
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational understanding of catalyst-controlled borylation of fluoroarenes: directed vs. undirected pathway†
Yu-hua Liu a and
Zhong-Jie Jiang*b
a and
Zhong-Jie Jiang*b
aSchool of Physics and Electronic Engineering, Guangzhou University, Guangzhou, 510006, China
bGuangzhou Key Laboratory for Surface Chemistry of Energy Materials, New Energy Research Institute, College of Environment and Energy, South China University of Technology, Guangzhou 510006, Guangdong, China. E-mail: eszjiang@scut.edu.cn
First published on 21st May 2020
Abstract
In this work, density functional theory (DFT) calculations are performed to understand the origin of the regioselective C–H borylation of aromatics catalyzed by Co(I)/iPrPNP and Ir(III)/dtbpy (4,4-di-tert-butyl bipyridine). The calculation results indicate that for the Co(I)/iPrPNP catalytic system, the undirected pathway is 2.9 kcal mol−1 more favoured over the directed pathway leading to ortho-to-fluorine selectivity. In contrast, for the Ir(III)/dtbpy catalytic system, the directed pathway is 1.2 kcal mol−1 more favoured over the undirected pathway bringing about ortho-to-silyl selectivity. For Co(I)/iPrPNP catalyzed borylation, the undirected pathway which involves steps of ortho-to-fluorine C–H oxidative addition, C–B reductive elimination, B–B oxidative addition, and B–H reductive elimination is favorable due to the electron deficient character of the ortho-to-fluorine C–H bond. For Ir(III)/dtbpy catalyzed borylation, the directed pathway consisting of Si–H oxidative addition, B–H reductive elimination, C–H oxidative addition, B–B oxidative addition, C–B reductive elimination, Si–H reductive elimination is favored over the undirected pathway attributed to the directing effect of the hydrosilyl group. The favourable undirected pathway (ortho-to-fluorine selectivity) for Co(I)/iPrPNP catalyzed borylation and the favourable directed pathway (ortho-to-silyl selectivity) for Ir(III)/dtbpy catalyzed borylation could explain well the experimentally observed ortho-to-fluorine borylation of hydrosilyl substituted fluoroarenes with cobalt catalyst (J. V. Obligacion, M. J. Bezdek and P. J. Chirik, J. Am. Chem. Soc., 2017, 139, 2825–2832) and ortho-to-silyl selectivity with iridium catalyst (T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534–7535).
Introduction
Transition metal catalyzed C–H borylation of aromatics has attracted considerable attention as it offers an alternative method to standard organic synthesis.1 However, different C–H bonds in reactants can result in different functionalization, leading to the formation of the resulting products with substantially different properties.2 Therefore, controlling the regioselectivity of the C–H borylation reaction is of great importance to obtain the desired synthetics. Generally, strategies for the regioselective C–H borylation of aromatics include the undirected reaction pathway, in which no directing groups are involved in the reaction,3 and directing group controlled regioselective borylation,3f–j where the reaction regioselectivity is well controlled by a directing group.Hartwig reported that the ortho-to-fluorine selectivity was controlled by steric hindrance in iridium/dtbpy catalyzed borylation of trisubstituted fluoroarenes4 (Scheme 1a) in which the product of borylated fluoroarene is especially essential for pharmaceutical chemistry.5 In contrast, Chatani reported platinum-NHC catalyzed C–H borylation of fluoroarenes, which afforded ortho-to-fluorine arylboronates with no steric protection.6 Similarly, Iwasawa reported the ortho-to-fluorine effect in platinum/PSiN catalyzed C–H borylation of arenes7 (Scheme 1b). Different from the steric controlled borylation by installing large group on the substrate or catalyst, introducing directing group bearing heteroatoms (e.g. N, O, S, Si) on the substrate is an effective way to achieve meta/para-to-fluorine C–H borylation.8–10 The interactions between directing groups and catalyst could bring one of the ortho-C–H bonds close to the metal center and promote its regioselective activation.9a,b For example, Iwasawa reported the iridium-picolylamine catalyzed ortho-to-amine borylation of fluoroarenes using dimethylamine as the directing group8 (Scheme 1c). Hartwig et al. reported the hydrosilyl directed borylation of fluoroarenes catalyzed by iridium/dtbpy favoring ortho-to-silyl selectivity10 (Scheme 2). In contrast, Obligacion et al. reported the same reaction catalyzed by cobalt/iPrPNP with ortho-to-fluorine selectivity11 (Scheme 2).
