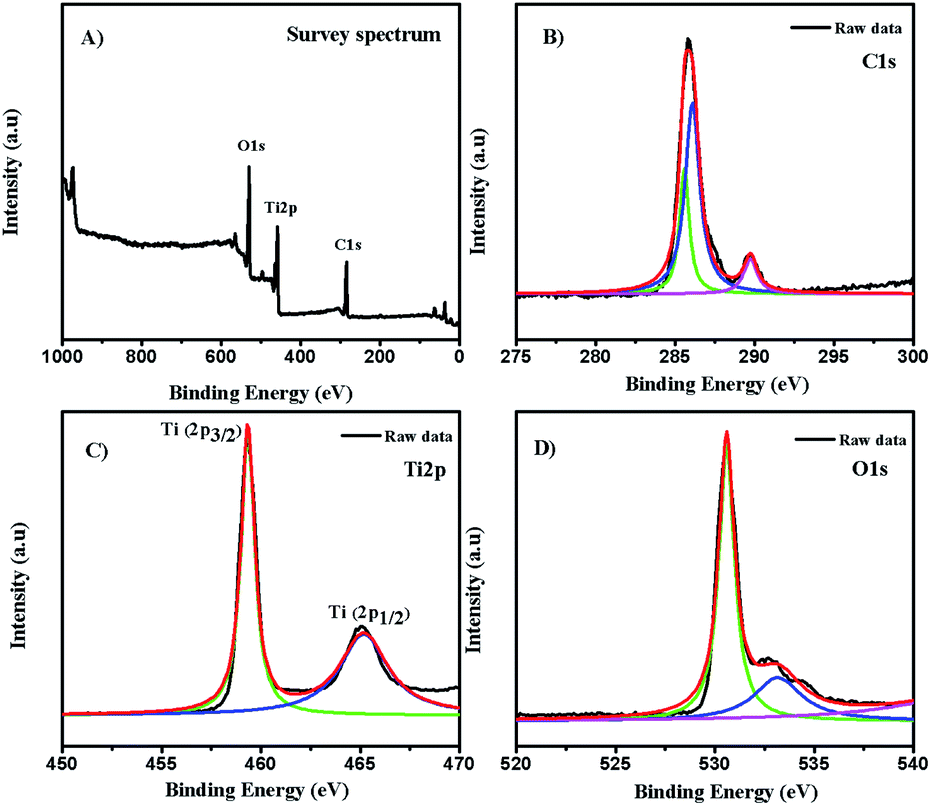
3.2. XPS analysis
The chemical composition and electronic state of the as-synthesized composite is shown in Fig. 2. The full survey spectrum (Fig. 2A) showed the presence of carbon, oxygen, and titanium. To further investigate the carbon states in the composite, the C1s spectrum (Fig. 2B) revealed the presence of two components at a binding energy of 285.8 eV and 289.79 eV, respectively. The major broad peak at 285.8 eV is typically assigned to contaminant hydrocarbons (C–C).38 The analysis showed two signature peaks for Ti species at 459.2 eV (Ti2p3/2) and 465.1 eV (Ti2p1/2), as displayed in Fig. 2C. The reported typical binding energy for Ti2p3/2in the titanium crystal ranges between 458.5 and 459.7 eV.39 A redshift was observed in the binding energy of the Ti2p3/2 peak of C-dot/TiNR, which occurred due to the formation of Ti3+ species.40 The Ti2p1/2 peak is much broader than Ti2p3/2 and confirmed the presence of hydrophilic groups on the surface of the C-dots, resulting in the good water solubility.41–43
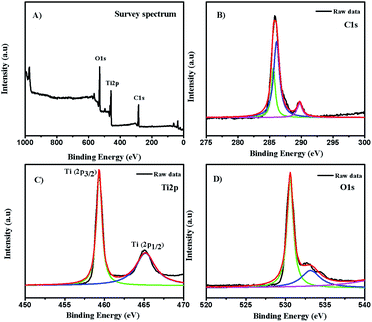 |
| | Fig. 2 XPS spectra of C-dots/TiNR. (A) Survey spectrum, (B) C1s, (C) Ti2p and (D) O1s. | |
The deconvolution of the O1s spectrum is presented in Fig. 2D. The binding energies of the components were 530.65 eV and 533.24 eV, which are associated with (Ti4+–O) and adsorbed H2O, which is in good agreement with the literature.44 Also, the component at 531.80 eV was unambiguously assigned to oxygen-bonded Ti3+.45
3.3. FTIR analysis and Raman spectroscopy
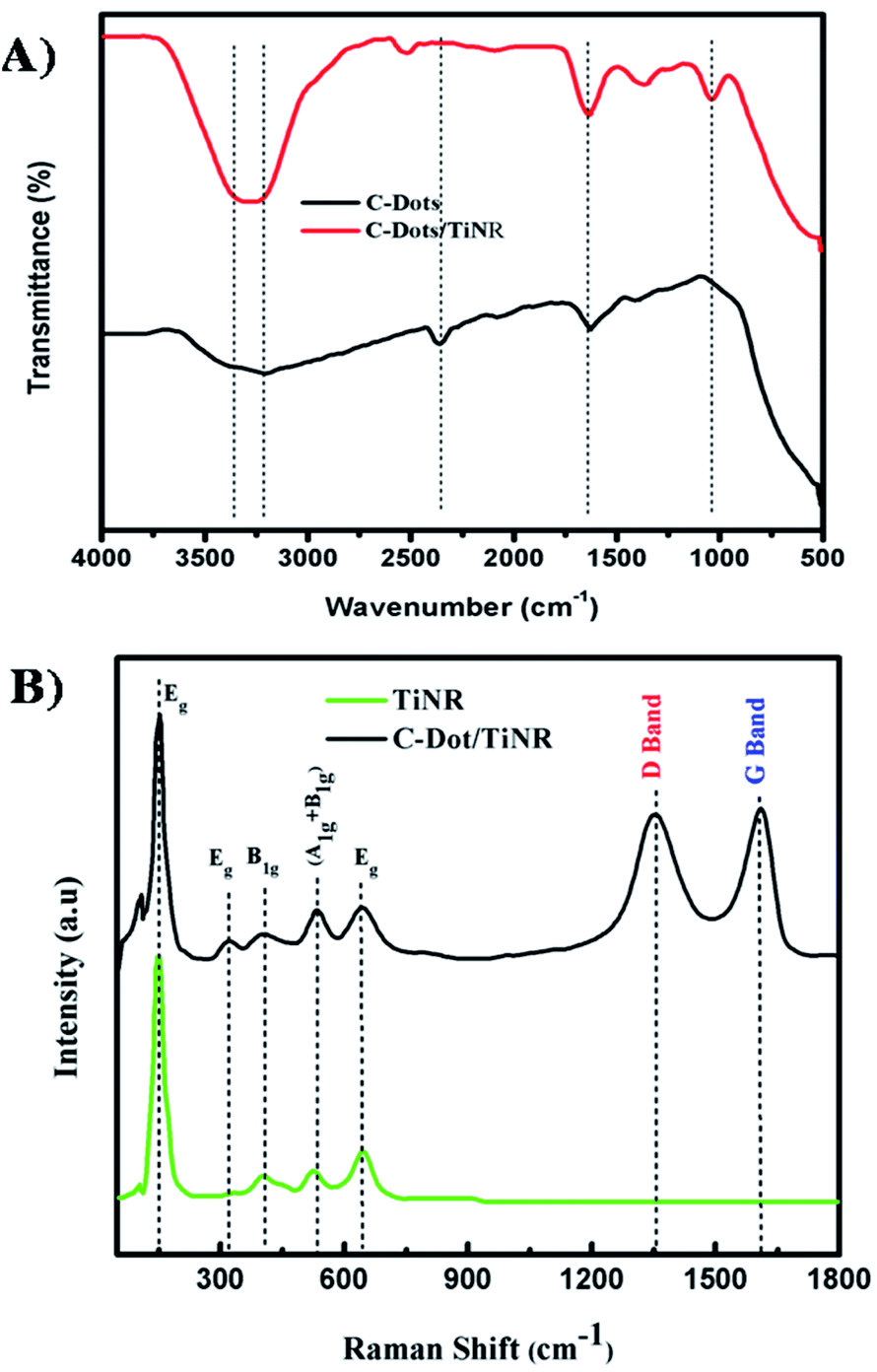
Fig. 3A shows the FTIR spectra of C-dots and C-dots/TiNR. Features in the region of 3200–3400 cm−1 and a sharp peak at 1630 cm−1(Ti–OH) appearing in C-dots and C-dots/TiNR are due to the adsorbed OH groups on the surface of the composite.46 Moreover, a sharp peak observed in C-dots at 2360 cm−1 was abruptly shifted to 2519 cm−1 in the case of C-dots/TiNR. The occurrence was due to the geometrical changes experienced by the nanorods upon the inclusion of C-dots. The peak for –CH3 at 1368 cm−1 represents the Ti–O modes present in C-dots/TiNR, whereas it was completely absent in the C-dots.47 The peak at 1040 cm−1 for C–O stretching represents the presence of anhydride compounds in the C-dots/TiNR composites.
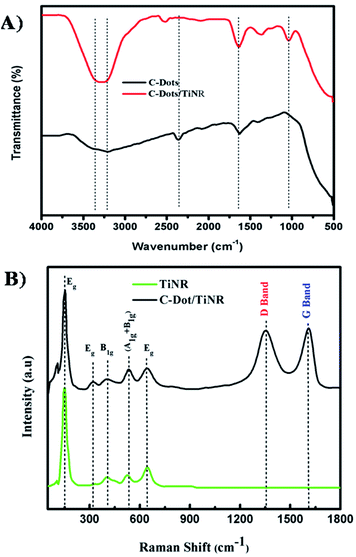 |
| | Fig. 3 (A) FTIR spectra of C-dots and C-dots/TiNR and (B) Raman spectra of the TiNR and C-dots/TiNR. | |
Raman analysis was carried out in the range of 100–1800 cm−1to further elucidate the phases present in C-dots/TiNR and is shown in Fig. 3B. Curve fitting was done in order to have proper alignment among the peaks. The features at 444 cm−1 and 609 cm−1 correspond to the rutile phase, which is thermodynamically stable.48 A strong intensity feature at 252 cm−1 appeared due to O–O interactions, based on residual valence electrons.49 Generally, anatase TiO2 has six Raman active modes (A1g + 2B1g + 3Eg) i.e., the peak at 197 cm−1 Eg(1), 312 cm−1 Eg(2), caused by the symmetric stretching vibrations of oxygen atoms in the O–Ti–O bond, 636 cm−1 Eg(3), 398 cm−1 B1g(1), 520 cm−1 A1g, and 520 cm−1 B1g(2), which were consistent with the present modes.50–53 The D (disordered) and G (graphitic phase of carbon) band occurring at 1352 cm−1 and 1610 cm−1 are associated with sp3 and sp2 hybridized carbon atoms, respectively. The existence of both the bands confirms the presence of C-dots.82 Here the ID/IG = 0.98 expresses the degree of functionalization of carbon nanomaterial that was reduced by the addition of C-dots, showing the increased graphite content.54–56,81 The obtained ratio confirmed the successful functionalization of the C-dots on to TiNR.
