DOI:
10.1039/D0RA02540B
(Paper)
RSC Adv., 2020,
10, 19482-19489
A fluorescence-based high-throughput screening method for determining the activity of diguanylate cyclases and c-di-GMP phosphodiesterases†
Received
19th March 2020
, Accepted 8th May 2020
First published on 21st May 2020
Abstract
The dinucleotide 3′,5′-cyclic diguanylic acid (c-di-GMP) is a critical second messenger found in bacteria. High cellular levels of c-di-GMP are associated with a sessile, biofilm lifestyle in many bacteria, which is associated with more than 70% of clinically resistant infections. Cellular c-di-GMP concentrations are regulated by diguanylate cyclases (DGCs) and phosphodiesterases (PDEs), which are responsible for the production and degradation, respectively, of c-di-GMP. Therefore, DGCs and PDEs might be attractive drug targets for controlling biofilm formation. In this study, a simple and universal high-throughput method based on a c-di-GMP-specific fluorescent probe for the determination of DGC and PDE activity was described. By using the proposed method, the c-di-GMP content in samples was rapidly quantified by measuring the fluorescence intensity in a 96-well plate by using a microplate reader. In addition, the probe molecule A18 directly interacted with the substrate c-di-GMP, and the method was not limited by the structure of enzymes.
Introduction
The dinucleotide 3′,5′-cyclic diguanylic acid (c-di-GMP) is a unique second messenger commonly found in bacteria.1–3 Recently, c-di-GMP has been receiving increasing attention because of its crucial role in attachment, motility, biofilm formation, and release of virulence factors as well as in bacterial life cycle operations and drug resistance.4,5 Effectively controlling the concentration of c-di-GMP in bacteria has become an active area of research in the development of novel antibacterial agents. In bacteria, two types of enzymes are responsible for the regulation of cellular c-di-GMP levels. Briefly, c-di-GMP monomers are synthesized by the catalytic synthesis of two GTP molecules by the action of diguanylate cyclases (DGCs) with a GGDEF domain. Furthermore, c-di-GMP is degraded by phosphodiesterases (PDEs) with EAl or HD-GYP domains, which specifically hydrolyze c-di-GMP into two molecules of GMP.6,7 An increasing number of studies have demonstrated that reducing the levels of c-di-GMP in bacterial cells by regulating the activity of these two classes of enzymes can directly affect drug resistance and biofilm formation by bacteria.8–11 Although the regulatory system based on c-di-GMP signaling molecules is extremely complex in bacteria, and there is also the possibility that several diguanylate cyclases and c-di-GMP phosphodiesterases are encoded in the genome of a bacterial cell, this leads to targeting enzymes is difficult because the inhibition or enhancement of a single enzyme is typically insufficient, it is obvious that the importance of finding new agents capable of regulating c-di-GMP level in bacteria is evident.12,13 These regulators can help explore of more details of the physiological processes, such as biofilm formation, drug resistance, affected by c-di-GMP signaling, which is a basis for implementing regulation and drug design based on this pathway.14,15
A convenient high-throughput screening method is a prerequisite for efficient screening of enzyme activity regulators. Although continuous efforts have been invested in the development of methods for detecting the activity of DGCs and PDEs in recent years, a universal method for in vitro high-throughput detection of the activity of DGCs and PDEs is not currently available.16 The direct and indirect enzymatic methods currently available for c-di-GMP quantification are summarized in Table 1, and the advantages and disadvantages of the methods as well as the types of enzymes that each method characterizes are compared. Among all currently developed detection methods, which include the use of HPLC-MS, circular dichroism biosensor, fluorescent analogs, and coupling colorimetric methods, only fluorescence resonance energy transfer (FRET) and methods based on substrate analog fluorescent probes can be used for high-throughput detection. However, these methods also exhibit a few drawbacks. In addition to the complex preparation process of FRET probes, both the aforementioned methods have highly specific detection principles; consequently, both methods are limited in their application scope and can only be used for certain types of enzymes. For example, the FRET method is only applicable to specific DGCs, and methods based on substrate analog fluorescent probes are only valid for PDEs, which limits the popularity in practical application of the aforementioned methods. Some other proposed methods have solved some problems; however, none of them meets the requirements of a method supporting high-throughput screening.
Table 1 Analytical methods available to measure enzymatic c-di-GMP synthesis and degradation
| Method |
High-throughput |
Advantage |
Disadvantage |
Enzyme analyzed |
Reference |
| DGC |
PDEa |
| All c-di-GMP phosphodiesterases listed in the table belong to the EAL-type of PDEs, unless specifically indicated in brackets. Qualitative attribution of enzymatic activity: catalytic parameters were not determined in the corresponding publication. |
| Fluorescent probe (microplate reader) |
Yes |
Simple operation low requirements on testing equipment |
Real-time monitoring is not possible |
WspR |
RocR |
Present work |
| Wide applicability |
| FRET (fluorescence resonance energy transfer) |
Yes |
Method innovation |
Need to build complex biosensors |
DgcA |
|
17,18 |
| Only applicable to specific enzymes |
| Fluorescent analogs of substrate |
Yes |
Simple operation |
Only valid for PDEs |
|
RocR |
19–21 |
| The universality of probe is not strong |
YybT |
| Substrate tag |
No |
Good accuracy |
Need to combine other testing methods and large-scale testing equipment |
|
|
22,23 |
| Circular dichroism |
No |
Real-time |
Operating instruments are expensive |
PleD |
RocR |
24 |
| Thin layer chromatography |
No |
Good sensitivity (<μM) |
Radiolabeled precursors required |
DosCb |
CC3396 |
25–27 |
| Enzyme inactivation step required before analysis |
All2874b |
PdeB |
| EnzChek™ Phosphate Assay Kit |
No |
Steady state kinetics |
Only valid for DGCs |
WspR |
|
28,29 |
| The upper monitoring limit is 5 μM min−1 |
XCC4471 |
| PleDb |
| Malachite Green |
NO |
Obvious phenomenon |
Complex pre-analysis processing |
PleDb |
|
30 |
| Be easily polluted by PI |
| Reverse-phase HPLC-MS |
No |
Classical analysis method |
Complex pre-analysis processing |
WspRb |
RavRb |
25,26,31–34 |
| PleD |
RocR |
| The precision of the experimental instrument is required |
AnapPleDb |
BlrP1 |
| HemDGC |
PA2567 |
| ThermoDGCb |
PdeR |
| |
RpfG |
| Exogenous luminescent group |
No |
Simple operation |
Only valid for PDEs |
|
MSDGC-1 |
35 |
| Chemical connection required |
In the present study, a method that is suitable for detecting both DGCs and PDEs by using our previously reported c-di-GMP-specific fluorescent probe A18 ((E)-2-(2-(1H-indol-3-yl)vinyl)-3-methylbenzo[d]thiazol-3-ium iodide) was developed.36 In addition to its universality, the proposed method is applicable to high-throughput screening of large quantities of samples by using a microplate reader. Simultaneously, a relatively simple detection procedure was established in the proposed method by using our optimized conditions. We determined the kinetics of two representative enzymes, namely WspR and RocR, by using the proposed method and found that the results were comparable with those obtained using the conventional HPLC detection method.