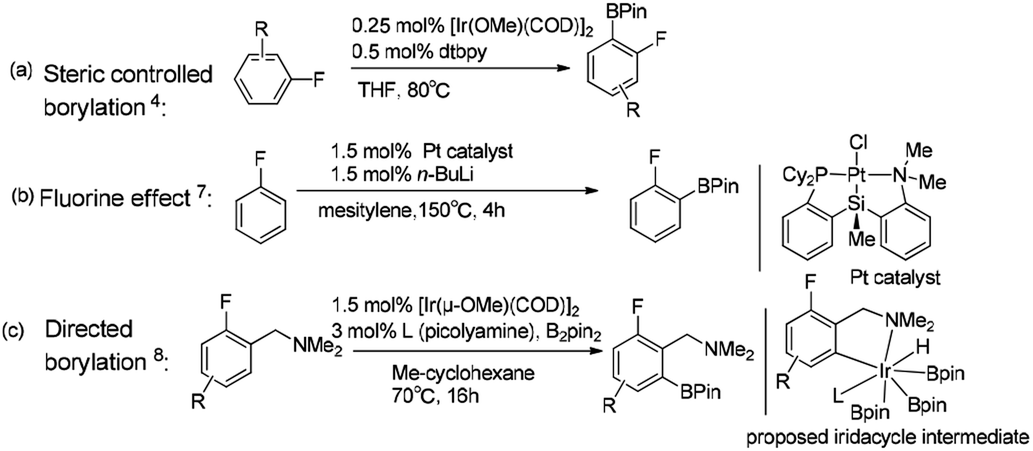 | ||
| Scheme 1 Complementary selectivity in transition metal catalyzed C(sp2)–H borylation of fluoroarenes.5,7,8 | ||
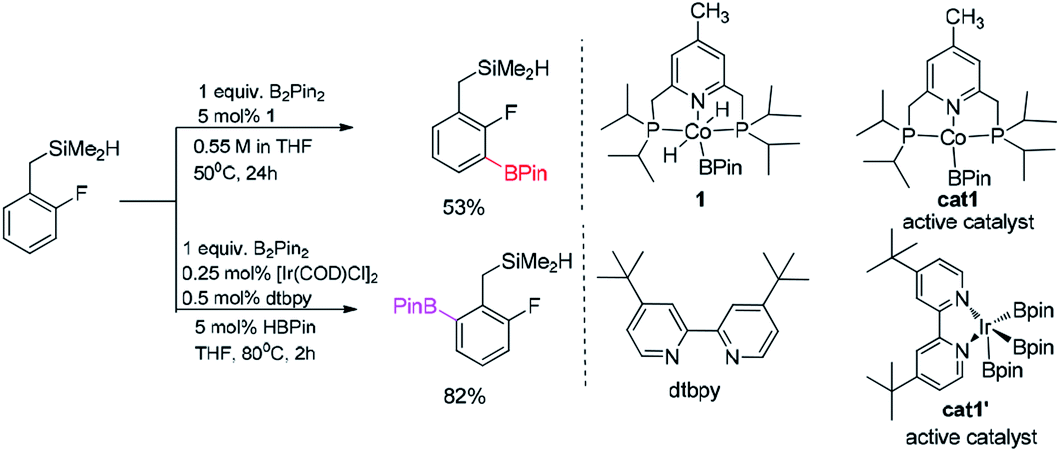 | ||
| Scheme 2 Computational model for borylation of hydrosilyl substituted fluoroarenes.11 | ||
The examples mentioned above clearly demonstrate that the C–H borylation of aromatics can proceed by the undirected or directed pathways with the iridium or cobalt-based catalysts.12,13 Recently, Hall's group had reported the cobalt pincer complex catalyzed regioselective borylation of aromatics13b (without directing group) while Sunoj's group reported the use of hydrosilyl (–SiR2H) as a directing group in the iridium-catalyzed regioselective borylation of the benzylic C–H bond.9a Although there have been some reports on the Si–Co and Si–Ir metal complex,3l,9a,12d and the directing effect of the silyl group has been identified in iridium catalyzed borylation of benzylic C–H bond,9a detailed mechanism study on the regioselectivity of the borylation reactions in Scheme 2 has not been reported. In the iridium and cobalt catalyzed borylation of hydrosilyl substituted fluoroarenes (Scheme 2), how about directing effect of hydrosilyl group for both the cobalt and the iridium catalyzed borylation? Is the directing effect always favourable? Based on these considerations, we studied the detailed mechanism of borylation of hydrosilyl substituted fluoroarenes catalyzed by iridium and cobalt by density functional theory (DFT) method. Specifically, our attention mainly focuses on the questions raised by experimental observations: (1) what are the details of the reaction pathways? (2) which step is the rate-determining step?
Results and discussion
We choose the reaction between hydrosilyl substituted fluoroarenes and B2pin2 as the model (Scheme 2). Activations of both ortho-to-fluorine C–H bond and ortho-to-silyl C–H bond by the cobalt catalyst 1 and Ir(III)/dtbpy are calculated to make comparison and explain the regiochemical preference (Scheme 2). Since the work by Patel et al.12a and Obligacion et al.13 have suggested that the most likely active catalysts for Co(I) and Ir(III) catalyzed borylation reactions are cat1 and cat1′, respectively, we discuss all the reaction pathways in the following sections with cat1 and cat1′ as the starting points, given that a cascade of steps along the reaction pathways is triggered by the active catalysts.According to the probable mechanisms of Ir(III) and Co(I) catalyzed borylation of aromatics proposed in literature,12a,13 the directing group controlled borylation catalyzed by iridium may proceed in a way that the iridium centre is brought close to the ortho-to-silyl C–H bond by the silyl group and induces the activation of ortho-to-silyl C–H bond to form a 5-membered cycle.12a In contrast, cobalt-catalyzed borylation starts with ortho-to-fluorine C–H oxidative addition13 and no participation of directing group. The adaption of reported mechanisms to our peculiar reaction system is shown in Fig. 1. The proposed pathways consist of the directed and undirected pathway. For undirected pathway (Fig. 1a), the cobalt catalyst 1 triggers a cascade of significant steps which involves: (i) B–B oxidation addition, (ii) H–B reductive elimination, (iii) C–H oxidation addition, and (iv) C–B reductive elimination. Alternatively, the undirected pathway may refer to the activation of the benzylic C–H bond which involves (Fig. 1b): (i) C–H oxidation addition, (ii) H–B reductive elimination, (iii) C–B reductive elimination, and (iv) B–B oxidation addition. While, directed pathway catalyzed by iridium is divided into six distinct steps (Fig. 1c): (i) Si–H oxidation addition to Ir center; (ii) H–B reductive elimination; (iii) C–H bond oxidation addition & H–B reductive elimination; (iv) B–B oxidation addition; (v) C–B bond formation; (v) H–B addition; (vi) Si–H elimination. There is also a possibility that both the directed and undirected pathways co-exist for the cobalt/iridium-catalyzed C–H activation process. Significant differences exist between the three catalytic cycles. The preference for which pathway depends on the nature of the catalyst such as the numbers of open coordination sites on the transition metal centre of catalyst and electronic effects of the catalyst. In the following section, we use the DFT method to calculate both directed and undirected C–H activation pathways to identify the energetically most favored pathway, with the aim of revealing the details of the mechanism.