3.4. UV-vis-DRS spectroscopy and PL spectroscopy
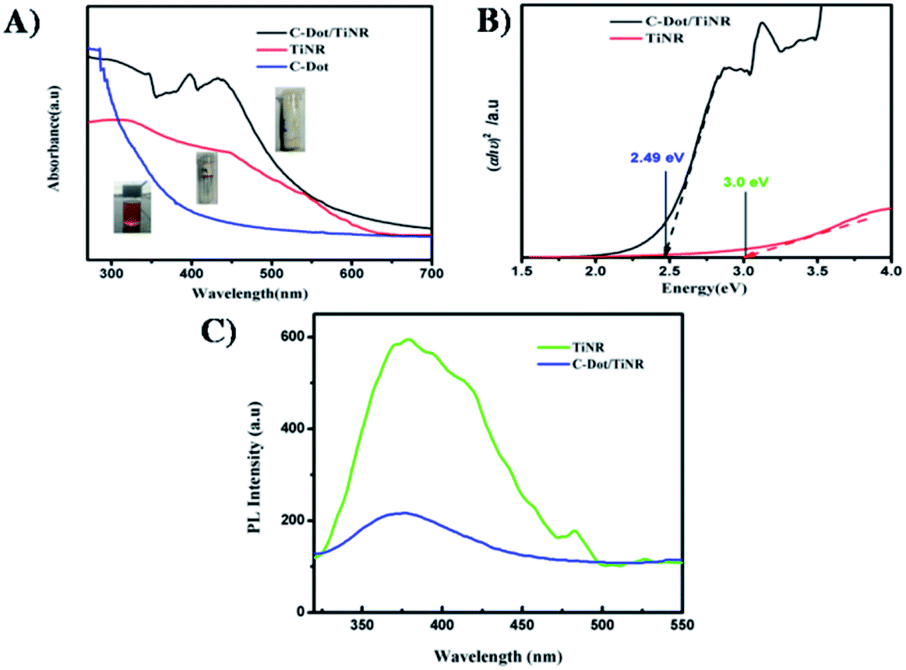
To gain further understanding of the photocatalytic mechanism, the light absorption properties and charge transfer separation of samples were studied. The samples were analyzed by UV-vis absorption spectroscopy and the obtained spectrum is reported Fig. 4A. The C-dots sample shows an absorbance edge at ca. 300 nm, corresponding to the characteristic band gap of TiO2 (3.0 eV) and was blueshifted with respect to TiNR. Whereas TiNR exhibited a wide range of absorbance towards the visible region (redshift), the composite with C-dots enhanced the absorbance significantly. Thus, the composite showed an extended absorption up to 600 nm. The existence of sp2 clusters within C-dots along with a carbon–oxygen sp3 matrix might yield a bandgap (Eg) that is consistent with the localization of electron–hole pairs observed in the photoluminescence properties.83 In Fig. 4B, (αhν)2 is plotted against hν, and the band gap energies were estimated by the Kubelka–Munk (K–M) eqn (5)–(7).57| | |
(F(R∞)hν)1/2 = B(hν − Eg)
| (5) |
| |
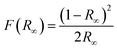 | (6) |
| |
 | (7) |
where R∞ is the reflectance of an infinitely thick specimen; Rstandard is the reflectance of TiO2; Rsample is the reflectance of the catalyst; h is the Planck constant; ν is the photon's frequency; B is a constant; Eg is the band gap energy. As shown in Fig. 4A and B, when the method was used directly, the intersection point of the linear fit and the x-axis gave the Eg values. The calculated band gap was found to be 2.49 eV, which is compared with the nominal band gap of standard TiO2 (∼3.2 eV). This reduction in the band gap occurs due to introduction of C-dots.58
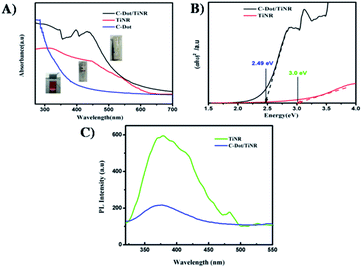 |
| | Fig. 4 (A) UV-visible absorption spectra of C-dot, TiNR and C-dot/TiNR, (B) band gap calculated from the Tauc plots for C-dot/TiNR and TiNR (C) photoluminescence spectra of TiNR and C-dots/TiNR excited (λex) at 300 nm. | |
Fig. 4C depicts the obtained photoluminescence (PL) spectra of the composite that reveals the electron–hole (e−/h+) mobility and its transfer onto the surface of metal oxides. Generally, it is well-known that the intensity of luminescence is directly proportional to the rate of the recombination of charge carriers.59 The as-obtained samples were excited at 300 nm. The spectrum was normalized among the wavelength vs. intensity to compare it with the sub-wavelength. PL contributions below the optical band gap are associated with electron–hole recombination in the presence of surface oxygen vacancies and defects.60 In the present investigation, it was found that the peak at 380 nm (∼3.2 eV) is due to the self-trapped interfacial electrons on the TiNR surface, which is featured in the visible region. On the other hand, a slight decrease in this feature was observed in the C-dots/TiNR, suggesting the efficient charge transfer occurred due to the C-dots. Moreover, the