Results and discussion
Specific interaction between the c-di-GMP–specific fluorescent probe A18 and c-di-GMP in the enzyme reaction solution
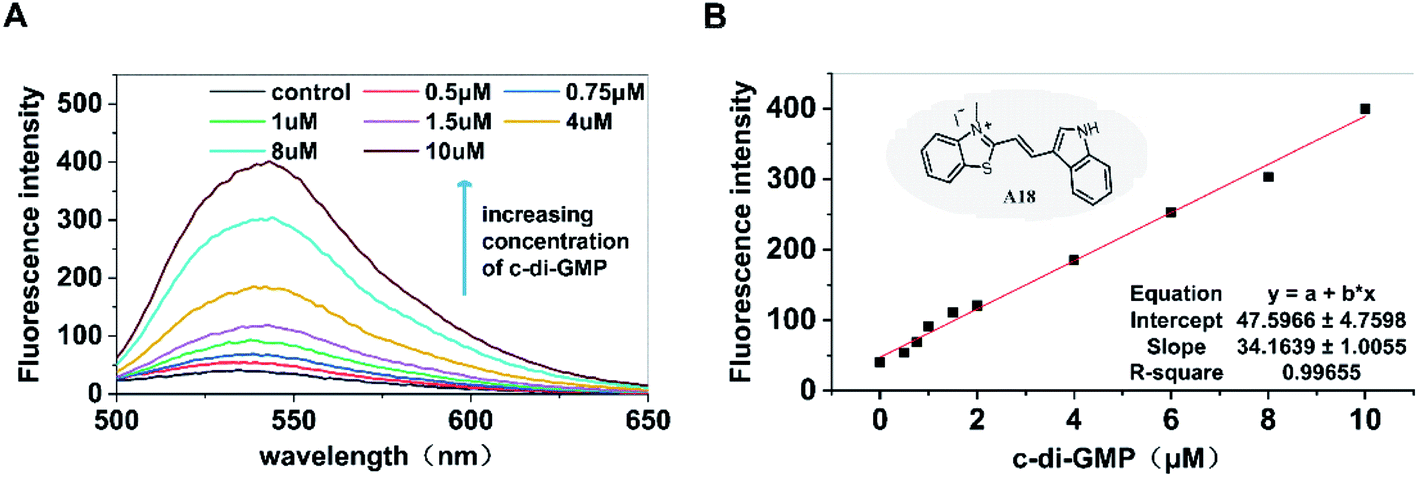
Our previous studies have provided adequate evidence indicating that the c-di-GMP–specific fluorescent probe A18 can induce c-di-GMP to form G-quadruplex and enhance the fluorescence of A18; this reaction is not exhibited by other nucleotides such as GTP and GMP.36 Based on the aforementioned phenomenon, in the present study, we investigated the possibility of development of a high-throughput screening method for determining the activity of PDEs and DGCs by determining c-di-GMP content by using the A18 probe. First, the fluorescence enhancement capability of probe A18 after interaction with c-di-GMP in incubation solutions containing PDEs and DGCs was explored. As shown in Fig. 1, after incubation for approximately 12 hours at −20 °C in the enzyme reaction solution (our previously reported detection system containing 2 mM MgCl2), in the absence of c-di-GMP, the fluorescence absorption of A18 was extremely low. However, with an increase in the concentration of c-di-GMP in the solution, the peak value (of fluorescence absorption) exhibited an obvious increasing trend. When the concentration of c-di-GMP in the solution was 10 μM, its peak value was approximately 10 times that of the pure probe. More importantly, we found that the intensity of the peak changed consistently with the concentration of c-di-GMP, and a linear relationship existed between the concentration of c-di-GMP and the intensity of the characteristic peaks (Fig. 1B). These result indicated the possibility of direct quantification of c-di-GMP present in the enzyme reaction solution by evaluating fluorescence intensity.