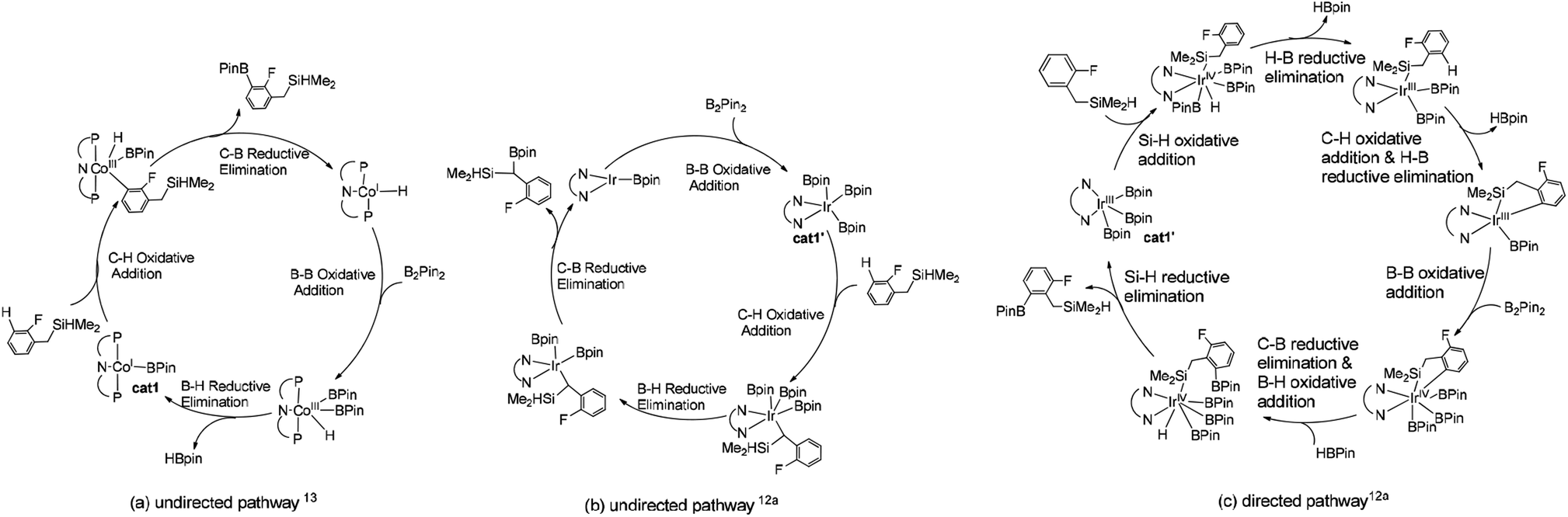 | ||
| Fig. 1 Potential mechanisms of (a) directed pathway for borylation of aromatic C–H bond13 and (b) undirected pathway for borylation of Si–H bond12a (c) directed pathway for borylation of benzylic C–H bond.12a | ||
Co(I)/iPrPNP catalyzed undirected and directed pathway
We have examined the geometric and energy features of each elementary step involved in the Co(I)/iPrPNP catalyzed directed and undirected pathways. The optimized structures of selected transition states (TS) and intermediates are given in Fig. 2, while the energy profiles are presented in Fig. 3. All the TS structures and Co(I) species studied show the tridentate coordination of the pincer ligand. The Co–N distance of cobalt pincer-complex cat1 (Fig. 2) is rather short, 2.00 Å.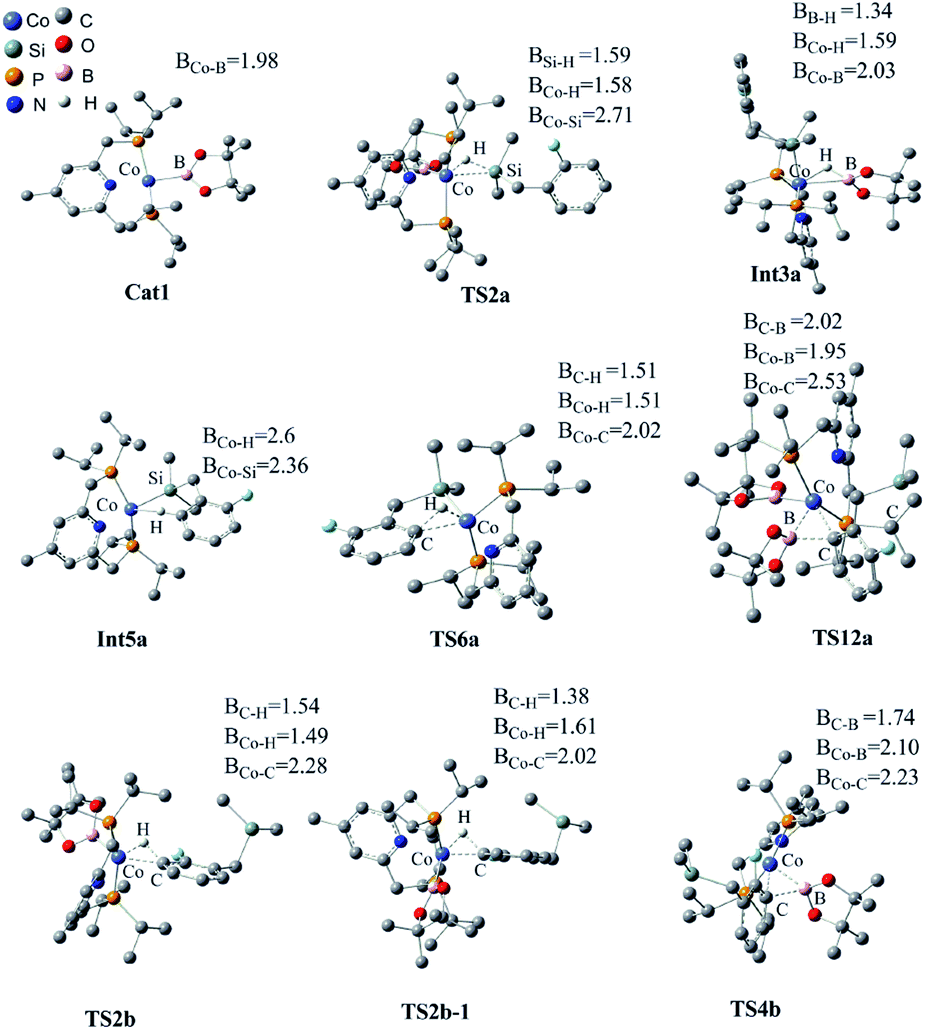 | ||
| Fig. 2 Optimized structures of the key transition states and intermediates in Co(I)/iPrPNP catalyzed borylation of silyl substituted fluoroarenes, along with the key bond distances in angstroms. Color code, C: dark gray, O: red, B: pink, H: light gray, N: dark blue, Co: light blue, Si: celeste, P: yellow. Irrelevant hydrogen atoms are omitted for clarity. | ||
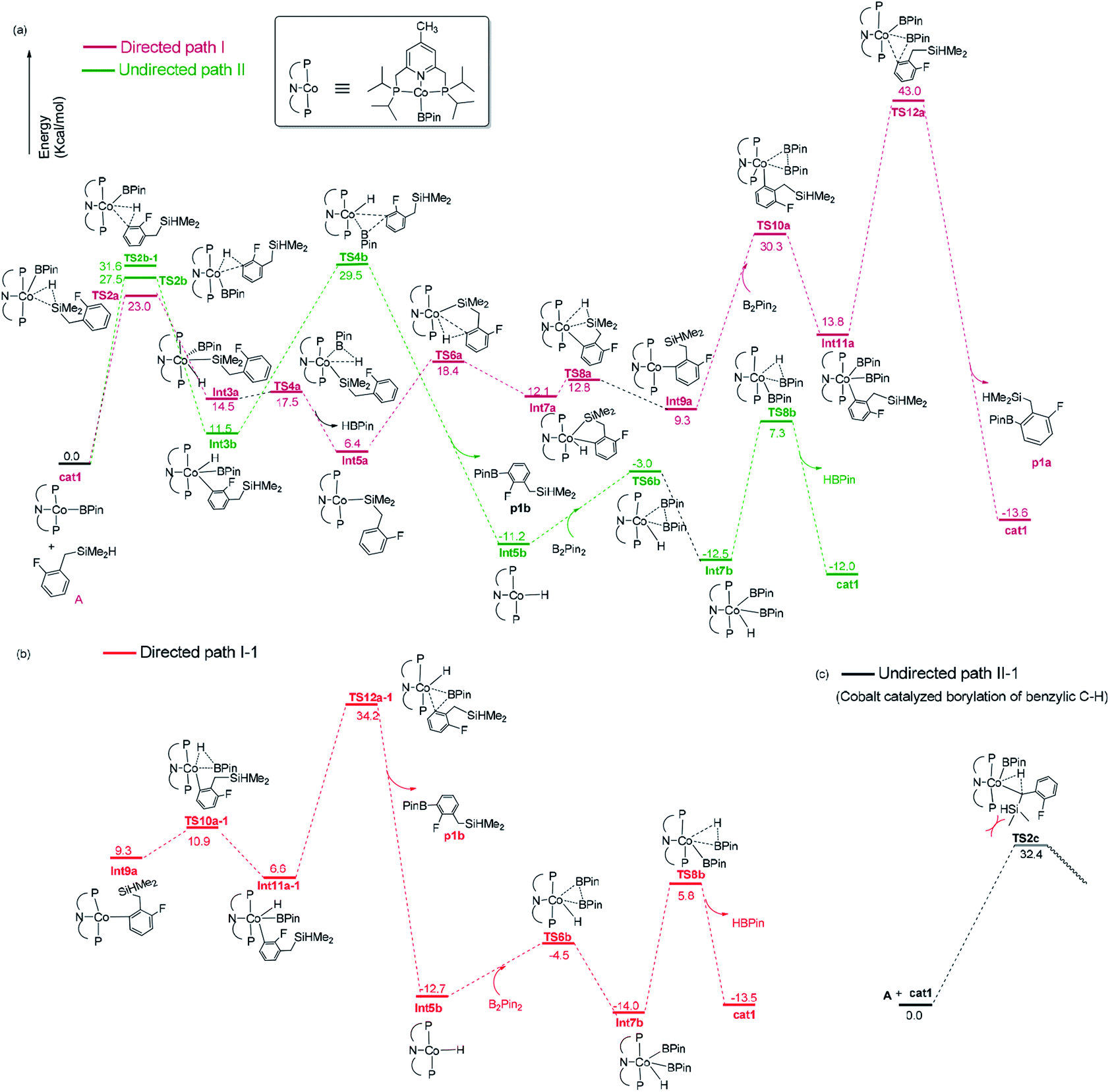 | ||
| Fig. 3 Gibbs free energy profile of directed path I and path I-1, undirected path II and path II-1 in the borylation of hydrosilyl substituted fluoroarenes catalyzed by Co(I)/iPrPNP. | ||
The directed pathway starts from silylation (Fig. 3a, dull red). The hydrogen of hydrosilyl group approaches cobalt centre from the position adjacent to boryl group affording Int3a via TS2a with a barrier of 23.0 kcal mol−1 (Fig. 3a). The comparison of geometry between TS2a and cat1 indicates that Si–H bond approaches to the axial position of the metal center, pushes the equatorial boryl ligands to the axial position (Fig. 2).14 The distance between the incoming hydride and the boron atom Int3a is 1.34 Å (Fig. 2) indicating a weak orbital interaction between the vacant