PL intensity of the C-dots/TiNR composites dampened effectively, compared with that for TiNR.61 Also, the chemical environment of Ti in the C-dots/TiNR was changed due to the strong interaction between the TiNR and C-dots, which results in the formation of Ti–O–C bonds. Hence, it can be concluded that the coupling of C-dots with TiNR is through the formation of Ti–O–C bonds.62 This analysis indicated that the prepared C-dots/TiNR should exhibit improved physical properties suitable for enhanced photocatalytic activity.
3.5. Photocatalytic degradation of methyl orange
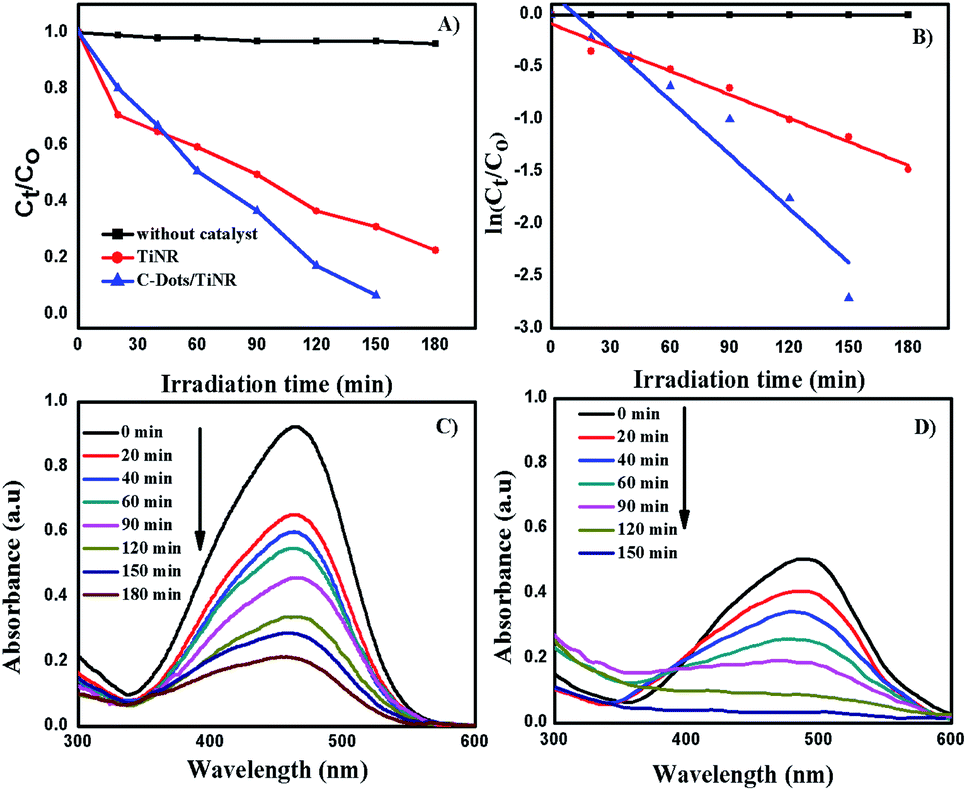
The solar photocatalytic activity of C-dots/TiNR nanoparticles was evaluated by degrading methyl orange (MO) and is shown in Fig. 5A and B. Fig. 5C and D show the obtained UV-vis spectra of the MO during the photocatalytic reaction. The composite exhibited the highest degradation of MO as compared to that of TiNR. The highest activity of the composite was attributed to the enhanced visible light utilization and low recombination of the charge carriers.63 The C-dots absorb visible light, and then emit at a shorter wavelength light (325–425 nm), which was utilized in the photocatalytic activity.64,65
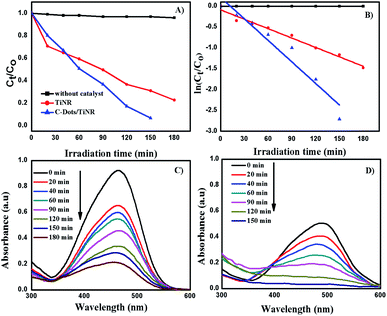 |
| | Fig. 5 Photocatalytic MO degradation under direct sunlight (A) plot of Ct/C0 vs. irradiation time, (B) ln(Ct/C0) vs. irradiation time, (C) TiNR and (D) C-dots/TiNR. | |
The dark reaction showed that almost all the mixtures reached adsorption equilibrium within 60 min. The kinetic parameters of the photocatalytic degradation of MO are in accordance with the pseudo-first order reaction kinetics and were obtained using the following equation:
where
C0 is the initial concentration at which the MO solution is adsorbed and equilibrated;
Ct is the concentration of the solution at time
t;
k is the reaction rate constant in min
−1. The value of
k is thus calculated from the slope of a plot of ln(
C0/
Ct)
versus t. The half-life time of the sample was calculated as:
66| |
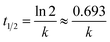 | (9) |
It was found that the MO degradation rate (min−1) by C-dot/TiNR is about 2.3 times that of TiNR, which further indicates that the introduction of C-dots improves the photocatalytic activity. The calculated half-life time is presented in Table 2, where the TiNR (solo) had a prolonged time of 92.15 min while the composite shortened the time by 51.93 min. The photocatalytic degradation of MO as observed from Fig. 5A shows that 78% of MO was degraded in 180 min by TiNR, whereas 93.36% MO was degraded by C-dots/TiNR in 150 min (Fig. 5B). In the inception phase, both the materials had an almost similar degradation percentage (35.15% for the solo TiNR and 32.98% for the composite) but as soon as the time increased, the composite dominated over the solo (63.36%) and reached a degradation percentage of 82.75% at the end of 2 h. At the end of 150 min, the MO degradation achieved a maximum of 93.36% by the composite while the solo TiNR took 3 h to reach a maximum degradation of 78%. This escalation in the degradation is attributed to the presence of C-dots, which significantly interacted with TiNR to enhance the absorption of visible light and suppress the recombination of charge carriers. Also, the control experiment clearly proved that the degradation of MO was attributed to the as-synthesized materials where the photolysis phenomenon was insignificant (∼5.8%).