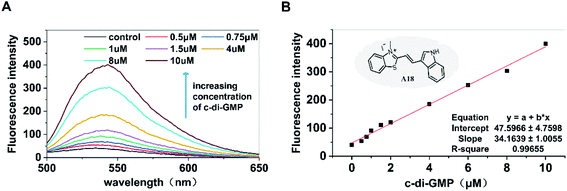 |
| | Fig. 1 (A) Dose-response curves for A18–c-di-GMP interaction. [A18] = 2.5 μM, buffer B: 10 mM Tris–HCl, 2 mM MgCl2, 1 M KCl pH 7.5. Ex = 485 nm, Em = 500–700 nm. Incubation by general method. Incubation temperature: −20 °C. Incubation time: 12 hours. (B) Relationship between the fluorescence intensity the corresponding c-di-GMP concentration. | |
Optimization of sample treatment conditions
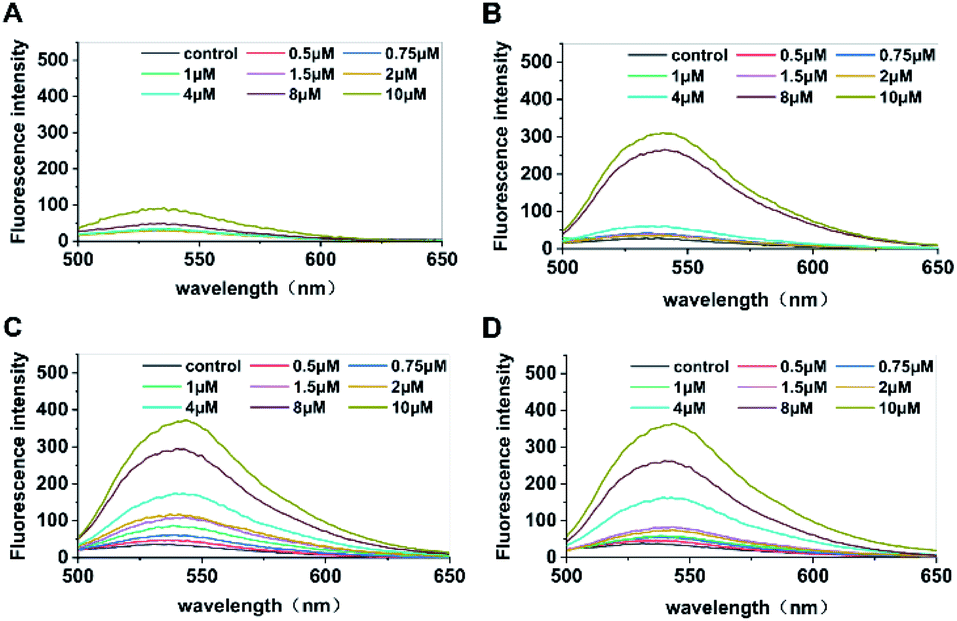
The enzyme was rapidly inactivated after the reaction, and c-di-GMP in the system was converted into its monomeric form to facilitate subsequent interaction between monomers and the fluorescent probe A18 to form G-quadruplexes. Hence, we replaced conventional water bath heating by microwave heating, which shortened the duration of preparation from the original 4 hours to less than 5 min; it also simplified the operation. Then, the optimal incubation time for the probe A18 and samples was obtained for different concentrations of standard c-di-GMP for determination of fluorescence at 1, 3, 5, and 7 hours after incubation. As shown in Fig. 2, the peaks of c-di-GMP samples increased gradually during incubation. In the first hour (Fig. 2A), fluorescence was not significantly enhanced in either high- or low-concentration samples. The peak values of high-concentration samples did not increase until 3 hours of incubation (Fig. 2B), whereas the peak value of low-concentration samples did not change considerably even at 3 hours of incubation. Until 5 hours of incubation (Fig. 2C), the peak values of samples with various concentrations did not reach stability and exhibit clear separation. We suspect that this phenomenon may have occurred in low-concentration samples because the probability of compound A18 capturing c-di-GMP monomers in low-concentration samples is much less than that in high-concentration samples; consequently, the number of effective collision between c-di-GMP and A18 will also be fewer than that in high-concentration samples. Simultaneously, we found that the intensity of the characteristic peak decreased slightly when the sample was excessively incubated (for approximately 7 hours) (Fig. 2D) compared with the peak value of incubation for 5 hours. Part of G-quadruplex with an unstable structure disintegrated with time, which may also have led to the aforementioned result. Therefore, to accurately quantify the content of c-di-GMP in the solution, the selection of sample incubation time is critical to ensure that the intensity of the characteristic peak accurately reflects the content of c-di-GMP in the solution. We separately analyzed results at 5 hours of incubation and found that not only the characteristic peak intensities of the samples of each concentration were satisfactorily separated but also that favorable results were obtained after fitting the linear relationship between the concentration and the intensity of characteristic peaks (see Fig. S2†).
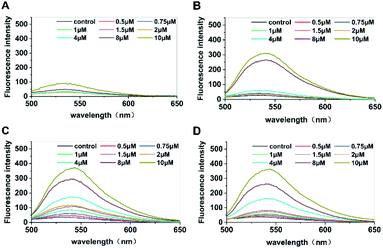 |
| | Fig. 2 Fluorescence intensity of samples containing different concentrations of c-di-GMP after incubation for (A) 1, (B) 3, (C) 5, and (D) 7 hours with the fluorescent probe A18. [A18] = 2.5 μM, buffer B: 10 mM Tris–HCl, 2 mM MgCl2, 1 M KCl pH 7.5. Ex = 485 nm, Em = 500–700 nm. The c-di-GMP was converted using microwave heating method before incubation. Incubation temperature: −20 °C. | |
Establishing a high-throughput detection method by measuring fluorescence intensity using a microplate reader
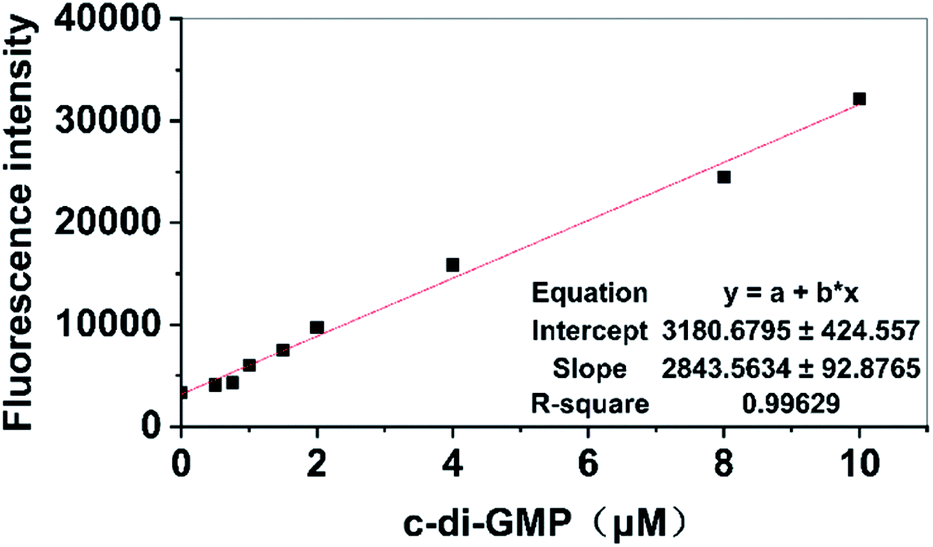
Previous studies on the development of new antibiofilm agents targeting PDEs or DGCs have demonstrated that preliminary screening is an extremely tedious task.5,37 A simpler method that can be used in high-throughput screening is urgently required. Although the principles of measuring fluorescence by using a microplate reader and fluorescence spectrophotometer are identical, microplate readers have the advantage of performing a high-throughput screening process by using a 96-well plate compared with a single measurement obtained using a fluorescence spectrophotometer; hence, we attempted to develop a high-throughput detection method by using a microplate reader for measurement. For the proposed method, we only needed to add A18 to the sample after addition of the compounds that were being screened for interaction with related DGCs or PDEs. Activity data of relevant DGCs or PDEs were indirectly obtained by calculating the content of c-di-GMP in the sample solution based on the fluorescence intensity measured using the microplate reader to determine whether the small molecular compound plays the expected role. The establishment of this method is based on the premise that the fluorescence value measured using the microplate reader can also display a linear relationship with the concentration of c-di-GMP in the sample. Therefore, different concentrations of a standard solution of c-di-GMP were used to test the concentration dependence of fluorescence intensity measured using the enzyme-labeled molecule. First, standard solutions that contained different concentrations of c-di-GMP were incubated for 5 hours with A18 after microwave heating. Then, 200 μL of each sample was drawn and added to a 96-well plate and determined using microplate reader with the emission wavelength of 540 nm and the excitation wavelength of 485 nm. Finally, the results were linearly fitted to the corresponding concentrations. As shown in Fig. 3, a satisfactory linear relationship was observed between the content of c-di-GMP in the sample solution and the fluorescence intensity. In the subsequent screening processes, we obtained the concentration of c-di-GMP in the corresponding sample solution by using the standard curve. The aforementioned characteristics of the proposed method make it a suitable high-throughput method for accurate screening of antibiofilm small molecule compounds targeting PDEs and DGCs based on fluorescence analysis by using microplate readers.