p-orbitals of the boron and the hydride.15,16 It is expected that Int3a first converts to Int5a to release vacant coordination sites via expulsion of a molecule of HBpin. The required energy for the removal of a molecular of HBpin from Int3a is only 3.0 kcal mol−1. The ortho-to-silyl aromatic C–H bond in Int5a is brought close to the cobalt center by silyl group, such that C–H could oxidatively add to cobalt center affording Int7a via a cyclic transition state TS6a (Fig. 2). At first glance, there is a possibility that B2pin2 oxidatively adds to the cobalt center of Int7a. However, attempts to find a corresponding TS is failed, presumably resulting from the unavailable coordination sites on the cobalt center. Subsequently, we investigate the possibility of Si–H bond reductive elimination from Int7a. The reductive elimination of Si–H bond from Int7a affords Int9a via TS8a. Given that there are partially open coordination sites at the cobalt center in Int9a, B2pin2 could add to the cobalt center to form Int11a. One of the boryl ligand in Int11a approaches the adjacent aromatic carbon to form C–B bond via TS12a (Fig. 2). Upon the C–B bond reductive elimination, the product p1a is afforded, and active catalyst cat1 is regenerated. Along with the energetic profile of the directed pathway (Fig. 3, dull red), the overall barrier for the C–B reductive elimination via TS12a is the largest (43.0 kcal mol−1), and can be considered as the rate determining step. Starting from int9a, in place of B2Pin2 oxidative addition, HBpin is also possible to add to the cobalt center following with the C–B reductive elimination and regeneration of cat1. The corresponding energetic profile is shown in Fig. 3, which shows an overall barrier of 34.2 kcal mol−1 (TS12a-1 in Fig. 3b). It is noteworthy that in this path the steps of the catalyst regeneration is the same as that in undirected path II (Fig. 3a, Int5b → cat1) and the steps before Int9a are the same as those in the directed path I (Fig. 3a, cat1 → Int9a).