Table 2 Calculated kinetics parameter on MO by solar photo degradation
| Photocatalyst |
Model |
k (min−1) |
R2 |
t1/2 (min) |
| TiNR |
First-order |
0.00752 |
0.97915 |
92.154 |
| C-Dots/TiNR |
First-order |
0.01723 |
0.93137 |
40.220 |
Fig. 5D shows the kinetic plot for the observed MO degradation with a pseudo-first order model. It was fit well by first order kinetics and under this condition the rate constant (k) was 0.00752 min−1 and 0.01723 min−1, respectively. The aromatic MO absorbs at 465 nm. Fig. 5C and D shows the MO absorbance over the degradation process. At each time interval, there is a clear decrease in the absorbance intensity, which verifies the degradation of the studied MO with an identical trend. The absorbance trend of the MO has a rising and falling pattern. The rising trend is attributed to the presence of polyaromatic rings present in MO where it starts to degrade, creating a mono-substituted aromatic that increases in intensity (300 > λ < 465). After 465 nm, it starts to decrease and similarly was observed in the band between 300 > λ < 350. Thus, this strongly indicates that the mineralization of MO to its final non-toxic form goes through a systematic pathway.67,68 In general, the concentration of the charge carriers is directly proportional to the amount of incident photons at a specific wavelength (visible light). Since the synthesized material is more susceptible to visible light rather than IR and NIR, most of the generation of charge carriers was attributed to visible light. Further, the quantum yield from the visible light is significantly less since only ∼42% of the solar light fraction could be utilized by the materials while the majority is unutilized. This cumulative phenomenon could contribute to a comparatively lesser efficiency of the composite. However, the performance of the composite was appreciable when compared with similar materials synthesized by other groups and is tabulated in Table 3.
Table 3 Comparison of present study with the literature for MO degradation utilizing various TiO2-based photocatalysta
| Photocatalyst |
Degradation (%)/time |
Source |
Ref. |
| The physical change in the degradation of MO is shown in Fig. S2 A and B. The fading of color from orange to colorless clearly validates its potential degradation rate. |
| Ag–MoO3–TiO2 |
97/300 min |
Sunlight |
69 |
| Nd-TiO2-SBA-15 |
94.1/140 min |
Xenon lamp |
32 |
| SnS2/TiO2 |
90.9/50 min |
Xenon arc lamp |
70 |
| Anatase TiO2 |
67.12/240min |
Xenon lamp |
71 |
| ZSM-5/ZnO/Ag |
90/120 min |
UV lamp |
72 |
| NiO:V2O5 |
88/70 min |
Sunlight |
73 |
| Cu–TiO2/ZnO |
83/60 min |
Xenon lamp |
74 |
| Fe2O3/TiO2 |
98/60 min |
Xenon |
75 |
| C–TiO2 |
91.89/240 min |
Xenon |
76 |
| Y3+–TiO2 |
99.8/70 min |
HP mercury lamp |
77 |
| TiO2/T-shirt |
98.6/30 min |
Sunlight |
78 |
| Au/TiO2 |
99/180min |
HP xenon lamp |
79 |
| TiO2 nanorods |
51/150 min |
Mercury lamp |
43 |
| C-Dots/TiNR |
93.3/150 min |
Sunlight |
Present study |
3.6. Detection of reactive oxidative species
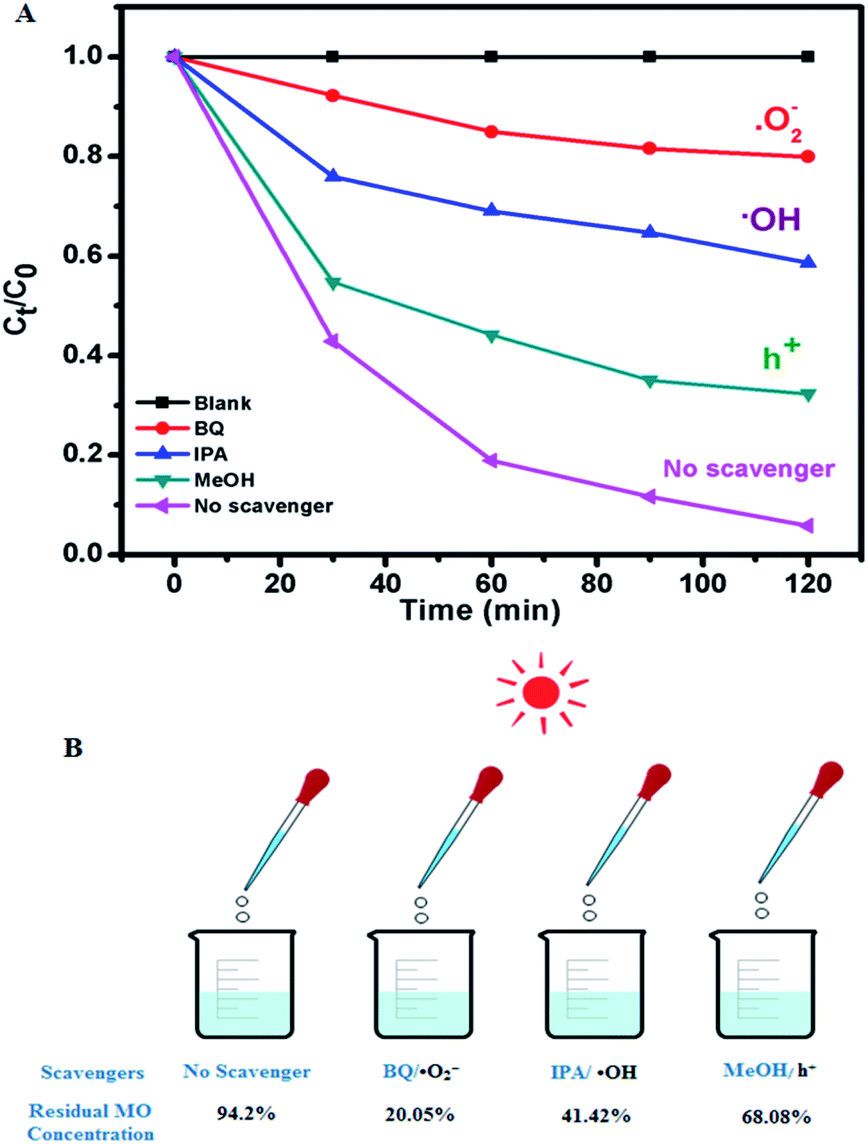