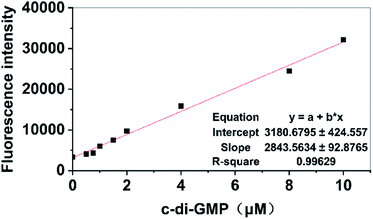 |
| | Fig. 3 Relationship between fluorescence intensity and concentration of c-di-GMP detected using a microplate reader. [A18] = 2.5 μM, buffer B: 10 mM Tris–HCl, 2 mM MgCl2, 1 M KCl pH 7.5. Ex = 485 nm, Em = 500–700 nm. Incubation by using the proposed method. Incubation temperature: −20 °C. Incubation time: 12 hours. | |
Probing enzymatic activity of DGCs and PDEs
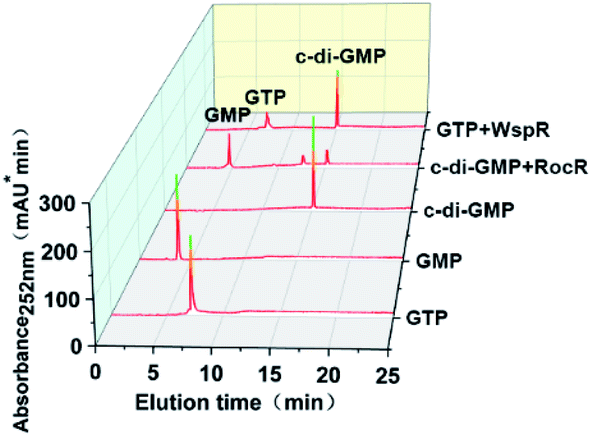
To further verify the feasibility of our proposed method, we measured the activity of two model enzymes, WspR, a well characterized DGC, and RocR, an EAL-type PDE by using this method. After expressing and purifying enzymes (ESI Fig. S1†), reversed-phase HPLC was first employed to qualitatively determine the activity of the proteins WspR and RocR. Standard solutions of c-di-GMP, GTP, and GMP were used to locate characteristic peaks to facilitate determination of WspR and RocR activity. As shown in Fig. 4, the characteristic peak of c-di-GMP was detected in the WspR incubation system, and the characteristic peak of GMP was detected in the RocR incubation system. This evidence satisfactorily indicated that the two enzymes that were expressed and purified were active. Simultaneously, we determined the peak areas of c-di-GMP standard solution with different concentrations by HPLC and plotted the standard curve according to the relationship between peak areas and concentrations (see Fig. S3†) for subsequent determination of concentrations and comparison.
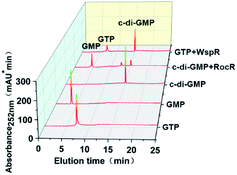 |
| | Fig. 4 HPLC spectra of pure GTP (250 μM), pure GMP (250 μM), pure c-di-GMP (100 μM), pure c-di-GMP (100 μM) with 1 μM RocR enzyme, and pure GTP (250 μM) with 1 μM WspR enzyme. The samples were allowed to react for 2 hours in reaction buffer B (10 mM Tris–HCl, 2 mM MgCl2, and 1 M KCl pH 7.5), followed by high-temperature inactivation treatment by microwave heating to stop the enzymatic reaction. The denatured protein was removed by filtration. Finally the samples were analyzed by HPLC. | |
Determining the dynamics of DGCs and PDEs
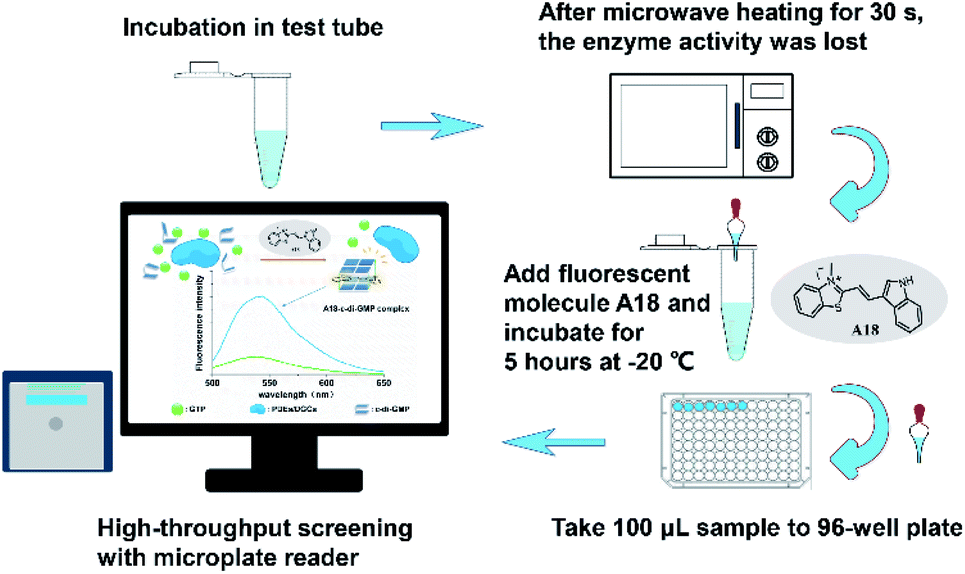
After confirming that each of the two purified model enzymes exhibited corresponding physiological activities, we used our proposed method for determining enzyme kinetics. Accordingly, samples from WspR and RocR enzyme reaction systems were drawn at different times. After microwave heating, the probe molecule A18 (at a final concentration of 2.5 μM) was added in to each sample. The samples were incubated at −20 °C for 5 hours. Subsequently, the fluorescence measurement was performed using a microplate reader (see Fig. 5).