Different from the directed pathway, the undirected pathway starts with the ortho-to-fluorine C–H activation. In this step, the ortho-to-fluorine hydrogen may approach cobalt center from the position adjacent to boryl group or from the location opposite to the boryl group to afford TS2b-1 and TS2b which requires an energy of 31.6 kcal mol−1 and 27.5 kcal mol−1, respectively. For the former approach, the carbon and boryl group is in an opposite position which is impossible for the subsequent C–B reductive elimination to afford the borylated product. So, we do not calculate the pathway along with the former. The intermediate Int3b is obtained via TS2b. The incoming hydride and boryl group of Int3b are situated in the equatorial position (Fig. 2). Then, the aromatic C–B is reductively eliminated from Int3b via TS4b, affording the product p1b. The overall barrier for the C–B reductive elimination is the largest (29.5 kcal mol−1) and is the rate-determining step (Fig. 3a). The catalyst cat1 is regenerated upon the following B–B oxidative addition and B–H reductive elimination, which require an energy of 8.2 kcal mol−1 and 19.8 kcal mol−1, respectively. In addition, we have calculated the pathway for Co(I)-catalyzed benzylic C–H borylation (Fig. 3c). As a result of the steric hindrance between the ligand and the silyl group, the barrier for the benzylic C–H activation (32.4 kcal mol−1) is too high to occur under the experimental condition (50 °C, 24 h). This calculation result is different from the hydrosilyl directed borylation of ortho-hydrosilyl benzene in which the borylation of the benzylic C–H is favorable over the aromatic C–H bond activation.9a
Obviously, the overall barrier in the undirect pathway (29.5 kcal mol−1, TS4b in Fig. 3a) for Co(I)/iPrPNP system is much lower than that of the direct pathway (43.0 kcal mol−1 barrier for TS12a in Fig. 3a or 34.2 kcal mol−1 barrier for TS12a-1 in Fig. 3b) We believe that the lower overall barrier of the undirected pathway than directed pathway can be ascribed to the electron-withdrawing of the fluorine atom ortho to the activated C–H bond, because the electron deficiency property of the transition state accelerates the C–B reductive elimination (18.0 kcal mol−1 of the barrier for TS4b vs. 29.2 kcal mol−1 of that for TS4a, Fig. 3a). Such a high barrier difference between the directed and undirected pathway (29.5 kcal mol−1 overall barrier for TS4b in the undirected pathway vs. 43.0 kcal mol−1 barrier for TS12a or 34.2 kcal mol−1 barrier for TS12a-1 in the directed pathway, Fig. 3) suggests that the Co(I)/iPrPNP catalyzed borylation favors the undirect pathway. With Co(I)/iPrPNP catalytic system, the calculated favorable undirect pathway (ortho-to-fluorine selectivity) is well consistent with the experimental observed ortho-to-fluorine regioselectivity which is reported in the literature11 (53%, Scheme 2), strongly suggesting the accuracy of our calculation. Additionally, the slightly high barrier of undirected pathway (29.5 kcal mol−1) also well explains the relatively low chemical yield in the cobalt catalyzed model reaction (53%).12 It is worth noting that a recent computational study has revealed that the cobalt–carbon bonds of the intermediate in the (iPrPNP)Co-catalyzed borylation could be strengthened by the ortho-fluorine atom, which gives an additional demonstration of the ortho-to-fluorine regioselectivity other than para- or meta-selectivity.13g
Ir(III) catalyzed undirected and directed pathway
After a brief study on the directed and undirected pathways catalyzed by Co(I)/iPrPNP, we turn to the similar pathways catalyzed by Ir(III)/dtbpy catalytic system. The energy and geometric features of each elementary step involved in the Ir(III)/dtbpy catalyzed directed and undirected pathways are shown in Fig. 4 and 5.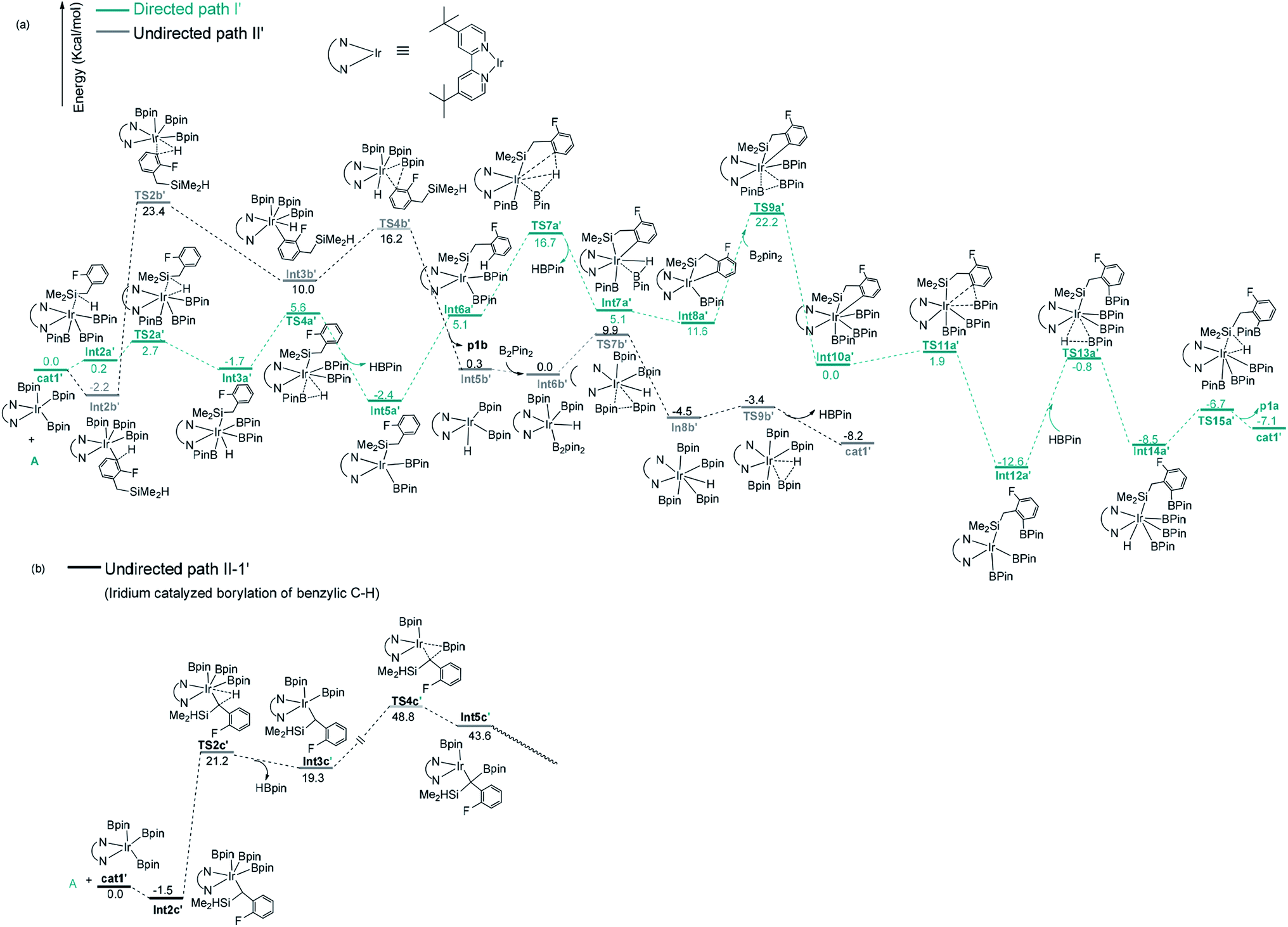 | ||