The role of charge carriers in the MO degradation by C-dot/TiNR was studied through charge trapping experiments using standard chemicals as scavengers and is presented in Fig. 6A and B. The results further substantiated the insignificant photolysis ability of MO under direct solar irradiation in the absence of catalysts. Further, the role of charge carriers is revealed, with superoxide (˙O2−) being the dominant among them all with a maximum formation by utilizing the electrons of the CB. Specifically, the photocatalytic activity of C-dots/TiNR was significantly suppressed under an inert atmosphere, indicating that the ˙O2− radicals originated strictly from the dissolved oxygen (namely, eqn (12)). Simultaneously, a good performance (52.78%) was also reported for IPA suggesting the role of ˙OH radical on degrading MO. It is obvious that ˙OH radicals were formed by holes reacting with hydroxyl anions (eqn (13)). However, a small amount of such radicals was also formed by a series of reactions triggered by the electrons on water molecules (eqn (10)–(17)). Notably, compared with IPA quencher and inert gas purging, MeOH as the scavenger of h+ had a weaker influence on the whole photodegradation reaction because the corresponding degradation ratio of MO exhibited a lesser degradation (26.12%), implying that the direct h+ oxidation could not contribute to the whole photocatalytic process. To some extent, it can be interpreted that the photogenerated VB holes are primarily involved in the formation of ˙OH radicals (eqn (13) and (17)). The contribution of these active species for MO degradation followed a sequence of ˙O2− > ˙OH > h+. The dominant role of superoxide and significant role of holes and ˙OH clearly clarifies the weaker recombination of the charge carriers i.e., electrons with holes. It is well-established that the C-dots played a crucial role in prolonging the lifetime of the charge carriers.
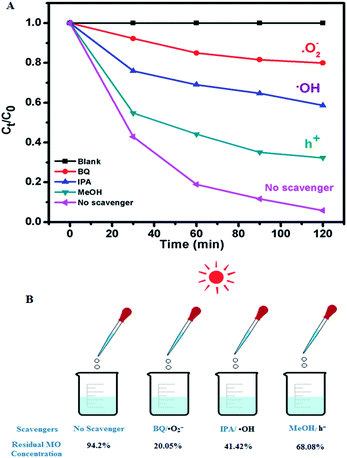 |
| | Fig. 6 (A) Radical species trapping experiments of photocatalytic degradation of MO over C-dots/TiNR under direct sunlight for 2 h and (B) the corresponding residual MO present. | |
3.7. Recycle test and TOC analysis
The solar photocatalytic degradation efficiency of C-dots/TiNR for five cycles was calculated and presented in Fig. S3.† The degradation was 92%, 89%, 83%, 78% and 76%, respectively, indicating that the as-synthesized composite possesses stability and reusability. One can observe that there is a significant decline in the degradation% after the 2nd cycle, which was attributed to the solar irradiation intensity on that specific day i.e., all the studies were not able to be carried out on the same day. Hence, it would be difficult to conclude that the associated surface phenomena were involved in the decline of the efficiency. Overall it is evident that the as-synthesized composite had robust stability with prolonged reusability. The solar photocatalytic study on MO presented in Fig. S2† is a bulk analysis where the decolorization was linked to the degradation. In order to overcome the uncertainty, a total organic carbon (TOC) analysis was carried out for the composite and is presented in Fig. S4.† The TOC removal rate (mineralization) of MO was in the increasing order of 18%, 27%, 44%, 58%, 65% and 74% within the reaction time of 150 min. This indicates that the reactive species were actively involved in mineralizing MO. The rate at which the MO was mineralized substantiates the lower formation of the intermediates and ultimate formation to CO2.