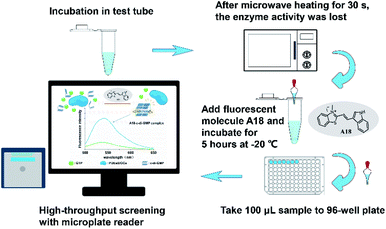 |
| | Fig. 5 Operation flow of a method for screening antibacterial agents targeting PDEs and DGCs with high throughput by using probe molecule A18. | |
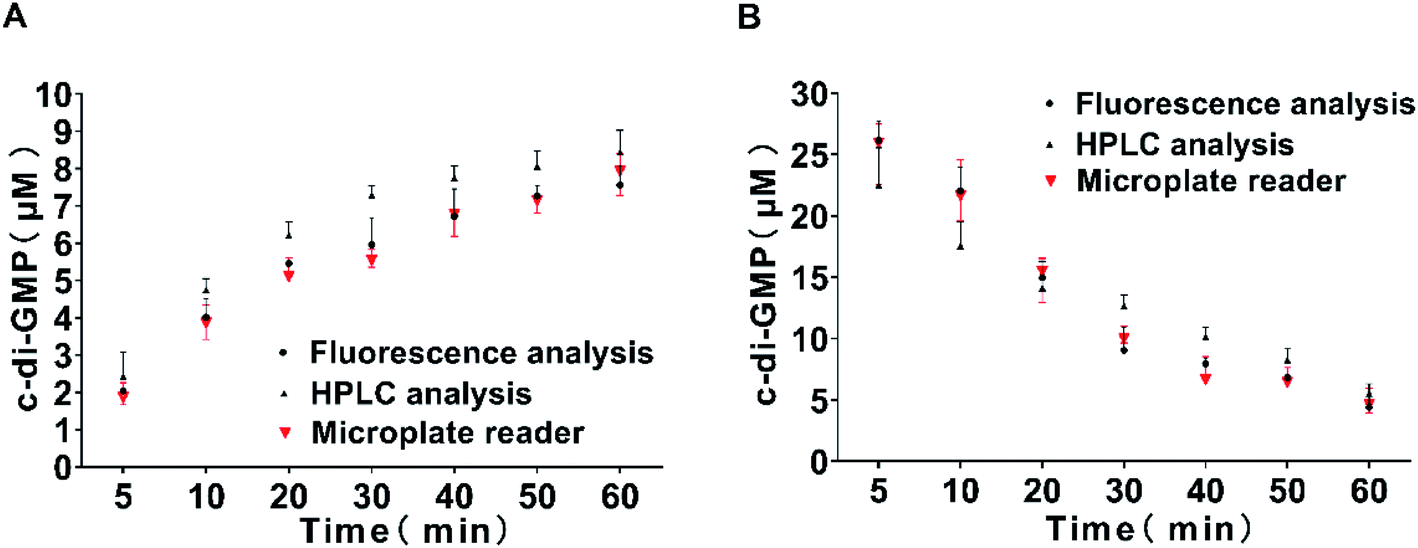
To evaluate the accuracy of the proposed method, we also took two aliquots of samples in parallel at each time point and measured through reversed-phase HPLC and fluorescence spectrophotometer respectively. Finally, quantitative analysis was performed using the standard curve previously obtained. The results are shown in Fig. 6. The measurement data obtained using the fluorescence spectrophotometer method and the microplate reader method were completely consistent, and the results obtained were comparable with those obtained through classical reversed-phase HPLC. Hence, the three methods exhibited a high degree of coincidence at high and low concentrations. These results indicate that our method of measuring the intensity of characteristic fluorescence peak by the microplate reader to quantify c-di-GMP is completely feasible and exhibited obvious advantages of simplicity and high-throughput screening. The current study not only requires an in-depth understanding of the mechanism controlling the turnover of c-di-GMP molecules but also detailed knowledge of regulation and elucidation of biochemical processes related to c-di-GMP synthesis and degradation. Simultaneously, the design of new antibiofilm small molecules targeting PDEs and DGCs has also attracted considerable scientific attention. Our proposed method is sufficiently simple ad accurate to be applied to the screening of small molecules.