| Fig. 4 Gibbs free energy profile of path I′ and path II′ in borylation of silyl substituted fluoroarenes with Ir(III)/dtbpy system. | ||
 | ||
| Fig. 5 Optimized structures of the key transition states and intermediates in Co(I)/iPrPNP catalyzed borylation of silyl substituted fluoroarenes, along with the key bond distances in angstroms. color code, C: dark gray, O: red, B: pink, H: light gray N: dark blue, Co: light blue, Si: celeste, P: yellow. Irrelevant hydrogen atoms are omitted for clarity. | ||
For the undirected path I′, the catalytic cycle starts with ortho-to-fluorine C–H activation of the substrate A to form Int3b′ via TS2b′ (a barrier of 25.6 kcal mol−1, Fig. 4a). The energy required for the subsequent C–B reductive elimination is 6.2 kcal mol−1 (TS4b′, Fig. 4a). After C–B reductive elimination, the intermediate Int6b′ is formed and coordinates with B2pin2 via TS7b′, which requires an energy of 9.9 kcal mol−1, a molecular HBpin is released via TS9b′, which require an energy of 1.1 kcal mol−1, and finally, the catalytic cycle is closed by the regeneration of the catalyst cat1′. The most stable intermediate in undirected path I′ is Int2b′. The intermediate Int5a′ in the directed path II′ is 0.2 kcal mol−1 more stable than Int2b′, and the “population” of Int2b′ will be marginally smaller than the population of Int5a′. The reaction may proceed from the intermediate Int5a′. Therefore, in a strict sense, the overall energy barrier within C–H approximation should be calculated as the energy difference between TS2b′ and Int5a′(25.8 kcal mol−1, Fig. 4a).
It is worth noting that the barrier for the initial ortho-to-fluorine C–H activation catalyzed by Ir(III)/dtbpy is lower than that catalyzed by Co(I)/iPrPNP (25.8 kcal mol−1 vs. 27.5 kcal mol−1) (TS2b′ in Fig. 4a vs. TS2b in Fig. 3a), which can be ascribed to the electron effects on the transition states of C–H activation. Comparing the transition states of the ortho-to-fluorine C–H activation in cobalt and iridium catalyzed borylation (TS2b, TS2b′), the Mulliken charge on the cobalt center of TS2b is negative while that on iridium of TS2b′ is positive (−0.517325 vs. 0.349631, Fig. 1, ESI†), which shows stronger electron withdrawing effect of iridium center than that of cobalt center. We propose that the electron withdrawing character of iridium center could facilitate the ortho-to-fluorine C–H activation, which may explain for the lower barrier of TS2b′ (Fig. 4a) than that of TS2b (Fig. 3a) (25.8 kcal mol−1 vs. 27.5 kcal mol−1).
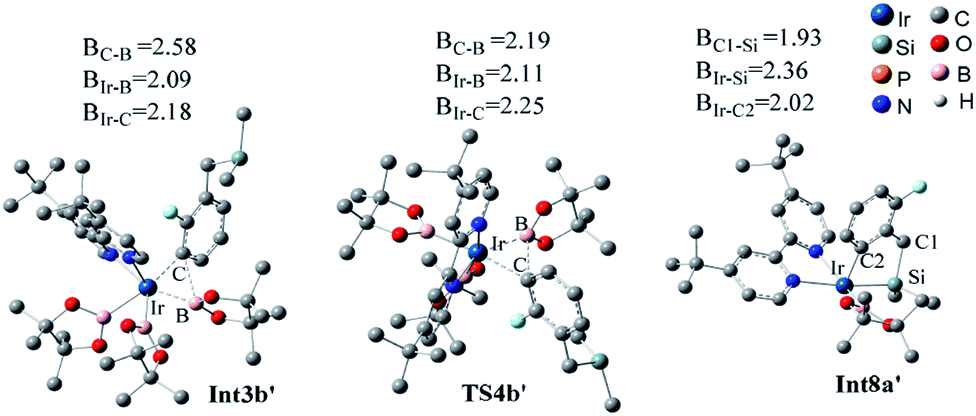
Additionally, as for iridium catalyzed directed path I′, the active catalyst cat1′ triggers the catalytic cycle by silylation, which only requires an energy of 2.7 kcal mol−1 (TS2a′). Given that the formed Int3a′ is saturated coordination,13a it is expected to exclude one molecular of HBpin prior to the C–H oxidative insertion. Upon release of one molecular HBpin, the following C–H oxidative insertion is accompanied by the release of another molecular HBpin which can be confirmed by IRC calculations. Then the intermediate Int8a′ is afforded, which has open coordination sites. One equivalent of B2pin2 thereafter oxidatively add to Int8a′, generating Int10a′ via TS9a′, with an overall barrier of 24.6 kcal mol−1. In the intermediate Int10a′, one boryl group is situated close to the ortho-to-silyl aromatic carbon (Fig. 4a). Then, C–B bond is formed via C–B reductive elimination from Int10a′, affording Int12a′ via TS11a′. Because intermediate Int12a′ has open coordination sites, one molecular HBpin is added to iridium center of Int12a′ to generate Int14a′ via TS13a′. The added HBpin provides hydrogen for the subsequent reductive elimination of the Si–H bond. Upon Si–H reductive elimination, the catalytic cycle is closed by affording the final product and regeneration of catalyst cat1′.