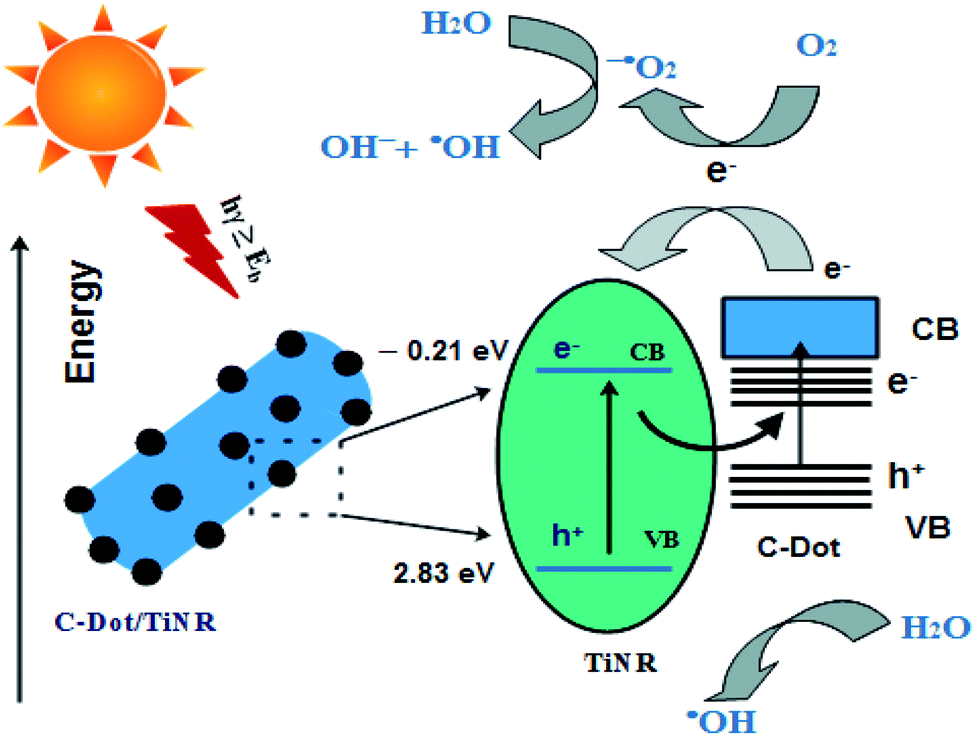
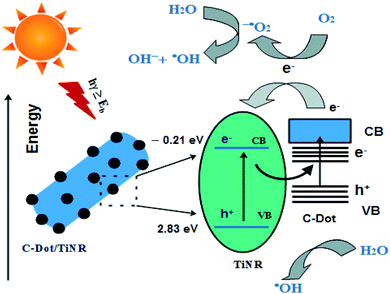
3.8. Mechanism
From the studied materials, photocatalysis and scavenging analyses, a mechanism is proposed and illustrated in Scheme 2. The C-dots/TiNR acts as a catalyst for generating charge carriers (e−/h+) under solar irradiation. The accumulated holes in the VB of TiNR with a more positive potential and electrons in the CB of C-dots with a more negative potential have a stronger oxidation and reduction capability to produce the active radicals. Here, C-dots mainly act as the electron reservoir, which helps reduce the recombination rate. The electrons present in the CB of C-dots get transferred to the CB of TiNR when it is under illumination. The formation of Ti–O–C bonds in the composite shifted the valence band (VB) edge upwards, resulting in a narrower band gap. Further, these single covalent bonds act as a conductive conduit for charge carriers.80 In more detail, the charge carriers react with the adsorbed O2 present on the surface of C-dots/TiNR and result in superoxide anions. These superoxide anions further form hydroxyl radicals in the reaction with holes. The adsorbed O2 combines with electrons and holes and forms hydrogen peroxide, which further dissociates into hydroxyl radicals and hydroxyl ions. Thus, the generated reactive species contribute to the degradation of MO.| | |
C-dots/TiNR + hν → C-dots/TiNR + hν(h+ + e−)
| (10) |
| | |
O2 + 2H+ + 2e− → H2O2
| (14) |
| | |
H2O2 + e− → OH˙ + OH−
| (15) |
| | |
h+/O2−˙ + MO → photodegradation products
| (17) |
 |
| | Scheme 2 Schematic showing the interaction of C-dots with TiNR. | |
Typically, the excited electron–hole would recombine at the interface of C-dots/TiNR. However, the mobilization of electrons from the CB of C-dots to the CB of TiNR allows for an excess availability of electrons, which overcomes the recombination phenomenon. This was also evident from the PL intensity, which suggested that the composite had a higher recombination rate of photogenerated electron–hole pairs. In spite of the higher recombination rate, the photocatalytic rate was high for the composite, which was consistent with the mobilization of the electrons from the C-dots.
4. Conclusion
C-Dots/TiNR was successfully synthesized by a two-step hydrothermal method with a good adhesion of C-dots on the TiNR surface. The various materials analyses demonstrated the robust photoharvesting ability of the prepared composite. The utilization of melon rind shows the robustness of a green carbon precursor source compared to conventional chemicals. The photocatalytic studies establish the ability of the composite to easily assimilate into complex water pollutant removal systems. Further, the role played by the ROS was demonstrated clearly while recycling tests proved the stability and durability of the composite. The study suggested that C-dots/TiNR can act as a potential long-lasting and cheaper photocatalyst towards water treatment applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The corresponding author is grateful to the Science and Engineering Research Board (SERB) and the Indian Institute of Technology (ISM) Dhanbad for the financial support received Early Career Research Award (ECR/2016/001400) and Faculty Research Scheme (FRS(109)/ESE/16-17) respectively.
Notes and references
- M. A. M. Adnan, N. M. Julkapli, M. N. I. Amir and A. Maamor, Int. J. Environ. Sci. Technol, 2019, 16, 547–566 CrossRef.
- K. Namratha, K. Byrappa, S. Byrappa, P. Venkateswarlu, D. Rajasekhar and B. K. Deepthi, J. Environ. Sci., 2015, 34, 48–255 CrossRef PubMed.
- J. Curtina, C. McInerney, B. O. Gallachóir, C. Hickey, P. Deane and P. Deeney, Renewable Sustainable Energy Rev., 2019, 116, 109402 CrossRef.
- L. Shi, Y. Shi, S. Zhuo, C. Zhang, Y. Aldrees, S. Aleid and P. Wang, Nano Energy, 2019, 60, 222–230 CrossRef CAS.
- S. Kumar, M. Nehra, A. Deep, D. Kedia, N. Dilbaghi and K. Kim, Renewable Sustainable Energy Rev., 2017, 73, 821–839 CrossRef CAS.
- X. Li, C. Tung and L. Wu, Nat. Rev. Chem., 2018, 2, 160–173 CrossRef CAS.
- I. S. Yunus, Harwin, A. Kurniawan, D. Adityawarman and A. Indarto, Environ. Technol. Rev., 2012, 1, 136–148 CrossRef CAS.
- S. Ishii, R. P. Sugavaneshwar and T. Nagao, J. Phy. Chem. C, 2016, 120, 2343 CrossRef CAS.
- J. Tian, Y. H. Leng, Z. H. Zhao, Y. Xia, Y. H. Sang, P. Hao, J. Zhan, M. C. Li and H. Liu, Nano Energy, 2015, 11, 419–427 CrossRef CAS.