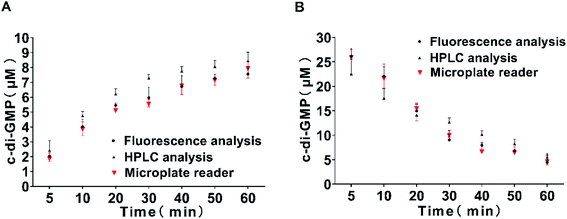 |
| | Fig. 6 Time course of the reaction catalyzed by the DGC WspR (A) and by the PDE RocR (B). Samples were drawn at 5, 10, 20, 30, 40, 50, and 60 min and the samples were subjected to reversed-phase HPLC (black circles), fluorescence spectrophotometry (black triangle), and microplate reader (red triangle). The assay was performed using a standard curve for quantification. All measurements were performed in triplicate. | |
Conclusion
In this study, a new high-throughput screening method for determining the activity of DGCs and PDEs based on fluorescence enhancement after addition of the probe molecule A18 and c-di-GMP activation was proposed. The fluorescence intensity exhibited a good linear relationship with the concentration of c-di-GMP in the solution when it is less than 20 μM (Fig. 1B and 3), and the detection limit of c-di-GMP for this assay is as low as 500 nM (Fig. 1A). By detecting the fluorescence intensity and analyzing the concentration of c-di-GMP in the solution, the catalytic activity of relevant enzymes could be indirectly obtained (Fig. 6); thus, the proposed method exhibited potential for screening of enzyme activity regulators. During the operation, after adding the tested compounds to the activated enzyme system, the content of c-di-GMP in the solution is detected at regular intervals, then whether the compound affect the enzyme can be analysed (see “Materials and methods” section). Compared with all the relevant detection methods we summarized above (Table 1), the most obvious advantage of our proposed method was that it could be used for high-throughput screening by using a microplate reader. Theoretically, only one well is required for detection of a compound molecule or a target enzyme, so the detection method can realize high-throughput detection by using a 96-well plate. Furthermore, the probe molecule directly interacted with the substrate molecule c-di-GMP; therefore, this method was not constrained by the structural specificity of any enzyme, and can theoretically be universally used to detect the activity of all c-di-GMP-regulating enzymes. But it should be reminded that in the actual application process, it needs to consider the feedback inhibition effect of the allosteric binding of the substrate c-di-GMP on the related c-di-GMP synthetases. On the other hand, this detection method will not only considerably simplify preliminary screening of potential enzyme activity regulators but will also be of high value in exploring enzymes that may be currently unknown but are crucial to the regulation of c-di-GMP. A detailed understanding of the mechanism and processes of these reactions is a prerequisite for designing and verifying effective enzyme inhibitors in the future. In short, the proposed method will play a critical strategic role in future research on c-di-GMP-related enzymes and screening of antibiofilm agents that target c-di-GMP signaling.
Materials and methods
Materials used in this study
C-di-GMP used in this study was purchased from Sigma-Innochem. GTP was purchased in Guangzhou Yike Co., Ltd. GMP was purchased from Beijing Hanlunda Technology Development Co., Ltd.
General methods
High-performance liquid chromatography (HPLC) analysis was performed on a 5C18-AR reverse phase column (nacalai tech) at room temperature on a LabFlow 4000 apparatus (LabService Analytica). Detection wavelength was 252 nm, and HPLC conditions: (A): 100 mM TEAA, (B): CH3CN, B%: 2−2%/3 min, 2–10%/15 min, 10–30%/25 min, 30–100%/30 min. 1H NMR and 13C NMR spectra were carried out using Bruker AM-400 spectrometer or Bruker AM-300 spectrometer with DMSO-d6 as the solvent and TMS as the internal standard. Electro-spray Ionization-Mass Spectrometer (ESI-MS) spectra were detected by TRACE METM spectrometer.
Synthesis and characterization of fluorescent probe A18
Synthesis of 2,3-dimethylbenzo[d]thiazol-3-ium iodide (2). 2-Methyl benzothiazole (0.76 mL, 6 mmol) was dissolved in 10 mL of acetonitrile solution. Methyl iodide (0.94 mL, 15 mmol) was added to the solution. Then the mixture was refluxed for 24 h under nitrogen atmosphere. After cooling, and the resulting precipitate was filtered, then washed with acetonitrile and diethyl ether to give a white solid 1.42 g yield 83%. 1H NMR (400 MHz, DMSO-d6) δ 8.49 (d, J = 8.1 Hz, 1H), 8.28 (d, J = 8.4 Hz, 1H), 7.85 (t, J = 7.8 Hz, 1H), 7.77 (t, J = 7.7 Hz, 1H), 4.23 (s, 3H), 3.23 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 177.45, 141.98, 129.70, 129.07, 128.45, 125.02, 117.26, 37.29, 18.40. ESI-MS (m/z):164[M − I]+.
Synthesis of (E)-2-(2-(1H-indol-3-yl)vinyl)-3-methylbenzo[d]thiazol-3-ium iodide (A18). A mixture of 2,3-dimethylbenzothiazolium iodide (150 mg, 0.52 mmol) and the indole-3-carbaldehyde (90 mg, 0.62 mmol) in methanol was refluxed for 12 h in the presence of pyridine (21 μL, 0.26 mmol). The reaction mixture was allowed to cool slowly to room temperature, and a dark solid was filtered off, washed with cold methanol and diethyl ether, then dried to afford orange solid A18, yield 74%. 1H NMR (300 MHz, DMSO-d6) δ 12.50 (s, 1H), 8.51–8.38 (m, 2H), 8.33 (d, J = 7.7 Hz, 1H), 8.29–8.23 (m, 1H), 8.13 (d, J = 8.3 Hz, 1H), 7.79 (t, J = 7.3 Hz, 1H), 7.69 (t, J = 7.5 Hz, 1H), 7.61–7.54 (m, 1H), 7.50 (d, J = 15.4 Hz, 1H), 7.38–7.30 (m, 2H), 4.27 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 172.30, 144.64, 142.31, 138.31, 138.00, 129.27, 127.79, 127.03, 125.19, 124.36, 124.26, 122.89, 121.43, 116.34, 114.69, 113.55, 106.14, 36.16. ESI-MS (m/z):291.1[M − I]+.
Fluorescence experiment
Fluorescence experiments were performed on PerkinElmer-LS 55 Fluorescence Spectrometer with 1 cm path length cuvette. Absorbance spectra were obtained on a ND2000C spectrophotometer with 1 cm path length cuvette. The Fluorescence instrument settings were chosen as follows: λex = 485 nm (slit 6 nm), λem = 500–700 nm (slit 6 nm). The measurements were carried out at 15 °C.
Microplate reader experiment
Microplate reader experiment were performed on Synergy H1 Hybrid Multi-Mode Reader. The parameters for fluorescence detection were set as follows: λex = 485 nm, λem = 540 nm. The measurements were carried out at room temperature.