In addition, we have calculated the pathway for Ir-catalyzed benzylic C–H borylation (Fig. 4b). The barrier for path II-1′ is much higher (48.8 kcal mol−1) than other pathways (25.8 kcal mol−1 for Ir catalyzed undirected path II′; 24.6 kcal mol−1 for Ir catalyzed directed path I′) and should be finally ruled out. Similar to Co(I)-catalyzed benzylic C–H borylation, it is different from the hydrosilyl directed borylation of ortho-hydrosilyl benzene in which the borylation of the benzylic C–H bond is favorable over the borylation of the aromatic C–H bond.9a Comparing the rate-determining step in the directed path I′ and undirected path II′ for the iridium catalyzed C–H borylation (TS9a′ vs. TS2b′), the directed pathway leading to ortho-to-silyl selectivity is 1.2 kcal mol−1 more favorable over undirected pathway leading to ortho-to-fluorine selectivity (24.6 kcal mol−1 barrier of TS9a′ in Fig. 4a vs. 25.8 kcal mol−1 barrier of TS2b′ in Fig. 4a).With Ir(III)/dtbpy catalytic system, the calculated favorable directed pathway (ortho-to-silyl selectivity) is also consistent with experimental observed ortho-to-silyl selectivity11 (82%, Scheme 2).
Conclusions
The mechanisms of regioselective borylation of hydrosilyl substituted fluoroarenes by Ir(III)/dtbpy and Co(I)/iPrPNP catalysts have been studied by DFT calculation. It shows that the borylation of hydrosilyl substituted fluoroarenes by Ir(III)/dtbpy exhibits an ortho-to-hydrosilyl selectivity and follows a directed reaction pathway consisting of steps: (i) Si–H oxidative addition (ii) H–B reductive elimination, (iii) C–H bond oxidative addition and H–B reductive elimination, (iv) B–B oxidative addition, (v) C–B formation, (vi) H–B oxidative addition and (vi) Si–H reductive elimination. The preference for ortho-to-hydrosilyl selectivity in Ir(III)/dtbpy catalyzed borylation can be attributed to the directing effect of hydrosilyl group. The borylation of hydrosilyl substituted fluoroarenes by Co(I)/iPrPNP, however, exhibits a high selectivity of ortho-to-fluorine and follows the undirected pathway which involving the steps: (i) C–H oxidative addition, (ii) C–B reductive elimination, (iii) B–B oxidative addition, and (iv) B–H reductive elimination. The preference for ortho-to-fluorine selectivity in Co(I)/iPrPNP catalyzed borylation is attributable to the acidity of ortho-to-fluorine C–H bond, while in the silyl-directed pathway, the high steric hindrance in the transition state of C–B reductive elimination leads to too high overall barrier to overcome. These calculation results well explain the experimental observation reported previously, strongly suggesting the accuracy of our calculation. The work present here therefore provides rational mechanistic insights into the origin of regioselective borylation of hydrosilyl substituted fluoroarenes. This will be helpful to well understand the underlying physics of the regioselective borylation of hydrosilyl substituted fluoroarenes and can be extended to the synthesis of other organic compounds with predictable regioselectivity by introducing suitable directing group, adjusting the available coordination sites on the catalyst, and/or modifying the electronic effects of the catalyst.Computational section
Geometry optimizations without symmetry restriction were carried out at the B3LYP17/BSI level, where BSI denotes the combination of the lanl2dz18 for Ir and Co, 6-31G(d)19 basis for other atoms. Frequency results were examined to confirm stationary points as transition states (only one imaginary frequency) or minima (no imaginary frequencies), and were also used to obtain zero-point energy-corrected enthalpies and free energies at 298.15 K and 1 atm. In addition, intrinsic reaction coordinate (IRC) analysis was conducted to confirm that the transition state connects the correct reactant and product on the potential energy surface.20 The energetic results were further improved by single-point energy calculations at ωB97XD21a/BSII level of theory, where BSII represents a basis set with SDD22 for Ir and Co and Def2TZVP21b basis set for other atoms. The solvent effects accounted by the SMD23 solvation model, using the experimental solvent tetrahydrofuran. Furthermore, natural population analysis (NPA)24 was performed at the same level by single point calculations. The Gibbs free energies obtained from the ωB97XD/BSII//B3LYP/BS1 level was discussed in this study, unless otherwise specified. All calculations are performed with the Gaussian 09 program package.25Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Funding for Youth from Guangzhou University. We are grateful for the helpful suggestion from Prof. Hui Gao at Guangzhou Medical University.References
- (a) D. Aguilar, L. Cuesta, S. Nieto, E. Serrano and E. P. Urriolabeitia, Curr. Org. Chem., 2011, 15, 3441–3464 CrossRef CAS; (b) L. Xu, G. H. Wang, S. Zhang, H. Wang, L. H. Wang, L. Liu, J. Jiao and P. F. Li, Tetrahedron, 2017, 73, 7123–7157 CrossRef CAS; (c) Z. Y. He, Y. Hu, C. G. Xia and C. Liu, Org. Biomol. Chem., 2019, 17, 6099–6113 RSC; (d) L. Kuehn, M. M. Huang, U. Radius and T. B. Marder, Org. Biomol. Chem., 2019, 17, 6601–6606 RSC; (e) Y. H. Liu, Z. J. Jiang and J. P. Chen, Org. Biomol. Chem., 2020 10.1039/D0OB00628A.
- (a) J. V. Obligacion, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 4133–4136 CrossRef CAS PubMed; (b) T. Furukawa, M. Tobisu and N. Chatani, Chem. Commun., 2015, 51, 6508–6511 RSC; (c) H. Zhang, S. Hagihara and K. Itami, Chem. Lett., 2015, 44, 779–781 CrossRef CAS; (d) T. Dombray, C. G. Werncke, S. Jiang, M. Grellier, L. Vendier, S. Bontemps, J. B. Sortais, S. Sabo-Etienne and C. Darcel, J. Am. Chem. Soc., 2015, 137, 4062–4065 CrossRef CAS PubMed; (e) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC; (f) C. Xue, Y. Luo, H. L. Teng, Y. L. Ma, M. Nishiura and Z. M. Hou, ACS Catal., 2018, 8, 5017–5022 CrossRef CAS; (g) C. R. K. Jayasundara, D. Sabasovs, R. J. Staples, J. Oppenheimer, M. R. Smith III and R. E. Maleczka Jr, Organometallics, 2018, 37, 1567–1574 CrossRef CAS.