- T. Hisatomi, K. Takanabe and K. Domen, Catal. Lett., 2015, 145, 95–108 CrossRef CAS.
- C. Zhang, Y. Li, D. Shuai, W. Zhang, L. Niu, L. Wang and H. Zhang, Chemosphere, 2018, 208, 84–92 CrossRef CAS PubMed.
- I. Khan, K. Saeed and I. Khan, Arab. J. Chem., 2019, 12, 908–931 CrossRef CAS.
- Q. Xiang, X. Ma, D. Zhang, H. Zhou, Y. Liao, H. Zhang, S. Xu, I. Levchenko and K. Bazaka, J. Colloid Interf. Sci., 2019, 556, 376–385 CrossRef CAS PubMed.
- M. Anjum, R. Miandad, M. Waqas, F. Gehany and M. A. Barakat, Arab. J. Chem., 2016, 12, 4897–4919 CrossRef.
- S. Y. Chai, Y. J. Kim and W. I. Lee, J Electroceram, 2006, 17, 909–912 CrossRef CAS.
- R. K. Mandal, S. Kundu, S. Sain and S. K. Pradhan, New J. Chem., 2019, 43, 2804–2816 RSC.
- G. Zhang, T. Zhang, B. Li, S. Jiang, X. Zhang, L. Hai, X. Chen and W. Wu, Appl. Surf. Sci., 2018, 433, 963–974 CrossRef CAS.
- G. Sanzone, M. Zimbone, G. Cacciato, F. Ruffino, R. Carles, V. Privitera and M. G. Grimaldi, Superlattices Microstruct., 2018, 123, 394–402 CrossRef CAS.
- B. Y. Lee, S. H. Park, S. C. Lee, M. Kang and C. H. Park andSJ.Choung, Korean J. Chem. Eng., 2003, 20, 812–818 CrossRef CAS.
- Z. Kang and S. T. Lee, Nanoscale, 2019, 11, 19214–19224 RSC.
- S. Feizpoor, H. Y. Aziz, I. Ahadzadeh and K. Yubuta, Adv. Powder Technol., 2019, 30, 1183–1196 CrossRef CAS.
- V. Roshni, S. Misra, M. K. Santra and D. Ottoor, J. Photochem. Photobiol., A, 2019, 373, 28–36 CrossRef CAS.
- B. Yousaf, G. Liu, Q. Abbas, R. Wang, H. Ullah, M. M. Mian, Amina and A. Rashid, RSC Adv., 2018, 8, 25983 RSC.
- W. Liu, Y. Gong, X. Li, C. Luo, C. Liu and Z. Chao, RSC Adv., 2019, 9, 3965–3971 RSC.
- Y. Li, X. Feng, Z. Lu, H. Yin, F. Liu and Q. Xiang, J. Colloid Interface Sci., 2018, 513, 866–876 CrossRef CAS PubMed.
- M. Semeniuk, Z. Yi, V. Poursorkhabi, J. Tjong, S. Jaffer, Z. H. Lu and M. Sain, ACS Nano, 2019, 13, 6224–6255 CrossRef CAS PubMed.
- M. T. Genc, G. Yanalak, G. Arslan and I. H. Patir, Mater. Sci. Semicond. Process., 2020, 109, 104945 CrossRef CAS.
- H. M. A. Al-Sayed and A. R. Ahmed, Ann. Agric. Sci., 2013, 58, 83–95 CrossRef.
- M. Yan, Y. Wu, Y. Yan, X. Yan, F. Zhu, Y. Hua and W. Shi, ACS Sustainable Chem. Eng., 2016, 4, 757–766 CrossRef CAS.
- C. Yun, L. Wang, Y. Zhou, Y. Wang, C. Li, L. Liang and X. Luo, Colloids Surf., A, 2020, 587, 124321 CrossRef.
- A. Patchaiyappan, S. Saran and S. P. Devipriya, Korean J. Chem. Eng., 2016, 33, 2107–2113 CrossRef CAS.
- L. Bai, S. Wang, Z. Wang, E. Hong, Y. Wang, C. Xia and B. Wang, Environ. Pollut., 2019, 248, 516–525 CrossRef CAS PubMed.
- J. H. Joshi, D. K. Kanchan, M. J. Joshi, H. O. Jethva and K. D. Parikh, Mater. Res., 2017, 93, 63–73 CAS.
- S. Muthukumaran and R. Gopalakrishnan, Opt. Mater., 2012, 34, 1946–1953 CrossRef CAS.
- A. B. Andrade, N. S. Ferreira and M. E. G. Valerio, RSC Adv., 2017, 7, 26839–26848 RSC.
- T. Ohta, K. Tanaka and H. Uchiki, Jpn. J. Appl. Phys., 2011, 50, 01BE16 CrossRef.
- H. Yin, Y. Wada, T. Kitamura, S. Kambe, S. Murasawa, H. Mori, T. Sakata and S. Yanagida, J. Mater. Chem., 2001, 11, 1694–1703 RSC.
- D. Xie, F. Wen, W. Yang, X. Li, Y. Leng, G. Wan, H. Sun and N. Huang, Acta Metall. Sin. (Engl. Lett.), 2014, 27, 239–244 CrossRef CAS.
- M. Yoshitake, A. Thananan, T. Aizawaki and K. Yoshihara, Surf. Interface Anal., 2002, 34, 698 CrossRef CAS.
- T. Ohno, T. Tsubota, M. Toyofukum and R. Inaba, Catal. Lett., 2004, 98, 255–258 CrossRef CAS.
- B. Erdem, R. A. Hunsicker, G. W. Simmons, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, Langmuir, 2001, 17, 2664–2669 CrossRef CAS.
- V. D. Chinh, A. Broggi, L. D. Palma, M. Scarcella, G. Speranza, G. Vilardi and P. N. Thang, J. Electron. Mater., 2018, 47, 2215–2224 CrossRef CAS.
- K. Santhi, M. Navaneethan, S. Harish, S. Ponnusamy and C. Muthamizhchelvan, Appl. Surf. Sci., 2020, 500, 144058 CrossRef CAS.
- G. Li, Z. Liu, Z. Zhang and X. Yan, Chin. J. Catal., 2009, 30, 37–42 CrossRef.
- J. Pouilleau, D. Devilliers, H. Groult and P. Marcus, J. Mater. Sci., 1997, 32, 5645 CrossRef CAS.
- T. Prakash, M. Navaneethan, J. Archana, S. Ponnusamy, C. Muthamizhchelvan and Y. Hayakawa, Mater. Lett., 2012, 82, 208–210 CrossRef CAS.
- S. Mugundan, G. Rajamannan, N. Viruthagiri, R. Shanmugam and P. Gobi, Appl. Nanosci., 2015, 5, 449–456 CrossRef CAS.