Expression and purification of WspR (37.9 kDa) and RocR (42.8 kDa) enzymes
The expression vector we used was synthesized by Shanghai Jierui Bio-Engineering Co., Ltd. the cloning vector used was PET28a, carrying 6X-cleavable His tag at its N-terminal, and finally introduced into E. coli cells BL21 (DE3) competent cells. The expression constructs was cultured in LB medium containing kanamycin (30 μg mL−1) in a shaking table at 37 °C until OD600 of 1.0. IPTG (0.3 mM) was added and cells were grown at 37 °C overnight. Cells were collected by centrifugation, resuspended in NiNTA buffer A (25 mM Tris–HCl [pH 8.2], 500 mM NaCl, 20 mM imidazole, and 5 mM 2-mercaptoethanol). Then lysozyme (1 mg mL−1) and protease inhibitor (0.5 μg mL−1) were added, incubate on ice for 20 min. Cells were lysed using a SCIENTZ-II D sonicator (SCIENTZ), the power was 200 W, the working time was 5 s, the interval was 10 s, and the total working time was 30 min. Then cell fragments were precipitated by 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g centrifugation, finally the clarified lysate was filtered through a filter membrane with a pore diameter of 0.45 μM, and finally the sample was loaded on a His Trap NiNTA column which has been balanced with NiNTA buffer A. The resin was then washed with NiNTA buffer A of 20 times the column volume, and proteins were eluted on a gradient from 20 to 500 mM imidazole in NiNTA buffer A over 20 column volumes. Finally, the obtained protein was desalted, concentrated to 4 mg mL−1, glycerol was added to 10% (v/v), aliquots were flash frozen in liquid nitrogen and stored at −80 °C. The purity were verified by SDS-polyacrylamide gel electrophoresis (SDS-PAGE, Fig. S1†).
000×g centrifugation, finally the clarified lysate was filtered through a filter membrane with a pore diameter of 0.45 μM, and finally the sample was loaded on a His Trap NiNTA column which has been balanced with NiNTA buffer A. The resin was then washed with NiNTA buffer A of 20 times the column volume, and proteins were eluted on a gradient from 20 to 500 mM imidazole in NiNTA buffer A over 20 column volumes. Finally, the obtained protein was desalted, concentrated to 4 mg mL−1, glycerol was added to 10% (v/v), aliquots were flash frozen in liquid nitrogen and stored at −80 °C. The purity were verified by SDS-polyacrylamide gel electrophoresis (SDS-PAGE, Fig. S1†).
Kinetic studies
To study the diguanylate cyclase activity of Pseudomonas aeruginosa WspR, 1 μM WspR enzyme solution were equilibrated in reaction buffer B (10 mM Tris–HCl, 2 mM MgCl2, 1 M KCl pH 7.5) overnight before activation with 0.1 mM BeCl2 and 10 mM NaF for 5 min at room temperature. Each reaction was run through a PD-10 desalting column to remove extra BeF3−, BeCl2, and NaF from the treated WspR protein in order to prevent it from inhibiting the interaction of compound A18 with c-di-GMP. After that, the diguanylate cyclase reaction was performed at 25 °C in the reaction buffer and was started by adding 100 μM GTP (antibacterial compounds also can be added at the same time). Next, the sample is processed by an improved method, and analyzed by fluorescence spectrum and microplate reader. In parallel, the reaction was followed under the same experimental conditions, the sample was boiled for 10 minutes to stop the enzymatic reaction, centrifuged and filtered to remove denatured precipitated proteins, and finally analyzed by reverse phase HPLC. Aliquots were taken out at different reaction times (5, 10, 20, 30, 40, 50 and 60 min), respectively, and analyzed by the above three different methods. All experiments were done in triplicate.
To study the phosphodiesterase activity of Pseudomonas aeruginosa RocR, 1 μM RocR enzyme solution were equilibrated in reaction buffer B (10 mM Tris–HCl, 2 mM MgCl2, 1 M KCl pH 7.5) overnight at room temperature. After that, the phosphodiesterase reaction was performed at 25 °C in the reaction buffer and was started by adding 30 μM c-di-GMP (antibacterial compounds also can be added at the same time). Next, the sample is processed by an improved method, and analyzed by fluorescence spectrum and microplate reader. In parallel, the reaction was followed under the same experimental conditions, the sample was boiled for 10 minutes to stop the enzymatic reaction, centrifuged and filtered to remove denatured precipitated proteins, and finally analyzed by reverse phase HPLC. Aliquots were taken out at different reaction times (5, 10, 20, 30, 40, 50 and 60 min), respectively, and analyzed by the above three different methods. All experiments were done in triplicate.
General preparation of sample before measurements assay
The c-di-GMP was added to the buffer B (10 mM Tris–HCl, 2 mM MgCl2, 1 M KCl pH 7.5) for mixing, heated in a 95 °C water bath for 5 minutes, then stop heating, allowed the sample to cool slowly to room temperature with the water, and kept it at room temperature for 10 min. A18 (2.5 μΜ) was then added to the mixture and incubated in the refrigerator at −20 °C overnight (about 12 h).
Improved preparation method before sample measurement
The c-di-GMP was added to the buffer B (10 mM Tris−Cl, 2 mM MgCl2, 1 M KCl pH 7.5) for mixing, put it in a microwave oven, heat it for about 30 seconds, and then slowly cooled back to room temperature. A18 (2.5 μΜ) was then added to the mixture and incubated in the refrigerator at −20 °C (about 5 h).
Conflicts of interest
The authors declare that there is no conflict of interest with any one about this manuscript.
Acknowledgements
This work was supported by the Pearl River S&T Nova Program of Guangzhou [201806010116]; and the Natural Science Foundation of Guangdong Province (2019A1515011801).
References
- U. Jenal, A. Reinders and C. Lori, Nat. Rev. Microbiol., 2017, 15, 271–284 CrossRef CAS PubMed.
- U. Jenal and J. Malone, Annu. Rev. Genet., 2006, 40, 385–407 CrossRef CAS PubMed.
- Z. X. Liang, Nat. Prod. Rep., 2015, 32, 663–683 RSC.
- M. Valentine and A. Filloux, Annu. Rev. Microbiol., 2019, 73, 387–406 CrossRef PubMed.
- G. Xu, S. Han, C. Huo, K. H. Chin, S. H. Chou, M. Gomelsky, G. Qian and F. Liu, Nucleic Acids Res., 2018, 46, 9276–9288 CrossRef CAS PubMed.
- M. Gentner, M. G. Allan, F. Zaehringer, T. Schirmer and S. Grzesiek, J. Am. Chem. Soc., 2012, 134, 1019–1029 CrossRef CAS PubMed.