- Sterically controlled selective borylation was reported, see: (a) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed; (b) S. Paul, G. A. Chotana, D. Holmes, R. C. Reichle, R. E. Maleczka, Jr and M. R. Smith III, J. Am. Chem. Soc., 2006, 128, 15552–15553 CrossRef CAS PubMed; (c) I. A. I. Mkhalid, D. N. Coventry, D. Albesa-Jove, A. S. Batsanov, J. A. K. Howard, R. N. Perutz and T. B. Marder, Angew. Chem., 2006, 118, 503–505 (Angew. Chem., Int. Ed., 2006, 45, 489–491) CrossRef; (d) M. A. Larsen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4287–4299 CrossRef CAS PubMed; (e) Y. Saito, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 5193–5198 CrossRef CAS PubMed . Directed selective borylation was reported, see:; (f) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712–717 CrossRef CAS PubMed; (g) L. Liu, G. H. Wang, J. Jiao and P. F. Li, Org. Lett., 2017, 19, 6132–6135 CrossRef CAS PubMed; (h) J. Thongpaen, T. E. Schmid, L. Toupet, V. Dorcet, M. Mauduit and O. Baslé, Chem. Commun., 2018, 54, 8202–8205 RSC; (i) S. I. Kallane and T. Braun, Angew. Chem., Int. Ed., 2014, 53, 9311–9315 CrossRef PubMed; (j) For selected examples: E. C. Keske, B. D. Moore, O. V. Zenkina, R. Wang, G. Schatte and C. M. Crudden, Chem. Commun., 2014, 50, 9883–9886 RSC ; For reviews:; (k) A. Ros, R. Fernandez and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC; (l) H. L. Ren, Y. P. Zhou, Y. P. Bai, C. M. Cui and M. Driess, Chem.–Eur. J., 2017, 23, 5663–5667 CrossRef CAS PubMed.
- D. W. Robbins and J. F. Hartwig, Org. Lett., 2012, 14, 4266–4269 CrossRef CAS PubMed.
- (a) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed; (b) P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed.
- T. Furukawa, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2015, 137, 12211–12214 CrossRef CAS PubMed.
- J. Takaya, S. Ito, H. Nomoto, N. Saito, N. Kirai and N. Iwasawa, Chem. Commun., 2015, 51, 17662–17665 RSC.
- A. J. Roering, L. V. A. Hale, P. A. Squier, M. A. Ringgold, E. R. Wiederspan and T. B. Clark, Org. Lett., 2012, 14, 3558–3561 CrossRef CAS PubMed.
- (a) C. Patel, V. Abraham and R. B. Sunoj, Organometallics, 2017, 36, 151–158 CrossRef CAS; (b) L. Zhang and D. C. Fang, J. Org. Chem., 2016, 81, 7400–7410 CrossRef CAS PubMed.
- T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534–7535 CrossRef CAS PubMed.
- J. V. Obligacion, M. J. Bezdek and P. J. Chirik, J. Am. Chem. Soc., 2017, 139, 2825–2832 CrossRef CAS PubMed.
- (a) C. Patel, V. Abraham and R. B. Sunoj, Organometallics, 2017, 36, 151–158 CrossRef CAS; (b) L. Zhu, X. T. Qi, Y. Z. Li, M. Duan, L. F. Zou, R. P. Bai and Y. Lan, Organometallics, 2017, 36, 2107–2115 CrossRef CAS; (c) J. Zhou, C. I. Lee and O. V. Ozerov, ACS Catal., 2018, 8, 536–545 CrossRef CAS; (d) Y. H. Liu, J. P. Chen, K. S. Zhan, Y. Q. Shen, H. Gao and L. M. Yao, RSC Adv., 2018, 8, 35453–35460 RSC.
- (a) J. V. Obligacion and P. J. Chirik, ACS Catal., 2017, 7, 4366–4371 CrossRef CAS PubMed; (b) J. V. Obligacion, H. Y. Zhong and P. J. Chirik, Isr. J. Chem., 2017, 57, 1032–1036 CrossRef CAS PubMed; (c) H. X. Li, J. V. Obligacion, P. J. Chirik and M. B. Hall, ACS Catal., 2018, 8, 10606–10618 CrossRef CAS PubMed; (d) T. M. Boller, J. M. Murphy, M. Hapke, T. Ishiyama, N. Miyaura and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 14263–14278 CrossRef CAS PubMed; (e) H. Tamura, H. Yamazaki, H. Sato and S. Sakaki, J. Am. Chem. Soc., 2003, 125, 16114–16126 CrossRef CAS PubMed; (f) G. P. Huang, M. Kalek, R. Z. Liao and F. Himo, Chem. Sci., 2015, 6, 1735–1746 RSC; (g) T. P. Pabst, J. V. Obligacion, É. Rochette, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 15378–15389 CrossRef CAS PubMed.
- E. E. Glendening, A. E. Reed, J. E. Carpenter, F. Weinhold, NBO (Natural Bond Orbital) 3.0, as implemented in GAUSSIAN 03, Gaussian Inc., Pittsburgh, PA, 2004 Search PubMed.
- Q. Li, C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 8755–8765 CrossRef CAS PubMed.
- P. C. Roosen, V. A. Kallepalli, B. Chattopadhyay, D. A. Singleton, R. E. Maleczka Jr and M. R. Smith III, J. Am. Chem. Soc., 2012, 134, 11350–11353 CrossRef CAS PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- (a) P. C. Hariharan and J. A. Pople, Theor. Chem. Acc., 1973, 28, 213–222 Search PubMed; (b) M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- (a) K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS; (b) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- (a) J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS; (b) A. E. Reed and F. J. Weinhold, Chem. Phys., 1985, 83, 1736–1740 CAS; (c) A. E. Reed, L. A. Curtis and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng,J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao,H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox,J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03428b |
| This journal is © The Royal Society of Chemistry 2020 |