- Y.-C. Yen, C.-C. Lin, P.-Y. Chen, W.-Y. Ko, T.-R. Tien, K.-J. Lin and R. Soc, Open Sci., 2017, 4, 161051 Search PubMed.
- F. D. Hardcastle, J. Arkansas Acad. Sci., 2011, 65, 9 Search PubMed.
- A. G. Ilie, M. Scarisoareanu, I. Morjan, E. Dutu, M. Badiceanu and I. Mihailescu, App. Surf. Sci., 2017, 417, 93–103 CrossRef CAS.
- X. Xue, W. Ji, Z. Mao, H. Mao, Y. Wang, X. Wang, W. Ruan, B. Zhao and J. R. Lombardi, J. Phys. Chem. C, 2012, 116, 8792–8797 CrossRef CAS.
- L. Jing, H. L. Tan, R. Amal, Y. H. Ng and K. N. Sun, J. Mater. Chem. A, 2015, 3, 15675–15682 RSC.
- T. Ohsaka, F. Izumi and Y. Fujiki, J. Raman Spectrosc., 1978, 7, 321 CrossRef.
- X. Miao, D. Qu, D. Yang, B. Nie, Y. Zhao, H. Fan and Z. Sun, Adv. Mater., 2018, 30, 1704740 CrossRef PubMed.
- H. C. Ho, K. Chen, T. Nagao and C. H. Hsueh, J. Phys. Chem. C, 2019, 123, 21103–21113 CrossRef CAS.
- A. Kouloumpis, E. Thomou, N. Chalmpes, K. Dimos, K. Spyrou, A. B. Bourlinos, I. Koutselas, D. Gournis and P. Rudolf, ACS Omega, 2017, 2, 2090–2099 CrossRef CAS PubMed.
- J. Ding, Z. Dai, F. Qin, H. Zhao, S. Zhao and R. Chen, Appl. Cat. B-Environ., 2017, 205, 281–291 CrossRef CAS.
- J. Zhang and S. Yu, Materials Today, 2016, 19, 382–393 CrossRef CAS.
- W. Gao, J. Lu, S. Zhang, X. Zhang, Z. Wang, W. Qin, J. Wang, W. Zhou, H. Liu and Y. Sang, Adv. Sci., 2019, 18, 1901244 CrossRef PubMed.
- J. P. Cassidy, J. A. Tan and K. L. Wustholz, J. Phys. Chem. C, 2017, 121, 15610–15618 CrossRef CAS.
- D. Syafei, S. Sugiarti, N. Darmawan and M. Khotib, Indones. J. Chem., 2017, 17, 37–42 CrossRef CAS.
- H. Yu, Y. Zhao, C. Zhou, L. Shang, Y. Peng, Y. Cao, L. Wu, C. Tunga and T. Zhang, J. Mater. Chem. A, 2014, 2, 3344–3351 RSC.
- X. Pan, M. Q. Yang, X. Fu, N. Zhang and Y. J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- P. Xia, M. Liu, B. Cheng, J. Yu and L. Zhang, ACS Sustain. Chem. Eng., 2018, 6, 8945–8953 CrossRef CAS.
- H. T. Li, X. D. He, Z. H. Kang, H. Huang, Y. Liu, J. L. Liu, S. Y. Lian, C. H. A. Tsang, X. B. Yang and S. T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430 CrossRef CAS PubMed.
- C. M. Kinley, M. Hendrikse, A. J. Calomeni, T. D. Geer and J. H. Rodjers Jr, Water, Air, Soil Pollut., 2018, 229, 167 CrossRef.
- T. Chen, Y. Zheng, J. M. Lin and G. Chen, J. Am. Soc. Mass Spectr., 2008, 19, 997–1003 CrossRef CAS PubMed.
- S. Al-Qaradawi and S. R. Salman, J. Photochem. Photobiol., A, 2002, 148, 161–168 CrossRef CAS.
- M. H. K. Neon and M. S. Islam, Environ. Nanotechnol. Monit. Manag., 2019, 12, 100244 Search PubMed.
- G. Dai, H. Qin, H. Zhou, W. Wang and T. Luo, Appl. Surf. Sci., 2018, 430, 488–495 CrossRef CAS.
- J. Dhanalakshmi and P. Padiyan, Mater. Res. Express, 2017, 4, 9 Search PubMed.
- Z. Vaezand and V. Javanbakht, J.Photochem.Photobiol. A: Chem., 2020, 388, 112064 CrossRef.
- S. A. Mohammed, L. A. Amouri, E. Yousif, A. A. Ali, F. Mabood, H. F. Abbas and S. Alyaqoobi, Heliyon, 2018, 4, e00581 CrossRef PubMed.
- M. Dorraj, M. Alizadeh, N. A. Sairi, W. J. Basirun, B. T. Goh, P. M. Woi and Y. Alias, Appl. Surf. Sci., 2017, 414, 251–261 CrossRef CAS.
- M. E. Hassan, Y. Chen, G. Liu, D. Zhu and J. Cai, J Water Process Eng, 2016, 12, 52–57 CrossRef.
- M. E. Hassan, L. Cong, G. Liu, D. Zhu and J. Cai, Appl. Surf. Sci., 2014, 294, 89–94 CrossRef CAS.
- N. Xinshu, L. Sujuan, C. Huihui and Z. Zianguo, J. Rare Earths, 2011, 29, 225–229 CrossRef.
- Y. Yang, C. Flatebo, J. Liang, P. Dong, J. Yuan, T. Wang, J. Zhang, W. Chen, J. Wu, P. M. Ajayan, L. Ci, Q. Li and J. Lou, Appl. Mater. Today, 2016, 3, 57–62 CrossRef.
- Y. Gao, X. B. Fan, F. Zhang, Q. S. Zhao, G. L. Zhang, F. B. Zhang and Y. Yi, Mater. Lett., 2014, 130, 1–4 CrossRef CAS.
- S. Umrao, S. Abraham, F. Theil, S. Pandey, V. Ciobota, P. K. Shukla, C. J. Rupp, S. Chakraborty, R. Ahuja, J. Popp, B. Dietzek and A. Srivastava, RSC Adv., 2014, 4, 59890–59901 RSC.
- H. Ming, Z. Ma, Y. Liu, K. Pan, H. Yu, F. Wang and Z. Kang, Dalton trans, 2012, 41, 9526–9531 RSC.
- J. Zhou, Z. Sheng, H. Han, M. Zou and C. Li, Mater. Lett., 2012, 66, 222–224 CrossRef CAS.
- G. Eda, Y. Y. Lin, C. Mattevi, H. Yamaguchi, H. A. Chen, I. S. Chen, C. W. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505–509 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03157g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*