- U. Romling, M. Y. Galperin and M. Gomelsky, Microbiol. Mol. Biol. Rev., 2013, 77, 1–52 CrossRef PubMed.
- C. A. Fux, J. W. Costerton, P. S. Stewart and P. Stoodley, Trends Microbiol., 2005, 13, 34–40 CrossRef CAS PubMed.
- N. Hoiby, T. Bjarnsholt, M. Givskov, S. Molin and O. Ciofu, Int. J. Antimicrob. Agents, 2010, 35, 322–332 CrossRef PubMed.
- R. Hengge, Philos. Trans. R. Soc., A, 2016, 371, 1707 Search PubMed.
- Q. Wei, S. Leclercq, P. Bhasme, A. M. Xu, B. Zhu, Y. H. Zhang, M. K. Zhang, S. W. Wang and L. Z. Ma, Appl. Environ. Microbiol., 2019, 85 DOI:10.1128/aem.01194-19.
- J. S. Madsen, O. Hylling, S. Jacquiod, S. Pécastaings, L. H. Hansen, L. Riber, G. Vestergaard and S. J. Sørensen, ISME J., 2018, 12, 2330–2334 CrossRef CAS PubMed.
- D. Cohen, U. Mechold, H. Nevenzal, Y. Yarmiyhu, T. E. Randall, D. C. Bay, J. D. Rich, M. R. Parsek, V. Kaever, J. J. Harrison and E. Banin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11359–11364 CrossRef CAS PubMed.
- M. W. Orr, G. P. Donaldson, G. B. Severin, J. X. Wang, H. O. Sintim, C. M. Waters and V. T. Lee, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, e5048–e5057 CrossRef CAS PubMed.
- C. Opoku-Temeng and H. O. Sintim, Sci. Rep., 2016, 6, 25445 CrossRef CAS PubMed.
- C. Opoku-Temeng, J. Zhou, Y. Zheng, J. Su and H. O. Sintim, Chem. Commun., 2016, 52, 9327–9342 RSC.
- M. Christen, C. Kamischke, H. D. Kulasekara, K. C. Olivas, B. R. Kulasekara, B. Christen, T. Kline and S. I. Miller, ChemBioChem, 2019, 20, 394–407 CrossRef CAS PubMed.
- M. Christen, H. D. Kulasekara, B. Christen, B. R. Kulasekara, L. R. Hoffman and S. I. Miller, Science, 2010, 328, 1295–1297 CrossRef CAS PubMed.
- J. Zhou, Y. Zheng, B. T. Roembke, S. M. Robinson, C. Opoku-Temeng, D. A. Sayre and H. O. Sintim, RSC Adv., 2017, 7, 5421–5426 RSC.
- S. Fernicola, I. Torquati, A. Paiardini, G. Giardina, G. Rampioni, M. Messina, L. Leoni, B. F. Del, R. Petrelli, S. Rinaldo, L. Cappellacci and F. Cutruzzolà, J. Med. Chem., 2015, 58, 8269–8284 CrossRef CAS PubMed.
- J. Zhou, D. A. Sayre, Y. Zheng, H. Szmacinski and H. O. Sintim, Anal. Chem., 2014, 86, 2412–2420 CrossRef CAS PubMed.
- B. I. Kazmierczak, Methods Mol. Biol., 2017, 1657, 279–283 CrossRef CAS PubMed.
- B. I. Kazmierczak, J. Mol. Biol., 2017, 1657, 23–29 CAS.
- V. Stelitano, A. Brandt, S. Fernicola, S. Franceschini, G. Giardina, A. Pica, S. Rinaldo, F. Sica and F. Cutruzzola, Nucleic Acids Res., 2013, 41, e79 CrossRef CAS PubMed.
- R. Paul, S. Weiser, N. C. Amiot, C. Chan, T. Schirmer, B. Giese and U. Jenal, Genes Dev., 2004, 18, 715–727 CrossRef CAS PubMed.
- R. Paul, S. Abel, P. Wassmann, A. Beck, H. Heerklotz and U. Jenal, J. Biol. Chem., 2007, 282, 29170–29177 CrossRef CAS PubMed.
- B. Christen, M. Christen, R. Paul, F. Schmid, M. Folcher, P. Jenoe, M. Meuwly and U. Jenal, J. Biol. Chem., 2006, 281, 32015–32024 CrossRef CAS PubMed.
- C. Y. Yang, K. H. Chin, M. L. C. Chuah, Z. X. Liang, A. H. J. Wang and S. H. Chou, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 997–1008 CrossRef CAS PubMed.
- N. De, M. Pirruccello, P. V. Krasteva, N. Bae, R. V. Raghavan and H. Sondermann, PLoS Biol., 2008, 6, e67 CrossRef PubMed.
- C. Chan, R. Paul, D. Samoray, N. Amiot, B. Giese, U. Jenal and T. Schirmer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17084–17089 CrossRef CAS PubMed.
- F. Yang, F. Tian, L. Sun, H. Chen, M. Wu, C. H. Yang and C. He, Mol. Plant-Microbe Interact., 2012, 25, 1361–1369 CrossRef CAS PubMed.
- Y. W. He, C. Boon, L. Zhou and L. H. Zhang, Mol. Microbiol., 2009, 71, 1464–1476 CrossRef CAS PubMed.
- F. Rao, S. Pasunooti, Y. L. Ng, W. C. Zhuo, L. Lim, A. W. X. Liu and Z. X. Liang, Anal. Biochem., 2009, 389, 138–142 CrossRef CAS PubMed.
- B. J. Laventie, J. Nesper, E. Ahrné, T. Glatter, A. Schmidt and U. Jenal, J. Visualized Exp., 2015, 97, e51404 Search PubMed.
- I. M. Sharma, T. Dhanaraman, R. Mathew and D. Chatterji, Biochemistry, 2012, 51, 5443–5453 CrossRef CAS PubMed.
- T. F. Xuan, J. Liu, Z. Q. Wang, W. M. Chen and J. Lin, Frontiers in Microbiology, 2020, 10, 3163 CrossRef PubMed.
- M. Valentini and A. Filloux, Annu. Rev. Microbiol., 2019, 73, 387–406 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02540b |
| ‡ Z. Q. W. and T. F. X. equally contributed to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Jing Lin
* and
Jing Lin